

Abstract
Juvenile papillomatosis of the breast, also known as Swiss cheese disease, is a rare and benign proliferative disorder affecting young women. These patients tend to have a strong family history of cancer. The lesion typically presents as a localized mass without sharp borders. Clinical presentation resembles that of a precancerous lesion. For this reason, JP is often misdiagnosed in the preoperative period. However postoperative histopathological examination reveals distinct microscopic features, such as duct papillomatosis, cysts and sclerosing adenosis, which confirm the diagnosis of juvenile papillomatosis. We report two cases of juvenile papillomatosis. Both cases were preoperatively diagnosed as benign proliferative lesions with fibrocystic changes. However, after surgical excision, histopathological examination showed juvenile papillomatosis. Interestingly, both patients had a strong family history of breast cancer in both the paternal and maternal line. More research is needed to assess the correlation between a family history of breast cancer and the juvenile papillomatosis.
Keywords: Breast, juvenile, papillomatosis
Introduction
Juvenile papillomatosis (JP) of the breast, also termed Swiss cheese disease, is a rare and benign disorder which predominantly occurs in females under the age of 30 year (1). In the preoperative period, JP is often misdiagnosed as fibroadenoma because of its clinical presentation. A family history of breast cancer has been identified in 33–58% of JP patients (2, 3). In addition, an increased risk of development of breast carcinoma during follow-up of patient diagnosed with JP has been observed. This report presents two cases of JP with a positive family history of breast cancer in both patients.
Case Presentations
Case 1
A 22-year-old woman was referred to our hospital with a self-palpated mass in the right breast. The patient showed a family history of breast cancer (Figure 1). The BRCA2 mutation was found on the parental side. The patient herself did not carry the mutated gene. A high frequency of cancer was also observed on the maternal side.
Figure 1.

Pedigree of the first case
By inspection, no abnormalities were identified. Physical examination found a non-tender, mobile, firm lump (30–40mm) in the lower, inner quadrant of the right breast. In the following months, changes in volume and consistency were observed with physical examination. Ultrasonography revealed a poorly-defined, heterogeneous, hypo-reflective zone (max. 30,9mm) with multiple cysts (Figure 2a). Pathological examination with core-needle-biopsy (CNB) (4×14G) showed ductal hyperplasia and diffuse hyperplastic tissue. The center of the lesion showed zones of elastoid connective tissue without glandular tissue, while at the edge of the lesion more glands appeared. There was no evidence for carcinoma. These findings led to the diagnosis of “a complex sclerosing lesion” post biopsy. MRI showed zones of fibrocystic changes (30×25×45mm) (Figure 3a, c). Consistent with the diagnosis of and pathognomonic for a complex sclerosing lesion, a distortion was visible in the center of the lesion. Additional mammography showed a cluster of microcalcifications (45×32mm) localized inferomedially in the right breast (Figure 4a, b). Due to the young age of the patient, only a mediolateral oblique view is obtained and no tomosynthesis is added.
Figure 2. a, b.

Ultrasonography. First case: a poorly-defined, heterogeneous, hypo-reflective zone (3,09cm) with multiple cysts (a). Second case: a hyporeflective mass (4.9 cm) with cystic changes (b)
Figure 3. a–d.

MRI. T2 weighted image first case: zones of fibrocystic changes, best seen on T2 weighted image (a). T2 weighted image second case (b), substraction image first case (c), Substraction image second case (d)
Figure 4. a, b.
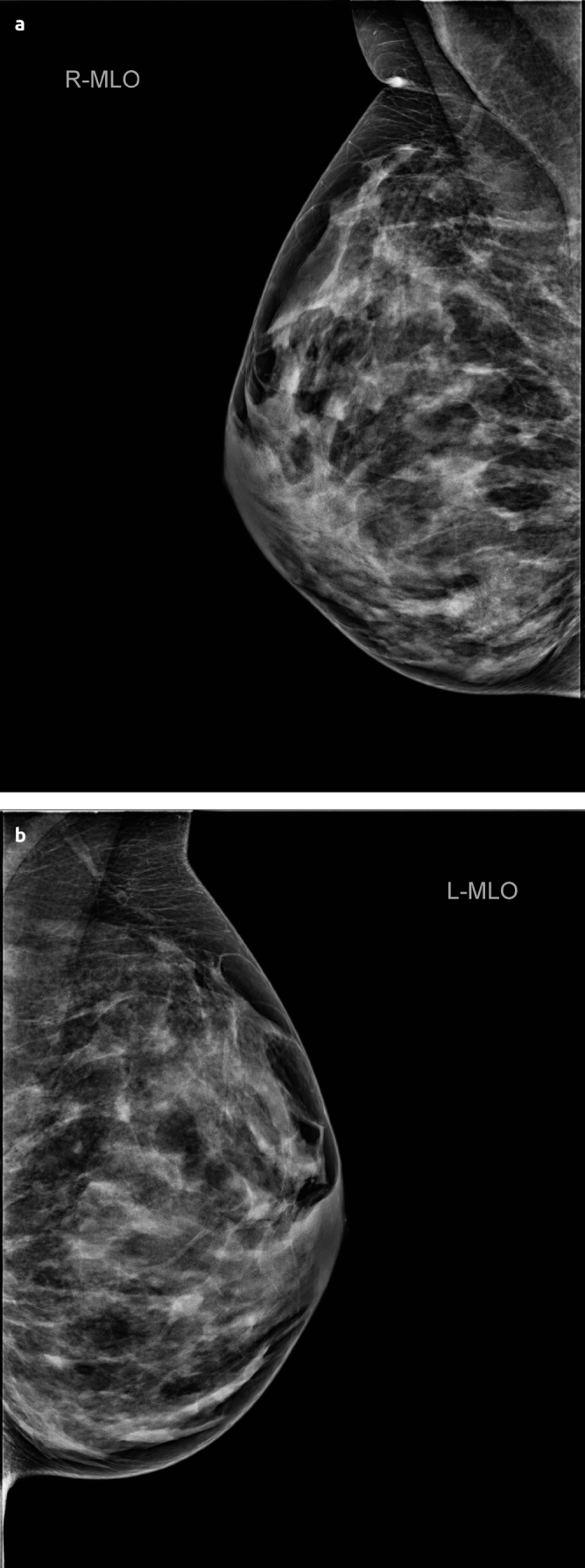
Mammography of first case. Right breast. A cluster of microcalcifications (size 45×32mm) inferomedial. No distortion visible (a). Left breast. Diffuse microcalcifications type Le Gal 2. No indication of abnormality (b)
The patient underwent breast-conserving-surgery under general anesthesia with complete excision of the lesion. Post-operative examination showed dilated ducts with epithelial hyperplasia, intraductal papillomas and apocrine and fibroadenomatoid changes. Consistent with the earlier CNB, sclerotic tissue in the core was observed. Based on these elements, the diagnosis of JP was made. Follow-up with ultrasonography every 6 months is recommended. Informed consent was obtained from the patient who participated in this case study.
Case 2
A 23-year-old woman was referred to our hospital. The patient presented with a mass in the left breast, which was first detected several years before at a solid diameter of 2cm. After her last pregnancy, the lesion had grown to a diameter of 5cm. The patient showed a family history of breast cancer. On the maternal side, several relatives had suffered from breast cancer. Age and number of these women is unknown. The grandmother on the paternal side had suffered from lung cancer. No information about genetic mutation is known. Physical examination showed a mobile nodule at the middle-upper-quadrant of the left breast. Ultrasonography showed a hypo-reflective mass (49mm) with cystic changes (Figure 3b, d). CNB showed adenosis with dilated ducts. There was no evidence for carcinoma. MRI revealed a highly contrast-enhancing mass (42×38×31mm) containing cysts. This was compatible with a zone of fibrocystic changes (Figure 4b, Figure 4d). The patient underwent surgical excision of the lesion. Post-operative examination revealed JP. We could not get informed consent because there was no communication with the patient.
Discussion and Conclusion
Rosen et al. (1) first described JP in 1980 after 32 cases with a multi-cystic nature of masses. The age at diagnosis of JP ranges from 10–48 with an average of 23 years old. Clinically, JP presents as a firm mass with dilated ducts and cysts on macroscopic examination. The size of the lesion ranges from 1–8cm (4). Histopathologic criteria for JP are as follows: duct papillomatosis, apocrine and non-apocrine cysts, papillary apocrine hyperplasia, sclerosing adenosis, and duct stasis.
The diagnosis of JP is often difficult, because of its clinical and anatomopathological resemblance to other, more common, benign lesions in this age group. Certain clinical features of fibroadenoma, such as young age and the presence of a palpable mass, are also seen in JP. Anatomopathologically, JP shows features of a complex sclerosing lesion for instance hyperplasia, adenosis and papillomatosis (5). Our first case emphasizes the difficulty of JP diagnosis. In this case a complex sclerosing lesion was diagnosed in the pre-operative period. Usually complex sclerosing lesions are found by routine mammography in asymptomatic middle-aged women (6). This was inconsistent with the findings in our young patient. The characteristics of JP as described earlier such as presence of a large, palpable mass and the patients’ age below 30 were in contrast to radial scar - consistent with our case. Postoperative examination revealed the lesion was in fact JP.
The presence of a family history of cancer in both cases described above is remarkable. Despite the high incidence of cancer, no mutations were found in either of the patients. In our first case, the BRCA2 mutation was found in the family, but had not been identified in the patient. In the maternal side of the first patient’s family, as well as in the second patients’ family, presence of cancer was abundant, but no abnormal germline mutation was found. This phenomenon raises several questions. The first matter we need to consider is the fact that JP could be hereditary. A family history of breast cancer is seen in a substantial amount of JP patients. Bazzocchi et al. (3) found a positive family relationship of 33% in these series. Rosen et al. (2) found an even greater association (58%). No studies have been published analyzing the correlation between the hereditary breast and ovarian cancer. The second matter we need to take into account is the absence of identified germline mutations in both cases. Considering these findings, JP is presumably due to a mutation in a not-yet-identified gene. More research is required to assess the genetic cause of JP and next generation sequencing might be the next step.
Two important factors that increase the risk of breast cancer in women with JP are the presence of proliferative breast disease and a positive family history. As patients with JP often have both of these risk factors, they are considered as high-risk-patients (2). Recommended treatment of JP is complete excision of the lesion followed by pathological confirmation. If no additional abnormalities are found, no other treatment is necessary. Incomplete excision invariably leads to recurrence (7). Intensive follow-up with ultrasonography for these high-risk cases is recommended.
Footnotes
Informed Consent: Written informed consent was obtained from patients who participated in this study.
Peer-review: Externally peer-reviewed.
Author Contributions: Concept - V.V.; Design - V.V.; Supervision - G.H., P.N.; Resources - M.K., P.N.; Materials - M.K., P.N.; Data Collection and/or Processing - V.V., T.L.; Analysis and/or Interpretation V.V.; Literature Search - V.V.; Writing Manuscript - V.V.; Critical Review - G.H., T.L.
Conflict of Interest: The authors have no conflicts of interest to declare.
Financial Disclosure: The authors declared that this study has received no financial support.
References
- 1.Rosen PP, Cantrell B, Mullen DL, DePalo A. Juvenile papillomatosis (Swiss cheese disease) of the breast. Am J Surg Pathol. 1980;4:3–12. doi: 10.1097/00000478-198004010-00001. [DOI] [PubMed] [Google Scholar]
- 2.Rosen PP, Kimmel M. Juvenile papillomatosis of the breast. A follow-up study of 41 patients having biopsies before 1979. Am J Clin Pathol. 1990;93:599–603. doi: 10.1093/ajcp/93.5.599. [DOI] [PubMed] [Google Scholar]
- 3.Bazzocchi F, Santini D, Martinelli G, Piccaluga A, Taffurelli M, Grassigli A, Marrano D. Juvenile papillomatosis (epitheliosis) of the breast. A clinical and pathologic study of 13 cases. Am J Clin Pathol. 1986;86:745–748. doi: 10.1093/ajcp/86.6.745. [DOI] [PubMed] [Google Scholar]
- 4.Kafadar MT, Anadolulu Z, Anadolulu Aİ, Tarini EZ. Juvenile Papillomatosis of the Breast in a Pre-Pubertal Girl: An Uncommon Diagnosis. Eur J Breast Health. 2018;14:51–53. doi: 10.5152/ejbh.2017.3828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mansel RE, Webster DJT, Sweetland HM, Hughes LE, Gower-Thomas K, Evans DGR. Hughes, Mansel & Webster’s Benign Disorders and Diseases of the Breast (Third Edition) Edinburgh: W.B. Saunders; 2009. CHAPTER 9 - Sclerosing adenosis, radial scar and complex sclerosing lesions; pp. 139–45. [Google Scholar]
- 6.Okita A, Ohsumi S, Takashima S, Okita R, Aogi K, Saeki T, Kurita A, Nishimura R. Non-palpable ductal carcinoma in situ (DCIS) with microinvasion arising in a radial scar presenting with spiculation alone on mammograms: a case report. Breast cancer. 2006;13:107–111. doi: 10.2325/jbcs.13.107. [DOI] [PubMed] [Google Scholar]
- 7.Lad S, Seely J, Elmaadawi M, Peddle S, Perkins G, Robertson S, Ibach K, Haggar F, Arnaout A. Juvenile papillomatosis: a case report and literature review. Clin Breast Cancer. 2014;14:e103–e105. doi: 10.1016/j.clbc.2014.03.003. [DOI] [PubMed] [Google Scholar]