

Supplemental Digital Content is available in the text
Keywords: coxsackievirus A6; hand, foot, and mouth disease; molecular epidemiology
Abstract
Background:
Enterovirus 71 (EV-A71) and Coxsackievirus A16 (CV-A16) are the most common causative agents causing hand, foot, and mouth disease (HFMD). However, coxsackievirus A6 (CV-A6), previously largely ignored, became the predominant pathogen in China in 2012. The objective of this study is to investigate the genetic characteristics and molecular epidemiology of HFMD caused by CV-A6 to guide the diagnosis and treatment of the disease, as well as disease prevention.
Material and methods:
A total of 138 suspected HFMD cases were enrolled in this study and analyses based on complete VP1 nucleotide sequences were performed to determine the evolutionary trajectory of emerging CV-A6.
Results:
Among 138 samples in Jiujiang, 125 (90.58%) were positive for enterovirus, the most frequently presented serotypes were CV-A6 (77, 61.60%), CV-A16 (28, 22.40%), EV-A71 (6, 4.80%) and untyped enteroviruses (14, 11.20%). Seventy-seven CV-A6 positive specimens were analyzed for the complete VP1 sequences by sequencing and 36 representative isolates were selected to perform nucleotide sequence similarity analysis. The results showed that 36 strains isolated from HFMD patients were clustered closely to the mainland China and were far from prototype strain CV-A6/Gdula (AY421764) and other international subtypes. Moreover, phylogenetic analysis of the VP1 gene revealed that 36 circulating strains were not significantly concentrated in one branch, but were widely distributed in each branch.
Conclusions:
Continuous surveillance of HFMD etiological agents other than EV-A71 and CV-A16 is necessary. CV-A6 is emerging as the most common pathogen causing HFMD. Closely monitoring the magnitude and trend of CV-A6 epidemic and the trend of pathogenic spectrum changes can provide scientific basis for this disease prevention and control to the department of disease control.
1. Introduction
Hand, foot, and mouth disease (HFMD) is a highly contagious illness caused by human enteroviruses, most commonly occurring in children 5 years old or younger.[1] It is characterized by fever along with vesicular exanthema on the hands and feet, oral mucosa and buttocks, and a rash with blisters.[2] Although HFMD is usually self-limiting disease, severe cases can cause serious complications, including encephalitis, meningitis, myocarditis, acute flaccid paralysis, pulmonary edema, and even lead to death.[3–5] Until now, there's still no effective treatment against this disease, nor effective prevention of severe cases. Enterovirus 71 (EV-A71) and coxsackievirus A16 (CV-A16) are the most common causative agents causing HFMD.[6] Other EVs, such as CV-A2, CV-A4, CV-A5, CV-A6, CV-A10, CVA12, and HEV-B virus (CV-A9 and echovirus 30 (E-30)), are also often associated with HFMD.[7–10] In the past few decades, this disease has been reported all over the world, especially in the Asian Pacific region.[11,12] In Mainland China, HFMD was categorized as a class “C” notifiable disease in May 2008, and nationwide surveillance has been performed since then. From 2008 to 2017, 17,712,562 probable cases of HFMD and 3501 deaths were reported in mainland China (http://www.chinacdc.cn/). During this period, the number of HFMD cases was the highest among all of the Chinese legally notifiable infectious diseases each year.
Jiujiang is one of the cities of Jiangxi province, located in central China. As part of national surveillance, both epidemiological and virological surveillance for HFMD have been carried out in Jiujiang since 2008 and have indicated a persistent HFMD epidemic. In the early stage of laboratory surveillance in Jiujiang, two serotypes of enterovirus EV-A71 and CV-A16 were regarded as the cause of severe and highly fatal HFMD from 2008 to 2010. However, clinical data from the Third People's Hospital of Jiujiang have shown that the detection ratio of non-EV-A71 and non-CV-A16 cases have indicated an upward trend, from about 28.60% in 2011 to 56.10% in 2012 and 62.39% in 2013, but information on the other EVs, including their epidemiological profiles, is very limited.[13] Since 2012, a newly emerged enterovirus (CV-A6) has rapidly replaced the original circulating strains EV-A71 and CV-A16, becoming the predominant pathogen responsible for HFMD in some areas in China.[14–18] Since then, CV-A6, which had been largely ignored, has spread in the mainland China and attracted considerable attention. Thus, in this study, we aimed to investigate the genetic characteristics and molecular epidemiology of HFMD caused by CV-A6 in Jiujiang over the period from November 2012 to April 2013. Analyses based on complete VP1 nucleotide sequences were performed to determine the evolutionary trajectory of emerging CV-A6.
2. Materials and methods
2.1. Specimen collection and processing
From November 2012 to April 2013, a total of 138 patients were diagnosed with HFMD by the Third People's Hospital of Jiujiang according to diagnostic criteria defined by the Ministry of Health of the People's Republic of China (http://www.moh.gov.cn/mohyzs/s3586/201004/46884.shtml).
2.2. RNA extraction and genotyping of enteroviruses
Viral RNA was extracted directly from clinical specimens using the TIANamp Virus RNA Kit (Tiangen Biotech Co., Ltd., Beijing, China) in accordance with the manufacturer's instructions and stored at −80°C until used. EV-A71 and CV-A16 were identified with reverse transcription-quantitative polymerase chain reaction (RT-qPCR) as previously described.[19] Meanwhile, for non-EV-A71/CV-A16 enterovirus, CVA6-specific RT-qPCR was performed by using commercial kit (Daan Gene Co., Ltd., Guangzhou, Guangdong, China). All tests were performed with the ABI StepOne plus Real-Time PCR system (Applied Biosystems, USA).
2.3. Amplifying the VP1 gene and sequencing
The complete VP1-encoding region of CV-A6 was amplified from some of the specimens, using the forward primer CV-A6-VP-F1 (AGAYACCCCCACTGAGGCTAA) and the reverse primer (GAGTGGCGAGATGTCGGTTTAC).[20,21] A reverse transcription PCR (RT-PCR) method was used to yield the desired target amplicons. First, cDNA was synthesized in a 20 μL volume reaction mixture using the RevertAid First Strand cDNA Synthesis Kit (Thermo Scientific, USA). The reaction was under these conditions: 42°C, 60 min; 70°C, 5 min. Second, the PCR of the VP1 gene was performed under the following conditions: 95°C for 3 min; 40 cycles of denaturation at 95°C for 15 s, annealing at 56°C for 25 s, and elongation at 72°C for 60 s; with a final extension step at 72°C for 10 min (Phanta Max Super-Fidelity DNA Polymerase Kit, Vazyme Biotech Co., Ltd, Nanjing, Jiangsu, China). Third, the amplified products were analyzed by electrophoresis on 1.0% agarose gel, purified using a commercial procedure (Tiangen Biotech Co., Ltd., Beijing, China) and subjected to DNA sequencing by Sunny Biotech Company (Shanghai, China). Finally, sequences were confirmed using BLAST in National Center for Biotechnology Information (NCBI).
2.4. Phylogenetic analysis
The sequences obtained in this study (n = 36) were submitted to NCBI and the GenBank accession numbers are MH544967–MH545001. Multiple-sequence alignments of CV-A6 was conducted using Clustal W. Phylogenetic trees based on the sequences of the VP1 gene (915 bp) were constructed by neighbor-joining method using MEGA 7.0 software. The bootstrap method with 1000 replicated datasets was used to perform the tests of phylogeny. Bootstrap values are indicated on the tree. All the reference strains used were retrieved from GenBank database (Supplementary Table S1).
2.5. Statistical analysis
Data were analyzed using the statistical software package, SPSS Statistics version 16.0 (IBM, USA).
2.6. Ethics statement
This study was in compliance with the Helsinki Declaration and was approved by the Human Research Ethics Committee of the Third People's Hospital of Jiujiang. Sample collection in this study was agreed by the patient's guardian with prior written informed consent.
3. Results
3.1. Patient demographics and enterovirus genotypes
From November 2012 to April 2013, a total of 138 suspected HFMD cases were enrolled in this study, which was comprised of 125 mild and 13 severe cases of HFMD. Amongst the HFMD cases, 86 cases (86/138, 62.32%) were males and 52 cases (52/138, 37.68%) were females, with a sex ratio of 1.65:1. The cases occurred in patients ranging from 2 months to 11 years of age. Approximately 96.37% (133/138) of cases occurred in children under the age of 5 years, especially in those <3 years of age (84.06%, 116/138 patients). Among 138 samples, 125 (90.58%) were enterovirus positive. Of the 125 enterovirus positive cases, the most frequently presented serotypes were CV-A6 (77, 61.60%), CV-A16 (28, 22.40%), EV-A71 (6, 4.80%) and untyped enteroviruses (14, 11.20%).
3.2. Nucleotide sequence similarity analysis of CV-A6 VP1 gene
The 915 bp VP1 region (2441 nt to 3355 nt, responding to CV-A6/Gdula-AY421764) nucleotide sequences of CV-A6 isolates were sequenced. A total of 77 samples were detected by sequencing and 36 representative isolates were selected to perform nucleotide sequence similarity analysis. The result of homology comparison in the VP1 genes of 36 CV-A6 strains from Jiujiang showed nucleic acid and deduced amino acid homology was 97.7%–99.9% and 98.4%–100%, respectively, but only 83.0%–83.6% and 94.1%–95.1% homology to CV-A6 prototype strain (CV-A6/Gdula, AY421764). The amino acid alignment analysis based on 36 CV-A6 strains complete amino acid sequences of the VP1 region (305AA) revealed that there were total 14 mutation sites. Among JJ12564, JJ12576, JJ12592, JJ13600 strain involved 3 mutations, 17 strains involved 1 or 2 mutations and 15 strains without mutation (base change, the amino acid that is coded for remains unchanged). At the174 amino acid position, 16 strains were V, and the other 20 strains were I (Fig. 1).
Figure 1.

Amino acid sequence difference in VP1 region of Jiujiang CV-A6 virus.
3.3. Phylogenetic analysis of CV-A6
To understand the evolution characteristics of CV-A6 strains from Jiujiang, we carried out a phylogenetic analysis based on 53 complete sequences of the VP1 region (915 bp) downloaded from GenBank. Complete VP1-gene sequences of CV-A6 from other parts around the world, including United States of America, France, Spain, Finland, Japan, India, Taiwan and other parts of China mainland (Fujian, Guangdong, Henan, Jiangsu, Shandong, Yunnan, Shanghai, Shanxi, Gansu, Zhejiang, Jiangxi, Xinjiang, Guizhou, Tianjin, Jilin, Liaoning, Hunan, Hubei, Sichuan, and Hebei), combined with the corresponding 36 CV-A6 sequences from Jiujiang, into one dataset (Supplementary Table S1). A phylogenetic dendrogram was constructed with the 89 CV-A6 VP1 sequences using the neighbor-joining method in MEGA 7.0.
These strains could be divided into four genotypic subgroups, lineage A, B, C and D, according to the criteria established for genotype EV-A71 by calculating genetic distance.[22,23] A difference of at least 15% in the entire VP1 region of CV-A6 strains was used to distinguish genotypes. Lineage A isolated in the USA in 1949 (Gdula) shared 80.8%–85.0% nucleotide identity to strains in other lineages. The CV-A6 strain isolated in Shandong province of mainland China in 1992 (SD/CHN/1992) formed an independent branch (Lineage B) displayed 82.2%–84.2% nucleotide identity to other CV-A6 strains. Lineage C included a Chinese strain (SD/CHN/1996) and an Indian strain (N-313), which showed 82.6%–85.9% genetic homology with strains in other lineages. Genotype D was divided into three sublineages D1-D3, the nucleotide identities within sublineages ranged from 89.0% to 99.8%. The phylogenetic analysis showed CV-A6 had continuously evolved and spread to other areas. As to genotype D, Cluster D1 was composed of strains isolated in Japan (1999, 2003, and 2009), Spain (2008) and France (2010). Cluster D2 was composed of strains isolated in Guangdong (2009), Shandong (2010), Shanghai (2011), Henan (2011), Jiangsu (2011), Gansu (2011), Shanxi (2011), Hebei (2011) of mainland China, and Japan (2009). Thirty-six Jiujiang CV-A6 strains isolated from 2012 to 2013 fell into cluster D3. Cluster D3 was composed of strains isolated in Shanghai (2010), Fujian (2011, 2013), Guangdong (2011, 2013), Henan (2012), Jiangsu (2012, 2013), Zhejiang (2013), Jilin (2013), Guizhou (2013), Xinjiang (2013), Liaoning (2013), Hebei (2013), Shanxi (2013, 2014), Sichuan (2014), Yunnan (2014), Hunan (2014), Tianjin (2014) Jiangxi (2014, 2015) of mainland China from 2008 to 2015, Taiwan (2009, 2010, and 2011), and Finland (2008), Spain (2008), France (2010), Japan (2011) (Figs. 2 and 3, Supplementary Table S2).
Figure 2.
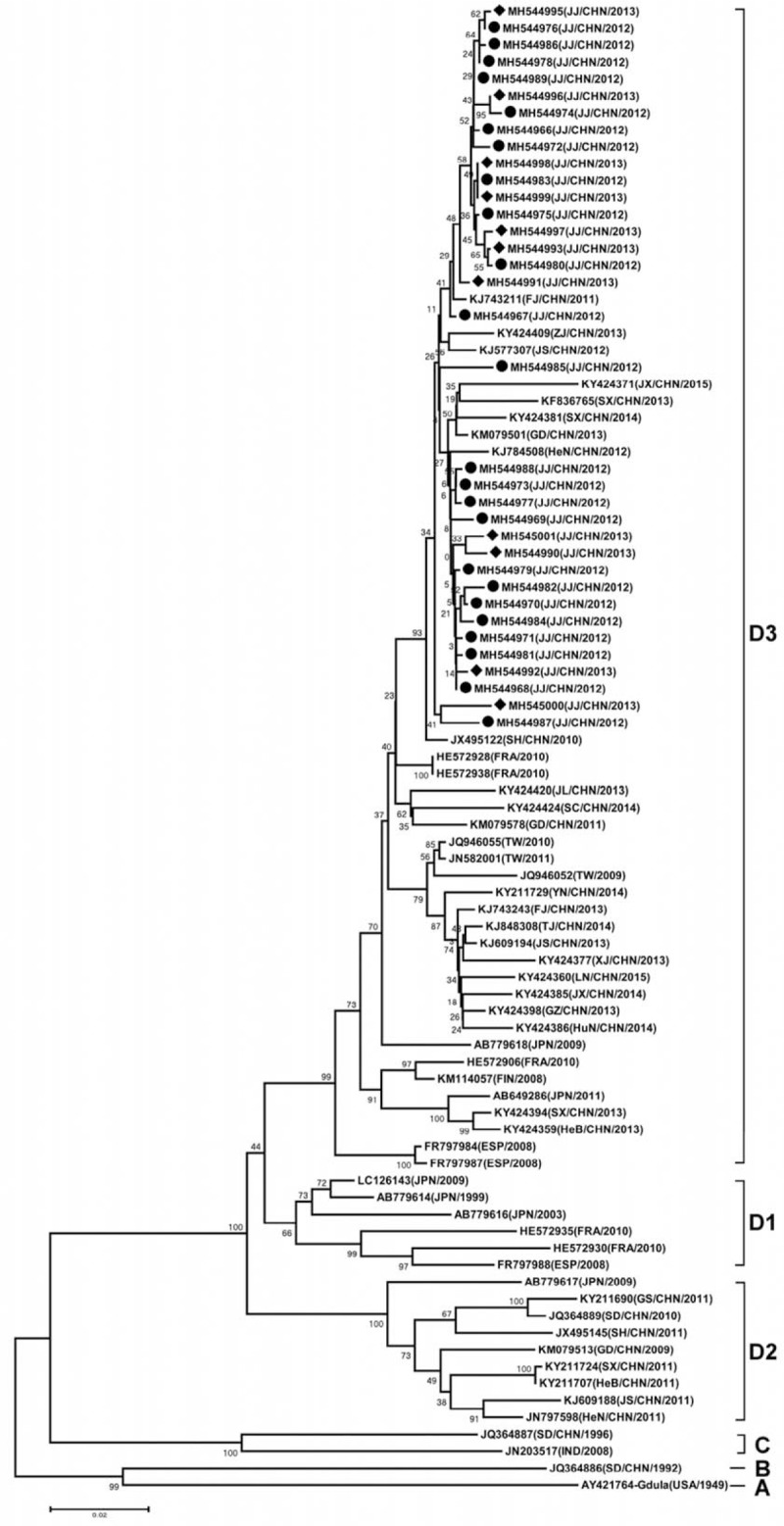
Phylogenetic tree of VP1 regions (2441–3355 nt according Gdula strain) of coxsackievirus A6 Strains. (•) indicates 2012 Jiujiang isolates; (♦) indicates 2013 Jiujiang isolates. The names of countries are abbreviated as CHN = China, ESP = Spain, FIN = Finland, FRA = France, JPN = Japan, IND = India, USA = United States. The Provinces in China are abbreviated as FJ = Fujian, GD = Guangdong, GS = Gansu, GZ = Guizhou, HeB = Hebei, HeN = Henan, HuB = Hubei, HuN = Hunan, JL = Jilin, JS = Jiangsu, JX = Jiangxi, LN = Liaoning, SC = Sichuan, SD = Shandong, SH = Shanghai, SX = Shanxi, TJ = Tianjin, TW = Taiwan, XJ = Xinjiang, YN = Yunnan, ZJ = Zhejiang.
Figure 3.
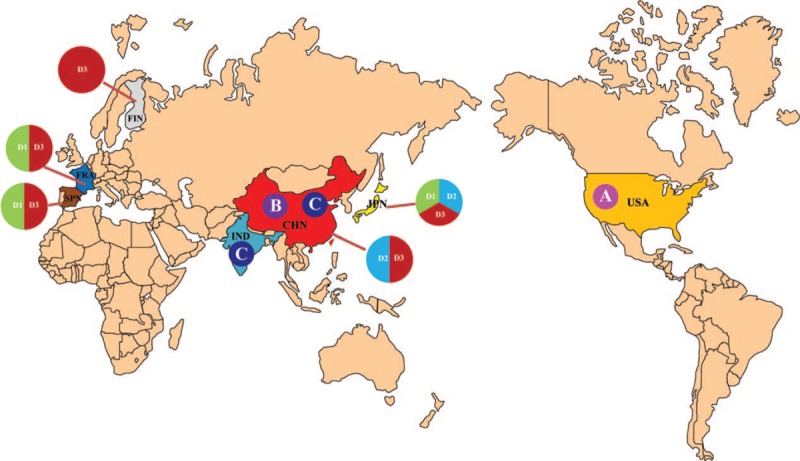
The geographic distribution of HFMD-associated CV-A6 strains in the world.
4. Discussion
As a common infectious disease, HFMD is a serious threat to public health concern worldwide, especially in Asian-Pacific region, and have caused numerous deaths every year in China. EV-A71 and CV-A6 are the primary causes of HFMD. However, other HEV-A pathogens have been found in sporadic cases of HFMD.[24,25] For example, CV-A6 has been associated with the major cause of several HFMD outbreaks in Finland in 2008, and then circulated in France, Spain and other European countries from 2009 to 2011.[26–28] In mainland China, outbreaks of HFMD caused by CV-A6 have also been frequently reported including Guangdong Province, Jilin Province and Beijing in 2013.[14,15,17] Together, these observations provide strong evidence of CV-A6 infections as a new and important cause of HFMD.
The incidence of HFMD has gradually increased in Jiujiang city. In our previous study, non-EV-A71 and non-CV-A6 replaced EV-A71 and CV-A16 as the main causative agent of HFMD, with a detection rate of 56.10% in Jiujiang in 2012.[13] This is the first HFMD outbreak caused by CV-A6 in Jiujiang area in the past few years. In this study, we analyzed the CV-A6 prevalence from November 2012 to April 2013 in Jiujiang area. The phylogenetic analysis showed CV-A6 had continuously evolved and spread to other areas. The phylogenetic analysis indicated that the CV-A6 strains were segregated into four genotypes (A, B, C, and D). At present lineage A, B, C were scarce. Genotype A contains a single strain the prototype strain of CV-A6 isolated in the USA in 1949 (Gdula). The first reported Chinese strain (SD/CHN/1992) formed an independent branch (Lineage B). Genotype C included a Chinese strain (SD/CHN/1996) and an Indian strain (N-313). Genotype D can be further subdivided into D1, D2, and D3 subgenotypes. It was very popular in many countries and regions, such as in France and Spain, co-circulated of genotypes D1 and D3. In Japan, genotypes D1–D3 were detected in 1999, 2003, 2009, 2010, and 2011. In mainland China, D2 and D3 co-circulated, but D3 was the major subgenotype. D2 subgenotype was prevalent in Guangdong, Shandong, Gansu, Shanxi, Hebei, Jiangsu, Henan, while D2 and D3 two subgenotypes co-circulated in Guangdong, Jiangsu, Shanxi, and Henan. The prevalence of the D1 subgenotype has not yet been found in China.
The VP1 coding region contains many important neutralization epitopes and was demonstrated to help identify EV serotypes. A phylogenetic dendrogram based on the entire VP1 capsid sequences of EV has been used for discrimination of genotypes; this approach is effective during temporal and geographical analysis of different outbreaks. So sequence of VP1 gene was selected to build phylogenetic tree. Phylogenetic tree analysis showed that 36 Jiujiang strains clustered closely to the mainland China (Fujian, Guangdong, Zhejiang, Jiangsu, Shanxi, and Henan) and were far from prototype strain CV-A6/Gdula and other international subtypes. The isolates of Jiujiang from November 2012 to April 2013 were in the cluster D3 of the phylogenetic tree, indicating that the CVA6 virus in this region has a single source with little variation. It is speculated that these epidemic strains are more adaptable to the local environment and have stronger transmission ability. In addition, the results showed that Jiujiang CV-A6 strains had not evolved independently, but coevolved with the CV-A6 strains in other provinces in mainland China. In addition, 36 circulating strains isolated from HFMD patients in Jiujiang were not significantly concentrated in one branch, but were widely distributed in each branch. It suggested that there were multiple similarly related CV-A6 propagation chains circulated in Jiujiang and it was consistent with domestic trends.
In a nutshell, CV-A6 is emerging as the most common pathogen causing HFMD in Jiujiang from November 2012 to April 2013. The results from phylogenetic tree showed that the genetic distance between Jiujiang CV-A6 isolates and the prototype and abroad strains was far, and the genetic distance was close to domestic isolates in China. However, there are fewer samples, shorter time, and mainly concentrated in one hospital in a city, so there may be some limitations. So far, CV-A6 has not been included in the routine testing of HFMD. As CV-A6 has been regarded as one of the major causative pathogens in HFMD epidemics, it is recommended that it should be included in routine testing of this highly contagious illness. The disease control department should closely monitor the magnitude and trend of CV-A6 epidemic, pay attention to the trend of pathogenic spectrum changes in HFMD, to provide scientific basis for this disease prevention and control.
Acknowledgments
This work was supported by Anhui Provincial Natural Science Research Project of University (KJ2018A0233, KJ2017A232, and KJ2016A472), Science and Technology projects of Health and Family Planning Commission of Jiangxi province (No. 20175562), Science and Technology Program of Anhui province (Key Laboratories project: 2017070503B037, 2016080503B035) and Natural Science Foundation of Bengbu Medical College (BYKY1769, BYKF1726).
Author contributions
Conceptualization: Xiaojing Wang.
Data curation: Tao Xu.
Funding acquisition: Hongtao Wang, Wenmin Yu, Tao Xu, Xiaojing Wang, Meiqun Sun.
Investigation: Wenmin Yu.
Project administration: Yuyun Li.
Software: Tao Xu.
Supervision: Yuyun Li.
Writing – Original Draft: Hongtao Wang.
Writing – Review & Editing: Meiqun Sun.
Hongtao Wang: 0000-0003-1720-7828.
Supplementary Material
Footnotes
Abbreviations: ABI = Applied biosystems, CV-A16 = Coxsackievirus A16, EV-A71 = Enterovirus 71, HFMD = Hand, Foot and Mouth Disease, NCBI = National Center for Biotechnology Information, RT-PCR = Reverse-transcription polymerase chain reaction.
HW and WY have contributed equally to this work.
The authors declare that they have no conflicts of interest.
Supplemental Digital Content is available for this article.
References
- [1].Wong SS, Yip CC, Lau SK, et al. Human enterovirus 71 and hand, foot and mouth disease. Epidemiol Infect 2010;138:1071–89. [DOI] [PubMed] [Google Scholar]
- [2].Lei X, Cui S, Zhao Z, et al. Etiology, pathogenesis, antivirals and vaccines of hand, foot, and mouth disease. Natl Sci Rev 2015;2:268–84. [Google Scholar]
- [3].Ho M, Chen ER, Hsu KH, et al. An epidemic of enterovirus 71 infection in Taiwan. N Engl J Med 1999;341:929–35. [DOI] [PubMed] [Google Scholar]
- [4].Wu Y, Yeo A, Phoon MC, et al. The largest outbreak of hand, foot and mouth disease in Singapore in 2008: the role of enterovirus 71 and coxsackievirus A strains. Int J Infect Dis 2010;14:1076–81. [DOI] [PubMed] [Google Scholar]
- [5].Renert-Yuval Y, Marva E, Weil M, et al. Coxsackievirus A6 polymorphic exanthem in Israeli children. Acta Dermato-venereologica 2016;96:546–9. [DOI] [PubMed] [Google Scholar]
- [6].Xing W, Liao Q, Viboud C, et al. Hand, foot, and mouth disease in China, 2008-12: an epidemiological study. Lancet Infect Dis 2014;14:308–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Hu YF, Yang F, Du J, et al. Complete genome analysis of coxsackievirus A2, A4, A5, and A10 strains isolated from hand, foot, and mouth disease patients in China revealing frequent recombination of human enterovirus A. J Clin Microbiol 2011;49:2426–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Yang F, Zhang T, Hu Y, et al. Survey of enterovirus infections from hand, foot and mouth disease outbreak in China, 2009. Virol J 2011;8: [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Pabbaraju K, Wong S, Chan EN, et al. Genetic characterization of a coxsackie A9 virus associated with aseptic meningitis in Alberta, Canada in 2010. Virol J 2013;10:93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Zhao YN, Jiang QW, Jiang RJ, et al. Echovirus 30, Jiangsu Province, China. Emerg Infect Dis 2005;11:562–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Chatproedprai S, Theanboonlers A, Korkong S, et al. Clinical and molecular characterization of hand-foot-and-mouth disease in Thailand, 2008-2009. Jpn J Infect Dis 2010;63:229–33. [PubMed] [Google Scholar]
- [12].Zhang Y, Zhu Z, Yang WZ, et al. An emerging recombinant human enterovirus 71 responsible for the 2008 outbreak of hand foot and mouth disease in Fuyang city of China. Virol J 2010;7:94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Xu HX, H WG, Chen CH, et al. Clinical characteristics and analysis of pathogen change with hand, foot and mouth disease in Jiujiang area in 2011-2013. Modern Med J (Chinese) 2014;42:731–6. [Google Scholar]
- [14].Han JF, Xu S, Zhang Y, et al. Hand, foot, and mouth disease outbreak caused by coxsackievirus A6, China, 2013. J Infect 2014;69:303–5. [DOI] [PubMed] [Google Scholar]
- [15].Lu J, Zeng H, Zheng H, et al. Hand, foot and mouth disease in Guangdong, China, in 2013: new trends in the continuing epidemic. Clin Microbiol Infect 2014;20:O442–5. [DOI] [PubMed] [Google Scholar]
- [16].Li JL, Yuan J, Yang F, et al. Epidemic characteristics of hand, foot, and mouth disease in southern China, 2013: coxsackievirus A6 has emerged as the predominant causative agent. J Infect 2014;69:299–303. [DOI] [PubMed] [Google Scholar]
- [17].Hongyan G, Chengjie M, Qiaozhi Y, et al. Hand, foot and mouth disease caused by coxsackievirus a6, Beijing, 2013. Pediatr Infect Dis J 2014;33:1302–3. [DOI] [PubMed] [Google Scholar]
- [18].Chen JF, Zhang RS, Ou XH, et al. The role of enterovirus 71 and coxsackievirus A strains in a large outbreak of hand, foot, and mouth disease in 2012 in Changsha, China. Int J Infect Dis 2014;28:17–25. [DOI] [PubMed] [Google Scholar]
- [19].Yu WM, Xu HX, Yin CC. Molecular epidemiology of human coxsackievirus A16 strains. Biomed Rep 2016;4:761–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Feng X, Guan W, Guo Y, et al. A novel recombinant lineage's contribution to the outbreak of coxsackievirus A6-associated hand, foot and mouth disease in Shanghai, China, 2012–2013. Sci Rep 2015;5: Article ID 10. 1038/srep11700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Tan X, Li L, Zhang B, et al. Molecular epidemiology of coxsackievirus A6 associated with outbreaks of hand, foot, and mouth disease in Tianjin, China, in 2013. Arch Virol 2015;160:1097–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Brown BA, Oberste MS, Alexander JP, Jr, et al. Molecular epidemiology and evolution of enterovirus 71 strains isolated from 1970 to 1998. J Virol 1999;73:9969–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Song Y, Zhang Y, Ji T, et al. “Persistent circulation of Coxsackievirus A6 of genotype D3 in mainland of China between 2008 and 2015,” Scientific Reports, 7, Article ID 5491, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Lu QB, Zhang XA, Wo Y, et al. Circulation of Coxsackievirus A10 and A6 in hand-foot-mouth disease in China, 2009–2011. PLoS ONE 2012;7:e52073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Cabrerizo M, Tarrago D, Muñoz-Almagro C, et al. Molecular epidemiology of enterovirus 71, coxsackievirus A16 and A6 associated with hand, foot and mouth disease in Spain. Clin Microbiol Infect 2014;20:O150–6. [DOI] [PubMed] [Google Scholar]
- [26].Osterback R, Vuorinen T, Linna M, et al. Coxsackievirus A6 and hand, foot, and mouth disease, Finland. Emerg Infect Dis 2009;15:1485–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Mirand A, Henquell C, Archimbaud C, et al. Outbreak of hand, foot and mouth disease/herpangina associated with coxsackievirus A6 and A10 infections in 2010, France: a large citywide, prospective observational study. Clin Microbiol Infect 2012;18:E110–8. [DOI] [PubMed] [Google Scholar]
- [28].Bracho MA, Gonza’lez-Candelas F, Valero A, et al. Enterovirus co-infections and onychomadesis after hand, foot, and mouth disease, Spain 2008. Emerg Infect Dis 2011;17:2223–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.