

Abstract
Efforts to precisely correct genomic mutations that underlie hereditary diseases for therapeutic benefit have advanced alongside the emergence and improvement of genome engineering technologies. These methods can be divided into two main classes: active nucleasebased platforms including the popular CRISPR/Cas9 system and oligo/polynucleotide strategies including triplex-forming oligonucleotides (TFOs), such as peptide nucleic acids (PNAs). These technologies have been successful in cell culture and in animals, but important challenges remain before these tools can be translated into the clinic; they must be effectively delivered to and taken up by specific cell types of interest, achieve correction levels in target cells that significantly ameliorate the disease phenotype, and demonstrate minimal off-target and toxicity effects. Here we review and compare the current strategies and non-viral delivery methods, mainly lipid and polymeric vehicles, proposed for genome editing of inherited disorders with a focus on in vivo delivery and efficacy. While the path to a safe and effective medical treatment may be arduous, the future outlook of therapeutic genome editing remains promising as long as precise technologies can be combined with efficient delivery.
Keywords: therapeutic genome editing, non-viral delivery, nuclease-mediated genome editing, oligonucleotide-mediated genome editing, CRISPR/Cas9, peptide nucleic acid
Introduction
Numerous diseases prevalent in the world today are linked to precise (often single) mutations in the genome, and their target sequences have been elucidated in large part due to the sequencing of the human genome [1]. Technologies that enable targeted editing of the genome are of interest due to their potential to cure or ameliorate thousands of diseases that have a genetic basis by replacing or correcting defective genes resulting from inherited disorders, ideally resulting in the recovery of normal gene function. These new therapeutic approaches are especially important for diseases with limited treatment options, such as haemophilia [2]. Gene editing approaches also offer advantages compared to other methods of ameliorating genetic diseases, such as transient gene therapy and RNA interference (RNAi), in that the mutation of interest is corrected at the root cause (i.e. genomic DNA), eliminating the need for continuous correction therapies.
There are several different technologies designed to edit DNA that take advantage of activating host DNA repair machinery by inducing DNA damage. Two broad categories are nuclease-based and oligo/polynucleotide-based approaches (Figures 1,2). Several nucleases have been developed to precisely edit the genome in mammalian cells: meganucleases, zinc finger nucleases (ZFNs) [3], transcription activator like effector nucleases (TALENs), and Cas9, the nuclease associated with Clustered Regularly Interspaced Short Palindromic Repeats (CRISPRs) [4–8]. Currently, the most widely used system is the bacteria-derived CRISPR/Cas9 platform, which differs from the other nucleases in that a single guide RNA (sgRNA) strand binds directly to the target sequence, leading the Cas9 protein to this site. The other nucleases achieve DNA binding via protein-DNA interactions. All these nucleases introduce genetic modifications in a similar manner—first by introducing double-stranded DNA breaks (DSBs) at specific loci, which subsequently results in the recruitment of endogenous DNA repair machinery (Figure 2). Host cell-mediated repair likely involves two pathways: non-homologous end joining (NHEJ), and homology-directed repair (HDR) [9]. During NHEJ, cells can insert or delete DNA fragments at the breakage site(s). If a template DNA strand containing the desired mutation correction is codelivered with the nuclease, the corrected sequence can be incorporated into the genome via homologous recombination and the HDR pathway.
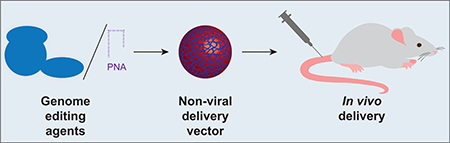
Figure 1.

Schematic summarizing non-viral strategies to deliver genome editing agents in vivo.
Figure 2. Schematic of gene editing machinery components that need to be encapsulated in delivery vehicles and their editing mechanisms.

The components required for nuclease-mediated genome editing are shown on the left (CRISPR/Cas9 system), and the components required for TFO-mediated genome editing are shown on the right (PNA system). Approximate sizes of each of the components are indicated. Shown below the editing system components are the mechanisms of site-specific gene correction for each type of editing agent.
Oligonucleotide and polynucleotide strategies use DNA oligomers or longer (>200bp) polymers to introduce modifications via homologous exchange in a sequence-specific region of host DNA [10]. This broad category includes several distinct strategies that involve introducing exogenous nucleic acids including short DNA fragments that are single- or double-stranded, single-stranded oligonucleotides, and triplex-forming oligonucleotides (TFOs). For example, small fragment homologous replacement (SFHR) is a therapy that falls under this category in which short double- or single-stranded DNA fragments (up to 1 kb) targeting a specific sequence are used to replace a homologous section of genomic DNA [11]. The sequence of the fragments is almost entirely identical to the endogenous one, except for the nucleotide base(s) needed to introduce the correction. Once inside a target cell, the short DNA fragments find their sequence homolog in the endogenous DNA. This technology is believed to take advantage of existing cellular DNA repair machinery, and in this case facilitate the exchange between the short fragments and their targets; although exact mechanisms remain unknown, it has been suggested that a combination of NHEJ and HDR may be involved [10]. Hybridization of the exogenous nucleotide is thought to occur when genomic DNA is transiently exposed during replication. Unmodified oligonucleotides are highly susceptible to degradation by nucleases, however, highlighting the need for effective delivery vehicles.
TFO technology is similar to the SFHR platform, in that it also takes advantage of endogenous DNA repair machinery, but different in that it involves two types of nucleic acids. Like, SFHR, the TFO platform requires a short DNA fragment (in this case single-stranded) containing the desired correction. To enhance gene editing, an additional oligonucleotide is delivered. The additional oligonucleotide has partial sequence homology with a polypurine- or polypyrimidine-rich region of genomic DNA adjacent to the correction site and is able to bind to the DNA and distort the helix to form an oligonucleotide-DNA triplex. Synthetic nucleic acids, particularly peptide nucleic acids (PNAs), are very effective as the co-delivered oligonucleotide due to their unique DNA-binding properties. PNAs consist of a charge-neutral peptide-like backbone and nucleobases enabling hybridization with DNA and RNA with high affinity. PNAs are generally 10–40 bases in length are designed to bind site-specifically to genomic DNA via strand invasion and form PNA/DNA/PNA triplexes by forming both Watson-Crick (WC) and Hoogsteen H-bonding with displacement of the non-bound DNA strand [12–14]. PNA/DNA/PNA triplexes recruit the cell’s endogenous DNA repair systems to initiate sitespecific modification of the genome when co-delivered with template DNA strands containing the desired sequence modification (Figure 2) [15]. PNAs have no intrinsic nuclease activity and stimulate endogenous repair when they bind tightly to their target site. Recent advances in PNA chemistry, such as the addition of miniPEG substitutions at the gamma position (γPNAs), have further improved the ability of these molecules to bind their target site [16, 17]. The evolution of PNAs as a therapeutic agent, particularly for genome editing, has recently been reviewed [18, 19].
There are several factors which will limit the potential of genome editing technologies as effective agents that can be administered directly in animals or humans: the ability to deliver the necessary correction templates and editing machinery into target cells in vivo, the efficiency with which the genome can be edited, and the likelihood of off-target editing. In this review, we focus on in vivo delivery platforms for these editing technologies and their recent success in this context primarily within the past two to five years. While it is sometimes possible to deliver the necessary components of the various genome editing technologies ex vivo to specific cell populations, which can then later be transplanted back into the body, this is not viable as a general strategy, as there are many cell types which are not amenable to manipulation outside of the body. Moreover, many common inherited disorders such as cystic fibrosis affect cells in organs throughout the body, which would all need to receive the relevant gene correction for complete reversal of disease. In the context of delivery, there are broad categories of in vivo vehicles that have been implemented, including viral and non-viral vectors. The main disadvantage of viral delivery is the potential to illicit an immune response and the lack of control over cargo release. In contrast, while non-viral delivery methods exhibit lower in vivo delivery efficiency, they can address the limitations of viral delivery such as toxicity and immunogenicity [20], carcinogenesis [21], and cargo encapsulation efficiency [22]. Further, they can be modified in various ways to target specific cell types and delivered in a multi-dose regimen [23]. Non-viral vectors are actively being pursued for gene therapy and genome editing, including lipid nanoparticles (NPs), liposomes, polymeric NPs, peptide conjugates, and cellderived membrane vesicles (CMVs) [23, 24]. Non-viral physical delivery methods such as microinjection and electroporation utilize physical energy to transfer editing agents to cells [25], but are more suitable for in vitro or ex vivo delivery and will not be discussed here. Ideal properties of non-viral delivery platforms include: the ability to deliver editing agents systemically with favorable biodistribution to target tissues and cell types, the effective encapsulation and release of cargo, and the promotion of therapeutic activity. In this review, we focus on non-viral delivery of nuclease- and oligonucleotide-based genome editing agents delivered locally and systemically in vivo (Figure 1). Recent advances are summarized in Table 1.
Table 1. Non-viral vectors used to deliver genome editing agents in vivo.
A summary of recent non-viral vectors used for local and systemic in vivo delivery of nuclease- and oligonucleotide-based editing agents with corresponding references.
| Local Delivery in vivo | |||||
|---|---|---|---|---|---|
| Nonviral vector | Target gene | Disease | Editing Agent | Form of editing agent | Reference |
| Turbofect(Polymer) | HPV Genome | Cervical cancer | TALEN | pDNA | [26] |
| DNA NCs (DNA/Polymer) | GFP | - | CRISPR/Cas9 | RNP | [27] |
| Polymer | Tumor suppressor genes | - | CRISPR/Cas9 | pDNA | [28] |
| Lipofectamine (Lipid) | EGFP | - | CRISPR/Cas9 | RNP | [29] |
| DNA-conjugated Gold/Polymer | DMD | DMD | CRISPR/Cas9 | RNP | [30] |
| Lipid | TMC1 | Hearing loss | CRISPR/Cas9 | RNP | [31] |
| Polymer | β-globin | - | PNA | PNA | [32] |
| Polymer | CFTR | Cystic fibrosis | PNA | PNA | [33] |
| Systemic Delivery in vivo | |||||
|---|---|---|---|---|---|
| Nonviral vector | Target gene | Disease | Editing Agent | Form of editing agent | Reference |
| Lipid | HBV DNA, PCSK9 | HBV infection, hypercholesterolemia | CRISPR/Cas9 | mRNA | [34] |
| Zwitterionic Amino Lipid | LoxP | - | CRISPR/Cas9 | mRNA | [35] |
| Lipid | TTR | transthyretin (TTR)- mediated amyloidosis | CRISPR/Cas9 | mRNA | [36] |
| Polymer | CCR5 | HIV | PNA | PNA | [37] |
| Polymer | β-globin | β-thalassemia | γPNA | γPNA | [17] |
Non-viral in vivo delivery of nuclease-based technologies
Nuclease-based editing technologies have shown great promise for gene editing in vitro and ex vivo, with genetic alterations reaching frequencies of over 50% in some cases [9]. However, exogenous nucleases have the potential to edit non-targeted regions of the genome [38]. In this respect, their activity in the body should ideally be temporary when delivered in vivo. Nuclease constituents can be delivered into cells in various formats. For example, with the CRISPR/Cas9 system, Cas9 can be delivered in the form of DNA, mRNA or protein along with the sgRNA and a DNA template with the desired sequence correction for site-specific modification (Figure 2). However, Cas9 DNA has the potential to integrate into host genomic DNA and result in aberrant nuclease expression. In this sense, the mRNA and protein forms of Cas9 are transient and therefore likely to produce fewer off-target effects and be less immunogenic. These forms of Cas9 also result in faster onset of editing, bypassing transcription. A drawback of Cas9 mRNA is poor stability, which affects both delivery strategies and editing efficiency. The delivery of active protein can also be challenging due to difficulties in developing delivery vehicles that do not inhibit enzymatic activity. Further, the Cas9 protein is large (~160 kDa) and positively charged, while sgRNA is negatively charged, making delivery of Cas9/sgRNA ribonucleoprotein (RNP) complexes difficult [39]. Moreover, once these foreign proteins enter their target cells, it also possible that sections of their peptide sequence will be presented on the cell surface to stimulate a cytotoxic T-cell-mediated immune response. The effective delivery of nuclease editing components in vivo into host cells remains a major challenge in part due to the difficulty of packaging multiple components into a single vector. A major advantage of non-viral cationic lipid or polymeric vehicles is that they readily load negatively charged nucleic acids (ex. mRNA, DNA) via electrostatic interactions, and subsequently release nucleic acid cargo upon cellular entry [40].
Local delivery in vivo
Local non-viral delivery in vivo of nuclease-based editing agents has been described using lipid and polymeric nanoparticles. Direct cervical delivery of TALEN plasmid targeting HPV-causing cervical cancer has been reported using the cationic polymer TurboFect system, after which tumor size was shown to decrease with no off-target mutations or signs of inflammation [26]. In another study, GFP disruption in a U20S-GFP tumor mouse model by Cas9/sgRNA was reported using intratumoral injection of DNA nanoclews (NCs). DNA NCs are nanoparticles that are partially complementary to sgRNA and coated with the cationic polymer polyethyleneimine (PEI). In this case, Cas9 RNP delivery resulted in 25% loss of GFP near the injection site [27]. PEI/Cas9 plasmid vehicles have also been used to target tumor suppressor genes in the mouse brain as a way to generate novel animal models. These were delivered by stereotactic injection into the cerebellum, but resulted in low viability and spatial accuracy [28]. Considering lipid-based delivery vehicles, Cas9 protein and sgRNA targeting EGFP encapsulated in cationic liposomes (Lipofectamine RNAiMAX) have been delivered to hair cells in the inner ear of a GFP reporter mouse model resulting in 13% loss of GFP near the injection site [29].
Non-viral vectors have recently been used to deliver CRISPR/Cas9 systems for the correction of monogenic disorders. For example, DNA-conjugated gold NPs complexed with cationic polymers to deliver donor DNA and Cas9 RNP have been used to treat Duchenne muscular dystrophy (DMD). After intramuscular injection alongside cardiotoxin, these NPs corrected the DMD-causing mutation at a frequency of 5.4% with reduced muscle fibrosis in a DMD mouse model [30]. Gao et al. used the Cas9 system for the treatment of autosomal dominant hearing loss using cationic lipids. The deafness associated allele in Tmc1 was targeted in a Beethoven mouse model by injection of lipid-RNP complexes into the cochlea of neonatal pups with a reported ~2% disruption of the dysfunctional Tmc1 allele [31].
Systemic delivery in vivo
Proof-of-concept studies on the non-viral systemic delivery of CRISPR/Cas9 components have improved the possibilities for this editing platform to treat genetic diseases. For example, lipid-like NPs have been used to deliver Cas9 mRNA and sgRNA to the liver to disrupt HBV DNA and pcsk9 for the treatment of hypercholesterolemia. These NPs were systemically delivered via tail vein injection in adult mice, resulting in decreased measurements of HBV viral loads and pcsk9 protein levels [34]. In another study, Cas9 mRNA and sgRNA designed for the LoxP gene were co-delivered intravenously using zwitterionic amino lipids, resulting in the expression of floxed tdTomato in liver, kidney, and lung and ~1.5–3.5% editing in hepatocytes [35]. Lipid NPs were also used to deliver Cas9 mRNA/sgRNA intravenously for the treatment of transthyretin (TTR)-mediated amyloidosis, enabling knockdown of the TTR gene in mice and rats with 70% reported editing across the liver [36]. Notably, only mRNA forms of Cas9 have been successfully delivered systemically in vivo via non-viral vectors.
Non-viral in vivo delivery of oligonucleotide-based technologies
A major advantage of the use of oligonucleotide-based genome editing is that the editing agents are considerably smaller than the agents used in nuclease-based technologies and more readily encapsulated into delivery vehicles. For example, PNA-mediated editing technology requires two components: a 10–40 nucleotide PNA molecule and a ~60 nucleotide ssDNA template (Figure 2). PNA/DNA editing agents also have the benefit of very low off-target effects due to the sequence specificity of PNA binding to genomic DNA. Further, the polyamide backbone of PNAs enhances stability and protects these molecules from degradation by nucleases and proteases. While there are no reports of in vivo editing by DNA oligonucleotides alone, polymeric delivery vehicles have recently been used to deliver PNA-based editing agents both locally and systemically.
Local delivery in vivo
Triplex-forming PNA and donor DNA encapsulated in NPs formulated with poly(lacticco-glycolic) (PLGA) acid, which is FDA-approved for various drug delivery applications, or a blend of PLGA and the cationic polymer poly(beta amino ester) (PBAE) have been used for in vivo delivery. Both NP types were used for intranasal administration of PNA and donor DNA to modify the human β−globin gene in an EGFP reporter mouse model [32]. Focusing on editing in the lung, Fields et al. demonstrated that PLGA/PBAE NPs resulted in increased editing (~0.2%), as measured by the percentage of EGFP positive cells, compared to PLGA NPs (~0.05%). The PLGA/PBAE NPs were further surface modified with the nuclear localization sequencecontaining cell-penetrating peptide MPG, which increased editing to ~0.4% of total lung cells. PLGA, PLGA/PBAE, and PLGA/PBAE/MPG NPs were also used to encapsulate PNA and donor DNA for the treatment of cystic fibrosis caused by the F508del mutation via intranasal delivery. Nasal epithelial cells in mice treated with PLGA/PBAE/MPG NPs exhibited up to ~5.7% correction of the dysfunctional allele, while approximately 1.2% of lung cells were edited [33].
Systemic delivery in vivo
Polymeric PLGA NPs have been used to deliver triplex-forming PNAs and donor DNA molecules to correct the human CCR5 gene in hematolymphoid cells in mice after systemic injection. This treatment resulted in 0.43% editing in hematopoietic cells in the spleen with minimal off-target effects [37]. In a more recent study, triplex-forming γPNAs and donor DNA oligonucleotides encapsulated in PLGA NPs were administered systemically via intravenous injection alongside hematopoietic stem cell factor (SCF) to treat a mouse model of β-thalassemia. Approximately 4% editing was achieved after four injections of NPs, which resulted in long-term phenotypic correction of the disease as measured by red blood cell morphology, hemoglobin levels, reticulocyte counts, and spleen architecture. Importantly, the editing frequency in this study is comparable to editing frequencies achieved thus far by CRISPR/Cas9 systems delivered systemically in vivo.
Challenges with in vivo delivery
While systemic delivery of genome editing agents is an ultimate goal for the treatment of many inherited disorders, there are many challenges and barriers that must be overcome for both the editing agents and the delivery vehicles encapsulating them. Among the most important challenges facing non-viral delivery are: designing delivery vehicles that effectively encapsulate the multiple components of editing platforms, stability of the delivery vehicle and its cargo in vivo, mitigating toxicity and immunogenicity, avoiding elimination and clearance, accumulation into therapeutically relevant tissues and organs, and achieving sufficient release of editing machinery in target organs and tissues to result in therapeutically relevant levels of editing.
Packaging of editing components into a single vector is a major challenge, particularly with nuclease-based systems where the nuclease DNA, mRNA, or protein can be very large. Moreover, depending on the form of the nuclease, the kinetics of DNA transcription, mRNA translation, and protein complex assembly must be optimized. Encapsulated components will also affect the characteristics of delivery vectors, such as size and surface properties, which will in turn affect their biodistribution and cargo release in vivo. Oligonucleotide-based approaches such as the triplex-forming PNA/DNA system have an advantage in this respect—PNA and DNA are much smaller in size and more readily encapsulated into non-viral vectors. Once administered, delivery vehicles and components of editing platforms need to remain stable before reaching their target organs and tissues. More fragile cargo such as mRNAs and proteins are at a higher risk of degradation, which may complicate vector design and formulation. Beyond packaging and stability, both the delivery vehicle and its cargo must be assessed for toxicity and immunogenicity regardless of the delivery method or type of editing technology as all delivery materials and editing agents are exogenous and foreign to the body. This is particularly important if a multi-dose treatment regimen is to be used. While lipid and polymeric NPs are considered less immunogenic than viral vectors, toxicity of the non-viral vector may be of concern. For example, cationic lipids and polymers are able to encapsulate substantial amounts of nucleic acid, but can also exhibit cytotoxicity [41].
The ability of non-viral vectors to evade clearance and accumulate in target tissues will depend heavily on the size and surface properties of the carriers. Generally, smaller vehicles, as long as they are large enough to avoid filtration by glomeruli in the kidney, will have a longer circulation time in the body and may have enhanced abilities to accumulate in hard-to-reach organs in vivo [42], for example reaching brain tissue by crossing the blood brain barrier (BBB). Biodistribution will be further dictated by the protein corona that forms around delivery vehicles upon entry into the bloodstream, which will depend on the biomaterials composing the non-viral vector as well as its cargo [43]. Adjusting these parameters to promote optimal biodistribution while maintaining high encapsulation efficiencies of editing agents will be especially challenging.
Once non-viral vectors encapsulating gene editing agents reach their target cells, they face the additional challenge of facilitating transport of all the required editing system components to the nucleus where editing takes place. The endocytic pathway is a major uptake mechanism for cells in which delivered agents become entrapped in endosomes and may encounter degradation in the lysosome. Here, delivery vehicles must facilitate endosomal escape [44]. While the proton sponge effect and membrane fusion are likely methods of endosomal escape for cationic polymers and lipid particles, respectively, a better understanding of the transport of nonviral carriers in the cell will be key to engineering new materials to overcome this barrier.
Conclusions and Future Outlook
The therapeutic potential of gene editing technologies have attracted tremendous interest and investment, and the development of effective delivery vehicles to aid in transporting these technologies to target sites in vivo is an important area of research. Indeed, the widespread use of editing technology is currently limited by transfection methods. Genome editing therapeutics, often composed of multiple components, present additional complexity for delivery platforms [45]. Moving forward, two main research areas need to be explored before gene editing technologies can be translated from research laboratories into clinical practice: optimizing the technologies themselves as well as developing safe and controlled delivery vehicles for various disease targets. At the same time, regulatory concerns regarding safety, efficacy, and quality control of these components must be addressed.
The detailed mechanisms by which the various technologies are able to edit the genome need to be elucidated and optimized to further improve upon editing efficiencies and the specificity of editing. Further, off-target activity of both nuclease and oligonucleotide-based techniques needs to be studied more thoroughly, requiring the development of new methods to deeply screen the genome for unwanted modifications. Genotoxicity assessments will need to involve a risk-benefit analysis of desirable corrections and other mutations. Gene editing technologies might be improved by the selection of more specific target sites. Further, in the case of Cas9-mediated editing, rational sgRNA design and the use of Cas9 nickases would likely alleviate unintentional modification. Additionally, “base editor” nuclease-inactivated Cas9s associated with deaminases could be used to correct point mutations and reduce the formation of indels near the target site [46]. PNA sequences currently designed using an iterative approach could also be improved with rational design strategies upon elucidation of their mechanism. Further chemical modifications to PNAs, for example additional backbone modifications and incorporation of nonstandard nucleobases, might also improve DNA-binding affinity and stimulation of host DNA repair [18].
Effective delivery vehicles remain a bottleneck for the successful clinical translation of editing technologies. A major effort is needed on the development of systemic delivery vehicles that are optimized in terms of molecular composition and route of administration with favorable biodistribution to target tissues and organs. Consideration should also be given to coordinating treatments with therapeutic windows in pathophysiological timelines. Non-viral vectors are promising for in vivo delivery and have already been successful in delivering both nucleasebased and oligonucleotide-based editing technologies and other gene therapies (Table 1). New lipids and polymers have been developed with the goal of improving nucleic acid delivery in vitro and in vivo, and these could prove useful for delivering editing agents. For example, poly(amine-co-ester) terpolymers have been used to successfully deliver pDNA in vivo [47, 48], and have recently been shown to effectively deliver TFO PNAs ex vivo (A.S. Piotrowski-Daspit et al., abstract 614, 21st Annual Meeting of American Society of Gene & Cell Therapy, Chicago IL, May 2018). Additionally, the incorporation of targeting moieties such as antibodies for specific cell types could be useful particularly for local delivery.
Overall, there is great therapeutic potential in genome engineering technologies. With improved design and delivery, new treatment options will someday be available for myriad of genetic diseases that are currently untreatable. Looking forward, expertise in editing technologies and biomaterials for the formation of delivery vehicles will need to be combined alongside regulatory considerations to achieve clinical translation from bench to bedside.
Highlights.
Both nuclease- and oligonucleotide-based genome editing agents stimulate endogenous DNA repair mechanisms.
Nuclease- and oligonucleotide-based editing agents differ in size and number of components, likely affecting encapsulation into non-viral delivery vectors.
Non-viral delivery vectors can be used to effectively deliver nuclease- and oligonucleotide-based genome editing technologies locally and systemically in vivo.
Challenges with in vivo non-viral delivery include: cargo encapsulation, toxicity/immunogenicity, rapid clearance, and accumulation of editing agents into target tissues.
Optimization of gene editing technologies and delivery vehicles for various disease targets are important future considerations alongside regulatory issues.
Acknowledgements
We thank members of the Saltzman and Glazer Laboratories for their insightful comments. This work was supported in part by grants from the US National Institutes of Health (NIH; AI112443 and HL125892). A.S.P. was supported in part by NIH National Research Service Awards (NRSAs) T32 (GM86287) and F32 (HL142144) Postdoctoral Fellowships.
Abbreviations:
- BBB
blood brain barrier
- CMV
cell-derived membrane vesicle
- CRISPR
Clustered Regularly Interspaced Short Palindromic Repeat
- DMD
Duchenne muscular dystrophy
- DSB
double-stranded DNA breaks
- HDR
homology-directed repair
- NHEJ
nonhomologous end joining
- NC
nanoclew
- NP
nanoparticle
- PACE
poly(amine-co-ester)
- PBAE
poly(beta amino ester)
- PEI
polyethyleneimine
- PLGA
poly(lactic-co-glycolic) acid
- PNA
peptide nucleic acid
- RNAi
RNA interference
- RNP
ribonucleoprotein
- SCF
stem cell factor
- SFHR
small fragment homologous replacement
- sgRNA
single guide RNA
- TALEN
transcription activator like effector nuclease
- TFO
triplex-forming oligonucleotide
- TTR
transthyretin
- WC
Watson-Crick
- ZFN
zinc finger nucleases
Footnotes
Conflicts of Interest
W.M.S and P.M.G are inventors on patents assigned to Yale University pertaining to PNA and NP-mediated gene editing. They have equity in and receive consulting fees from Trucode Gene Repair, Inc.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
Note: Annotated references are marked as *of special interest or **of outstanding interest.
- 1.Lander ES. Initial impact of the sequencing of the human genome. Nature 2011, 470(7333):187–97. [DOI] [PubMed] [Google Scholar]
- 2.Callaghan MU, Sidonio R, Pipe SW. Novel therapeutics for hemophilia and other bleeding disorders. Blood 2018. [DOI] [PubMed] [Google Scholar]
- 3.Miller JC, Holmes MC, Wang J, Guschin DY, Lee YL, Rupniewski I, et al. An improved zinc-finger nuclease architecture for highly specific genome editing. Nat Biotechnol 2007, 25(7):778–85. [DOI] [PubMed] [Google Scholar]
- 4.Gasiunas G, Barrangou R, Horvath P, Siksnys V. Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc Natl Acad Sci U S A 2012, 109(39):E2579–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337(6096):816–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, et al. Multiplex Genome Engineering Using CRISPR/Cas Systems. Science 2013, 339(6121):819–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, et al. RNA-Guided Human Genome Engineering via Cas9. Science 2013, 339(6121):823–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cho SW, Kim S, Kim JM, Kim J-S. Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nat Biotech 2013, 31(3):230–2. [DOI] [PubMed] [Google Scholar]
- 9.Sander JD, Joung JK. CRISPR-Cas systems for editing, regulating and targeting genomes. Nat Biotech 2014, 32(4):347–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sargent RG, Kim S, Gruenert DC. Oligo/Polynucleotide-Based Gene Modification: Strategies and Therapeutic Potential. Oligonucleotides 2011, 21(2):55–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Luchetti A, Malgieri A, Sangiuolo F. Small Fragment Homologous Replacement (SFHR): sequence-specific modification of genomic DNA in eukaryotic cells by small DNA fragments. Methods Mol Biol 2014, 1114:85–101. [DOI] [PubMed] [Google Scholar]
- 12.Egholm M, Buchardt O, Christensen L, Behrens C, Freier SM, Driver DA, et al. PNA hybridizes to complementary oligonucleotides obeying the Watson-Crick hydrogenbonding rules. Nature 1993, 365(6446):566–8. [DOI] [PubMed] [Google Scholar]
- 13.Nielsen PE, Egholm M, Berg RH, Buchardt O. Sequence-selective recognition of DNA by strand displacement with a thymine-substituted polyamide. Science 1991, 254(5037):1497–500. [DOI] [PubMed] [Google Scholar]
- 14.Faruqi AF, Egholm M, Glazer PM. Peptide nucleic acid-targeted mutagenesis of a chromosomal gene in mouse cells. Proc Natl Acad Sci U S A 1998, 95(4):1398–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rogers FA, Vasquez KM, Egholm M, Glazer PM. Site-directed recombination via bifunctional PNA-DNA conjugates. Proc Natl Acad Sci U S A 2002, 99(26):16695–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bahal R, Quijano E, McNeer NA, Liu Y, Bhunia DC, López-Giráldez F, et al. Single-Stranded γPNAs for In Vivo Site-Specific Genome Editing via Watson-Crick Recognition. Curr Gene Ther 2014, 14(5):331–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. **.Bahal R, Ali McNeer N, Quijano E, Liu Y, Sulkowski P, Turchick A, et al. In vivo correction of anaemia in β-thalassemic mice by γPNA-mediated gene editing with nanoparticle delivery. Nat Commun 2016, 7:13304 Thus study demonstrated the use of γPNAs for nuclease-free editing upon systemic delivery in vivo in a β-thalassemia mouse model. γPNA-mediated editing achieved significant amelioration of disease symptoms with no off-target effects. Polymeric NPs encapsulating γPNA and donor DNA cargo were delivered via IV injection. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Quijano E, Bahal R, Ricciardi A, Saltzman WM, Glazer PM. Therapeutic Peptide Nucleic Acids: Principles, Limitations, and Opportunities. Yale J Biol Med 2017, 90(4):583–98. [PMC free article] [PubMed] [Google Scholar]
- 19.Ricciardi AS, Quijano E, Putman R, Saltzman WM, Glazer PM. Peptide Nucleic Acids as a Tool for Site-Specific Gene Editing. Molecules 2018, 23(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bessis N, GarciaCozar FJ, Boissier MC. Immune responses to gene therapy vectors: influence on vector function and effector mechanisms. Gene Ther 2004, 11 Suppl 1:S10–7. [DOI] [PubMed] [Google Scholar]
- 21.Baum C, Kustikova O, Modlich U, Li Z, Fehse B. Mutagenesis and oncogenesis by chromosomal insertion of gene transfer vectors. Hum Gene Ther 2006, 17(3):253–63. [DOI] [PubMed] [Google Scholar]
- 22.Thomas CE, Ehrhardt A, Kay MA. Progress and problems with the use of viral vectors for gene therapy. Nat Rev Genet 2003, 4(5):346–58. [DOI] [PubMed] [Google Scholar]
- 23.Yin H, Kanasty RL, Eltoukhy AA, Vegas AJ, Dorkin JR, Anderson DG. Non-viral vectors for gene-based therapy. Nat Rev Genet 2014, 15(8):541–55. [DOI] [PubMed] [Google Scholar]
- 24.van Dommelen SM, Vader P, Lakhal S, Kooijmans SA, van Solinge WW, Wood MJ, et al. Microvesicles and exosomes: opportunities for cell-derived membrane vesicles in drug delivery. J Control Release 2012, 161(2):635–44. [DOI] [PubMed] [Google Scholar]
- 25.Wells DJ. Gene therapy progress and prospects: electroporation and other physical methods. Gene Ther 2004, 11(18):1363–9. [DOI] [PubMed] [Google Scholar]
- 26.Hu Z, Ding W, Zhu D, Yu L, Jiang X, Wang X, et al. TALEN-mediated targeting of HPV oncogenes ameliorates HPV-related cervical malignancy. J Clin Invest 2015, 125(1):425–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sun W, Ji W, Hall JM, Hu Q, Wang C, Beisel CL, et al. Self-assembled DNA nanoclews for the efficient delivery of CRISPR-Cas9 for genome editing. Angew Chem Int Ed Engl 2015, 54(41):12029–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zuckermann M, Hovestadt V, Knobbe-Thomsen CB, Zapatka M, Northcott PA, Schramm K, et al. Somatic CRISPR/Cas9-mediated tumour suppressor disruption enables versatile brain tumour modelling. Nat Commun 2015, 6:7391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zuris JA, Thompson DB, Shu Y, Guilinger JP, Bessen JL, Hu JH, et al. Cationic lipidmediated delivery of proteins enables efficient protein-based genome editing in vitro and in vivo. Nat Biotechnol 2015, 33(1):73–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. *.Lee K, Conboy M, Park HM, Jiang F, Kim HJ, Dewitt MA, et al. Nanoparticle delivery of Cas9 ribonucleoprotein and donor DNA in vivo induces homology-directed DNA repair. Nat Biomed Eng 2017, 1(11):889–901. Gold NPs conjugated to DNA and complexed with PEI are used to deliver Cas9 RNPs to correct DMD via local injection. Editing of disease-causing point mutations is achieved by local intramuscular delivery of these NPs in vivo, resulting in reduced muscle fibrosis. This is one of the first studies using non-viral gold/polymer delivery vehicles to locally deliver Cas9 components in vivo to correct a genetic disease. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. *.Gao X, Tao Y, Lamas V, Huang M, Yeh W-H, Pan B, et al. Treatment of autosomal dominant hearing loss by in vivo delivery of genome editing agents. Nature 2017, 553:217 The deafness-associated Tmc1 gene is targeted by Cas9 RNPs encapsulated in lipid NPs via local delivery to the cochlea of Beethoven mice. Following treatment, enhanced acoustic startle responses and 1.8% Tmc1Bth allele disruption were observed. This is the first study to use lipid NPs for local Cas9 delivery to correct a disease-causing mutation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fields RJ, Quijano E, McNeer NA, Caputo C, Bahal R, Anandalingam K, et al. Modified Poly(lactic-co-glycolic acid) Nanoparticles for Enhanced Cellular Uptake and Gene Editing in the Lung. Adv Healthc Mater 2015, 4(3):361–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McNeer NA, Anandalingam K, Fields RJ, Caputo C, Kopic S, Gupta A, et al. Nanoparticles that deliver triplex-forming peptide nucleic acid molecules correct F508del CFTR in airway epithelium. Nat Commun 2015, 6:6952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. *.Jiang C, Mei M, Li B, Zhu X, Zu W, Tian Y, et al. A non-viral CRISPR/Cas9 delivery system for therapeutically targeting HBV DNA and pcsk9 in vivo. Cell Res 2017, 27(3):440–3. Lipid-like NPs are used to deliver Cas9 mRNA and sgRNA to treat HBV infection and hypercholesterolemia systemically via IV injection in adult animals. Target genes were HBV DNA and the pcsk9 gene, which is a drug target for hypercholesterolemia. These NPs exhibited delivery specific to the liver and decreased HBV viral loads and pcsk9 protein levels. This was among the first studies to non-virally deliver Cas9 editing components systemically in vivo. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Miller JB, Zhang S, Kos P, Xiong H, Zhou K, Perelman SS, et al. Non-viralCRISPR/Cas gene editing in vitro and in vivo enabled by synthetic nanoparticle co-delivery of Cas9 mRNA and sgRNA. Angew Chem Int Ed Engl 2017, 56(4):1059–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. *.Finn JD, Smith AR, Patel MC, Shaw L, Youniss MR, van Heteren J, et al. A Single Administration of CRISPR/Cas9 Lipid Nanoparticles Achieves Robust and Persistent In Vivo Genome Editing. Cell Rep 2018, 22(9):2227–35. Cas9 mRNA and sgRNA encapsulated in lipid NPs are systemically delivered to the liver to treat TTR-mediated amyloidosis. A single treatment resulted in >97% knockdown of serum TTR levels. Additionally, editing levels were shown to be stable over one year. This study demonstrates the use of lipid NPs for Cas9 delivery in vivo, resulting in clinically relevant editing levels for a genetic disorder. [DOI] [PubMed] [Google Scholar]
- 37.McNeer NA, Schleifman EB, Cuthbert A, Brehm M, Jackson A, Cheng C, et al. Systemic delivery of triplex-forming PNA and donor DNA by nanoparticles mediates sitespecific genome editing of human hematopoietic cells in vivo. Gene Ther 2013, 20(6):658–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cameron P, Fuller CK, Donohoue PD, Jones BN, Thompson MS, Carter MM, et al. Mapping the genomic landscape of CRISPR-Cas9 cleavage. Nat Methods 2017, 14(6):600–6. [DOI] [PubMed] [Google Scholar]
- 39.Zuris JA, Thompson DB, Shu Y, Guilinger JP, Bessen JL, Hu JH, et al. Efficient Delivery of Genome-Editing Proteins In Vitro and In Vivo. Nat Biotechnol 2015, 33(1):7380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shim G, Kim D, Park GT, Jin H, Suh S-K, Oh Y-K. Therapeutic gene editing: delivery and regulatory perspectives. Acta Pharmacol Sin 2017, 38:738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li L, He ZY, Wei XW, Gao GP, Wei YQ. Challenges in CRISPR/CAS9 Delivery: Potential Roles of Nonviral Vectors. Hum Gene Ther 2015, 26(7):452–62. [DOI] [PubMed] [Google Scholar]
- 42.Hoshyar N, Gray S, Han H, Bao G. The effect of nanoparticle size on in vivo pharmacokinetics and cellular interaction. Nanomedicine (Lond) 2016, 11(6):673–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bertrand N, Grenier P, Mahmoudi M, Lima EM, Appel EA, Dormont F, et al. Mechanistic understanding of in vivo protein corona formation on polymeric nanoparticles and impact on pharmacokinetics. Nat Commun 2017, 8(1):777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Selby LI, Cortez-Jugo CM, Such GK, Johnston APR. Nanoescapology: progress toward understanding the endosomal escape of polymeric nanoparticles. Wiley Interdiscip Rev Nanomed Nanobiotechnol 2017, 9(5). [DOI] [PubMed] [Google Scholar]
- 45.Maggio I, Goncalves MA. Genome editing at the crossroads of delivery, specificity, and fidelity. Trends Biotechnol 2015, 33(5):280–91. [DOI] [PubMed] [Google Scholar]
- 46.Komor AC, Kim YB, Packer MS, Zuris JA, Liu DR. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 2016, 533:420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhou J, Liu J, Cheng CJ, Patel TR, Weller CE, Piepmeier JM, et al. Biodegradable poly(amine-co-ester) terpolymers for targeted gene delivery. Nat Mater 2011, 11:82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang J, Cui J, Deng Y, Jiang Z, Saltzman WM. Multifunctional Poly(amine-co-esterco-orthoester) for Efficient and Safe Gene Delivery. ACS Biomater Sci Eng 2016, 2(11):2080–9. [DOI] [PMC free article] [PubMed] [Google Scholar]