

Abstract
Development of tamoxifen resistance remains a tremendous challenge for the treatment of estrogen-receptor (ER)-positive breast cancer. Small GTPases of the Ras superfamily play crucial roles in intracellular trafficking and cell signaling, and aberrant small-GTPase signaling is implicated in many types of cancer. In this study, we employed a targeted, quantitative proteomic approach that relies on stable-isotope labeling by amino acids in cell culture (SILAC), gel fractionation, and scheduled multiple-reaction-monitoring (MRM) analysis, to assess the differential expression of small GTPases in MCF-7 and the paired tamoxifen-resistant breast cancer cells. The method displayed superior sensitivity and reproducibility over the shotgun-proteomic approach, and it facilitated the quantification of 96 small GTPases. Among them, 13 and 10 proteins were significantly down- and up-regulated (with >1.5-fold change), respectively, in the tamoxifen-resistant line relative to in the parental line. In particular, we observed a significant down-regulation of RAB31 in tamoxifen-resistant cells, which, in combination with bioinformatic analysis and downstream validation experiments, supported a role for RAB31 in tamoxifen resistance in ER-positive breast-cancer cells. Together, our results demonstrated that the targeted proteomic method constituted a powerful approach for revealing the role of small GTPases in therapeutic resistance.
Graphical Abstract
Breast cancer is the most prevalent cancer among women worldwide. According to the American Cancer Society, 268 000 new breast-cancer cases and 41 400 deaths were estimated in 2018 in the United States.1 As a highly heterogeneous disease, breast cancer can be categorized into three major subtypes: estrogen-receptor-α-positive (ER-positive) breast cancer, human-epidermal-growth-factor-receptor-2 (HER2)-amplified (HER2-positive) breast cancer, and triplenegative breast cancer (TNBC). Among them, the ER-positive subtype remains the most prevalent and diverse, accounting for approximately 80% of diagnosed cases of breast cancer.2 In this respect, antiestrogen drugs including tamoxifen and fulvestrant are frequently used, and tamoxifen remains the standard frontline endocrine therapy complementary to surgery. However, approximately half of patients who receive tamoxifen as the first-line therapy for recurrent diseases do not respond to the treatment because of intrinsic resistance. For those patients who initially respond to the drug, development of resistance is a major cause of treatment failure.3
Small GTPases of the Ras family are crucial regulators of intracellular trafficking and can mediate a wide range of biological events.4 Several small GTPases play important roles in breast-cancer progression, including RAB2A (tumorigenesis),5 RAB27B (migration and invasion),6 RAB31 (proliferation and metastasis),7 and RND1 (tumorigenesis and invasion).8 Defective endocytic pathways arising from down-regulation of small GTPases RAB5, RAC1, and RHOA were found in squamous-cell carcinomas (SCCs) carrying resistance to chemotherapeutic drugs such as cisplatin.9 Cellular pathways that involve small-GTPase signaling, including the Ras–Raf1–MAPK pathway, the Rac1–PAK1 pathway, and the Cdc42-mediated redox pathway, have also been shown to mediate responses to tamoxifen in breast-cancer cells.10–12 Therefore, we reason that a systematic interrogation of small GTPases involved in modulating tamoxifen resistance may offer a better understanding of the mechanisms of resistance in tamoxifen therapy.
Mass-spectrometry-based proteomic methods have been widely employed for studying drug resistance and for discovering novel biomarkers and therapeutic targets in breast cancer.13–15 Building upon the previously reported method of gel fractionation followed by liquid chromatography–tandem mass spectrometry (LC-MS/MS),16,17 we recently developed a targeted, quantitative proteomic approach, relying on stableisotope labeling by amino acids in cell culture (SILAC), gel fractionation, and LC-MS/MS in scheduled multiple-reactionmonitoring (MRM) mode, for high-throughput profiling of small GTPases.18 In the present study, we employed this method to comprehensively investigate alterations in the expression of small GTPases during the development of tamoxifen resistance in a pair of matched wild-type (WT)–tamoxifen-resistant (TamR) breast-cancer cell lines (i.e., MCF-7/WT and MCF-7/TamR). The quantitative proteomic data and cell-based assays with the use of two ER-positive cell lines (i.e., MCF-7 and T47D), together with bioinformatic analysis of publicly available data, led to the discovery of a novel role for RAB31 in modulating acquired tamoxifen resistance.
EXPERIMENTAL SECTION
Cell Culture.
MCF-7 cells were purchased from ATCC (#HTB-22). The tamoxifen-resistant variant of MCF-7 cells (MCF-7/TamR) was generously provided by Dr. Guandi Wang (Xavier University). The T47D cells were kindly provided by Dr. Ameae Walker (University of California, Riverside). The cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetalbovine serum (FBS; Invitrogen-Gibco) and penicillin– streptomycin (100 IU/mL) at 37 °C in an atmosphere with 5% CO2. The MCF-7/TamR cells were continuously cultured in the above-described medium containing 0.10 μM (Z)-4-hydroxytamoxifen (4-OHT, Sigma-Aldrich) for at least 6 months to allow them to develop resistance to the drug with an IC50 of ∼5 μM 4-OHT.13
For other experiments, 4-OHT was dissolved in ethanol at a concentration of 10 mM and stored at −20 °C. For SILAC experiments, [13C6,15N2]-l-lysine and [13C6]-l-arginine (Cambridge Isotopes Inc.) or their unlabeled counterparts were added to SILAC DMEM medium depleted of l-lysine and l-arginine (Thermo Scientific Pierce) until their final concentrations reached 0.398 and 0.798 mM, respectively, to yield “heavy” and “light” media. Cells were cultured in the “heavy” SILAC DMEM medium for at least 6 cell doublings to ensure complete incorporation of heavy-isotope-labeled amino acids.
Sample Preparation and Scheduled LC-MRM Analysis.
To assess the differential expression of small GTPases in wild-type (WT) and tamoxifen-resistant (TamR) MCF-7 cells, we conducted SILAC-based, quantitative proteomic experiments with forward- and reverse-labeling strategies. Briefly, we combined lysates of light-labeled WT cells and heavy-labeled TamR cells in a 1:1 ratio in the forward-labeling experiments. The reverse-labeling experiment was conducted in the opposite way. The mixed cell lysates (100 μg in total) were loaded onto a 10% SDS-PAGE gel and separated by electrophoresis. The gel bands corresponding to the molecular-weight range of 15–37 kDa were cut, reduced with 20 mM dithiothreitol, alkylated with 55 mM iodoacetamide, and digested in gel with trypsin at an enzyme–protein ratio of 1:100. After tryptic digestion, the peptide mixtures were desalted and subjected to LC-MRM analyses. All LC-MRM experiments were performed on a TSQ vantage triple-quadruple mass spectrometer (Thermo Scientific) coupled with an EASY-nLC II system (Thermo Scientific). The samples were automatically loaded onto a 4 cm trapping column (150 μm i.d.) packed with ReproSil-Pur 120 C18-AQ resin (5 μm in particle size and 120 Å in pore size, Dr. Maisch GmbH HPLC) at 3 μL/min. The trapping column was coupled to a 20 cm fused silica analytical column (75 μm i.d.) packed with ReproSil-Pur 120 C18-AQ resin (3 μm in particle size and 120 Å in pore size, Dr. Maisch GmbH HPLC). The peptide mixtures were then separated using a 157 min linear gradient of 2–35% acetonitrile in 0.1% formic acid at a flow rate of 300 nL/min. The spray voltage was set as 1.8 kV. Ions were isolated in both Q1 and Q3 using 0.7 fwhm resolution, for which the cycle time and dwell time were 5 s and 10 ms, respectively. The optimal collisional energy (CE) for each targeted peptide was calculated using a linear equation specific to the TSQ Vantage instrument and the precursor mass-to-charge ratio (m/z) according to the default setting in Skyline.19
To enable high-throughput quantitative analysis, we applied a previously developed scheduled LC-MRM method,18 in which the mass spectrometer was programmed to acquire the MS/MS of the precursor ions for a limited number of peptides in each 6 min retention-time (RT) window. The MRM data for all targeted peptides were manually inspected to ensure correct peak picking. In this regard, the dot-product (dotp) value has to exceed 0.80.20 In addition, the iRT value represents an intrinsic property (i.e., hydrophobicity) of a peptide; hence, a substantial deviation of measured RT from that projected from the linear plot of RT over iRT signals a false-positive detection.
Data Availability.
A complete list of the processed data is provided in Supplementary Table S1. The Skyline MRM library (including the transition list for quantification and the iRT file) for the human small-GTPase proteome and the raw files obtained from the LC-MRM analyses of small GTPases for the WT and TamR MCF-7 cells were deposited into PeptideAtlas with the identification number PASS01267 (http://www.peptideatlas.org/PASS/PASS01267).
Cell-Proliferation Assay.
The proliferation of cells under OHT treatment was evaluated using a Cell Counting Kit 8 (CCK-8, Dojindo Laboratories). Briefly, MCF-7/WT, T47D and MCF-7/TamR cells were seeded in a 96-well flatbottomed microplate (3000 cells/well) in complete growth medium (100 μL/well) for 24 h. The cells were then incubated with or without various concentrations of 4-OHT for 5 days in the dose-dependent experiments or with 1 μM 4-OHT in the time-dependent experiments, and ethanol was used as the vehicle control. At the end of treatments, 10 μL of the CCK-8 dye was added to each well, and the cells were incubated at 37 °C for 4 h prior to using the Synergy H1 Hybrid Multi-Mode Microplate Reader (BioTek Instruments) for measuring the absorbance at 450 nm.
Colony-Formation Assay.
MCF-7/WT or MCF-7/TamR cells were cultured in complete growth medium. The cells were seeded at a density of 2000 cells/well in 2 mL of medium in six-well plates and allowed to adhere overnight. The next day, the cells were treated with 1 μM 4-OHT, and an equal volume of ethanol was used as a vehicle control. The cells were then allowed to grow until colonies reached >50 cells per colony for the control group (approximately 10 to 14 days). Colonies were then fixed with glutaraldehyde for 30 min, stained with crystal violet (0.1% in 20% methanol) for 30 min, and washed. Colony numbers were determined manually. The experiments were conducted in triplicate, and the data represent means ± standard errors of the means.
RESULTS AND DISCUSSION
Application of a High-Throughput LC-MRM Assay for Studying Acquired Tamoxifen Resistance.
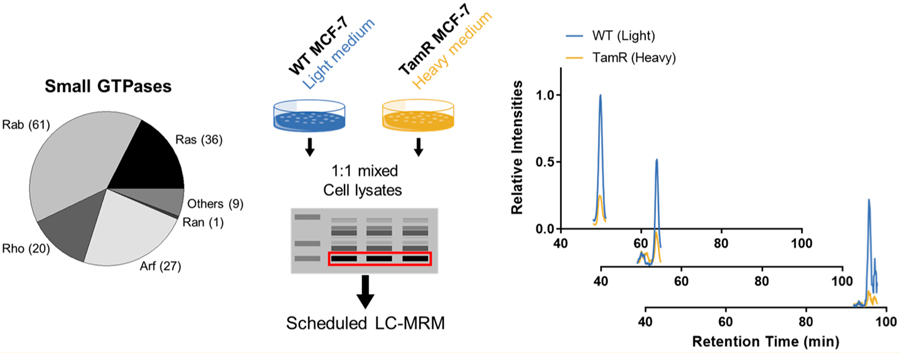
We set out to explore the alterations of small GTPases during the development of tamoxifen resistance in ER-positive breast cancer. To this end, we employed our recently developed scheduled-MRM-based targeted proteomic method18 to assess, in high-throughput, the reprogramming of the small-GTPase proteome during development of tamoxifen resistance. The method involved metabolic labeling of MCF-7 cells and isogenic cells that are resistant to tamoxifen using SILAC, SDS-PAGE for the enrichment of proteins in the molecular-weight range of 15–37 kDa (the molecular weights for ∼95% of small GTPases fall in this range), in-gel tryptic digestion, and LC-MS/MS analysis of the resulting tryptic peptides in scheduled MRM mode (Figure 1A).
Figure 1.

Targeted, quantitative analysis of small GTPases in tamoxifen resistance. (A) A schematic diagram showing the workflow of the targeted, quantitative proteomic analysis, relying on metabolic labeling with SILAC, SDS-PAGE fractionation, and scheduled LC-MRM analysis. (B) Heatmap showing the differential expression of small GTPases in MCF-7/WT and MCF-7/TamR cells. The log2 ratios of TamR-cell levels to WTcell levels obtained from two forward- and one reverse-SILAC-labeling experiments (F1 and F2: forward experiments, R1: reverse experiment) are shown. As indicated by the scale bar, the red and blue bars designate the small GTPases that are up- and down-regulated, respectively, by at least 1.5-fold in the drug-resistant cells compared with in the parental MCF-7 cells. (C) Bar chart showing substantially up-regulated (>1.5-fold) and down-regulated (>1.5-fold) small GTPases quantified from three LC-MRM experiments.
To obtain reliable quantification results, we conducted SILAC experiments in triplicate, with two sets of forward labeling and one set of reverse labeling. The method facilitated the quantification of a total of 96 small GTPases (Figure 1B), among which 13 and 10 proteins were significantly down- and up-regulated (with >1.5-fold changes), respectively, in the drug-resistant MCF-7 cells relative to the parental line (Figure 1C). In this vein, we chose a cutoff of a 1.5-fold change on the basis of the average relative standard deviation (RSD; 14%, Table S1) for all the quantified small GTPases. The method facilitated the coverage of approximately 65% of the human small-GTPase proteome in two LC-MRM runs.
We found that several RAB small GTPases were down-regulated (e.g., RAB27B, RAB30, RAB31, and RAB32), whereas several others were up-regulated (e.g., RAB7A, RAB18, and RAB6B), in tamoxifen-resistant cells (Figure 1C). Figure S1 displays the extracted-ion chromatograms (XICs) for several differentially expressed small GTPases, including ARL3 (ARF subfamily), RHOF (RHO subfamily), RAB30 (RAB subfamily), and RRAS2 (RAS subfamily). Notably, we observed a ∼2-fold up-regulation of RRAS2 protein in the tamoxifen-resistant MCF-7 cells. In this vein, RRAS2 is known to promote primary tumorigenesis and late steps of metastasis in breast-cancer cells, and it can also contribute to increased resistance to tamoxifen.21–23 Hence, our method validated the differential expression of a small GTPase that was previously shown to be involved with tamoxifen resistance.
There have been no literature precedents about the functions of ARL3, RHOF, and RRAS in drug resistance in breast cancer. Nevertheless, ARL3 was suggested to be transcriptionally regulated by ER-related mechanisms.24 In addition, RRAS inhibits the proliferation, migration, and cellcycle progression of cultured breast-cancer cells.25 RHOF plays an important role in controlling the formation of filopodia, which may contribute to proliferation, invasion, and formation of micrometastases of cancer cells. RHOF, however, enhances the resistance of pancreatic cancer to gemcitabine through regulation of the epithelial-to-mesenchymal transition.26 The potential roles of ARL3, RRAS, and RHOF in modulating tamoxifen resistance warrant future investigation.
To assess the performance of our MRM-based method, we also analyzed the same samples by employing LC-MS/MS in data-dependent-acquisition (DDA) mode. As shown in Figure 2A, DDA analyses only led to the identification of 51 and 45 small GTPases in the two forward SILAC samples (F1 and F2, respectively), and 44 small GTPases in the reverse SILAC sample (R). In stark contrast, the LC-MRM approach led to substantially higher coverage of the small-GTPase proteome (Figure 2A). As noted, the quantification was based on three independent LC-MRM experiments, which included two forward- and one reverse-SILAC-labeling experiments, and the small GTPases reported were reproducibly quantified in all three replicates, with the mean RSD being 14% (Table S1). Therefore, these results demonstrated that the scheduled LC-MRM method outperformed the shotgun proteomic approach in terms of reproducibility and sensitivity. Additionally, we observed an excellent linear fit (R2 = 0.9238) for the log2-transformed SILAC ratios of all the quantified small GTPases obtained from one forward- and one reverse-SILAC-labeling experiment (Figure 2B), which again underscored the excellent reproducibility of the method. With respect to retention-time (RT) scheduling, all of the 10 standard peptides derived from BSA exhibited an excellent linear fit (R2 = 0.9996) between the observed RTs and the iRT values in the library (Figure 2C). Highly reliable and reproducible RT prediction was also reflected by the superb linearity for the RTs observed for the small-GTPase peptides in different replicates (Figure 2D,E).
Figure 2.
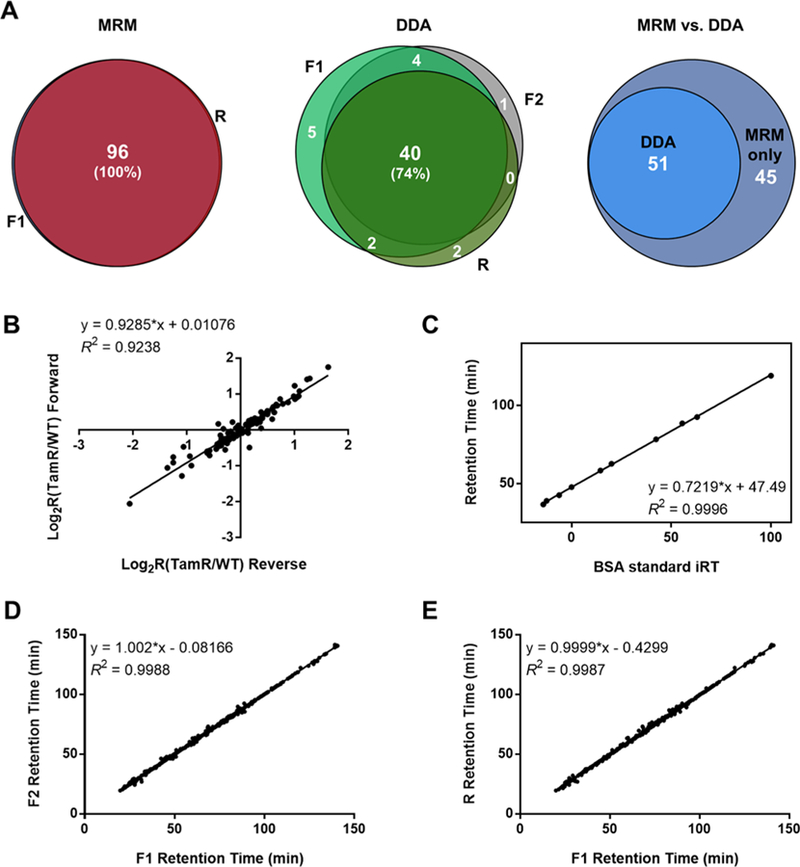
Performances of the scheduled LC-MRM method for targeted, quantitative analysis of differential expression of small GTPases in tamoxifen resistance. (A) Venn diagrams displaying the overlap between quantified small GTPases in the forward- and reverse-SILAC-labeling experiments, as obtained from MRM analyses and DDA analyses, respectively, and comparison of the performances of the two methods. (B) Correlation between the log2-transformed SILAC ratios (log2R) obtained from one forward- and one reverse-SILAC-labeling experiment with a relatively high correlation coefficient (R2 = 0.9238). (C) Correlation between BSA-standard iRT values and measured RT values with a very high correlation coefficient (R2 = 0.9996). (D) Measured RTs obtained in two forward-labeling reactions (F1 vs F2) with a very high correlation coefficient (R2 = 0.9988). (E) Measured RTs obtained in one forward- and one reverse-labeling reactions (F1 vs R) with a very high correlation coefficient (R2 = 0.9987).
To achieve confident identification of targeted peptides, we manually processed the LC-MRM data to ensure the coelution of the MRM transitions with the dot-product (dotp) value being >0.8, as calculated by Skyline.20 The dotp of a peptide was based on the correlation for the ratio of the MRM peak intensities observed versus those in the library MS/MS acquired from shotgun proteomic experiments. Shown in Figure 3A are the representative XICs displaying nearly identical retention times for different MRM transitions monitored for each of the three distinct peptides of RAB31 with high dotp values (>0.95). Relative quantification was achieved by integrating the areas of the peaks found in the XICs acquired from the LC-MRM analyses (Figure 3B shows the MRM traces acquired from the forward- and reverse-SILAC-labeling experiments). The down-regulation of RAB31 protein in the tamoxifen-resistant line was further confirmed by Western-blot analysis (Figures 3C and 2D). In addition to RAB31, we also validated the differential expression of RAB27A and RAB27B proteins by Western-blot analysis (Figure S2), supporting that the LC-MRM method is capable of accurately profiling the differential expression of small GTPases.
Figure 3.

RAB31 is down-regulated in tamoxifen-resistant MCF-7 cells. (A) Representative MRM traces for three transitions monitored for each of the three unique tryptic peptides derived from RAB31: FHSLAPMYYR (y8, y6, and y5), QDSFYTLK (y6, y4, and y3), and GSAAAVIVYDITK (y7, y6, and y5). (B) Extracted-ion chromatograms (XICs) for the quantification of the three peptides in panel (A) in one forward- and one reverse-SILAC-labeling experiment. (C) Validation of the differential expression of RAB31 in MCF-7/WT and MCF-7/TamR cells by Western-blot analysis. (D) Comparison of quantification results obtained from LC-MRM (Peptides #1, #2, and #3 refer to FHSLAPMYYR, QDSFYTLK and GSAAAVIVYDITK, respectively, as shown in panel A) and Western-blot analyses (n = 3). The p values were calculated by using a paired, two-tailed Student’s t test (**p < 0.01). Error bars represent standard deviations.
Prognostic Values of RAB31 in Breast Cancer Revealed by Bioinformatic Analyses.
As discussed above, our LC-MRM results confirmed a known protein target that promotes acquired tamoxifen resistance: RRAS2. We next sought to further examine the roles of other differentially expressed small GTPases in breast cancer by performing Kaplan–Meier survival analysis for significantly up- and down-regulated proteins shown in Figure 1C. Particularly, for down-regulated proteins, lower expression levels of ARL3, RHOF, and RAB30 are significantly associated with poor relapse-free survival (RFS, Figure S3A–C). In contrast, higher RRAS2-mRNA levels are significantly correlated with poor RFS (Figure S3D). For patients who received endocrine therapy (tamoxifen only), increased RAB31 expression predicts better outcomes, whereas for those without endocrine therapy, RAB31 does not serve as an effective indicator for RFS (Figure S4A). Notably, among those genes that exhibited significantly altered protein abundance in tamoxifen resistance, only the mRNA expression of RAB31 is significantly correlated with tamoxifen efficacy in ER-positive breast-cancer patients, but not that of ARL3, RAB30, RRAS2, or RHOF (Figures S3 and S4B). We also assessed the prognostic values of RAB31 stratified by ER status and found only a significant correlation between RAB31 expression and survival in ER-positive but not ER-negative breast-cancer patients (Figure S4C,D).
We also interrogated several Gene Expression Omnibus (GEO) data sets for Kaplan–Meier survival analysis and differential-mRNA-expression analysis: GSE3494 (Karolinska Institute, n = 236),27 GSE4922 (Genome Institute of Singapore, n = 347),28 GSE6434 (Baylor College of Medicine, n = 24),29 GSE42568 (Dublin City University, n = 115),30 GSE24460 (National Cancer Institute, n = 52),31 and GSE26495 (Emory University). The GSE6434, GSE26495, and GSE24460 data sets revealed significantly decreased levels of RAB31 in docetaxel-, tamoxifen-, and doxorubicin-resistant MCF-7 cell lines, respectively (Figure 4A–C). In this respect, docetaxel and doxorubicin are other small-molecule drugs commonly used in chemotherapy against breast cancer. Therefore, these results suggest that RAB31 down-regulation could be a common event in drug-resistant MCF-7 cells. For survival analysis, lower RAB31 expression was significantly associated with poorer RFS and overall survival (OS) in the GSE42568 cohort (Figure 4D,E). Meanwhile, the same trend holds for the disease-specific survival (DSS) in the GSE3494 data set (Figure 4F). Notably, in the GSE4922 cohort, RAB31 is significantly down-regulated in the most metastatic grade III tumors as compared with in grade I tumors (Figure 4G), supporting a potential role for RAB31 in breast-cancer progression.32 Taken together, these results suggest that RAB31 may serve as a potential biomarker for predicting acquired tamoxifen resistance in breast-cancer patients.
Figure 4.
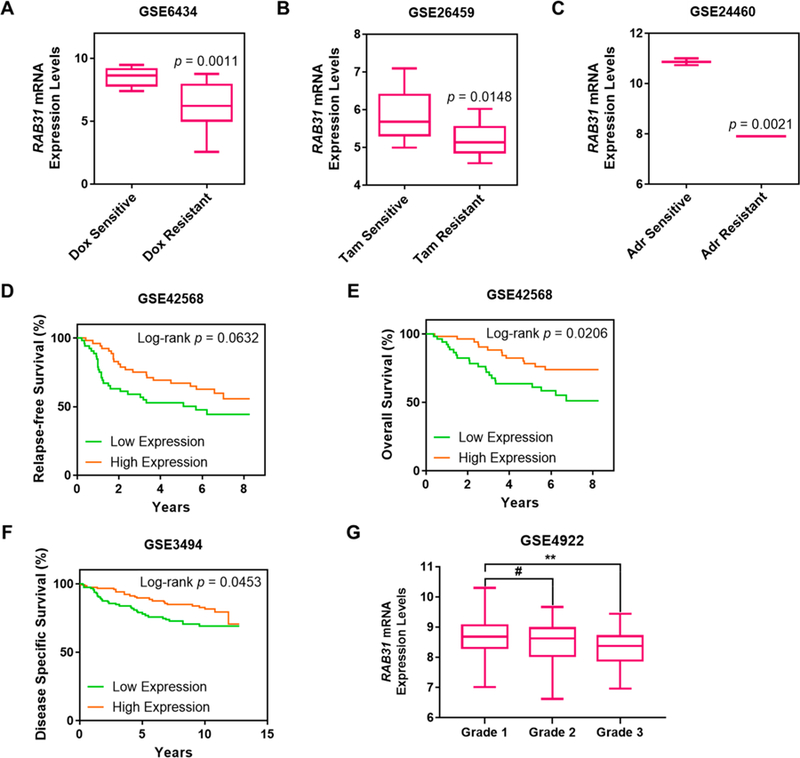
Bioinformatic analysis revealing RAB31 as a potential predictive marker for tamoxifen resistance. (A–C) Differential-expression analysis for RAB31 in (A) docetaxel-, (B) tamoxifen-, and (C) doxorubicin-resistant MCF-7 cell lines in three data sets, GSE6434, GSE26495, and GSE24460, respectively (Dox: docetaxel, Tam: tamoxifen, Adr: adriamycin or doxorubicin). (D–G) Kaplan–Meier survival analyses for (D) relapse-free survival (RFS) in the GSE42568 data set, (E) overall survival (OS) in the GSE42568 data set, (F) disease-specific survival (DSS) in the GSE3494 data set, (G) differential RAB31 expression in different grades of breast-cancer progression in the GSE4922 data set. The patient population was stratified by median RAB31-mRNA-expression levels. The p values for the Kaplan–Meier curves were calculated by using the logrank test. The p values for the box-whisker plots were calculated by using an unpaired, two-tailed Student’s t test (#p > 0.05, *p < 0.05, **p < 0.01).
Correlation of RAB31 Expression with ER Status and Breast-Cancer Subtypes.
To elucidate the association between RAB31 expression and breast-cancer subtypes, we next interrogated a large and comprehensive breast-cancer patient cohort: The Cancer Genome Atlas Breast Invasive Carcinoma (TCGA-BRCA). Our results showed that RAB31-mRNA-expression levels were significantly correlated with the mRNA-expression levels of the ESR1 gene, which encodes ERα, in the TCGA-BRCA cohort (Figure S5A). In addition, the same correlations could be observed for the GSE4922 and GSE23988 data sets (Figure S5B,C). Aside from ER status, we extended the expression analysis to elucidate the association between RAB31-mRNA levels and different molecular subtypes of breast cancer: luminal-A, luminal-B, HER2-enriched, basal-like, and normal-like breast cancer.33 As shown in Figure S5D, the mRNA-expression levels of RAB31 were significantly lower in the basal subtype and the HER2-enriched subtype than in the luminal-A subtype. Clinical data demonstrate reduced responses to endocrine therapy in tumors with HER2 amplification. Suppressed expression of RAB31 might therefore be correlated with reduced ER expression and the HER2-overexpressing and resistant phenotypes.
RAB31 Knockdown Rendering MCF-7 and T47D Cells More Resistant to 4-OHT Treatment.
The above results showed that diminished RAB31 expression confers poor prognosis for ER-positive breast-cancer patients, especially for those who received tamoxifen as a first-line therapy. Hence, we reason that RAB31 may be associated with tamoxifen efficacy and therefore may play a role in tamoxifen resistance. To further examine the role of RAB31 in tamoxifen resistance, we used lentiviral transduced shRNA to enable a stable knockdown of RAB31 in MCF-7 cells. Western-blot analysis showed that two separate shRNA sequences gave rise to 60–70% depletions of RAB31 protein compared with that from the control shRNA sequence (shScramble) in MCF-7 cells (Figure S6A,B). We next performed cell-proliferation assay to assess the effects of RAB31 knockdown on tamoxifen response in MCF-7 cells. We found that, upon treatment with 1 μM (Z)-4-hydroxytamoxifen (4-OHT), an active metabolite of tamoxifen, genetic depletion of RAB31 led to significantly higher cell viability at 72 and 96 h but not at 24 or 48 h (Figure S6C,D). Moreover, RAB31 knockdown led to higher resistance to 4-OHT treatment in a broad range of doses (Figure S6E). Collectively, the results support that loss of RAB31 could modulate tamoxifen response in MCF-7 cells.
To further substantiate the above findings, we assessed how shRNA-mediated depletion of RAB31 in T47D cells alters the sensitivity of these cells toward tamoxifen. Consistent with our hypothesis, diminished expression of RAB31 in both MCF-7 and T47D cells led to increased proliferation rates and elevated resistance toward tamoxifen (Figure S7A–C). In addition, RAB31 knockdown gave rise to augmented resistance to tamoxifen in T47D cells (Figure S7D,E). Taken together, the above results support the role of RAB31 in tamoxifen efficacy in two ER-positive breast-cancer cell lines: MCF-7 and T47D.
Sensitization of MCF-7/TamR Cells to 4-OHT Treatment by RAB31 Overexpression.
We next examined how a gain of function of RAB31 modulates tamoxifen sensitivity by ectopically overexpressing the RAB31 gene in tamoxifen-resistant MCF-7/TamR cells. A cell-proliferation assay showed that RAB31-overexpressing MCF-7/TamR cells exhibited elevated sensitivity toward 4-OHT treatment as compared with that of the empty vector group in a dose- and time-dependent manner (Figure 5A,B). In agreement with the cell-proliferation assay, a colony-formation assay showed that ectopic expression of RAB31 confers significantly increased tamoxifen sensitivity for the MCF-7/TamR cells (Figure 5C,D). In summary, our results substantiated the role of RAB31 in suppressing acquired tamoxifen resistance in MCF-7/TamR cells.
Figure 5.
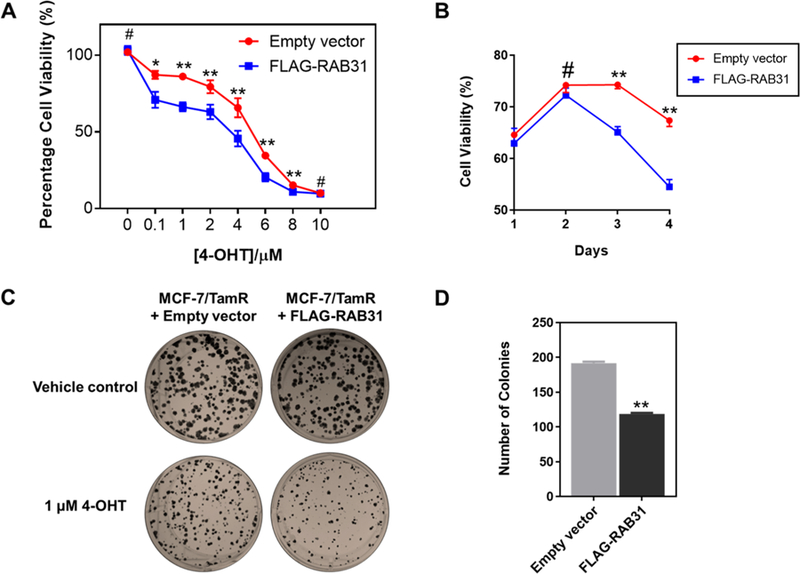
Ectopic expression of RAB31 led to elevated tamoxifen sensitivity. (A) Cell-proliferation assay for MCF-7/TamR cells after treatment with different doses of 4-OHT. (B) Cell-proliferation assay for MCF-7/TamR cells after treatment with 4-OHT (1 μM) for different periods of time. (C) Colony-formation assay for MCF-7/TamR cells expressing empty vector or FLAG-RAB31. (D) Quantification results for the colony-formation assay. The p values were calculated by using a paired, two-tailed Student’s t test (#p > 0.05, *p < 0.05, **p < 0.01). Error bars represent standard errors of means.
CONCLUSIONS
In the present study, we applied a targeted, quantitative proteomic method to assess the differential expression of small GTPases in paired wild-type and tamoxifen-resistant MCF-7 cells. The method provided high-throughput, accurate, and reproducible quantifications of the relative expression levels of >90 small GTPases in the paired cell lines. Among them, 10 and 13 small GTPases were up- and down-regulated by at least 1.5-fold in the drug-resistant cells compared with the parental MCF-7 cells. In this context, it is worth noting that our current targeted proteomic approach relies on SILAC labeling, which is not applicable to clinical specimens (e.g., biological fluids and tumor-tissue samples) from breast-cancer patients that manifest de novo or acquired tamoxifen resistance. Nevertheless, the targeted proteomic method can be adapted for handling clinical specimens with the use of heavy-isotope-labeled synthetic peptides, and such an approach is currently being explored in our laboratory.
Combined with bioinformatic analyses, we identified RAB31 as a novel predictive marker for acquired tamoxifen resistance. Through the use of two ER-positive cell lines (i.e., MCF-7 and T47D), we uncovered, for the first time, a role of RAB31 in modulating tamoxifen sensitivity. The functions of RAB31 were also explored in other cancer types.34 RAB31 can localize to endocytic compartments and functions in the post-Golgi, endocytic, or exocytic trafficking of the epidermal-growth-factor receptor (EGFR) in A431 and HeLa cells.35 Silencing of RAB31 inhibited the endocytic trafficking of the ligand-bound EGFR to late endosomes and its subsequent degradation. It is also worth noting that increased levels of receptor tyrosine kinases, including EGFR and HER2, can directly alter the cellular response to tamoxifen.36 Therefore, we reason that down-regulation of RAB31 in MCF-7/TamR cells may perturb EGFR trafficking, thereby influencing drug resistance.
In addition to receptor-tyrosine-kinase signaling, ER-associated proteins are essential players in tamoxifen resistance. Some suggest elevated ESR1-mRNA levels in tamoxifen-resistant ER-positive breast-cancer cells.22 However, several lines of evidence demonstrated suppressed yet functional ER-regulated signaling pathway in MCF-7/TamR cells, as reflected by down-regulated ESR1 expression.13,37–39 Notably, RAB31 is among the 11 genes that are robustly overexpressed in ER-positive breast-carcinoma samples,40 which is in line with the observation that the promoter region of the RAB31 gene harbors an ER-responsive element.41 On the basis of these findings, we reason that down-regulated RAB31 in tamoxifen-resistant MCF-7 cells may arise from ER-modulated molecular adaptations.
Taken together, we showed, for the first time, that targeted, quantitative proteomics, in combination with bioinformatics, provided novel insights into the roles of small GTPases in acquired tamoxifen resistance in breast cancer. It can be envisaged that the method can also be employed for understanding the therapeutic resistance of other types of cancer.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the National Institutes of Health (R01 CA210072 to Y.W.), and M.H. was supported in part by an NRSA Institutional Training Grant (T32 ES018827). The authors would like to thank Dr. Guandi Wang for sharing with us the MCF-7/TamR cell line and Dr. Ameae Walker for sharing with us the T47D cell line.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.analchem.8b04526.
Supplementary experimental section; oligodeoxyribonucleotide sequences used for the construction of shRNA plasmids using the pLKO.1 lentiviral vector; LC-MRM for the quantification of the relative levels of ARL3, RHOF, RAB30, and RRAS2 proteins in the paired MCF-7/WT and MCF-7/TamR cells; LC-MRM and Western-blot analyses for the quantification of the relative levels of RAB27A and RAB27B proteins in the paired MCF-7/WT and MCF-7/TamR cells; represen-tative Kaplan–Meier survival curves for the implications of ARL3, RHOF, RAB30, and RRAS2-mRNA expression in breast-cancer patients; breast-cancer-patient outcomes predicted by RAB31 expression; correlation of RAB31 expression with ER status and breast-cancer subtypes; increased tamoxifen resistance conferred by RAB31 knockdown; and elevated tamoxifen resistance and proliferation rates from RAB31 knockdown (PDF) Complete list of all small-GTPase proteins and peptides quantified in the scheduled LC-MRM analysis from two sets of forward and one set of reverse SILAC experiments (XLSX)
Accession Codes
The Skyline files for the MRM spectral library containing the MS/MS spectra and iRT information for all targeted small-GTPase peptides and 10 standard BSA peptides are available at PeptideAtlas (http://www.peptideatlas.org/) with the identifier PASS01267.
Notes
The authors declare no competing financial interest.
REFERENCES
- (1).Siegel RL; Miller KD; Jemal A Ca-Cancer J. Clin 2018, 68, 7–30. [DOI] [PubMed] [Google Scholar]
- (2).Dai X; Chen A; Bai Z Sci. Rep 2015, 4, 6566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Meijer D; van Agthoven T; Bosma PT; Nooter K; Dorssers LC Mol. Cancer Res 2006, 4, 379–386. [DOI] [PubMed] [Google Scholar]
- (4).Takai Y; Sasaki T; Matozaki T Physiol. Rev 2001, 81, 153–208. [DOI] [PubMed] [Google Scholar]
- (5).Luo ML; Gong C; Chen CH; Hu H; Huang P; Zheng M; Yao Y; Wei S; Wulf G; Lieberman J; Zhou XZ; Song E; Lu KP Cell Rep 2015, 11, 111–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Hendrix A; Maynard D; Pauwels P; Braems G; Denys H; Van den Broecke R; Lambert J; Van Belle S; Cocquyt V; Gespach C; Bracke M; Seabra MC; Gahl WA; De Wever O; Westbroek WJ Natl. Cancer Inst 2010, 102, 866–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Grismayer B; Solch S; Seubert B; Kirchner T; Schafer S; Baretton G; Schmitt M; Luther T; Kruger A; Kotzsch M; Magdolen V Mol. Cancer 2012, 11, 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Okada T; Sinha S; Esposito I; Schiavon G; Lopez-Lago MA; Su W; Pratilas CA; Abele C; Hernandez JM; Ohara M; Okada M; Viale A; Heguy A; Socci ND; Sapino A; Seshan VE; Long S; Inghirami G; Rosen N; Giancotti FG Nat. Cell Biol 2015, 17, 81–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Shen DW; Su A; Liang XJ; Pai-Panandiker A; Gottesman MM Br. J. Cancer 2004, 91, 270–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).McGlynn LM; Kirkegaard T; Edwards J; Tovey S; Cameron D; Twelves C; Bartlett JM; Cooke TG Clin. Cancer Res 2009, 15, 1487–1495. [DOI] [PubMed] [Google Scholar]
- (11).Gonzalez N; Cardama GA; Comin MJ; Segatori VI; Pifano M; Alonso DF; Gomez DE; Menna PL Cell. Signalling 2017, 30, 154–161. [DOI] [PubMed] [Google Scholar]
- (12).Chen HY; Yang YM; Stevens BM; Noble M EMBO Mol. Med 2013, 5, 723–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Zhou C; Zhong Q; Rhodes LV; Townley I; Bratton MR; Zhang Q; Martin EC; Elliott S; Collins-Burow BM; Burow ME; Wang G Breast Cancer Res 2012, 14, R45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Hengel SM; Murray E; Langdon S; Hayward L; O’Donoghue J; Panchaud A; Hupp T; Goodlett DR J. Proteome Res 2011, 10, 4567–4578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Umar A; Kang H; Timmermans AM; Look MP; Meijervan Gelder ME; den Bakker MA; Jaitly N; Martens JW; Luider TM; Foekens JA; Pasa-Tolic L Mol. Cell. Proteomics 2009, 8, 1278–1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Halvey PJ; Ferrone CR; Liebler DC J. Proteome Res 2012, 11, 3908–3913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Zhang CC; Li R; Jiang H; Lin S; Rogalski JC; Liu K; Kast JJ Proteome Res 2015, 14, 967–976. [DOI] [PubMed] [Google Scholar]
- (18).Huang M; Qi TF; Li L; Zhang G; Wang Y Cancer Res 2018, 78, 5431–5445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).MacLean B; Tomazela DM; Shulman N; Chambers M; Finney GL; Frewen B; Kern R; Tabb DL; Liebler DC; MacCoss MJ Bioinformatics 2010, 26, 966–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Kawahara R; Bollinger JG; Rivera C; Ribeiro AC; Brandao TB; Leme AFP; MacCoss MJ Proteomics 2016, 16, 159–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Rokavec M; Schroth W; Amaral SM; Fritz P; Antoniadou; Glavac D; Simon W; Schwab M; Eichelbaum M; Brauch H Cancer Res 2008, 68, 9799–9808. [DOI] [PubMed] [Google Scholar]
- (22).Mendes-Pereira AM; Sims D; Dexter T; Fenwick K; Assiotis I; Kozarewa I; Mitsopoulos C; Hakas J; Zvelebil M; Lord CJ; Ashworth A Proc. Natl. Acad. Sci. U. S. A 2012, 109, 2730–2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Larive RM; Moriggi G; Menacho-Marquez M; Canamero; de Alava E; Alarcon B; Dosil M; Bustelo XR Nat. Commun 2014, 5, 3881. [DOI] [PubMed] [Google Scholar]
- (24).Zhou X; Shi T; Li B; Zhang Y; Shen X; Li H; Hong G; Liu C; Guo Z PLoS One 2013, 8, No. e70017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Song J; Zheng B; Bu X; Fei Y; Shi S Oncol. Rep 2014, 31, 2776–2784. [DOI] [PubMed] [Google Scholar]
- (26).Yang RM; Zhan M; Xu SW; Long MM; Yang LH; Chen W; Huang S; Liu Q; Zhou J; Zhu J; Wang J Cell Death Dis 2017, 8, No. e3129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Miller LD; Smeds J; George J; Vega VB; Vergara L; Ploner A; Pawitan Y; Hall P; Klaar S; Liu ET; Bergh J Proc. Natl. Acad. Sci. U. S. A 2005, 102, 13550–13555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Ivshina AV; George J; Senko O; Mow B; Putti TC; Smeds J; Lindahl T; Pawitan Y; Hall P; Nordgren H; Wong JE; Liu ET; Bergh J; Kuznetsov VA; Miller LD Cancer Res 2006, 66, 10292–10301. [DOI] [PubMed] [Google Scholar]
- (29).Chang JC; Wooten EC; Tsimelzon A; Hilsenbeck SG; Gutierrez MC; Tham YL; Kalidas M; Elledge R; Mohsin S; Osborne CK; Chamness GC; Allred DC; Lewis MT; Wong H; O’Connell PJ Clin. Oncol 2005, 23, 1169–1177. [DOI] [PubMed] [Google Scholar]
- (30).Clarke C; Madden SF; Doolan P; Aherne ST; Joyce H; O’Driscoll L; Gallagher WM; Hennessy BT; Moriarty M; Crown J; Kennedy S; Clynes M Carcinogenesis 2013, 34, 2300–2308. [DOI] [PubMed] [Google Scholar]
- (31).Calcagno AM; Salcido CD; Gillet JP; Wu CP; Fostel JM; Mumau MD; Gottesman MM; Varticovski L; Ambudkar SV J. Natl. Cancer Inst 2010, 102, 1637–1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Dalton LW; Pinder SE; Elston CE; Ellis IO; Page DL; Dupont WD; Blamey RW Mod. Pathol 2000, 13, 730–735. [DOI] [PubMed] [Google Scholar]
- (33).Liu MC; Pitcher BN; Mardis ER; Davies SR; Friedman PN; Snider JE; Vickery TL; Reed JP; DeSchryver K; Singh B; Gradishar WJ; Perez EA; Martino S; Citron ML; Norton L; Winer EP; Hudis CA; Carey LA; Bernard PS; Nielsen TO; et al. NPJ Breast Cancer 2016, 2, No. 15023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Chua CE; Tang BL J. Cell Mol. Med 2015, 19, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Chua CE; Tang BL J. Biol. Chem 2014, 289, 12375–12389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Jin K; Kong X; Shah T; Penet MF; Wildes F; Sgroi DC; Ma XJ; Huang Y; Kallioniemi A; Landberg G; Bieche I; Wu X; Lobie PE; Davidson NE; Bhujwalla ZM; Zhu T; Sukumar S Proc. Natl. Acad. Sci. U. S. A 2012, 109, 2736–2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Liang YK; Zeng; Xiao YS; Wu Y; Ouyang YX; Chen M; Li YC; Lin HY; Wei XL; Zhang YQ; Kruyt FA; Zhang GJ Cancer Lett 2017, 386, 65–76. [DOI] [PubMed] [Google Scholar]
- (38).Shah N; Jin K; Cruz LA; Park S; Sadik H; Cho S; Goswami CP; Nakshatri H; Gupta R; Chang HY; Zhang Z; Cimino-Mathews A; Cope L; Umbricht C; Sukumar S Cancer Res 2013, 73, 5449–5458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Kim S; Lee J; Oh SJ; Nam SJ; Lee JE Oncol. Rep 2015, 34, 1613–1619. [DOI] [PubMed] [Google Scholar]
- (40).Abba MC; Hu Y; Sun H; Drake JA; Gaddis S; Baggerly K; Sahin A; Aldaz CM BMC Genomics 2005, 6, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Jin C; Rajabi H; Pitroda S; Li A; Kharbanda A; Weichselbaum R; Kufe D PLoS One 2012, 7, No. e39432. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
A complete list of the processed data is provided in Supplementary Table S1. The Skyline MRM library (including the transition list for quantification and the iRT file) for the human small-GTPase proteome and the raw files obtained from the LC-MRM analyses of small GTPases for the WT and TamR MCF-7 cells were deposited into PeptideAtlas with the identification number PASS01267 (http://www.peptideatlas.org/PASS/PASS01267).