

Abstract
Organismal fitness demands proper response to neutralize the threat from infection or injury. At the mammalian intestinal epithelium barrier, the inflammasome coordinates an elaborate tissue repair response marked by the induction of anti-microbial peptides, wound-healing cytokines, and reparative proliferation of epithelial stem cells. The inflammasome in myeloid and intestinal epithelial compartments exerts these effects in part, through maintenance of a healthy microbiota. Disease-associated mutations and elevated expression of certain inflammasome sensors have been identified. In many cases, inhibition of inflammasome activity has dramatic effects on disease outcome in mouse models of experimental colitis. Here, we discuss recent studies on the role of distinct inflammasome sensors in intestinal homeostasis and how this knowledge may be translated into a therapeutic setting.
Keywords: ASC, CASPASE-1, CASPASE-8, IL-1β, IL-18, intestine, gasdermin, wound healing
Inflammation: the antidote to restoring tissue homeostasis
Homeostatic balance is critical for the health of multi-cellular organisms and is under threat during infection and traumatic tissue injury. The cellular and tissue response to these insults is critical for the health and survival of the organism. This has drawn many investigators to study the underlying mechanisms that help restore this homeostatic balance. Inflammation is a host response defined by immune cells infiltrating affected tissues. It has a central role in mediating host defense against pathogens, tissue repair and restoration of homeostasis. Immune effectors and specialized stromal cells at epithelial surfaces produce cytokines and anti-microbial defensins to orchestrate tissue repair and to minimize opportunistic infections, respectively. Recent studies indicate that the pro-inflammatory cytokines IL-1β and IL-18 have critical roles in tissue homeostasis in the intestinal epithelium. Their expression is controlled by the inflammasome, a multi-component Moreover, dysregulation of inflammasome activity and IL-1β expression have been implicated in the pathogenesis of inflammatory bowel diseases (IBD)[2]. Accordingly, therapeutic agents targeting certain inflammasome receptors and IL-1β have shown promise in mouse models of colitis[3, 4]. Thus, the potential clinical relevance in IBD and other inflammatory diseases has fueled recent advances in inflammasome biology, a topic explored in this review.
Tissue homeostasis in the intestinal epithelium
In addition to its role in nutrient breakdown and uptake, the mammalian intestinal epithelium functions as a critical barrier against pathogens and microbiota. This barrier function is achieved by the coordinated action of immune effectors, which include T and B lymphocytes, intra-epithelial lymphocytes (IELs), mononuclear phagocytes (MNPs), and innate lymphoid cells (ILCs), that populate the epithelial layer and the underlying lamina propria (Fig. 1)[5]. These cells are often organized into specialized gut-associated lymphoid tissues (GALT) such as in Peyer’s patches[5]. Due to the constant exposure to food antigens and microbes in the lumen, a sophisticated homeostatic system is needed to balance inflammation and tolerance in order to avoid inflammatory pathology, as well as microbial invasion. For example, M cells in Peyer’s patches efficiently capture luminal antigens through transcytosis for antigen presentation[5]. In addition to immune effectors, specialized secretory cells in the intestinal lining such as goblet cells and Paneth cells secrete mucus and anti-microbial peptides (AMPs) respectively, which help fend off pathogenic microbes[5]. Moreover, commensal bacteria that reside in the intestinal lumen also contribute to tissue homeostasis through production of metabolites such as short chain fatty acids, which facilitate the functional development of immune effectors[6]. In turn, microbial metabolites are sensed by G-protein coupled receptors (GPR) such as GPR43 in colonic epithelial cells, which subsequently stimulate potassium efflux and NLRP3 inflammasome activation[7, 8]. As discussed below, inflammasome activation plays critical roles in the maintenance of a healthy and balanced microbiome. These observations have revealed an interplay between host and microbe, shaping the intestinal microbiota. Indeed, the importance of a healthy microbiota is highlighted by the association between intestinal dysbiosis and inflammatory bowel diseases (IBD)[9]. This is biologically relevant because breakdown of any of these homeostastic mechanisms can lead to susceptibility to infection or contribute to injury to the intestinal epithelium, chronic inflammation, or other detrimental pathologies, including cancer[10].
Figure 1. The multifaceted mechanisms that maintain mammalian intestinal epithelium integrity.
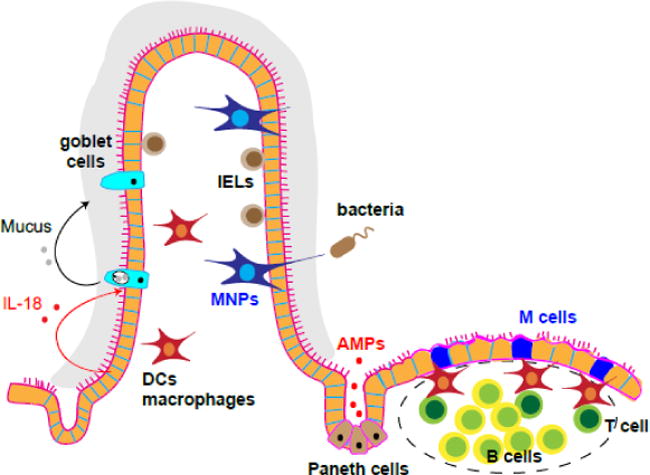
The intestine is populated by many different types of immune effectors including T cells, B cells, intra-epithelial lymphocytes (IELs), mononuclear phagocytes (MNPs), dendritic cells (DCs) and macrophages. These cells can organize into special lymphoid structures such as Peyer’s patches in the lamina propria. M cells in these gut-associated lymphoid tissues (GALT) and MNPs both sample antigens in the lumen as a first line defense against disease or injury. Moreover, goblet cell and Paneth cells secret mucus and anti-microbial peptides (AMPs) respectively to minimize pathogen invasion.
The Inflammasome: architecture, mechanism and biological effects on tissue homeostasis
Inflammasome Architecture
Inflammasomes are intracellular molecular protein scaffolds that govern the cleavage of the IL-1 family of cytokines and lead to an inflammatory form of cell death termed pyroptosis[11]. The inflammasome also triggers necroptosis, a lytic form of inflammatory cell death[12–15]. While the inflammasome is primarily known to mediate host defense against pathogens, recent evidence from mouse models in which specific inflammasome sensors were inhibited shows that it also plays critical roles in intestinal tissue homeostasis[16]. In addition to maintenance of tissue homeostasis at quiescence, the inflammasome also senses acute injury through damage-associated molecular patterns (DAMPs) released from dying cells in the intestine to trigger the tissue repair response[17]. These activities can be mediated by distinct inflammasome sensors such as NLRP3 in hematopoietic and stromal cells in the mammalian intestine[16, 18].
The basic architecture of inflammasome complexes consists of an upstream receptor or sensor from the nucleotide-binding domain and a leucine rich repeat containing protein (NLR), the AIM2 (absent in melanoma 2)-like receptor, tripartite motif (TRIM) proteins, an adaptor protein called ASC (apoptosis-associated speck like protein containing caspase recruitment domain (CARD)), and the cysteine protease CASPASE-1[1] (Fig. 2). Inflammasomes are considered to be in an inhibitory state under resting conditions[1, 11]. A variety of signals of self or foreign origins can relieve this inhibition, leading to the recruitment of ASC via pyrin domain-mediated homotypic interactions. Subsequently, activation of the inflammasome sensors trigger prion-like polymerization of the adaptor molecule ASC and CASPASE-1 via homotypic interaction of the CARD domain, leading to proximity-induced activation of CASPASE-1[19–21]. CASPASE-1-mediated cleavage then facilitates the generation of mature IL-1β and IL-18, given that these cytokines are initially generated as proforms. In some cases, CASPASE-8 can also promote IL-1β and IL-18 maturation[22–24]. Since both IL-1β and IL-18 lack signal peptides, it has been suggested that cell death through pyroptosis or necrosis is critical for secretion of these cytokines from the cells[25]. In support of this model, ligands that activate the inflammasome, and pyroptosis, trigger concomitant release of IL-1β[25]. However, this view has recently been challenged, as imaging analysis with single cell and isolated liposomes indicates that IL-1β can be released from living dendritic cells and macrophages[26, 27].
Figure 2. Inflammasomes and intestinal homeostasis.
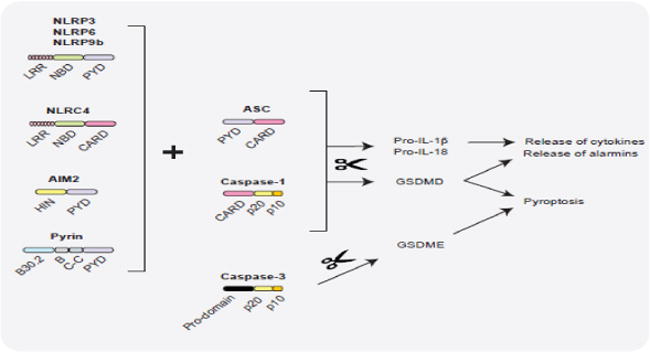
A wide variety of signals of host and microbial origins are sensed by distinct inflammasome receptors leading to the assembly of ASC- CASPASE-1 complexes and the maturation of CASPASE-1. Enzymatically, active CASPASE-1 activates pro-IL-1β, pro-IL-18, and gasdermin D (GSDMD) by proteolytic processing. GSDMD executes lytic cell death via plasma membrane perforation and also facilitates IL-1β, IL-18 and alarmin release. All these outcomes of inflammasome activation impact the functions of intestinal epithelial cells (IELs) and lamina propria immune cells to varying extents, and thus, orchestrate intestinal homeostasis in a context-dependent fashion. In addition, the apoptosis caspase CASPASE-3 can cleave GSDME in response to chemotherapy to trigger cancer cell pyroptosis. It is however unclear if GSDME-induced pyroptosis participates in intestinal homeostasis. LRR: Leucine rich repeats, NBD: nucleotide binding domain, PYD: pyrin domain, CARD: caspase activation and recruitment domain, HIN: HIN-200 domain.
Inflammasome and Tissue Homeostasis
Both IL-1β and IL-18 act on a variety of innate and adaptive immune cells to regulate their differentiation, activation and mobilization to local tissues[28]. They also target non-hematopoietic cells such as endothelial and epithelial cells to stimulate vascular functions and tissue repair[28]. While IL-1β expression is usually stimulus-driven, IL-18 is expressed constitutively in intestinal tissues in mice[29–31]. Intestinal epithelial cells (IECs) are the major producers of constitutively expressed IL-18[32]. Steady state expression of IL-18 in turn, contributes to intestinal barrier function through the maintenance of goblet cells (the major producers of mucus in the intestine), and is also critical for maintenance of a healthy microbiota (Fig. 1)[33]. Consistent with the role for IL-18 in intestinal homeostasis, disease-associated polymorphisms in human IL18, IL18 receptor 1 (IL18R1) and IL18 receptor accessory protein (IL18RAP) have been found in Crohn’s disease[2, 34]. In addition, mice lacking IL-18 or the IL-18 receptor have been reported to be more susceptible to dextran sodium sulfate (DSS), a colitogenic agent that causes intestinal injury and inflammation[35]. Indeed, a radiation chimera experiment using Il18−/− and Il18r1−/− mice showed that IL-18 restricts inflammation-driving Th17 cell differentiation and can promote regulatory T cell-mediated inhibition of inflammation[35]. This indicates that although IL-18 is normally considered to be an innate cytokine, it also regulates adaptive immune cells at the epithelial surface. Moreover, in response to tissue damage, IL-18 expression can be further elevated and the tissue repair-associated cytokine IL-22 has a critical role in stimulating both steady state and induced expression of IL-18[36]. For example, infection of Il22−/− mice with the colitis-inducing bacteria Citrobacter rodentium was shown to compromise IL-18 expression, leading to more severe weight loss and colitis in the animals[36]. This indicates that IL-22 is a key driver for IL-18 expression. Interestingly, IL-18 can also promote expression of IL-22, as in the case of Toxoplasma gondii infection in mice[36]. In contrast to IL-18, IL-1β is transiently induced in response to infection or injury[29–31]. While transient IL-1β expression can stimulate protective responses through induction of repair-associated cytokines such as IL-22[31], sustained IL-1β exposure can also lead to detrimental pathology. For instance, in mouse models of chemically-induced colitis, failure to properly repair intestinal injury has been shown to lead to sustained IL-1β expression, ultimately increasing the susceptibility to inflammation-induced colon cancer[10]. Thus, this is a recurring theme: tissue homeostasis can be achieved with the appropriate level and duration of cytokine exposure[37].
Inflammasome Activation and Pyroptosis
Another outcome of inflammasome activation is cell death by pyroptosis. Pyroptosis is characterized by loss of plasma membrane integrity, osmotic swelling, membrane rupture, and lysis[1]. Recently, two independent studies employing ENU mutagenesis and genome-wide Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)/Cas9 screens in mice and mouse macrophages identified Gasdermin D (GSDMD) as the executioner of pyroptosis[38, 39]. GSDMD, highly conserved in mouse and humans, belongs to the gasdermin family of proteins that also include GSDMA, GSDMB, GSDMC, and GSDME[40]. GSDMD-N has a strong affinity for phosphatidylinositol lipid species found on the inner leaflet of the plasma membrane[41, 42]. The N-terminal domain of GSDMD (GSDMD-N) possesses pore-forming activity, but is normally inhibited by the C-terminal domain (GSDMD-C)[38, 39]. Specifically, when inserted into the plasma membrane, GSDMD-N undergoes oligomerization and forms pores with a 10 – 15 nm inner diameter; extensive pore formation by GSDMD results in osmotic membrane rupture and cell death[27]. Of note, by using glycine to prevent membrane rupture, a recent report showed that GSDMD pores can facilitate the release of mature IL-1β from living macrophages without inducing cell death[26, 27]. Active CASPASE-1 and CASPASE-11 (and CASPASE-4 and CASPASE-5 in humans) liberate GSDMD-N from GSDMD-C by cleaving the linker region between the two domains[38, 39, 41–43]. Moreover, certain chemotherapy drugs such as topotecan, etoposide and cisplatin can stimulate cancer cell pyroptosis via CASPASE 3-mediated cleavage of the related GSDME[44]. In addition to IL-1 family cytokine secretion and pyroptosis, CASPASE-1 activation can also trigger the unconventional release of DAMPs or alarmins such as S100A8, that also lack the signal sequence for secretion[45]. To date, a role for GSDMD or pyroptosis in intestinal homeostasis has not been investigated. However, GSDMD-deficient mice are viable and could be useful in deciphering the role of GSDMD in intestinal homeostasis[39]. The challenge remains being able to distinguish the impact of pyroptotic versus non-pyroptotic effects of GSDMD in intestinal dysbiosis. Nonetheless, collectively, these recent findings illustrate the important concept that machinery that controls pyroptosis and other related cell death modes such as necroptosis, can exacerbate inflammation through cell death-independent cytokine expression and secretion. These two responses provide powerful synergy to orchestrate an effective response to restore homeostasis[46]. Below, we discuss recent progress on the role of different inflammasome sensors in intestinal homeostasis.
The NLRC4 inflammasome
NLRC4 and AIM2 inflammasome receptors survey the intracellular milieu for the presence of microbial components. Each of these inflammasome sensors are activated by distinct triggers. The bacterial type III secretion system (T3SS) machinery and flagellin (a bacterial locomotive component), elicit NLRC4 inflammasome assembly in the cytosol[47–49]. The ability of a single NLRC4 protein to sense multiple bacterial proteins is conferred by a family of nucleotide-binding oligomerization domain-like receptor family apoptosis inhibitory proteins (NAIP) in mice[50, 51]; NAIP1 binds to the needle protein of the T3SS, NAIP2 binds to the basal rod component of T3SS, and NAIP5 as well as NAIP6 sense flagellin[52, 53]. However, humans only have one NAIP protein, capable of recognizing both T3SS components and flagellin[54, 55]. A recent cryo-electron microscopy study revealed that multiple copies of NAIP bind to the ligand to induce an open conformation in NLRC4, leading to the formation of the mouse NLRC4 inflammasome complex[56]. As bacterial pathogens inject virulence proteins through the T3SS to manipulate host cell responses, NLRC4 surveillance of T3SS is critical for anti-bacterial host defense.
Recent studies also showed that the NAIP-NLRC4 inflammasome mediates protection against Salmonella Typhimurium in rodents[32]. Expulsion of bacteria from infected IECs into the intestinal lumen was reported to be compromised in Naip1-6−/−, Casp1/11−/− (caspase deficient) and Nlrc4−/− mice, leading to increased bacterial load in the intraepithelial region[57]. Furthermore, this NAIP-NLRC4 response was independent of IL-1β, IL-18 and GSDMD[57, 58]. This is a surprising and significant finding because it is the first example in which inflammasome regulates immunity independent of its two most well-known functions, namely IL-1 family processing and pyroptosis. By imaging murine intestinal organoid cultures when using a loxP-STOP-loxP Nlrc4 transgene that restored Nlrc4 expression in a tissue-specific manner in cells from Nlrc4−/− mice, one study demonstrated that NLRC4 could stimulate actin rearrangement to promote Salmonella expulsion from infected IECs[58]. Furthermore, examination of mice deficient in CASPASE-1 and CASPASE-8 revealed that NLRC4 could mediate these effects through both caspases[58]. This appears to be one of many scenarios where CASPASE-8 can functionally replace CASPASE-1 to stimulate inflammasome activity. These results also highlight the importance of IEC-intrinsic inflammasome signaling in intestinal homeostasis and indicate that the inflammasome can promote immunity and intestinal homeostasis independently from its cytokine processing function. Of relevance, two gain-of-function NLRC4 mutations, namely T337S and V341A substitutions, lead to the constitutive activation of NLRC4, which has been linked to autoinflammatory enterocolitis in humans[59, 60]. Consequently, these findings underscore the critical role of the NLRC4 inflammasome in intestinal homeostasis.
The AIM2 inflammasome
Double stranded DNA (dsDNA) is normally excluded from the cytosol, but can enter the cytosol during invasion with intracellular bacteria, DNA viruses, or in response to cellular trauma that causes nuclear DNA leakage into the cytosol[61]. In these situations, cytosolic dsDNA acts as a platform to oligomerize AIM2 via the C-terminal HIN domain, triggering the assembly of AIM2-ASC-caspase-inflammasome (Fig. 2)[62–64]. Recent evidence indicates that AIM2 has critical functions in regulating intestinal inflammation. For example, expression of AIM2 protein and mRNA, and the mRNA expression of the related inflammasome sensor Interferon gamma inducible protein 16 (IFI16) is increased in patients with inflammatory bowel disease[65]. Moreover, Aim2−/− mice have been reported to exhibit decreased basal antimicrobial peptide expression, a likely cause for dysbiosis in these animals[66]. In this study, increased expression of basal IL-22 binding protein (IL-22BP) by colonic dendritic cells – antagonizing the tissue repair-associated cytokine IL-22 – was noted relative to wild type mice. These changes also led to reduced IL-1β and IL-18 expression and increased sensitivity to DSS-induced colitis in Aim2−/− mice relative to wild type mice. However, the severe colitis in Aim2−/− mice was ameliorated with intraperitoneal administration of recombinant IL-18, indicating that inflammasome cleavage activity was responsible for AIM2-mediated protection from colitis[66, 67]. These results indicate that AIM2 can promote intestinal homeostasis through maintenance of steady state expression of IL-18.
Chemotherapy targets rapidly induce dividing cells towards a cell death pathway; as such, the intestinal epithelium is susceptible to chemotherapy-induced damage. For instance, in response to the chemotherapeutic agent irinotecan, massive quantities of self-DNA have been shown to be released in the intestine in humans and mice, stimulating AIM2 activation and promoting IL-1β and IL-18 secretion. These events in turn, lead to intestinal inflammation and diarrhea[68]. As such, A, deficiency has been documented to protect mice from ionizing irradiation-induced gastrointestinal disease[69] rendering the animals susceptible to inflammation-induced colorectal cancer[70]. Accordingly, supporting the notion that AIM2 has a critical role in restricting inflammation-induced colorectal cancer, AIM2 mutations and microsatellite instability have been detected in human small intestine adenocarcinomas[71, 72], and loss of AIM2 has been associated with poor survival in colorectal cancer in human patients[73]. Hence, AIM2 may be a key player in the maintenance of intestinal tissue homeostasis (Fig. 3A). Whether AIM2 exerts its effects in the intestine through microbiota balance is a subject that warrants further investigations.
Figure 3. Distinct inflammasome receptors can mediate mammalian tissue repair and homeostasis in a context-dependent manner.

A. Chemotherapy causes damage of the intestinal epithelium and release of DNA, which stimulates the AIM2 inflammasome. IL-1β release in turn can activate the expression of IL-22 by immune cells such as innate lymphoid cells (ILCs) to initiate the tissue repair program.
B. Intestinal epithelium injury caused by other chemical insults can activate the NLRP3 inflammasome in both intestinal epithelial cells and immune cells such as mononuclear phagocytes (MNPs). Increased expression of IL-1β and IL-18 can stimulates a two-fold response: IL-1β can induce IL-22 expression in ILCs to initiate tissue repair program. At the same time, IL-18 can inhibit the expression of IL-22BP, thus further increasing the efficacy of IL-22-induced tissue repair.
C. In a homeostatic state, microbial metabolites help shape the healthy intestinal tissue milieu by stimulating NLRP6-mediated tonic expression of IL-18. This tonic level of IL-18 can promote the production of anti-microbial peptides (AMPs) by Paneth cells and mucus production by goblet cells to form a protective layer against commensal bacteria and invading pathogens.
The NLRP3 inflammasome
Changes in the intracellular environment can be a cue for the activation of certain inflammasome receptors, and NLRP3 is a prime example of such a receptor[1]. NLRP3 is activated by an expanding number of stimuli including, but not limited to, pore-forming toxins, endogenous danger-associated molecular patterns such as uric acid and ATP, and crystal/particulate matters such as silica and alum[1]. The diverse chemical nature of these triggers suggests that NLRP3 is not a direct sensor of these agents. Instead, these stimuli appear to converge on interconnected cellular events such as depletion of intracellular potassium, destabilization of organelles such as lysosomes, and mitochondrial reactive oxygen species (ROS), all of which have been implicated in the activation of NLRP3 in mice and humans[74]. The Never In Mitosis Gene A (NIMA)-related expressed kinase 7 (NEK7) is a serine-threonine kinase involved in the cell cycle, recently identified as an essential component of NLRP3 activation through a N-ethyl-N-nitrosourea (ENU) mutagenesis screen in mice, as well as through a genome-wide CRISPR/Cas9 screen and a proteomic screen, both in mouse macrophages[75–77]. The N-terminal leucine-rich repeat (LRR) domain of NLRP3 has been shown to interact with NEK7, thus promoting the oligomerization of the NLRP3-ASC complex[75]. These results indicate that unlike other inflammasome complexes, the NLRP3 inflammasome contains an additional component critical for its activity.
NLRP3 and Metabolism
Emerging evidence suggests that the perturbation of cellular metabolic processes can be critical for NLRP3 inflammasome assembly in mouse macrophages, as in the case of glycolysis[78–80]. Glycolysis metabolizes glucose to generate ATP, pyruvate to fuel the tricarboxylic acid (TCA) cycle, and other intermediates for the pentose phosphate pathway. The glycolytic pathway has been associated with inflammatory responses of both innate and adaptive cell types. For instance, hexokinase, localized on the mitochondrial outer membrane, is a critical glycolytic enzyme responsible for the phosphorylation of glucose to glucose-6-phosphate[79]. On the one hand, the displacement of hexokinase from the mitochondrial outer membrane by N-acetylglucosamine, or the accumulation of glucose-6-phosphate, can lead to NLRP3 activation in mouse macrophages[79]. Similarly, elevated intracellular levels of citrate (a TCA cycle intermediate), can also trigger NLRP3 activation by impairing the glycolytic cycle in mouse macrophages[79]. On the other hand, hexokinase 1 has also been shown to positively regulate the NLRP3 inflammasome, as evidenced from hexokinase 1 silencing, which was found to suppresses NLRP3 activation in mouse macrophages[80]. Similarly, genetic and pharmacological inhibition of pyruvate kinase isoform M2 (PKM2), an enzyme that catalyzes dephosphorylation of PEP in glycolysis, has been found to impair the activation of the NLRP3 inflammasome[81]. Fatty acid metabolism has also been linked to the NLRP3 inflammasome. Specifically, fatty acid synthesis can promote the priming steps of NLRP3 activation, such as by leading to the expression of Nlrp3 and Il1b genes[82]. Moreover, wild type mice treated with a NOX4 inhibitor, or Nox4−/− mice, have been found to exhibit reduced expression of the fatty acid oxidation enzyme carnitine palmytoyltransferase 1A, leading to reduced NLRP3 activation in mouse macrophages.[78]. Taken together, it is evident that alterations in cellular metabolism can impinge on the activity of the NLRP3 inflammasome.
NLRP3 and Intestinal Homeostasis
Since the NLRP3 inflammasome can respond to many different signals, it might not be surprising if it played a key role in intestinal homeostasis. However, as in the case of many other inflammasome regulators, the role of NLRP3 in intestinal inflammation is controversial, with data supporting both beneficial and disease-driving roles. For instance, several studies have reported that NLRP3-deficient mice are susceptible to DSS-induced colitis or to induction of colorectal cancer (using the pro-carcinogen azoxymethane (AOM) and DSS mouse models)[18, 83, 84]. Another study indicated that Nlrp3−/− mice developed a more severe colitis than controls in response to Citrobacter rodentium infection[85]. Moroever, consistent with a role for NLRP3 in restricting injury-induced intestinal inflammation, Pycard−/− (ASC-deficient) and Casp1−/− mice were shown to succumb to more severe DSS-induced colitis and were more prone to inflammation-associated colorectal cancer than wild type mice[84].
However, in contrast to these reports, there is evidence that supports a disease-driving role for NLRP3 in the intestine. A few studies have indicated that Nlrp3−/− and Il1b−/− mice are more protected against DSS-induced colitis than wild type mice[86, 87]. In fact, the majority of reports suggest that NLRP3 inhibition ameliorates colitis in different mouse models. For instance, glybenclamide (aka glyburide) – which blocks potassium efflux and therefore NLRP3 inflammasome activation—was shown to significantly reduce colitis in Il10−/− mice as well as the expression of cytokines such as TNF, IL-6, IL-1β and IL-17 in intestinal tissue explants derived from Crohn’s disease patients relative to controls[88]. Accordingly, NLRP3 expression has correlated with disease severity in Crohn’s disease patients[88, 89]. Also, disease-driving mutations in the autophagy adaptor ATG16L1 have been documented in human IBD patients[90, 91], and loss of ATG16L1 in mouse macrophages has been shown to compromise NLRP3 inflammasome activation[92].
Furthermore, radiation chimera experiments in mice in which the wild type hematopoietic compartment was replaced with Nlrp3−/− donor cells, have shown that NLRP3-mediated IL-1β production in the murine hematopoietic cell compartment can mediate protection against AOM/DSS-induced colorectal cancer[84]. A subsequent study using diphtheria toxin-mediated depletion of chemokine CCR2+ cells revealed that CCR2+ monocytes could be a major source of NLRP3-driven IL-1β production[86]. These results indicated that NLRP3 could function in both hematopoietic and stromal compartments to mediate intestinal homeostasis.
In addition to IL-1β, NLRP3 can also regulate intestinal injury and tissue repair through an IL-18-IL-22BP axis. IL-22BP antagonizes IL-22, whose transient expression is critical for tissue repair[93]. IL-22BP is constitutively expressed by conventional dendritic cells in the intestinal lamina propria[94]. In response to tissue injury, its expression decreases[93]. Specifically, in a mouse model of DSS-induced colitis, intestinal tissue damage resulted in stimulation of Nlrp3 and expression of IL-18 in the intestine, in turn inhibiting IL-22BP expression[93]. Thus, sensitivity to tissue damage in colitic Nlrp3-deficient mice has been attributed to dysregulated IL-1β and IL-18 expression (Fig. 3B)[93]. Collectively, the role of NLRP3 in intestinal homeostasis is not clear-cut and warrants further study. It is possible that the presence or absence of certain pathobionts in different animal facilities might contribute to the discrepant results observed in Nlrp3−/− mice for different experimental colitis studies[86, 95]. One possible explanation for the increased susceptibility of Nlrp3−/− mice to intestinal injury and inflammation could be due to altered microbiota; indeed, co-housing Nlrp3−/− mice with wild type mice has been found to restore a normal response of to DSS-induced colitis[96].
The Pyrin, NLRP6, and NLRP9b Inflammasomes
Pyrin
Pyrin, which is a tripartite motif (TRIM) protein, forms inflammasome complexes with ASC and CASPASE-1 by a distinct mechanism: instead of detecting bacterial components directly like the NAIP-NLRC4 inflammasome, pyrin senses bacterial virulence factor-induced modifications of host proteins, similarly to the ‘guard’ mechanism in plants[97]. Specifically, Rho GTPase modifications by bacterial toxins such as Clostridium difficile toxin B (TcdB) and pertussis toxin-mediated ADP-ribosylation activate the pyrin inflammasome[98, 99]. A recent study showed that in response to DSS, Pyrin-deficient mice exhibited increased intestinal permeability and became more susceptible to DSS-induced colitis and colitis-induced colorectal cancer relative to controls[100]. IL-18 expression was also reduced in Pyrin-deficient mice, and intravenous administration of recombinant IL-18 reversed the intestinal permeability and susceptibility of Pyrin-deficient mice to DSS-induced colitis[100].
NLRP6, and NLRP9b
Unlike many other inflammasome sensors, which are expressed in hematopoietic and stromal cells, NLPR6 is mainly expressed in colonic myofibroblasts and IECs in mice. NLRP6 can regulate the expression of IL-18 and anti-microbial peptides, as well as the secretion of mucus by goblet cells in mice[101, 102]. Moreover, the NLRP6 inflammasome can be activated by low molecular weight metabolites such as taurine, pinitol, sebacate, and undecanedioate derived from colonic bacteria in mice[103, 104]. As such, NLRP6 has been hypothesized to play a key role in determining colonic microbiota composition (Fig. 3C)[103, 105]. Consistent with this model, Nlrp6−/− mice were originally reported to be highly susceptible to DSS-induced colitis and AOM/DSS-induced tumorigenesis[106]. However, this view has been recently challenged, as several reports have shown that these effects could be attributed to the presence of certain pathobionts in specific mouse colonies[107–109]. Specifically, co-housed wild type and Nlrp6−/− littermates have been found to harbor comparable microbiota compositions, and thus, to exhibit similar sensitivity to chemically-induced colitis[107, 109]. Nonetheless, patients with ulcerative colitis do exhibit reduced epithelial NLRP6 expression[110], and NLRP6 has been shown to confer protection against colitis in IL-10-deficient mice[111], suggesting that NLRP6 may exert microbiota-independent effects to maintain intestinal tissue integrity. In addition to homeostatic control, NLRP6 has also been implicated in antiviral immunity against enteric viruses such as encephalomyocarditis virus and norovirus[92]. In this case, NLRP6 engages the RNA helicase Dhx15 to induce type I and type III interferon production in response to oral infection with encephalomyocarditis virus in mice[112]. Hence, while it is unlikely that NLRP6 regulates microbiota balance, it appears to have a significant function in gut immunity and homeostasis.
Laslty, the NLRP9b inflammasome has also been implicated in viral recognition: a recent study in mice demonstrated that the NLRP9b inflammasome in IECs recognized viral double-stranded RNA via the RNA helicase Dhx9 and mediates resistance to rotavirus[113]. Indeed, IEC-specific deletion of NLRP9b compromised the ability of mice to contain rotavirus infection[113]. Taken together, these results indicate that distinct inflammasome sensors may have specialized functions in recognition of commensal and pathogenic bacteria and viruses.
The Noncanonical inflammasome
Besides CASPASE 1-associated inflammasomes, recent work has identified a novel role for CASPASE 11 as a non-canonical inflammasome. Specifically, both CASPASE 11 in mice and CASPASE 4 and CASPASE 5 in humans can function as intracellular receptors for lipopolysaccharide (LPS)– an abundant endotoxin of Gram-negative bacteria – and are activated by direct binding to LPS[114–117]. LPS gains access to the cytosol when bacteria breach the phagosome or via outer membrane vesicles secreted by bacteria[118, 119]. CASPASE 11 activation is triggered by LPS binding via the CARD domain, which leads to activation of the NLRP3 inflammasome and pyroptosis[115]. Besides LPS, CASPASE 11 also senses certain endogenous lipid species such as oxidized phospholipids (1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphorylcholine) that are liberated during oxidative stress and tissue damage[26]. Similar to CASPASE 1, CASPASE 11 plays a protective role in DSS-induced colitis in mice[120, 121]. Of note, although co-housing Casp11−/− mice with wild type mice has been shown to normalize the microbiota of the former, it fails to rescue the hyper-sensitivity of Casp11−/− mice to DSS-induced colitis[120, 121]. In this model, IL-1β and IL-18 expression do not seem to be ablated, but rather are elevated in Casp11−/− mice. These results seem to indicate that CASPASE 1 and CASPASE 11 can confer protection against injury-induced inflammation in the intestine via distinct mechanisms, although further testing is needed to better elucidate the biological impact of these findings in the context of intestinal inflammation.
Modifiers of inflammasome activity in intestinal homeostasis
Receptor interacting protein kinase 2 (RIPK2) has been reported to transduce signals from the intracellular pattern recognition receptor NOD2 to confer protection against Crohn’s disease[122]. In addition, recent work has shown that the related kinase RIPK3 – an essential mediator of necroptosis—can also regulate intestinal homeostasis by modulating the NLRP3 inflammasome and inflammasome-independent processing of pro-IL-1β in mice[30]. In response to DSS-induced intestinal tissue injury in mice, RIPK3 was deemed able to stimulate the optimal production of IL-1β and IL-23 by colonic dendritic cells, which in turn induced the expression of the repair-associated cytokine IL-22 by innate lymphoid cells[30]. As a result, Ripk3−/− mice were highly susceptible to inflammation-induced colorectal cancer[10]. Another study demonstrated that in mouse bone marrow derived dendritic cells, RIPK3 could promote IL-1β processing and secretion through inflammasome-associated CASPASE 1 as well as CASPASE 8[24]. Furthermore, since mouse dendritic cells that express kinase inactive RIPK3 retained this activity, necroptosis was found to be dispensable and RIPK3 function in this situation was surmised to be distinct from necroptosis[24]. This necroptosis-independent function of RIPK3 was also found to be mediated by a unique population of CD11c+ mononuclear phagocytes in the mouse intestinal lamina propria, since cell sorting of these cells revealed that RIPK3 deficiency significantly impaired Il1b and Il23 expression[29]. RIPK3- and CASPASE 8-dependent inflammasome activation and pro-IL-1β processing have also been observed in mouse dendritic cells, macrophages and human monocytes, as well as in a mouse model of α-hemolysin-producing Escherichia coli-induced sepsis[12, 23, 123–125]. However, it is noteworthy that increased sensitivity to DSS-induced colitis in Ripk3−/− mice were not observed in another study[126]. Since separately-housed Ripk3−/− mice exhibit an altered microbiota compared to wild type littermates[30], the discrepant results might be attributed to colony-specific microbiota differences as well as to the dosage and duration of DSS used in the two different studies, although this remains to be further determined. And, although the precise mechanism by which RIPK3 promotes inflammasome activity has not been elucidated, these results nonetheless highlight the possibility that inflammasome activity can be modulated by accessory factors in a cell type- and context-dependent manner.
Concluding remarks
Emerging experimental evidence now shows that the inflammasome has critical roles in intestinal homeostasis beyond host defense against pathogens. Although it is still unclear whether disease-associated mutations or polymorphisms in the inflammasome sensors can drive human IBD[127], mounting evidence in mouse models suggests that inhibition of NLRP3 might prove to be a potentially effective therapeutic tool in human IBD, pending further investigation. Since other inflammasome sensors have also been shown to participate in the disease process in mouse models of colitis, one pressing challenge will be to interrogate if targeting inflammasome sensors, modifiers of inflammasome activity, or the pyroptosis effector GSDMD, might also yield putative therapeutic benefits in the clinic (Box 1). However, this is not a straight-forward task, as it is increasingly evident that commensal bacteria and the metabolites they produce can influence inflammasome activity and vice versa[7, 8] (see Outstanding Questions). Thus, it will be important to develop and refine bioinformatics tools and model systems to enable the precise determination of how distinct microbiota can modulate the behavior and activity of specific inflammasome sensors; such approaches may facilitate getting one step closer to achieving targeted treatments for human IBD.
Box 1: Clinician’s Corner.
-
–
Cytokines are key drivers of intestinal injury, inflammation and tissue repair. However, they also have a role in maintaining tissue homeostasis. While transient induction of cytokines in response to acute injury usually initiates a beneficial wound healing program, sustained injury can lead to chronic elevation of inflammatory mediators and damaging pathology. Hence, cytokines that normally exert beneficial effects in tissue homeostasis can drive detrimental disease if the magnitude and duration of their activity is not properly calibrated.
-
–
Are any of the Inflammasome receptors viable targets in inflammatory bowel diseases? Several classes of NLRP3 inhibitors have been developed[3, 4]. These inhibitors have shown efficacy in mouse models of multiple sclerosis and several other inflammatory diseases. However, their efficacy in intestinal inflammation has not been tested.
-
–
Instead of directly targeting the core components of the inflammasome, one therapeutic strategy may be to target modulators of inflammasome activity. In this regard, inhibitors against RIPK1, a related kinase to RIPK3, are currently been tested in pre-clinical trials for inflammatory diseases including inflammatory bowel disease[128]. This class of inhibitors may harbor a potential added advantage of inhibiting multiple inflammatory pathways.
-
–
Canakinumab is a humanized anti-IL-1β antibody being used to treat autoinflammatory disorders such as Muckle-wells syndrome. It is also being tested as a therapy in rheumatoid arthritis, obstructive pulmonary disease and Type I and Type 2 diabetes[129, 130]. Given the role of IL-1β in intestinal inflammation, Canakinumab and other IL-1β antagonists might consitute promising therapeutic tools for treating colitis and colitis-associated colorectal cancer, but this remains to be determined.
Highlights.
Tissue homeostasis in the intestine requires the coordinate action between immune effectors, stromal cells and the commensal microbiota.
Distinct inflammasome sensors mediate the crosstalk between immune effectors, intestinal epithelial cells and the microbiota to maintain intestinal homeostasis.
The cytokines IL-1β and IL-18 have key functions in the maintenance of homeostatic balance in the intestine.
Whether cytokines promote or inhibit pathological inflammation is determined by the strength and duration of their activity.
Outstanding questions.
-
–
Do the different inflammasome sensors/receptors have functions beyond IL-1 family cytokine cleavage and pyroptosis that can account for their differential roles in antimicrobial defense and tissue homeostasis in the intestine?
-
–
Mouse models of experimental colitis are highly sensitive to the microbiome. This has led to disparate observations from different laboratories regarding inflammasome roles even when genetically identical mice have been used. Improved models and practices that minimize the impact of environmental effects such as the influence of microbiota on inflammasome activity are essential to achieve a proper understanding of the molecular mechanisms that regulate mammalian intestinal homeostasis.
-
–
How do modifiers such as RIPK3 regulate the activity of the NLRP3 inflammasome in tissue homeostasis?
Acknowledgments
We apologize to our colleagues whose work we cannot cite here due to space limitation. This work is supported by NIH grants to F.K. Chan (AI 119030) and V. Rathinam (AI119015), and the Charles Hood Child Health Research Award (V. Rathinam). F.K. Chan is a senior scholar of the Crohn’s and Colitis Foundation of America.
Glossary
- Alarmins
Factors or cytokines produced by cells in response to trauma that stimulate an inflammatory reaction
- ASC
An adaptor that contains a pyrin domain and a CARD domain whose function includes linking the inflammasome receptor to caspase 1 to facilitate their polymerization and activation
- Caspases
Cysteine proteases that recognizes and cleave tetrapeptide consensus sequences after an aspartic acid
- Commensal microbiota
Bacteria that reside in the lumen of the intestine and which contribute to intestinal homeostasis through the production of specific small molecule metabolites
- CRISPR/Cas9 screen
Loss-of-function genetic screen that uses a modified bacterial defense mechanism to introduce inactivating base insertions or deletions in genes using an RNA guide (gRNA)
- Damage-associated molecular patterns (DAMPs)
Intracellular molecules normally sequestered in healthy cells, but are released to the extracellular milieu upon cell injury or cell death
- ENU mutagenesis screen
A genetic screen that inactivates genes using the alkylating agent N-ethyl-N-nitrosourea to modify bases in DNA sequences
- Gasdermin D (GSDMD)
The effector molecule that executes cell death by pyroptosis. Upon cleavage by caspase 1, the N-terminus of gasdermin D forms pores on the plasma membrane to mediate pyroptosis
- Goblet cells
Specialized secretory cells that produce the mucus lining the lumen side of the intestinal epithelium
- Gut-associated lymphoid tissues (GALT)
Regions in the intestine lamina propria in which lymphocytes, myeloid cells and other specialized stromal cells organize together to maximize injury- and microbe-sensing
- Inflammasome
macromolecular complexes that mediate processing of IL-1 family cytokines and a lytic form of cell death called pyroptosis
- Intestinal epithelium
A monolayer of stromal cells that functions as a physical barrier between the intestinal lumen and the soft tissues underneath it
- IL-1β and IL-18
Pro-inflammatory cytokines that mediates intestinal tissue homeostasis and acute injury-induced tissue repair
- Irinotecan
A topoisomerase inhibitor widely used in the treatment of colon cancer
- Lamina propria
The connective tissue layer underneath the intestinal epithelium where immune effectors reside
- Microfold cells (M cells)
Specialized cells whose main function is to survey and capture antigens in the intestinal lumen for transport and presentation to immune cells in the lamina propria
- Microsatellite instability
The condition of genetic instability caused by defects in DNA mismatch repair
- Muckle-wells syndrome
A rare autosomal dominant disease caused by mutation in CIAS1, which encodes the protein cryopyrin. It belongs to a group of conditions collectively referred to as periodic fever syndrome. Disease manifestation includes sensorineural deafness, recurrent hives, episodic fever and joint pain
- Inflammatory bowel disease (IBD)
A group of inflammatory conditions affecting the small and large intestine. Major types of IBD include Crohn’s disease and ulcerative colitis
- Intestinal dysbiosis
Imbalance of the normal, healthy microbial composition in the intestine
- Intra-epithelial lymphocytes (IELs) and innate lymphoid cells (ILCs) and mononuclear phagocytes (MNPs)
Distinct groups of specialized immune effectors in the lamina propria with distinct functions in tissue homeostasis and immune defense against pathogens. They mainly exert their effects through cytokine expression
- Necroptosis
A pro-inflammatory form of cell death marked by plasma membrane leakage and mediated by the kinase RIPK3
- NLRP3 inflammasome
A macro-molecular complex that cleaves pro-IL-1β and pro-IL-18 into the mature cytokines. It senses a diverse array of stimuli that culminate in potassium efflux and its activation
- Non-canonical inflammasome
pathway characterized by the presence of caspase-4 and caspase-5 in humans and caspase-11 in rodents and is involved in innate immune sensing of cytosolic LPS
- Paneth cells
Specialized secretory cells found at the base of the intestinal crypts that produce anti-microbial peptides to maintain a balanced microbiota
- Peyer’s patches
Specialized lymphoid follicles found mainly in the small intestine. It is a major form of GALT
- Prion-like polymerization
Pathogenic prion proteins convert or polymerize normal prion protein to cause disease-driving prion protein aggregation. During inflammasome activation, a similar process takes place in which the inflammasome adaptor ASC polymerizes CASPASE 1 via the CARD
- Pyroptosis
A lytic programmed cell death characterized by loss of plasma membrane integrity, release of inflammatory intracellular contents as well as cell swelling, and mediated by gasdermins
- Quiescence
A quiet state in which normal activity and function of the intestine is not perturbed by injury or infection
- Regulatory T cells (Tregs)
Specialized effector T cells whose main function is to maintain immune homeostasis by preventing immune response against self-antigens
- RIPK3
A serine/threonine kinase that drives inflammation through necroptosis and cell death-independent cytokine expression
- Th17 cells
Specialized CD4+ effector T lymphocytes defined by the production of the inflammatory cytokine IL-17 and have critical roles in host defense against extracellular pathogens and multiple forms of inflammatory diseases
- Tissue Homeostasis
A state of equilibrium in which the body keeps the structural and functional integrity of tissues and organs intact
- Transcytosis
A form of cellular transport in which molecules are transported from one end of the cell to the other. In the case of M cells, transcytosis mediates transport of antigens from the lumen side of the epithelium to the lamina propria for presentation to sentinel immune effectors such as dendritic cells
- Type III secretion system (T3SS)
A bacterial multi-protein syringe-needle like secretion apparatus with which bacteria deliver an array of virulence factors into host cells
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Rathinam VA, Fitzgerald KA. Inflammasome Complexes: Emerging Mechanisms and Effector Functions. Cell. 2016;165(4):792–800. doi: 10.1016/j.cell.2016.03.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Opipari A, Franchi L. Role of inflammasomes in intestinal inflammation and Crohn’s disease. Inflamm Bowel Dis. 2015;21(1):173–81. doi: 10.1097/MIB.0000000000000230. [DOI] [PubMed] [Google Scholar]
- 3.Youm YH, et al. The ketone metabolite beta-hydroxybutyrate blocks NLRP3 inflammasome-mediated inflammatory disease. Nat Med. 2015;21(3):263–9. doi: 10.1038/nm.3804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Coll RC, et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat Med. 2015;21(3):248–55. doi: 10.1038/nm.3806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mowat AM, Agace WW. Regional specialization within the intestinal immune system. Nat Rev Immunol. 2014;14(10):667–85. doi: 10.1038/nri3738. [DOI] [PubMed] [Google Scholar]
- 6.Maslowski KM, et al. Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature. 2009;461(7268):1282–6. doi: 10.1038/nature08530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vieira AT, et al. A Role for Gut Microbiota and the Metabolite-Sensing Receptor GPR43 in a Murine Model of Gout. Arthritis Rheumatol. 2015;67(6):1646–56. doi: 10.1002/art.39107. [DOI] [PubMed] [Google Scholar]
- 8.Macia L, et al. Metabolite-sensing receptors GPR43 and GPR109A facilitate dietary fibre-induced gut homeostasis through regulation of the inflammasome. Nat Commun. 2015;6:6734. doi: 10.1038/ncomms7734. [DOI] [PubMed] [Google Scholar]
- 9.Sartor RB, Wu GD. Roles for Intestinal Bacteria, Viruses, and Fungi in Pathogenesis of Inflammatory Bowel Diseases and Therapeutic Approaches. Gastroenterology. 2017;152(2):327–339e4. doi: 10.1053/j.gastro.2016.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Moriwaki K, et al. Border Security: The Role of RIPK3 in Epithelium Homeostasis. Front Cell Dev Biol. 2016;4:70. doi: 10.3389/fcell.2016.00070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.de Zoete MR, et al. Inflammasomes. Cold Spring Harb Perspect Biol. 2014;6(12):a016287. doi: 10.1101/cshperspect.a016287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kang S, et al. Caspase-8 scaffolding function and MLKL regulate NLRP3 inflammasome activation downstream of TLR3. Nat Commun. 2015;6:7515. doi: 10.1038/ncomms8515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gutierrez KD, et al. MLKL Activation Triggers NLRP3-Mediated Processing and Release of IL-1beta Independently of Gasdermin-D. J Immunol. 2017;198(5):2156–2164. doi: 10.4049/jimmunol.1601757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Conos SA, et al. Active MLKL triggers the NLRP3 inflammasome in a cell-intrinsic manner. Proc Natl Acad Sci USA. 2017;114(6):E961–E969. doi: 10.1073/pnas.1613305114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang X, et al. MLKL and FADD Are Critical for Suppressing Progressive Lymphoproliferative Disease and Activating the NLRP3 Inflammasome. Cell Rep. 2016;16(12):3247–3259. doi: 10.1016/j.celrep.2016.06.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zaki MH, et al. The Nlrp3 inflammasome: contributions to intestinal homeostasis. Trends Immunol. 2011;32(4):171–9. doi: 10.1016/j.it.2011.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cummings RJ, et al. Different tissue phagocytes sample apoptotic cells to direct distinct homeostasis programs. Nature. 2016;539(7630):565–569. doi: 10.1038/nature20138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zaki MH, et al. The NLRP3 inflammasome protects against loss of epithelial integrity and mortality during experimental colitis. Immunity. 2010;32(3):379–91. doi: 10.1016/j.immuni.2010.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lu A, et al. Unified polymerization mechanism for the assembly of ASC-dependent inflammasomes. Cell. 2014;156(6):1193–1206. doi: 10.1016/j.cell.2014.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cai X, et al. Prion-like polymerization underlies signal transduction in antiviral immune defense and inflammasome activation. Cell. 2014;156(6):1207–1222. doi: 10.1016/j.cell.2014.01.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Franklin BS, et al. The adaptor ASC has extracellular and ‘prionoid’ activities that propagate inflammation. Nat Immunol. 2014;15(8):727–37. doi: 10.1038/ni.2913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vince JE, et al. Inhibitor of apoptosis proteins limit RIP3 kinase-dependent interleukin-1 activation. Immunity. 2012;36(2):215–27. doi: 10.1016/j.immuni.2012.01.012. [DOI] [PubMed] [Google Scholar]
- 23.Kang TB, et al. Caspase-8 blocks kinase RIPK3-mediated activation of the NLRP3 inflammasome. Immunity. 2013;38(1):27–40. doi: 10.1016/j.immuni.2012.09.015. [DOI] [PubMed] [Google Scholar]
- 24.Moriwaki K, et al. A RIPK3-caspase 8 complex mediates atypical pro-IL-1beta processing. J Immunol. 2015;194(4):1938–44. doi: 10.4049/jimmunol.1402167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cullen SP, et al. Diverse Activators of the NLRP3 Inflammasome Promote IL-1beta Secretion by Triggering Necrosis. Cell Rep. 2015;11(10):1535–48. doi: 10.1016/j.celrep.2015.05.003. [DOI] [PubMed] [Google Scholar]
- 26.Zanoni I, et al. An endogenous caspase-11 ligand elicits interleukin-1 release from living dendritic cells. Science. 2016;352(6290):1232–6. doi: 10.1126/science.aaf3036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Evavold CL, et al. The Pore-Forming Protein Gasdermin D Regulates Interleukin-1 Secretion from Living Macrophages. Immunity. 2017 doi: 10.1016/j.immuni.2017.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Palomo J, et al. The interleukin (IL)-1 cytokine family–Balance between agonists and antagonists in inflammatory diseases. Cytokine. 2015;76(1):25–37. doi: 10.1016/j.cyto.2015.06.017. [DOI] [PubMed] [Google Scholar]
- 29.Moriwaki K, et al. Distinct Kinase-Independent Role of RIPK3 in CD11c+ Mononuclear Phagocytes in Cytokine-Induced Tissue Repair. Cell Rep. 2017;18(10):2441–2451. doi: 10.1016/j.celrep.2017.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moriwaki K, et al. The necroptosis adaptor RIPK3 promotes injury-induced cytokine expression and tissue repair. Immunity. 2014;41(4):567–78. doi: 10.1016/j.immuni.2014.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Longman RS, et al. CX(3)CR1(+) mononuclear phagocytes support colitis-associated innate lymphoid cell production of IL-22. J Exp Med. 2014;211(8):1571–83. doi: 10.1084/jem.20140678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nordlander S, et al. NLRC4 expression in intestinal epithelial cells mediates protection against an enteric pathogen. Mucosal Immunol. 2014;7(4):775–85. doi: 10.1038/mi.2013.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nowarski R, et al. Epithelial IL-18 Equilibrium Controls Barrier Function in Colitis. Cell. 2015;163(6):1444–56. doi: 10.1016/j.cell.2015.10.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gao SJ, et al. Interleukin-18 genetic polymorphisms contribute differentially to the susceptibility to Crohn’s disease. World J Gastroenterol. 2015;21(28):8711–22. doi: 10.3748/wjg.v21.i28.8711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Harrison OJ, et al. Epithelial-derived IL-18 regulates Th17 cell differentiation and Foxp3(+) Treg cell function in the intestine. Mucosal Immunol. 2015;8(6):1226–36. doi: 10.1038/mi.2015.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Munoz M, et al. Interleukin-22 induces interleukin-18 expression from epithelial cells during intestinal infection. Immunity. 2015;42(2):321–331. doi: 10.1016/j.immuni.2015.01.011. [DOI] [PubMed] [Google Scholar]
- 37.Sonnenberg GF, et al. Border patrol: regulation of immunity, inflammation and tissue homeostasis at barrier surfaces by IL-22. Nat Immunol. 2011;12(5):383–90. doi: 10.1038/ni.2025. [DOI] [PubMed] [Google Scholar]
- 38.Kayagaki N, et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature. 2015;526(7575):666–71. doi: 10.1038/nature15541. [DOI] [PubMed] [Google Scholar]
- 39.Shi J, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. 2015;526(7575):660–5. doi: 10.1038/nature15514. [DOI] [PubMed] [Google Scholar]
- 40.Kovacs SB, Miao EA. Gasdermins: Effectors of Pyroptosis. Trends Cell Biol. 2017;27(9):673–684. doi: 10.1016/j.tcb.2017.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu X, et al. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature. 2016;535(7610):153–8. doi: 10.1038/nature18629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ding J, et al. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature. 2016;535(7610):111–6. doi: 10.1038/nature18590. [DOI] [PubMed] [Google Scholar]
- 43.He WT, et al. Gasdermin D is an executor of pyroptosis and required for interleukin-1beta secretion. Cell Res. 2015;25(12):1285–98. doi: 10.1038/cr.2015.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang Y, et al. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature. 2017;547(7661):99–103. doi: 10.1038/nature22393. [DOI] [PubMed] [Google Scholar]
- 45.Keller M, et al. Active caspase-1 is a regulator of unconventional protein secretion. Cell. 2008;132(5):818–31. doi: 10.1016/j.cell.2007.12.040. [DOI] [PubMed] [Google Scholar]
- 46.Linkermann A, et al. Regulated cell death and inflammation: an auto-amplification loop causes organ failure. Nat Rev Immunol. 2014;14(11):759–67. doi: 10.1038/nri3743. [DOI] [PubMed] [Google Scholar]
- 47.Sutterwala FS, et al. Immune recognition of Pseudomonas aeruginosa mediated by the IPAF/NLRC4 inflammasome. J Exp Med. 2007;204(13):3235–45. doi: 10.1084/jem.20071239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Miao EA, et al. Pseudomonas aeruginosa activates caspase 1 through Ipaf. Proc Natl Acad Sci USA. 2008;105(7):2562–7. doi: 10.1073/pnas.0712183105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Miao EA, et al. Innate immune detection of the type III secretion apparatus through the NLRC4 inflammasome. Proc Natl Acad Sci USA. 2010;107(7):3076–80. doi: 10.1073/pnas.0913087107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kofoed EM, Vance RE. Innate immune recognition of bacterial ligands by NAIPs determines inflammasome specificity. Nature. 2011;477(7366):592–5. doi: 10.1038/nature10394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhao Y, et al. The NLRC4 inflammasome receptors for bacterial flagellin and type III secretion apparatus. Nature. 2011;477(7366):596–600. doi: 10.1038/nature10510. [DOI] [PubMed] [Google Scholar]
- 52.Rauch I, et al. NAIP proteins are required for cytosolic detection of specific bacterial ligands in vivo. J Exp Med. 2016;213(5):657–65. doi: 10.1084/jem.20151809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhao Y, et al. Genetic functions of the NAIP family of inflammasome receptors for bacterial ligands in mice. J Exp Med. 2016;213(5):647–56. doi: 10.1084/jem.20160006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yang J, et al. Human NAIP and mouse NAIP1 recognize bacterial type III secretion needle protein for inflammasome activation. Proc Natl Acad Sci USA. 2013;110(35):14408–13. doi: 10.1073/pnas.1306376110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kortmann J, et al. Cutting Edge: Inflammasome Activation in Primary Human Macrophages Is Dependent on Flagellin. J Immunol. 2015;195(3):815–9. doi: 10.4049/jimmunol.1403100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tenthorey JL, et al. The structural basis of flagellin detection by NAIP5: A strategy to limit pathogen immune evasion. Science. 2017;358(6365):888–893. doi: 10.1126/science.aao1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sellin ME, et al. Epithelium-intrinsic NAIP/NLRC4 inflammasome drives infected enterocyte expulsion to restrict Salmonella replication in the intestinal mucosa. Cell Host Microbe. 2014;16(2):237–248. doi: 10.1016/j.chom.2014.07.001. [DOI] [PubMed] [Google Scholar]
- 58.Rauch I, et al. NAIP-NLRC4 Inflammasomes Coordinate Intestinal Epithelial Cell Expulsion with Eicosanoid and IL-18 Release via Activation of Caspase-1 and -8. Immunity. 2017;46(4):649–659. doi: 10.1016/j.immuni.2017.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Canna SW, et al. An activating NLRC4 inflammasome mutation causes autoinflammation with recurrent macrophage activation syndrome. Nat Genet. 2014;46(10):1140–6. doi: 10.1038/ng.3089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Romberg N, et al. Mutation of NLRC4 causes a syndrome of enterocolitis and autoinflammation. Nat Genet. 2014;46(10):1135–1139. doi: 10.1038/ng.3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Di Micco A, et al. AIM2 inflammasome is activated by pharmacological disruption of nuclear envelope integrity. Proc Natl Acad Sci USA. 2016;113(32):E4671–80. doi: 10.1073/pnas.1602419113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hornung V, et al. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature. 2009;458(7237):514–8. doi: 10.1038/nature07725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rathinam VA, et al. The AIM2 inflammasome is essential for host defense against cytosolic bacteria and DNA viruses. Nat Immunol. 2010;11(5):395–402. doi: 10.1038/ni.1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fernandes-Alnemri T, et al. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature. 2009;458(7237):509–13. doi: 10.1038/nature07710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Vanhove W, et al. Strong Upregulation of AIM2 and IFI16 Inflammasomes in the Mucosa of Patients with Active Inflammatory Bowel Disease. Inflamm Bowel Dis. 2015;21(11):2673–82. doi: 10.1097/MIB.0000000000000535. [DOI] [PubMed] [Google Scholar]
- 66.Ratsimandresy RA, et al. The AIM2 inflammasome is a central regulator of intestinal homeostasis through the IL-18/IL-22/STAT3 pathway. Cell Mol Immunol. 2017;14(1):127–142. doi: 10.1038/cmi.2016.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hu S, et al. The DNA Sensor AIM2 Maintains Intestinal Homeostasis via Regulation of Epithelial Antimicrobial Host Defense. Cell Rep. 2015;13(9):1922–36. doi: 10.1016/j.celrep.2015.10.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lian Q, et al. Chemotherapy-induced intestinal inflammatory responses are mediated by exosome secretion of double-strand DNA via AIM2 inflammasome activation. Cell Res. 2017;27(6):784–800. doi: 10.1038/cr.2017.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hu B, et al. The DNA-sensing AIM2 inflammasome controls radiation-induced cell death and tissue injury. Science. 2016;354(6313):765–768. doi: 10.1126/science.aaf7532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Man SM, et al. Critical Role for the DNA Sensor AIM2 in Stem Cell Proliferation and Cancer. Cell. 2015;162(1):45–58. doi: 10.1016/j.cell.2015.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Schulmann K, et al. HNPCC-associated small bowel cancer: clinical and molecular characteristics. Gastroenterology. 2005;128(3):590–9. doi: 10.1053/j.gastro.2004.12.051. [DOI] [PubMed] [Google Scholar]
- 72.Michel S, et al. Coding microsatellite instability analysis in microsatellite unstable small intestinal adenocarcinomas identifies MARCKS as a common target of inactivation. Mol Carcinog. 2010;49(2):175–82. doi: 10.1002/mc.20587. [DOI] [PubMed] [Google Scholar]
- 73.Dihlmann S, et al. Lack of Absent in Melanoma 2 (AIM2) expression in tumor cells is closely associated with poor survival in colorectal cancer patients. Int J Cancer. 2014;135(10):2387–96. doi: 10.1002/ijc.28891. [DOI] [PubMed] [Google Scholar]
- 74.Vanaja SK, et al. Mechanisms of inflammasome activation: recent advances and novel insights. Trends Cell Biol. 2015;25(5):308–15. doi: 10.1016/j.tcb.2014.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.He Y, et al. NEK7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature. 2016;530(7590):354–7. doi: 10.1038/nature16959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Schmid-Burgk JL, et al. A Genome-wide CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats) Screen Identifies NEK7 as an Essential Component of NLRP3 Inflammasome Activation. J Biol Chem. 2016;291(1):103–9. doi: 10.1074/jbc.C115.700492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Shi H, et al. NLRP3 activation and mitosis are mutually exclusive events coordinated by NEK7, a new inflammasome component. Nat Immunol. 2016;17(3):250–8. doi: 10.1038/ni.3333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Moon JS, et al. NOX4-dependent fatty acid oxidation promotes NLRP3 inflammasome activation in macrophages. Nat Med. 2016;22(9):1002–12. doi: 10.1038/nm.4153. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 79.Wolf AJ, et al. Hexokinase Is an Innate Immune Receptor for the Detection of Bacterial Peptidoglycan. Cell. 2016;166(3):624–636. doi: 10.1016/j.cell.2016.05.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Moon JS, et al. mTORC1-Induced HK1-Dependent Glycolysis Regulates NLRP3 Inflammasome Activation. Cell Rep. 2015;12(1):102–115. doi: 10.1016/j.celrep.2015.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 81.Xie M, et al. PKM2-dependent glycolysis promotes NLRP3 and AIM2 inflammasome activation. Nat Commun. 2016;7:13280. doi: 10.1038/ncomms13280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Moon JS, et al. UCP2-induced fatty acid synthase promotes NLRP3 inflammasome activation during sepsis. J Clin Invest. 2015;125(2):665–80. doi: 10.1172/JCI78253. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 83.Zaki MH, et al. IL-18 production downstream of the Nlrp3 inflammasome confers protection against colorectal tumor formation. J Immunol. 2010;185(8):4912–20. doi: 10.4049/jimmunol.1002046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Allen IC, et al. The NLRP3 inflammasome functions as a negative regulator of tumorigenesis during colitis-associated cancer. J Exp Med. 2010;207(5):1045–56. doi: 10.1084/jem.20100050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Song-Zhao GX, et al. Nlrp3 activation in the intestinal epithelium protects against a mucosal pathogen. Mucosal Immunol. 2014;7(4):763–774. doi: 10.1038/mi.2013.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Seo SU, et al. Distinct Commensals Induce Interleukin-1beta via NLRP3 Inflammasome in Inflammatory Monocytes to Promote Intestinal Inflammation in Response to Injury. Immunity. 2015;42(4):744–55. doi: 10.1016/j.immuni.2015.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Bauer C, et al. Colitis induced in mice with dextran sulfate sodium (DSS) is mediated by the NLRP3 inflammasome. Gut. 2010;59(9):1192–9. doi: 10.1136/gut.2009.197822. [DOI] [PubMed] [Google Scholar]
- 88.Liu L, et al. The Pathogenic Role of NLRP3 Inflammasome Activation in Inflammatory Bowel Diseases of Both Mice and Humans. J Crohns Colitis. 2017;11(6):737–750. doi: 10.1093/ecco-jcc/jjw219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lazaridis LD, et al. Activation of NLRP3 Inflammasome in Inflammatory Bowel Disease: Differences Between Crohn’s Disease and Ulcerative Colitis. Dig Dis Sci. 2017;62(9):2348–2356. doi: 10.1007/s10620-017-4609-8. [DOI] [PubMed] [Google Scholar]
- 90.Hampe J, et al. A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat Genet. 2007;39(2):207–11. doi: 10.1038/ng1954. [DOI] [PubMed] [Google Scholar]
- 91.Rioux JD, et al. Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat Genet. 2007;39(5):596–604. doi: 10.1038/ng2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Saitoh T, et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature. 2008;456(7219):264–8. doi: 10.1038/nature07383. [DOI] [PubMed] [Google Scholar]
- 93.Huber S, et al. IL-22BP is regulated by the inflammasome and modulates tumorigenesis in the intestine. Nature. 2012;491(7423):259–63. doi: 10.1038/nature11535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Martin JC, et al. Interleukin-22 binding protein (IL-22BP) is constitutively expressed by a subset of conventional dendritic cells and is strongly induced by retinoic acid. Mucosal Immunol. 2014;7(1):101–13. doi: 10.1038/mi.2013.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ayres JS, et al. Lethal inflammasome activation by a multidrug-resistant pathobiont upon antibiotic disruption of the microbiota. Nat Med. 2012;18(5):799–806. doi: 10.1038/nm.2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Bauer C, et al. Protective and aggravating effects of Nlrp3 inflammasome activation in IBD models: influence of genetic and environmental factors. Dig Dis. 2012;30(Suppl 1):82–90. doi: 10.1159/000341681. [DOI] [PubMed] [Google Scholar]
- 97.Lorang J, et al. Tricking the guard: exploiting plant defense for disease susceptibility. Science. 2012;338(6107):659–62. doi: 10.1126/science.1226743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Xu H, et al. Innate immune sensing of bacterial modifications of Rho GTPases by the Pyrin inflammasome. Nature. 2014;513(7517):237–41. doi: 10.1038/nature13449. [DOI] [PubMed] [Google Scholar]
- 99.Dumas A, et al. The inflammasome pyrin contributes to pertussis toxin-induced IL-1beta synthesis, neutrophil intravascular crawling and autoimmune encephalomyelitis. PLoS Pathog. 2014;10(5):e1004150. doi: 10.1371/journal.ppat.1004150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Sharma D, et al. Pyrin Inflammasome Regulates Tight Junction Integrity to Restrict Colitis and Tumorigenesis. Gastroenterology. 2017 doi: 10.1053/j.gastro.2017.11.276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wlodarska M, et al. NLRP6 inflammasome orchestrates the colonic host-microbial interface by regulating goblet cell mucus secretion. Cell. 2014;156(5):1045–59. doi: 10.1016/j.cell.2014.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Birchenough GM, et al. A sentinel goblet cell guards the colonic crypt by triggering Nlrp6-dependent Muc2 secretion. Science. 2016;352(6293):1535–42. doi: 10.1126/science.aaf7419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Levy M, et al. Microbiota-Modulated Metabolites Shape the Intestinal Microenvironment by Regulating NLRP6 Inflammasome Signaling. Cell. 2015;163(6):1428–43. doi: 10.1016/j.cell.2015.10.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Prochnicki T, Latz E. Inflammasomes on the Crossroads of Innate Immune Recognition and Metabolic Control. Cell Metab. 2017;26(1):71–93. doi: 10.1016/j.cmet.2017.06.018. [DOI] [PubMed] [Google Scholar]
- 105.Elinav E, et al. NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell. 2011;145(5):745–57. doi: 10.1016/j.cell.2011.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Normand S, et al. Nod-like receptor pyrin domain-containing protein 6 (NLRP6) controls epithelial self-renewal and colorectal carcinogenesis upon injury. Proc Natl Acad Sci USA. 2011;108(23):9601–6. doi: 10.1073/pnas.1100981108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Lemire P, et al. The NLR Protein NLRP6 Does Not Impact Gut Microbiota Composition. Cell Rep. 2017;21(13):3653–3661. doi: 10.1016/j.celrep.2017.12.026. [DOI] [PubMed] [Google Scholar]
- 108.Galvez EJC, et al. Shaping of Intestinal Microbiota in Nlrp6- and Rag2-Deficient Mice Depends on Community Structure. Cell Rep. 2017;21(13):3914–3926. doi: 10.1016/j.celrep.2017.12.027. [DOI] [PubMed] [Google Scholar]
- 109.Mamantopoulos M, et al. Nlrp6- and ASC-Dependent Inflammasomes Do Not Shape the Commensal Gut Microbiota Composition. Immunity. 2017;47(2):339–348e4. doi: 10.1016/j.immuni.2017.07.011. [DOI] [PubMed] [Google Scholar]
- 110.Alipour M, et al. Mucosal Barrier Depletion and Loss of Bacterial Diversity are Primary Abnormalities in Paediatric Ulcerative Colitis. J Crohns Colitis. 2016;10(4):462–71. doi: 10.1093/ecco-jcc/jjv223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Seregin SS, et al. NLRP6 Protects Il10−/− Mice from Colitis by Limiting Colonization of Akkermansia muciniphila. Cell Rep. 2017;19(10):2174. doi: 10.1016/j.celrep.2017.05.074. [DOI] [PubMed] [Google Scholar]
- 112.Wang P, et al. Nlrp6 regulates intestinal antiviral innate immunity. Science. 2015;350(6262):826–30. doi: 10.1126/science.aab3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Zhu S, et al. Nlrp9b inflammasome restricts rotavirus infection in intestinal epithelial cells. Nature. 2017;546(7660):667–670. doi: 10.1038/nature22967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kayagaki N, et al. Non-canonical inflammasome activation targets caspase-11. Nature. 2011;479(7371):117–21. doi: 10.1038/nature10558. [DOI] [PubMed] [Google Scholar]
- 115.Shi J, et al. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature. 2014;514(7521):187–92. doi: 10.1038/nature13683. [DOI] [PubMed] [Google Scholar]
- 116.Kayagaki N, et al. Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science. 2013;341(6151):1246–9. doi: 10.1126/science.1240248. [DOI] [PubMed] [Google Scholar]
- 117.Hagar JA, et al. Cytoplasmic LPS activates caspase-11: implications in TLR4-independent endotoxic shock. Science. 2013;341(6151):1250–3. doi: 10.1126/science.1240988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Aachoui Y, et al. Caspase-11 protects against bacteria that escape the vacuole. Science. 2013;339(6122):975–8. doi: 10.1126/science.1230751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Vanaja SK, et al. Bacterial Outer Membrane Vesicles Mediate Cytosolic Localization of LPS and Caspase-11 Activation. Cell. 2016;165(5):1106–1119. doi: 10.1016/j.cell.2016.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Demon D, et al. Caspase-11 is expressed in the colonic mucosa and protects against dextran sodium sulfate-induced colitis. Mucosal Immunol. 2014;7(6):1480–91. doi: 10.1038/mi.2014.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Oficjalska K, et al. Protective role for caspase-11 during acute experimental murine colitis. J Immunol. 2015;194(3):1252–60. doi: 10.4049/jimmunol.1400501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Yang S, et al. Pellino3 ubiquitinates RIP2 and mediates Nod2-induced signaling and protective effects in colitis. Nat Immunol. 2013;14(9):927–36. doi: 10.1038/ni.2669. [DOI] [PubMed] [Google Scholar]
- 123.Greve AS, et al. P2X1, P2X4, and P2X7 Receptor Knock Out Mice Expose Differential Outcome of Sepsis Induced by alpha-Haemolysin Producing Escherichia coli. Front Cell Infect Microbiol. 2017;7:113. doi: 10.3389/fcimb.2017.00113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Lawlor KE, et al. RIPK3 promotes cell death and NLRP3 inflammasome activation in the absence of MLKL. Nat Commun. 2015;6:6282. doi: 10.1038/ncomms7282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Gaidt MM, et al. Human Monocytes Engage an Alternative Inflammasome Pathway. Immunity. 2016;44(4):833–46. doi: 10.1016/j.immuni.2016.01.012. [DOI] [PubMed] [Google Scholar]
- 126.Newton K, et al. RIPK3 deficiency or catalytically inactive RIPK1 provides greater benefit than MLKL deficiency in mouse models of inflammation and tissue injury. Cell Death Differ. 2016;23(9):1565–76. doi: 10.1038/cdd.2016.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Lewis GJ, et al. Genetic association between NLRP3 variants and Crohn’s disease does not replicate in a large UK panel. Inflamm Bowel Dis. 2011;17(6):1387–91. doi: 10.1002/ibd.21499. [DOI] [PubMed] [Google Scholar]
- 128.Harris PA, et al. Discovery of a First-in-Class Receptor Interacting Protein 1 (RIP1) Kinase Specific Clinical Candidate (GSK2982772) for the Treatment of Inflammatory Diseases. J Med Chem. 2017;60(4):1247–1261. doi: 10.1021/acs.jmedchem.6b01751. [DOI] [PubMed] [Google Scholar]
- 129.Cabrera SM, et al. Interleukin-1 antagonism moderates the inflammatory state associated with Type 1 diabetes during clinical trials conducted at disease onset. Eur J Immunol. 2016;46(4):1030–46. doi: 10.1002/eji.201546005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Choudhury RP, et al. Arterial Effects of Canakinumab in Patients With Atherosclerosis and Type 2 Diabetes or Glucose Intolerance. J Am Coll Cardiol. 2016;68(16):1769–1780. doi: 10.1016/j.jacc.2016.07.768. [DOI] [PMC free article] [PubMed] [Google Scholar]