

Abstract
Antibiotics with novel mechanisms of action are desperately needed to combat the increasing rates of multidrug-resistant infections. Bacterial pantothenate kinase (PanK) has emerged as a target of interest to cut off the biosynthesis of coenzyme A. Herein we report the results of an in vitro high-through-put screen of over 10000 small molecules against Bacillus anthracis PanK, as well as a follow-up screen of hits against PanK isolated from Pseudomonas aeruginosa and Burkholderia ceno-cepacia. Nine hits are structurally categorized and analyzed to set the stage for future drug development.
Keywords: antibiotic resistance, Bacillus, high-throughput screening, pantothenate kinase, Pseudomonas
Antibiotic resistance has been identified as a critical threat to human health, prompting a 2013 Centers for Disease Control report on resistance threats[1] and a 2014 executive order to combat antibiotic resistance.[2] As current drugs become less effective at treating infections, innovative molecular strategies are needed to prevent our return to a pre-antibiotic era. Although new molecular scaffolds that are less prone to resistance are needed, the core principles of finding “magic bullet” drugs, molecules that can selectively target the infecting bacteria and not the host,[3] are still very much alive. The identification and evaluation of appropriate targets for this purpose requires interdisciplinary drug discovery efforts at the intersection of chemistry and biology
A potential strategy receiving increased attention is inhibiting the production of coenzyme A (CoA), a cofactor that is critical in the Krebs cycle and fatty acid metabolism and is essential for all living organisms.[4] In the biosynthetic pathway for CoA, the first committed step is the phosphorylation of pan-tothenate (vitamin B5) into 4’-phosphopantothenate with ATP by the dimeric enzyme pantothenate kinase (PanK) (Figure 1 A). Type II PanK is generally used by eukaryotes for CoA production, whereas bacteria generally rely on Type I or Type III PanK. Genome analysis as well as protein expression indicate that Type III PanK is structurally conserved across many bacteria.[5] Furthermore, while the monomeric Type II and Type III PanK structures are similar, differing loop regions cause distinct tertiary dimer structures and an altered conformation of the substrate binding pockets (Figure 1 B,C).[6] The essential nature of the enzyme, ubiquity of the structure across bacterial species, and dissimilarity from its host counterpart make PanK an appealing target for inhibition of CoA biosynthesis as a novel antibiotic mechanism. We set out to evaluate this strategy in vitro with a high-throughput screening platform.
Figure 1.
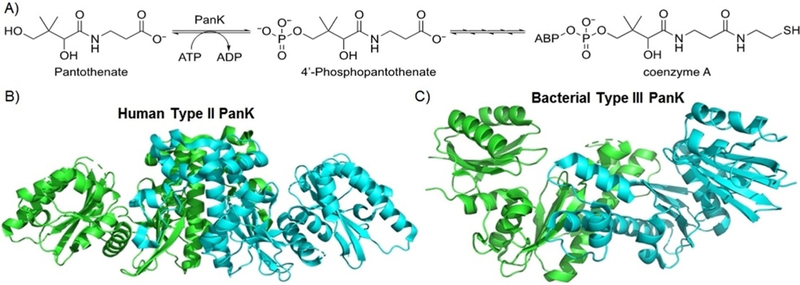
A) Biosynthesis of coenzyme A (CoA), beginning with the first committed step, the conversion of pantothenate into 4’-phosphopantothenate by pantothenate kinase (PanK). ABP = adenosine 3’,5’-bisphosphate. B) Crystal structure of eukaryotic Type II PanK dimer from Homo sapiens. C) Crystal structure of bacterial Type III PanK from Pseudomonas aeruginosa.
The goal of this study was to conduct a high-throughput screen (HTS) of compounds against three different bacterial PanK enzymes and thus identify new antibiotics to target both Gram-positive and Gram-negative bacteria. We chose the enzyme from Bacillus anthracis (BaPanK), a bio-weaponized organism and the causative agent of anthrax,[7] for initial testing. Hits were further tested against the enzymes from Pseudomonas aeruginosa (PaPanK) and Burkholderia cenocepacia (BcPanK), two organisms associated with lung infections in immunocompromised patients.[8] Structure-function analysis of active compounds and the inferred enzyme conformation have revealed four major scaffolds which share broad structural features of H-bonding moieties and heteroatom-containing π systems.
A collection of 10 240 compounds from the Molecular Library Small Molecule Repository (https://pubchem.ncbi.nlm.-nih.gov/) were tested twice at 30 μm for inhibition of BaPanK. Z’ values calculated from 128 positive and 128 negative control values on each plate were consistently > 0.8 (average = 0.92) indicating that the screen was robust and capable of detecting active compounds when tested at a single concentration. The data from the two separate assay runs (8 × 1536-well plates, each containing 1280 test compounds) showed good reproducibility (Pearson’s correlation = 0.77; Figure 2). Using the mean percent inhibition ± 3×SD of the combined test data as the activity threshold (12.5% inhibition), 79 compounds were identified with replicate activity in both runs of the assay (Figure 2, grey shaded area).
Figure 2.
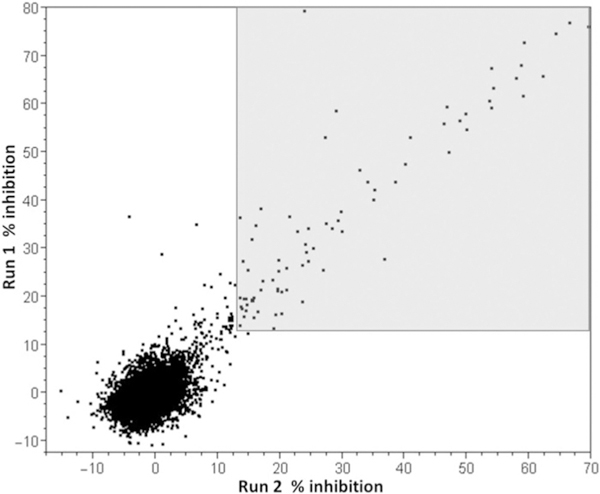
Correlation plot of replicate data from two separate inhibition assays against BaPanK. The area shaded in light grey denotes compounds with replicate percent inhibition above the activity threshold of 12.5%.
A secondary screen was performed on 62 compounds that were commercially available. Fresh samples of each compound were retested at 10 concentrations against BaPanK as well as enzymes from P. aeruginosa (PaPanK) and B. cenocepacia (BcPanK) and in a counter-screen assay in which enzyme was omitted and exogenous ADP added. Thirty-two of these compounds were not active when retested for concentration-re-sponse and were therefore considered false positives from the screen. One compound (1) shows cross reactivity with BaPanK and PaPanK with IC50 values of 12.55 and 84.39 pm, respectively. Three compounds (2, 3, and 9) appear to be the most active against BaPanK, exhibiting concentration-dependent inhibition (IC50 values < 13 μm) with little or no activity against the other enzymes or in the counter-screen. Five compounds (4–8) show moderate specific activity against BaPanK, with IC50 values between 39 and 80 μm. None of the compounds inhibited BcPanK with an IC50 < 100 μm (Supporting Information Table 1).
The structures of the active compounds fit into four core scaffolds (Figure 3): thioxothiazolidinonyl acetic acid (1-3), bi- thiazolamine (4, 5), p-phenol/pseudophenol (6, 7), and thiotria-zole (8, 9). Among these, the thioxothiazolidinonyl acetic acids possess the most potent inhibitory activity and the only compound with cross-activity in P. aeruginosa. While compound 9, a thiotriazole, appears to possess a lower IC50 value than any thioxothiazolidinonyl acetic acid, it also shows greater activity in the counter-screen, and thus may be a result of some additional nonspecific activity.
Figure 3.
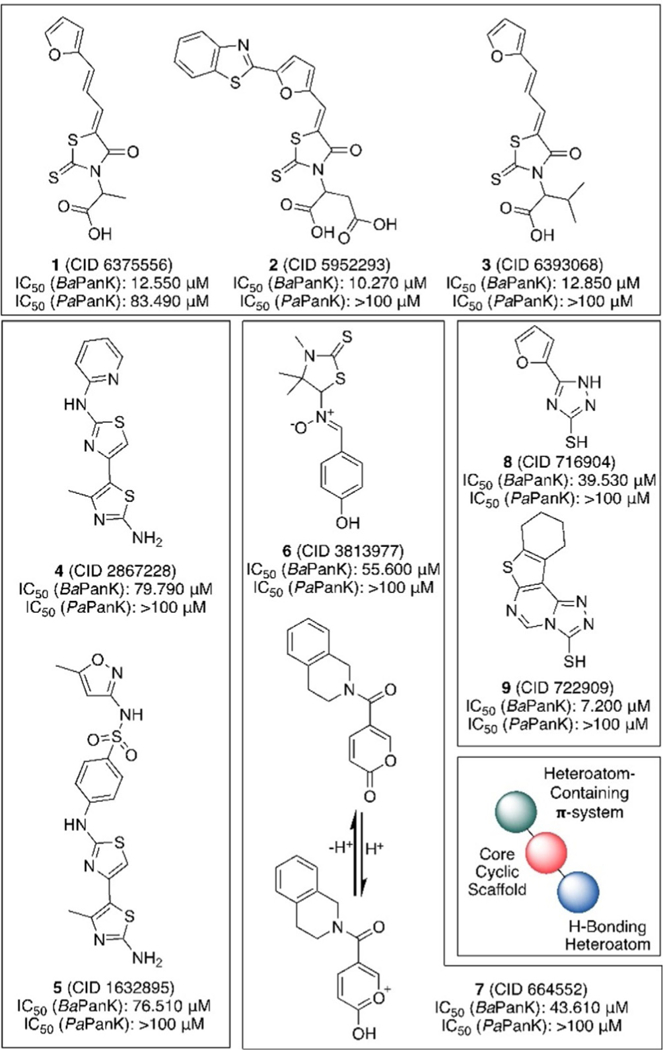
Structures of hits against BaPanK, grouped by structural similarity, with IC50 values against BaPanK and PaPanK listed below. All compounds were inactive against BcPanK and the counter-screen.
Broadly, all of the active compounds can be viewed as having a free heteroatom H-bonding moiety (OH, SH, or NH2) directly attached to or positioned near a heterocycle (the exception being the phenyl ring in compound 6) with an aromatic resonance structure. Additionally, these scaffolds have, positioned opposite the H-bonding moiety, heterocycle-containing π systems either attached directly to or nearby the core scaffold. It is likely that the scaffolds, while structurally quite different at first glance, use these chemically analogous structural motifs to create tight noncovalent interactions with the PanK binding pocket. Based on the crystal structure of Thermotoga maritima PanK in complex with 4’-phosphopantothenate and ADP,[9] we hypothesized that the inhibitors occupy the ATP binding site with the H-bond donor toward the polar residues facing the pantothenate binding site and the conjugated heterocyclic systems near the nucleoside-interacting aromatic residues to engage in π-stacking interactions.
While Type III PanK is generally considered to have large structural similarity across bacteria, the finding that the inhibitors we recognized here lacked activity against P. aeruginosa and B. cenocepacia PanK suggests key differences. Indeed, computational sequence alignment of publicly available crystal structures for Type III PanK from these microbes shows good overall overlap (Figure 4A), which is reflected in the pantothenate binding site displaying near perfect alignment of all major secondary structural features (α-helices, β-sheets, loop regions; Figure 4B). In contrast, the nucleotide binding site shows good overlap between the Gram-negative PaPanK and BcPanK but a significant alteration in the substrate-interacting loop region (Figure 4C). Together with the inhibition data, these results support our hypothesis that the HTS hits act by blocking the ATP binding site of BaPanK and explain the general lack of activity against PaPanK and BcPanK. Compounds 1 and 3 have nearly the exact same structure, with the former bearing a methyl group near the H-bonding site and the latter bearing an isopropyl; however, 1 has inhibitory activity against PaPanK and 3 does not. This indicates that steric bulk at this position is detrimental to binding in this enzyme pocket. This rudimentary structure-activity relationship should be considered in the development of future analogues.
Figure 4.
A) Computed structural alignment of PanK crystal structures from Bacillus anthracis (magenta), Pseudomonas aeruginosa (grey), and Burkholderia cen- ocepacia (cyan). Pictured substrates (pantothenate, adenosine monophosphate, and imidodiphosphoric acid) are from the crystal structure of B. cenocepacia. B) Aligned structures of the pantothenate binding site, showing strong overlap. C) Aligned structures of the nucleotide binding site, showing a significant structural change in BaPanK relative to PaPanK and BcPanK, potentially explaining differential inhibition.
New antibiotic scaffolds with novel mechanisms of action that are less prone to the development of resistance are desperately needed to supplement traditional therapeutics. The biosynthesis of coenzyme A, specifically the phosphorylation of pantothenate by Type III PanK (in contrast to human Type II PanK), is critical to bacteria and thus an antibiotic target of interest.[4] Herein we describe a high-throughput screen in search for small-molecule inhibitors of PanK enzymes from three infectious organisms: B. anthracis, P. aeruginosa, and B. cenocepacia. We identified nine molecules with moderate to good activity against BaPanK, with one having moderate activity against PaPanK. The molecules fall into four basic chemical scaffolds, which further share broad chemical characteristics with one another of H-bonding and π-stacking. This may play a role in PanK affinity and hint at an ATP binding pocket related mechanism of action, which is supported by alignment of crystal structures from the three organisms. Not only will these data aid in the development of our HTS hits as potential narrow-spectrum therapeutics against B. anthracis (and/or other Gram-positives), but the structural information may allow the rational design of Gram-negative or broad-spectrum PanK inhibitors.
Future development of the HTS hits will require whole-cell activity screens against B. anthracis along with a variety of Gram-positive and Gram-negative microbes. These investigations will determine the ability of these compounds to overcome issues of membrane permeability and intracellular degradation. In parallel, the toxicity of the scaffolds against human cells must be assessed; in principle, the structural heterogeneity between human and bacterial PanK should result in low cross-activity against host CoA biosynthesis, but this must be confirmed by in vitro inhibition assays of Type II PanK and human cell lines. Previously identified pantothenate mimics developed as inhibitors of Staphalococcus aureus Type III PanK were found to not strongly inhibit the growth of human HepG2 liver cells despite strong structural similarity to endogenous pantothenate,[10] lending credence to this strategy. Organic synthesis of analogues along with biochemical validation of the proposed mechanism will allow structure-function analysis and form the basis of the development of more potent func-tionalized derivatives. The newly identified activity of these scaffolds has opened up opportunities for drug development on a promising antibiotic target, Type III PanK, in the ongoing fight against drug-resistant infections.
Experimental Section
Enzyme purification:
The Type III PanKs from B. anthracis (BaPanK) and P. aeruginosa (PaPanK) were expressed and purified as previously described.[6,7] For PaPanK, the expression plasmid was generously provided by Dr. Suzanne Jackowski (St. Jude Children’s Research Hospital). To overexpress and purify the B. cenocepacia J2315 PanK enzyme, the gene (BCAL0693) was cloned with an N-terminal His6 tag. The pLIC-His + BcPanK expression plasmid was created using a codon-optimized pantothenate kinase gene from B. cenocepacia J2315 (BCAL0693) that was purchased from Genscript and was supplied in the pUC57 vector. The gene was amplified and inserted into the pLIC-His vector, following a protocol adapted from Stols et al.[11] BcPanK was expressed in 3.2 L of BL21(DE3)/pLIC-His + BcPanK grown in Studier’s ZYM-5052 auto-induction media[12] at 30 °C. Cells were harvested, washed with 50 mM potassium phosphate (pH 7.0), and frozen at −80°C. Harvested cells were resuspended in buffer A (50 mM sodium phosphate, pH 8.0, 300 mM NaCl) supplemented with 0.5 mM 4-(2-ami-noethyl)benzenesulfonyl fluoride and lysed by two passes through an Avestin EmulsiFlex-C5 homogenizer. After centrifugation at 40000 × g for 45 min, nucleic acids were removed from the supernatant by addition of 2% (w/v) streptomycin sulfate, followed by centrifugation. After addition of 25 mM imidazole to the cleared extract, it was loaded onto a 25 mL Cobalt-Sepharose column (GE Healthcare). The column was washed with buffer A containing 25 mM imidazole, and BcPanK was eluted by increasing the imidazole concentration to 500 mM. The eluted protein was concentrated to 4 mL before loading onto a 250 mL Superose 12PG column (GE Healthcare) equilibrated with 20 mM HEPES/NaOH (pH 7.0), 300 mM NaCl, 5% glycerol, and 1 mM TCEP. The 74 mg yield of protein was concentrated to 8 mL (9.25 mgmL−1) and frozen in aliquots at −80 °C.
To characterize BcPanK, standard PanK activity assays were carried out in 100 mM HEPES/NaOH (pH 7.5), 10 mM MgCl2, 60 mM NH4Cl, and 2 mM PEP. The 96-well microtiter assays (200 μL volume) also contained 0.5 mM NADH, 2.75 units of lactate dehydrogenase, and 2 units of pyruvate kinase. Pantothenate was varied from 10 to 500 μM. ATP concentration was varied from 2 to 25 mM. Preliminary kinetic analysis for the purified recombinant BcPanK gave KM(Pan) = 71 μM, KM(ATP)≈13 mM, and Vmax = 2.8 s−1 at pH 7.5 and 25°C.
High-throughput pantothenate kinase assay:
ADP Hunter Plus Assay Kit (DiscoverX) is a biochemical assay to measure the accumulation of ADP, a product of kinase enzyme activity. ADP Hunter Plus is specifically designed for high-throughput screening of kinase inhibitors. The assay uses coupled enzymatic reactions to produce hydrogen peroxide in direct proportion to the amount of ADP produced. In the presence of peroxidase the hydrogen peroxide converts ADHP (10-acetyl-3,7-dihydroxyphenoxazine) into fluorescent resorufin. Fluorescence intensity was measured in a PerkinElmer Envision plate reader using excitation/emission wavelengths of 530/590 nm. The assay was used here in an HTS 1536-well format (Corning black 1536-well plates were purchased from Thermo Fisher Scientific) to measure pantothenate kinase activity.
Pantothenate kinase assays were run at room temperature in a total volume of 2.5 μL in 1536-well microplates. The kinase reaction was performed under the following conditions: Assay Buffer: 15 mM HEPES (pH 7.4), 20 mM NaCl, 1 mM EGTA, 0.02% Tween 20, 10 mM MgCl2, and 0.1 mgmL−1 BGG (bovine-γ-globulins) 10 mM NH4Cl, and 2% DMSO. Substrate Mix: 0.5 mM D-pantothenate and 0.25 mM ATP in Assay Buffer. Enzyme Mix: 20 μg mL−1 PanK in Assay Buffer. Equal volumes of Enzyme Mix and Substrate were mixed together to initiate the kinase reaction in optimized conditions. Assay Buffer without enzyme was used as the background mix; 1.25 μL of Substrate Mix and 1.25 μL of Enzyme Mix (or Assay Buffer without enzyme for the background control) were added to the wells with a BioRaptr FRD (Beckman Coulter). Enzymatic reactions were carried out for 2 h and then stopped by adding 1.25 μL of ADP Hunter Solution A and 2.5 μL of ADP Hunter Solution B. Following a 30-min incubation period the increase in fluorescence 530/590 was read on an EnVision Multilabel Reader (PerkinElmer).
For IC50 determinations of the test compounds,[13] plates were predrugged using a Labcyte ECHO555 Liquid Handler. Inhibitor con-centration-response curves were analyzed by fitting data to the four-parameter logistic equation (sigmoidal dose-response curve with variable slope) from which IC50 values were calculated.
Enzyme structural alignment:
Crystal structures of BaPanK (PDB ID: 2H3G, DOI: 10.2210/pdb2H3G/pdb), PaPanK (PDB ID: 2F9T, DOI: 10.2210/pdb2F9T/pdb), and BcPanK (PDB ID: 5B8H, DOI: 10.2210/pdb5B8H/pdb) were acquired from the RCSB Protein Data Bank. Structural alignment was conducted with PyMOL Version 2.1.1. Outlier rejection parameters are as follows: Cycles = 5, Cutoff = 2.0.
Supplementary Material
Acknowledgements
The expression plasmid for PaPanK was generously provided by Dr. Suzanne Jackowski at St. Jude Children’s Research Hospital (Memphis, TN, USA). This work was funded by the Cystic Fibrosis Foundation (CLAIBO14G0), the US National Institutes of Health (NIH NIGMS GM119426, NCI CA098468), and the National Science Foundation (NSF CHE1755698).
Footnotes
Supporting information and the ORCID identification number(s) for the author(s) of this article can be found under: https://doi.org/10.1002/cmdc.201800652.
Supporting Information: A table of IC50 curves for initial hits is available in the Supporting Information.
Conflict of interest
The authors declare no conflict of interest.
References
- [1].Antibiotic Resistance Threats in the United States 2013, US Department of Health and Social Services, Centers for Disease Control and Prevention (CDC): https://www.cdc.gov/drugresistance/pdf/ar-threats-2013-508.pdf (accessed November 22, 2018).
- [2].Obama B, Combatting Antibiotic-Resistant Bacteria, Office of the Press Secretary, Washington DC (USA), 2014. [Google Scholar]
- [3].Gradmann C, Dynamis 2011, 31, 305–321. [DOI] [PubMed] [Google Scholar]
- [4].Spry C, Kirk K, Saliba KJ, FEMS Microbiol. Rev. 2008, 32, 56–106. [DOI] [PubMed] [Google Scholar]
- [5].Yang K, Strauss E, Huerta C, Zhang H, Biochemistry 2008, 47, 1369–1380. [DOI] [PubMed] [Google Scholar]
- [6].Hong BS, Yun MK, Zhang YM, Chohnan S, Rock CO, White SW, Jackowski S, Park HW, Leonardi R, Structure 2006, 14, 1251–1261. [DOI] [PubMed] [Google Scholar]
- [7].Nicely NI, Parsonage D, Paige C, Newton GL, Fahey RC, Leonardi R, Jackowski S, Mallett TC, Claiborne A, Biochemistry 2007, 46, 3234–3245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Coutinho H, Falcâo-Silva VS, Gonçalves G, Int. Arch. Med. 2008, 1, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Yang K, Eyobo Y, Brand LA, Martynowski D, Tomchick D, Strauss E, Zhang H, Bacteriol J. 2006, 188, 5532–5540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Choudhry AE, Mandichak TL, Broskey JP, Egolf RW, Kinsland C, Begley TP, Seefeld MA, Ku TW, Brown JR, Zalacain M, Ratnam K, Antimicrob. Agents Chemother. 2003, 47, 2051–2055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Stols L, Gu M, Dieckman L, Raffen R, Collart FR, Donnelly MI, Protein Expr. Purif. 2002, 25, 8–15. [DOI] [PubMed] [Google Scholar]
- [12].Studier FW, Protein Expr. Purif. 2005, 41, 207–234. [DOI] [PubMed] [Google Scholar]
- [13].Austin CP, Brady LS, Insel TR, Collins FS, Science 2004, 306, 1138–1139. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.