

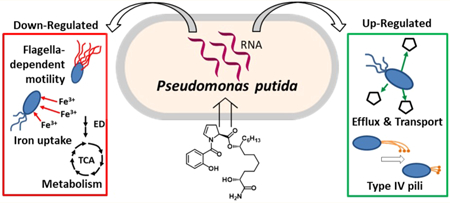
Abstract
Promysalin, a secondary metabolite produced by P. putida RW10S1, is a narrow-spectrum antibiotic that targets P. aeruginosa over other Pseudomonas spp. P. putida KT2440, a nonproducing strain, displays increased swarming motility and decreased pyoverdine production in the presence of exogenous promysalin. Herein, proteomic and transcriptomic experiments were used to provide insight about how promysalin elicits responses in PPKT2440 and rationalize its species selectivity. RNA-sequencing results suggest that promysalin affects PPKT2440 by (l) increasing swarming in a flagella-independent manner; (2) causing cells to behave as if they were experiencing an iron-deficient environment, and (3) shifting metabolism away from glucose conversion to pyruvate via the Entner–Doudoroff pathway. These findings highlight nature’s ability to develop small molecules with specific targets, resulting in exquisite selectivity.
Keywords: Pseudomonas putida, RNA-Seq, promysalin, surface motility, iron, metabolism
Graphical Abstract
Bacterial antibiotic resistance is arguably one of the most pressing issues facing modern society. Increasing resistance among human bacterial pathogens leads to persistent infections that are difficult to treat, can be fatal, and cause an increased economic burden on health care.1 Fear of promoting and contributing to antimicrobial resistance in pathogens has impacted the use of antibiotics in agriculture and the approaches used to control plant pathogens. Diseases caused by bacterial plant pathogens result in low crop yields, lower food/nutritional quality, and large economic losses.2 However, regulatory policy appropriately prevents the use of antibiotics, except streptomycin and oxytetracycline, for the control of plant diseases because of the potential to select for a vast environmental reservoir of antibiotic-resistant genes.3,4 Consequently, the discovery and development of new and effective control agents for both human- and plant-pathogenic bacteria remain challenges in medicinal, agricultural, and environmental sciences.
The rhizosphere has become a rich area of study for the identification of new methods to combat human and plant pathogens.5 Natural products produced by microorganisms in the rhizosphere are seen as a largely untapped resource of novel antimicrobial agents. Competition for resources in this biologically diverse ecosystem leads to the production of secondary metabolites able to inhibit or facilitate the growth of other microorganisms. In 2011, a secondary metabolite, promysalin ((–)−1), was isolated from the rhizosphere- inhabiting bacteria Pseudomonas putida, strain RW10S1.6,7 Promysalin conferred increased surface motility and colonization of the producing strain while exhibiting narrow-spectrum specificity, inhibiting the growth of various pseudomonads with no activity seen against other proteobacteria.6 The structure and the genetics behind the biosynthesis of promysalin have been elucidated; however, the mechanisms involved in stimulating surface colonization and motility in the producing strain and antagonism in susceptible strains (both P. putida) are largely unknown.6,8–10 Interestingly, promysalin’s biosynthetic gene cluster is flanked by genes that encode for enzymes participating in the tricarboxylic (TCA) cycle and are syntenic in the Pseudomonas spp. Additionally, it was found that the expression of the biosynthetic gene cluster is regulated by GacA, a regulator that modulates social behavior, the production of virulence factors, the production of antimicrobial metabolites, and surface motility.6
Over the past 3 years, our laboratory (W.M.W.) has been interested in understanding the basis of promysalin’s narrow- spectrum activity. Figure 1 highlights our key findings leading up to this work. In brief, after completing the first total synthesis of promysalin, we found that the small molecule repressed the fluorescence of P. putida KT2440 (PPKT2440), presumably by the inhibition of pyoverdine production, and also promoted swarming on solid agar (up to 1.0%).10 In a follow-up study, we identified the key moieties responsible for biological activity and also found that promysalin was able to weakly bind iron, although at the time it was unclear if this ability is biologically relevant.9 Most recently, we identified succinate dehydrogenase (Sdh) as the target of promysalin in P. aeruginosa.11 Taken together, we sought to leverage these findings in combination with mechanism-based approaches to better understand the complexities of its narrow-spectrum activity.
Figure 1.
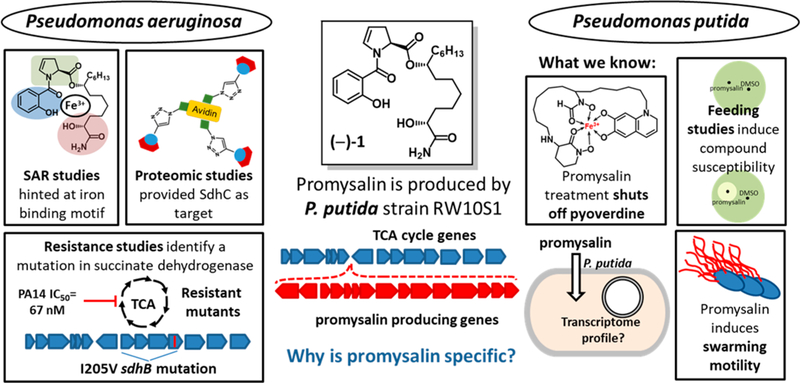
Previous biological effects of promysalin, (–)−1, on pseudomonads (PA14 and PPKT2440).
We initially focused on implementing proteomic methods for target identification. Affinity-based protein profiling (AfBPP) was used to determine proteomic targets of small molecules by covalent modification. Previous efforts in our group utilized a promysalin derivative bearing a photoaffinity probe ((–)−2) to identify Sdh as the biological target in P. aeruginosa (PAO1 and PA14).11 On the basis of these initial successes, we sought to leverage the same chemical probe in AfBPP studies with PPKT2440. Three samples were prepared, and in all cases, the promysalin photoaffinity probe ((–)−2) was incubated with PPKT2440 and irradiated to thereby covalently attach the probe to the nearest protein. Following cross-linking, the cells were lysed, reacted with biotin-azide, and enriched on avidin beads. Trypsin digest and dimethyl-isotope labeling permitted the analysis of enriched proteins via LC–MS/MS (Figure 2). After proteomic analysis, whereby 2 controls were used to eliminate nonspecific binders (see Figure S1 for full details), 45 enriched proteins were identified. This was in contrast to our previous experiments on PAO1 and PA14 where SdhC was the only statistically significant hit. Although SdhC was identified in this assay, many other potential targets were also found, including the most prominent hit, a xenobiotic reductase, PP_1478 (Figure 2, Table S1). As promysalin is not toxic to PPKT2440 but elicits a multitude of biological effects, we speculated that the molecule could elicit an antibiotic resistance response whereby it is either altered or expelled via efflux.
Figure 2.
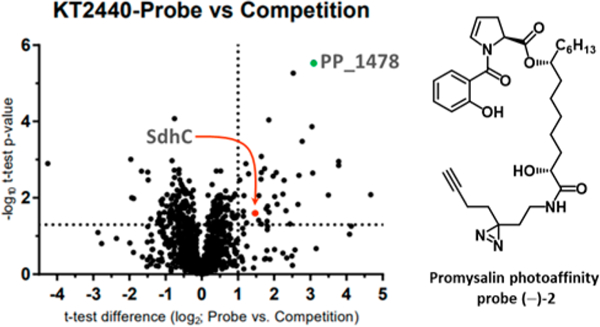
Volcano plot of photoaffinity probe (–)−2 and competition experiment. Results in the upper right quadrant are statistically significant. Notable hits are highlighted. (See the SI for the full list.)
RNA sequencing has provided useful insights into the mechanisms of antibiotic susceptibility and resistance as well as insights into the putative mode of action of molecules.12,13 Therefore, we performed a global transcriptome analysis (RNA- Seq) on bacterial cells that were treated with promysalin or DMSO as the control to better understand how promysalin is affecting cellular processes. Since phenotypic differences can be seen after 24 h on a swarming medium between cells treated with promysalin and those with the DMSO control, we chose to evaluate transcriptional changes at this physiologically relevant time point. A total of 109 540 045 reads were acquired for the 3 promysalin-treated samples, and 151 278 351 total reads were acquired for the control samples (Table S2). DESeq2 was used to identify those genes differentially expressed between the two conditions. An analysis of the RNA-Seq data revealed that 455 genes were differentially expressed between the promysalin-treated and control samples using an FDR of 0.05 and a fold-change cutoff of 1.5-fold (433 down-regulated and 22 up-regulated). The complete list of genes that showed differential expression is shown in the Supporting Information.
Our previous studies suggested that promysalin enhanced surface motility, even when used with agar concentrations that typically inhibit swarming (>0.5%).6,10 Swarming motility typically requires flagella and biosurfactant; therefore, we were surprised to find that eight genes were down-regulated (PP_4359 (fliL), PP_4370 (fliE), PP_4372 (fleS), PP_4377, PP_4378 (fliC), PP_4386 (flgF), PP_4391 (flgB), and PP_4393) that encode proteins involved in flagella-dependent motility. We hypothesized that the decreased expression of genes involved in flagella assembly would result in reduced swimming in the presence of promysalin. To determine if promysalin influenced flagella, we evaluated swimming motility with 0.3% agar media (Figure S2). In contrast to the swarming phenotype observed when PPKT2440 is treated with 100 μM promysalin, we see reduced swimming as compared to DMSO at the same concentration, suggesting that flagella production or function is inhibited in the presence of promysalin (Figure S3). To determine if flagella production was influenced by the presence of promysalin, Western blot was performed using cells that were grown in the presence or absence of promysalin. No observable differences in FliC could be detected (Figure S4). It is possible that the slight differences observed in the transcriptional profiles (2-fold) do not equate to differences at the protein level. Interestingly, among the genes up-regulated by the presence of promysalin, one gene, PP_3481, is annotated as an uncharacterized protein but has sequence similarity to the pilus assembly protein, PilO, and encodes a protein that belongs to the PilN superfamily, suggesting that this protein functions in type IV pili assembly. Taken together, the swimming results further support that promysalin inhibits flagella motility and the induced swarming phenotype is flagella-independent.
Swarming motility can be influenced by the availability of iron. We recently confirmed that promysalin binds iron, albeit quite weakly, and questioned its ability to act as a siderophore.9 We found that promysalin had a widespread effect on the expression of genes predicted to be involved in iron binding or homeostasis, influencing the expression of 15 genes in this category (Table S3). The PPKT2440 genome has three loci predicted to encode bacterioferritin genes: PP_0482 (bfr-I), PP_1082 (bfr-II), and PP_4856. Bacterioferritins are proteins that oxidize Fe2+ to Fe3+ for Fe3+ storage when excess iron is available.14 In the current study, the expression of all three bfr genes decreased in promysalin-treated cells (Figure 3a), suggesting that the cells are experiencing iron-limiting conditions even though they are being grown in iron-rich media. Under iron-limited conditions, many proteins that use iron as a cofactor, such as cytochromes involved in electron transport, are often down-regulated due to the lack of iron availability needed for protein fUnction.15,16 Our RNA-Seq data showed that 11 genes predicted to encode iron-binding proteins were down-regulated in response to promysalin (Figure S5 and Table S3). Two of these, PP_4324 and PP_4325 (ccmD and ccmC, respectively), encode part of the cytochrome C maturation (ccm) system that is involved in heme transport and cytochrome C biosynthesis.17,18 PP_0489 (fdoG) and PP_5212 encode iron-binding metabolic enzymes, formate dehydrogenase, and an oxidoreductase, respectively. The remaining seven genes in this category are annotated as cytochrome subunits or cytochrome-type proteins involved in electron transport. Taken together, these findings suggest that promysalin-treated cells are experiencing an iron-deficient environment. While it is tempting to speculate that the iron- binding properties of promysalin allow it to sequester iron and plunge the cells into iron-limited conditions, it must be noted that we did not observe an increased expression of genes involved in the biosynthesis of siderophores, a hallmark of bacteria that are exposed to iron-limiting conditions. In fact, previous studies from our laboratory have shown that pyoverdine production is specifically inhibited by promysalin in PPKT2440.
Figure 3.
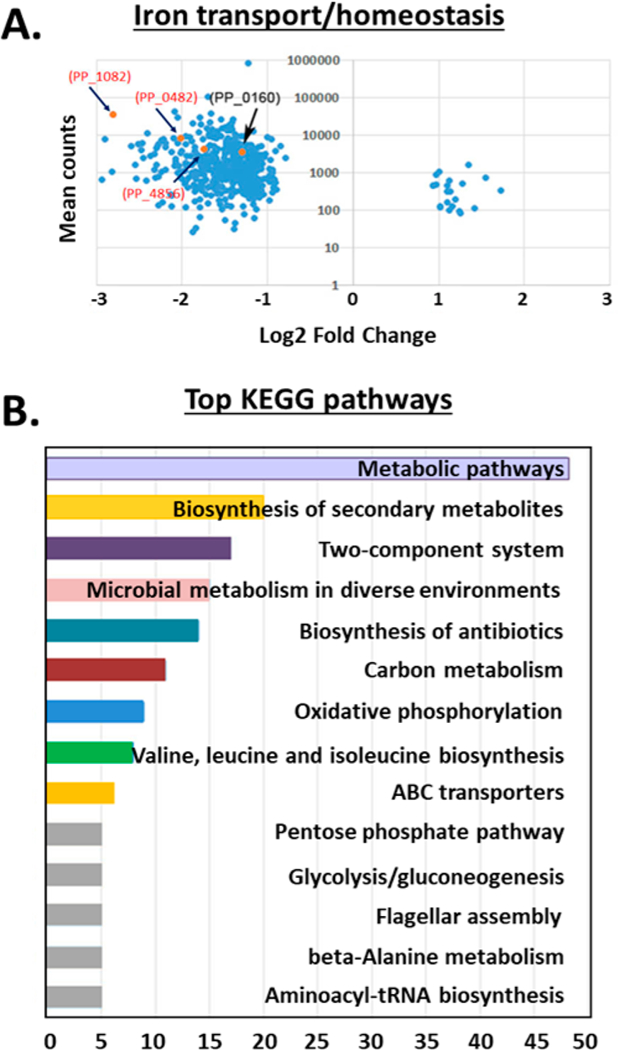
(A) Volcano plots showing the mean counts (y axis) and fold change (log 2, x axis) of genes involved in iron transport. Points colored orange indicate those genes predicted to encode proteins involved in iron binding, iron transport, or iron homeostasis. (B) KEGG pathway analysis representing the breakdown of biological pathways identified in promysalin-treated cells on the basis of their putative functions, assigned using protein function databases. For each KEGG pathway, the bar shows the fold enrichment of the pathway. Analysis was performed by a Fisher exact test. Significantly enriched KEGG pathways (p < 0.05) are presented.
On the basis of our inconclusive proteomic results, we postulated that promysalin might be removed by efflux from the bacteria before it can properly engage its target. Seven of the genes that are up-regulated in response to promysalin encode putative transporters, four of which are efflux transporters and three of which are predicted to be involved in the uptake of nutrients. Among the efflux transporters, two genes, PP_2429 and PP_3520, are likely involved in the transport of amino acid analogs as they are similar to threonine/homoserine/homoserine lactone efflux genes. PP_2429 belongs to the LysE family of transporters. This family of transporters catalyzes the export of amino acids, lipids, and heavy metal ions.19 They play important roles in ionic homeostasis and protection from excessive heavy metals and metabolites in the cell. The remaining two efflux transporter genes, PP_1266 and PP_2651, are annotated as encoding a fusaric acid-resistant protein and a major facilitator superfamily transporter, respectively. PP_1266 is also similar to multi-drug-resistant efflux transporters. Major facilitator superfamily transporters can function as drug efflux pumps to confer resistance to a variety of toxic compounds and are the largest family of multidrug transporters.20 Although changes in gene expression do not always correlate with phenotypic differences, the up-regulation of genes encoding proteins involved in multidrug resistance suggests a mechanism for resistance to promysalin via export.
The iron regulatory network is one of the central control mechanisms of cell metabolism. We identified Sdh as the biological target in PAO1 and PA14. On the basis of the high levels of homology of the enzyme between PA and PPKT2440, we suspected that differences in metabolic capacity or preference for TCA intermediates may account for the insusceptibility. It is known that in PPKT2440 the primary catabolic pathway for the conversion of glucose to pyruvate is the Entner–Doudoroff (ED) pathway rather than the more common Embden–Meyerhof–Parnas (EMP) pathway due to a lack of the 6-phosphofructokinase enzyme. In rich media such as TSB, the ED pathway is the central metabolic pathway for the catabolism of glucose. Metabolites produced in the pathway are precursors for both the pentose phosphate pathway (PPP) and the TCA cycle, where the PA target is located (Figure 4). Our RNA-seq data revealed that the expression of genes involved in several steps in the ED pathway are down-regulated (Figure 4). This suggests that P. putida cells exposed to promysalin are using alternative metabolic pathways. This is additionally supported both by the up-regulation of transporters involved in the uptake of nutrients (PP_1418, PP_2418, and PP_2543 (gabp-II)) and our previous finding that promysalin is toxic to PPKT2440 when grown in succinate-supplemented minimal media.11
Figure 4.
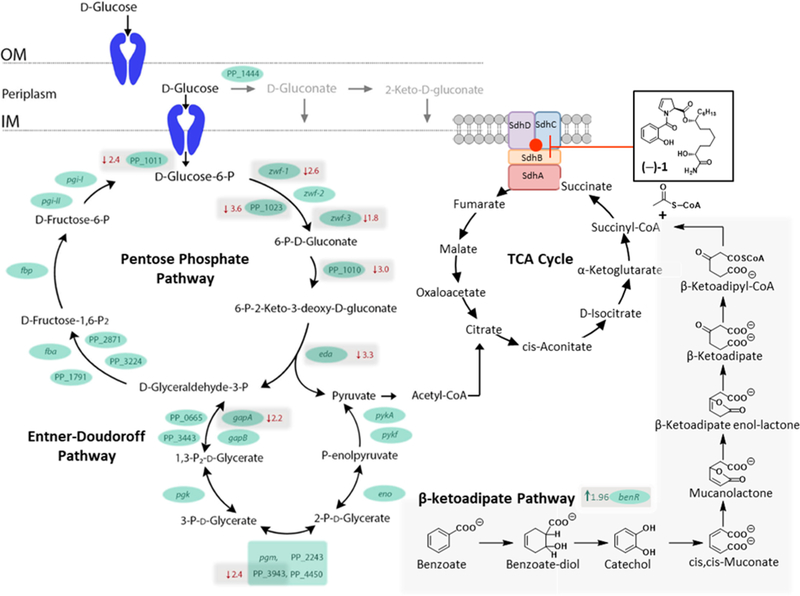
Representation of glucose catabolism in PPKT2440. Shown are reactions that take place in the periplasmic space as well as the catabolic reactions that take place in the cytoplasm. (Blue cartoons represent membrane transporters.) The genes encoding the enzymes involved in each of the steps are indicated. OM represents the outer membrane; IM represents the inner membrane. If a gene demonstrated differential expression between PPKT2440 cells treated with promysalin and those treated with DMSO (control), then the fold change is indicated in red. Arrows beside the fold change indicate the direction of fold change.
The expression of aruC (a gene involved in arginine metabolism), PP_1418 (tctC) (a tricarboxylate transporter), and benR (a transcriptional regulator that activates the transcription of genes involved with benzoate metabolism) were also up-regulated (Supporting Information) upon promysalin exposure, lending evidence to the activation of alternative metabolic pathways. In related bacteria, TctC is induced during catabolite repression and has been implicated in citrate and isocitrate import, indicating the switch to alternative routes of metabolism during promysalin treatment.21,22 PP_3159 (benR) encodes a protein with similarity to the XylS family of transcriptional regulators, and there are several orthologs with this group. This family of transcriptional activators regulates genes involved in carbon metabolism via the β-ketoadipate pathway, stress response, and pathogenesis.23 Increased expression of benR suggests an increase in the utilization of the β-ketoadipate pathway. It is intriguing to postulate that PPKT2440 is able to digest promysalin by down- regulating genes en route to the TCA cycle and up-regulating the peripheral pathways to avoid toxicity, as these pathways are commonly used to catabolize xenobiotics. In a similar fashion, PPKT2440 has been shown to combat the toxicity of trinitrotoluene (TNT), a nitroaromatic compound, by up- regulating genes encoding a xenobiotic reductase pathway, XenR, in addition to the up-regulation of efflux pumps.24 This finding, in particular, is relevant as the major hit in our initial proteomic assay was PP_1478, a putative xenobiotic reductase, lending credence that it might be the preferred target within PPKT2440. While it certainly may not be the only aspect at play, modifications in metabolism indicate that other bacteria may identify promysalin as a xenobiotic and as such modify their functions for its removal.
PP_2430 and PP_3022 belong to the AraC family of transcriptional regulators. These proteins are widespread among bacteria and regulate the transcription of genes having myriad functions including carbon metabolism and the stress response.25,26 Additionally, AraC regulators also function as negative regulators which could play a regulatory role in the down-regulation of genes in promysalin-treated cells.27
Using the Kyoto Encyclopedia of Genes and Genomes (KEGG), we assigned the differentially expressed genes from our RNA- Seq study to functional pathways (http://www.genome.jp/kegg). Out of 456 genes, 91 had significant matches to 1 or more KEGG pathways and were collectively assigned to 282 functional pathways. The top 14 pathways assigned are shown in Figure 3b, with the most frequently assigned pathways overall being “metabolic pathways”, indicating that these pathways are important in the cellular response to promysalin. Recently, it was reported that promysalin can inhibit the growth of Gram-positive bacteria, albeit at much higher (and likely not physiological) concentrations, and the mechanism of action is proposed to involve membrane damage.28 Our results here do not indicate that any compensatory mechanisms are being triggered in response to potential cell membrane damage, in contrast to this previous report.
In addition to our previous findings listed in Figure 1, we wanted to further verify the gene expression trends seen in the RNA-Seq data. We tested the expression of selected genes via qRT-PCR and compared the expression in promysalin-treated samples to that of control samples (Figure S6). PP_0160 and PP_0861 are predicted TonB-dependent siderophore receptors and PiuA orthologs, respectively. We found the changes in expression measured by qRT-PCR to be consistent with the differential expression trends from our RNA-Seq experimental data. The expression of PP_0160 was down (30-fold) in the promysalin-treated samples, and PP_0861 was not differentially expressed. Additionally, we chose to test the expression of PP_1082, a predicted iron ion transporter, and PP_4378 (fliC), important for flagellar-dependent motility, both down-regulated in our RNA-Seq data. We observed the expression of PP_1082 and fliC to be decreased in promysalin-treated samples compared to the control. Finally, we looked at PP_0372 (aruC), involved in ornithine metabolism and up-regulated in our RNA-Seq data, and the expression of PP_0372 was increased (approximately 5-fold). Collectively, the results of the qRT-PCR data showed consistent trends with our RNA-Seq data.
Our findings add to the discoveries of others who have reported on transcriptional changes in PPKT2440 to various small-molecule stressors. Dominguez-Cuevas et al. reported that when PPKT2440 is exposed to toluene (a stress) they redirect priorities of the cell to more vital tasks.29 For example, upon exposure PPKT2440 increases the expression of a number of genes that encode chaperones and genes involved in general metabolism. Flagella and chemotaxis genes were shown to be repressed under this type of stress.29 Similarly, upon exposure to promysalin we observed differences in the expression of genes related to heat shock, motility, metabolism, and iron acquisition. However, these systems are down- regulated in the presence of promysalin, suggesting that the bacteria are not experiencing a stressful condition. We did, however, observe a decreased expression of genes related to motility, iron homeostasis, and the repression of genes involved in primary metabolism, which is consistent with a strategy to redirect resources to save energy.29
In natural environments, competition between bacteria for resources can lead to the production of compounds designed to promote the survivability of the producing species and/or inhibit the growth of competing species. Often, these natural products are secondary metabolites with a very narrow distribution among species and have a variety of functions including cell signaling, interspecies or even interkingdom communication, chemotaxis, and antimicrobial activity.30 In the rhizosphere, bacteria experience extreme competition for the limited resources in the soil, and an array of biologically active compounds have been isolated from the bacteria that reside in this niche that can inhibit the growth of (bacteriostatic) or kill (bactericidal) other nonproducing species of bacteria.5,31
Herein we utilize transcriptomics to gain a deeper understanding of how promysalin functions in the rhizosphere. We utilized differential gene expression data to rationalize our previously observed biological phenotypes (Figure 5). Key findings related specifically to the mechanism of action of promysalin include the following: (l) compound-induced swarming is likely not flagellar-dependent, (2) a response to an “iron-limited” environment, (3) up-regulation of efflux and nutrient transport, and (4) significant changes to metabolic flux.
Figure 5.
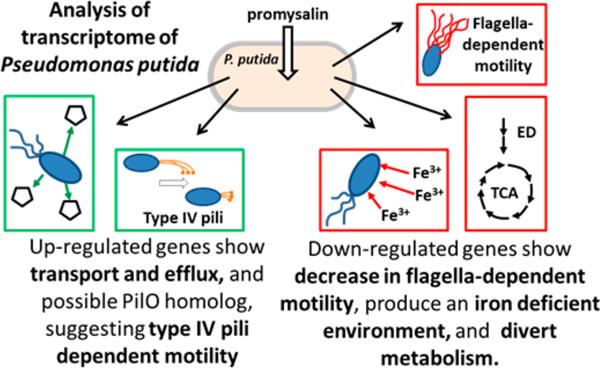
Summary of biological effects elucidated via the transcriptome profiling of PPKT2440 cells treated with promysalin.
Taken together, our results suggest that PPKT2440 is able to tolerate promysalin exposure by taking a two-prong approach. First, it up-regulates efflux and nutrient transporter genes, presumably minimizing promysalin exposure while simultaneously compensating for metabolic changes. Evaluating the expression of efflux transporters in P. aeruginosa or expressing efflux transporters from P. putida in P. aeruginosa may provide insight into other possible resistance mechanisms. Second, it redistributes metabolic flux to avoid Sdh, which in turn triggers a localized iron-limited response. However, this effect is quite surprising as no difference in gene expression was observed for siderophore biosynthesis genes (and, in particular, pyoverdine) between cells exposed to promysalin or left untreated, which one would expect for such a response. It is possible that the metabolic rerouting that occurs to avoid Sdh deemphasizes the importance of Fe3+, which in turn limits pyoverdine production in an effort to maximize resources and is ultimately responsible for this localized response. These findings complement our earlier work and add credence to the hypothesis that promysalin elicits its narrow-spectrum activity via targeting primary metabolism in Pseudomonas spp.
MATERIALS AND METHODS
Bacterial Strains and Culture Conditions.
P. putida KT2440 was grown in trypticase soy broth (TSB) (Becton, Dickson and Company) and on TSB supplemented with 0.5% agar at 28 °C or r.t., respectively. A 50 μM total concentration of promysalin (synthesized as previously described9,10) dissolved in 10% DMSO or a 10% DMSO solution without promysalin was used to supplement the media as indicated.
Affinity-Based Protein Profiling.
Preparation of Samples.
TSB medium (100 mL) was inoculated 1:100 from an overnight culture of P. putida KT2440 and incubated (30 °C, 200 rpm) until OD600 = 1.0. The cultures were centrifuged (5 min, 6000g), washed with 0.5 volume of PBS, and resuspended in the same amount of PBS. Per sample, 20 mL of washed culture was incubated (30 min, 200 rpm) with either the promysalin probe (3 μM) or the inactive promysalin probe (3 μM) or for competition experiments with promysalin (30 μM, 10 min preincubation) and the promysalin probe (3 μM). The samples were poured into Petri dishes (90 mm) and irradiated for 20 min (365 nm, Philips TL-D BLB 18W) without lids. The cultures were centrifuged (10 min, 6000g) and washed with the same amount of PBS. The pellets were resuspended in 1 mL of PBS, transferred to a 1.5 mL tube, centrifuged again, and stored at –80 °C.
Click Chemistry.
Pellets were resuspended in 150 μL of lysis buffer with EDTA-free protease inhibitors (PBS with 0.5% SDS and 1% Triton 100) and lysed using sonication (2 × 15 s, 60% intensity) with cooling on ice. The protein concentration was adjusted to 1 mg/mL using the BCA assay. Each sample (500 μL) was used for the CuAAC reaction. All reagents for the click reaction were premixed and added to the samples at the indicated final concentrations: 100 μM biotin-azide (10 mM stock in DMSO), 1 mM CuSO4 (50 mM stock in ddH2O), 1 mM TCEP (50 mM stock in ddH2O), and 100 μM TBTA (10 mM stock in DMSO). Samples were incubated at r.t. for 1 h. EDTA (10 μL, 500 mM) was added, and the whole sample was then transferred to a 15 mL tube containing 3 mL of cold acetone (–80 °C). Proteins were allowed to precipitate overnight at –80 °C.
Pulldown and Digestion.
The precipitate was centrifuged (10 min, 18 000g) and washed twice with 1 mL of cold methanol (–80 °C) with resuspension (5 s in an ultrasonic bath) and centrifugation steps (10 min, 18 000g) in between. All pellets were air-dried for 30 min at r.t. The washed pellets were resuspended in 500 μL of PBS (containing 0.2% SDS and 1 mM DTT) and centrifuged (20 min, 17 000g), and the supernatant was transferred in low-binding tubes containing 50 μL of washed avidin agarose beads and incubated (2 h, r.t.) with gentle rotation. Avidin beads were washed five times with 700 μL of PBS (containing 0.2% SDS). After incubation, beads were washed (2 × 200 μL PBS + 0.2% SDS; 3 × 200 μL 4 M urea in PBS; 3 × 200 μL 50 mM TEAB) and reconstituted in 50 μL of 50 mM TEAB, and 2 μL of 250 mM DTT was added, followed by incubation on a shaker (30 min, 55 °C, 600 rpm). Beads were washed with 50 mM TEAB followed by the addition of 50 μL of TEAB and 2 μL of 500 mM iodoacetamide with incubation on a shaker in the dark (30 min, 25 °C, 600 rpm). After two more washes with TEAB, beads were reconstituted in 50 μL of 50 mM TEAB and 2 μL of trypsin (0.5 μg/μL in 50 mM acetic acid) and incubated overnight (37 °C and 750 rpm).
Desalt and Dimethyl Label.
The beads were centrifuged, the supernatant was transferred in a new low-bind tube, and digestion was stopped by the addition of 0.7 μL of formic acid (FA). Beads were washed two more times with 50 μL of 0.1% FA in 50 mM TEAB, and supernatants were combined.
Desalting of the samples was conducted on 50 mg SepPak C18 columns (Waters). Columns were equilibrated with 1 mL of acetonitrile (ACN), 1 mL of elution buffer (80% ACN, 0.5% FA), and 3 mL of an aqueous 0.5% FA solution. The acidified samples were loaded by gravity flow, washed five times with 1 mL of 0.5% FA, and then labeled five times with 1 mL of the respective dimethyl labeling agents (light (L): 30 mM NaBH3CN, 0.2% CH2O, 45 mM sodium phosphate buffer, pH 7.5; medium (M): 30 mM NaBH3CN, 0.2% CD2O, 45 mM sodium phosphate buffer, pH 7.5; heavy (H): 30 mM NaBD3CN, 0.2% 13CD2O, 45 mM sodium phosphate buffer, pH 7.5). Column-bound peptides were washed two more times with 1 mL of 0.5% FA and then eluted two times with 250 μL of elution buffer. Each sample (900 μL) was combined in a 15 mL tube, frozen in liquid nitrogen, and lyophilized. Prior to LC-MS/MS measurement, samples were dissolved in 40 μL of 1% FA and filtered with 0.22 μm ultrafree centrifugal filters (Merck) equilibrated with 300 μL of 1% FA. The filtrate was transferred into MS vials and queued for LC–MS/MS measurement.
LC/MS Data analysis.
Each sample (4 μL) was injected into the LC-MS/MS system. Samples were analyzed via HPLC–MS/MS using an UltiMate 3000 nano HPLC system (Dionex, Sunnyvale, CA, USA) equipped with an Acclaim C18 PepMap100 75 μm i.d. × 2 cm trap and an Acclaim Pepmap RSLC C18 separation column (75 μm i.d. × 50 cm) in an EASY-spray setting coupled to a Thermo Fischer LTQ Orbitrap Fusion (Thermo Fisher Scientific Inc., Waltham, MA, USA). Samples were loaded on the trap and washed with 0.1% TFA (at 5 μL/ min) and then transferred to the analytical column and separated using a nonlinear 115 min gradient from 5% A to 32% B and then in 10 min to 90% B followed by another 10 min at 90% B (at a 300 nL/min flow rate) (buffer A: H2O with 0.1% FA; buffer B: MeCN with 0.1% FA). The LTQ Orbitrap Fusion was operated in 3 s top speed data-dependent mode. Full scan acquisition was performed in the orbitrap at a resolution of 120 000 and an AGC target of 2e5 in the scan range of 300–1500 m/z. Monoisotopic precursor selection and dynamic exclusion for 60 s were enabled. Precursors with charge states of 2–7 and intensities greater than 5e3 were selected for fragmentation. Isolation was performed in the quadrupole using a window of 1.6 m/z. Precursors were collected for an AGC target of 1e4 for a maximum injection time of 50 ms with “inject ions for all available parallelizable time” enabled. Fragments were generated using higher-energy collisional dissociation (HCD, 30% normalized collision energy) and detected in the ion trap at a rapid scan rate. Internal calibration was performed using the ion signal of fluoranthene cations (EASY-IC).
Raw files were analyzed using MaxQuant software (version 1.5.3.8) with the Andromeda search engine. The search included the carbamidomethylation of cysteines as a fixed modification and the oxidation of methionines and acetylation of protein N-termini as variable modifications. Light, medium, and heavy labels (of lys and N-term) were set according to the samples, and the number of maximum labeled AAs was set to 4. Trypsin was specified as the proteolytic enzyme with N-terminal cleavage to proline and two missed cleavages allowed. The precursor mass tolerance was set to 4.5 ppm (main search), and the fragment mass tolerance was set to 0.5 Da. Searches were performed against the Uniprot database for Pseudomonas putida KT2440 (taxon identifier 160488, including isoforms). The second peptide identification option was enabled. False discovery rate determination was carried out using a decoy database, and thresholds were set to 1% FDR at both peptide-spectrum match and protein levels. I = L, requantification, and match between runs (0.7 min match and 20 min alignment time windows) options were enabled.
Statistical analysis was performed with Perseus software (version 1.5.2.6). Putative contaminants, reverse sequences, and those identified only by site hits were filtered out. Ratios were converted to logarithms (log2) and z-score normalized (within one replicate). Statistical evaluation was performed using a one-sample t-test (both-sided; Benjamini-Hochberg FDR 0.05).
RNA Extraction and Library Preparation.
For RNA extraction, P. putida KT2440 was grown in TSB media overnight at 28 °C. Overnight cultures were adjusted to an OD600 of 0.3–0.5 and spotted onto TSB plates supplemented with 0.5% agar and 50 μM promysalin. Control plates were supplemented with 0.5% agar and 10% DMSO solution. Inoculated plates were incubated at r.t. for 24 h. After 24 h, cells were harvested into TSB media using sterile swabs. Cells were pelleted via centrifugation and then resuspended in RNA lysis buffer (Zymo Research, Irvine, CA).
RNA was isolated using the ZYMO Research Quick-RNA MiniPrep kit followed by incubation with DNase I (Thermo Fisher Scientific, Waltham, MA) and subsequently purified using the RNA Clean and Concentrator-5 kit (Zymo Research). All procedures were carried out using specified manufacturer’s protocols. During the purification step, large RNAs (>200 bases) were retained and small RNAs (17–200 bases) were removed. The concentration of RNA samples was determined using a Qubit spectrofluorometer, and the integrity of the RNA was assessed at the Cornell Institute of Bioinformatics Genomics Facility using an AATI Fragment Analyzer. mRNA was enriched using the Ribo-Zero rRNA removal kit (Illumina, San Diego, CA) for Gram-negative bacterial RNA, and the rRNA-depleted samples were purified using the Zymo RNA Clean and Concentrator-5 kit.
RNA-Seq libraries were prepared with 10 to 50 ng of each rRNA-depleted sample using the Script-Seq V2 library preparation kit (Illumina) per the manufacturer’s instructions. Single-end barcodes were added to each separate sample for multiplexing using Illumina’s Index Set 1 primer set. Libraries were purified using the Agencourt AMPure XP kit (Beckman Coulter, Brea, CA), and sequenced by the Cornell Genomics Facility using the Illumina HiSeq sequencer. Three libraries were prepared for each condition (promysalin-treated and control). The six libraries were multiplexed and sequenced in a single lane of a flow cell.
RNA-Seq Data Analysis.
Fastq-MCF from GitHub commit 27a4809 of the Expression Analysis package29,30 with default parameters was used to trim the HiSeq adapter and low-quality sequences from the raw Illumina reads. Bowtie2 2.2.931 was used to align the trimmed reads against the P. putida KT2440 genome (NC_002947.4). Custom scripts were used to remove ambiguous reads and make sinister and naive profiles for RNA- seq data as described in Filiatrault et al.32 DeSeq2 1.14.133 was run with a false-discovery rate (FDR) cutoff of 0.05 to assign fold changes to genes.
Bioinformatics Analysis.
Gene annotations and functional assignments were carried out using public databases. Annotations for each of the differentially expressed genes were assigned using the pseudomonas genome database (pseudomonas.com). Functional pathways were assigned using the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway assignments downloaded from http://pseudomonas.com. Gene ontology predictions were made for genes having a (log 2)-fold change in expression of between –0.9 and 0.9 using the web-based tool EggNOG 4.5 (http://eggnog-mapper.embl.de), which assigns genes to functional categories based on orthologous groups. All uncharacterized genes were compared with the NCBI nonredundant nucleic acid database (NT), the NCBI nonredundant protein database (NR http://www.ncbi.nlm.nih.gov/), the Swiss-Prot database (http://www.expasy.ch/sprot), and the Conserved Domain Database (https://www.ncbi.nlm.nih.gov/Structure/cdd/cdd.shtml).
Swimming Motility Assay.
The medium used for assays was TSB that contained 0.3% (w/v) agar and DMSO (control) or promysalin. Swim plates were inoculated with bacteria grown overnight on TSB plates containing 1.5% agar. Bacteria were resuspended in TSB and a sterile needle was used to inoculate the swim plates. Plates were kept at r.t. for 20–24 h and then photographed. Two independent assays were performed with two technical replicates for each independent assay.
Swarming and Swimming Assays.
Pseudomonas putida strain KT2440 was grown overnight on Trypticase soy broth (TSB) agar plates. The swarming medium consisted of TSB with an agar concentration of 0.5% (w/v) and a 100 μM final concentration of promysalin or an equal volume of 10% DMSO. Swarming ability was assessed by spotting 2 μL of a cell suspension of overnight-grown cells onto the surface of the swarming medium and evaluating surface spreading after 20 h at r.t. Swimming plates contained 0.3% agar (w/v) and a 100 μM final concentration of promysalin or an equal volume of 10% DMSO. Swimming ability was assessed by stabbing the swimming medium with an inoculating needle previously dipped into a suspension of overnight-grown cells. Swimming motility was visualized after 20 h at r.t. Each assay was performed in duplicate. (Note: Specifically 1.8 mL of swarming or swimming medium was added to the well, and then either 200 μL of 1 mM promysalin (to give a final concentration of 100 μM) or 200 μl of 10% DMSO (final concentration of 1%) was added. The medium was allowed to solidify for 1 h before inoculating with bacteria.)
Western Blot.
P. putida KT2440 was grown overnight (O/N) in either DMSO (control) or promysalin. Cells were pelleted and prepared in SDS-PAGE sample buffer. SDS-PAGE (4–20% gradient TBX gel; Bio-Rad) was performed, and proteins were transferred to a PVDF membrane, blocked in 5% nonfat milk in phosphate-buffered saline (PBS), and then incubated with monoclonal antibody mabg-flapa (InvivoGen) diluted to 1:1000 in PBS. The blot was washed three times for 5 min each with phosphate-buffered saline solution and then incubated for 1 h with goat-antimouse IgG secondary HRP conjugate (Invitrogen) diluted to 1:10 000 in PBS. The blot was washed with PBS and then developed using 3,3ʹ5,5ʹ- tetramethylbenzidine (TMB) substrate for membranes (Sigma).
Reverse Transcription Quantitative PCR (qRT-PCR).
Overnight cultures of P. putida KT2440 were adjusted to an OD600 of 0.3 and spotted onto TSA plates with 0.5% agar, supplemented with 50 μM promysalin (experimental), or TSB plates with 0.5% agar, supplemented with 50 μM of 10% DMSO solution (control). Plates were allowed to dry on the benchtop and then incubated at r.t. for 24 h. After 24 h, cells were harvested from plates using sterile cotton swabs and homogenized in 600 μL of TRI reagent (Zymo Research). RNA was purified using the Direct-zol RNA purification kit (Zymo Research) following the suggested protocol. Eluted RNA was treated with Ambion DNase I RNase-free enzyme (Thermo Fisher Scientific) and purified using the RNA Clean and Concentrator-5 kit (Zymo Research). One hundred nanograms of total RNA was reverse transcribed in a thermocycler using the iScript cDNA synthesis kit (Bio-Rad, Hercules, CA) according to the manufacturer’s instructions. Real-time PCR was performed by using the My IQ5 sequence detection system (Bio-Rad) and iQ SYBR green Supermix (Bio-Rad) according to the manufacturer’s protocols. The resulting threshold cycle (CT) values were calculated using the My IQ5 software, and relative changes in gene expression were calculated using the ΔΔCT method described in ABI User Bulletin No. 2 (Applied Biosystems, 2001). For each experiment, the CT values of each gene tested were normalized to the CT values of the ribosomal rpsL (PP_0449) reference gene.
Supplementary Material
ACKNOWLEDGMENTS
The U.S. Department of Agriculture (USDA) is an equal opportunity provider and employer. Mention of trade names or commercial products in this publication is solely for the purposes of providing specific information and does not imply recommendation or endorsement by the USDA. This study was supported by the National Institutes of General Medical Sciences (GM119426, W.M.W.), the National Science Foundation (CHE-1755698, W.M.W.), and Temple University (University Fellowship, C.E.K.).
ABBREVIATIONS
- AfBPP
affinity-based protein profiling
- DMSO
dimethyl sulfoxide
- ED
Entner–Doudoroff pathway
- EDTA
ethylene-diaminetetraacetic acid
- EMP
Embden–Meyerhof–Parnas pathway
- FA
formic acid
- FDR
false discovery rate
- HPLC
high-performance liquid chromatography
- KEGG
Kyoto Encyclopedia of Genes and Genomes
- LC
liquid chromatography
- MS/MS
tandem mass spectrometry
- O/N
overnight
- P.aeruginosa
Pseudomonas aeruginosa
- PA
Pseudomonas aeruginosa
- PBS
phosphate-buffered saline
- P. putida
Pseudomonas putida
- PP
Pseudomonas putida
- PPP
pentose phosphate pathway
- qRT-PCR
quantitative reverse-transcription polymerase chain reaction
- RNA
ribonucleic acid
- RNA-seq
ribonucleic acid sequencing
- rpm
revolutions per minute
- r.t.
room temperature
- Sdh
succinate dehydrogenase
- spp.
species
- TCA cycle
tricarboxylic acid cycle
- TSB
trypticase soy broth
- TEAB
tetraethylammonium bromide
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsinfec-dis.8b00041.
AfBPP-enriched proteins; workflow of AfBPP; RNA sequencing reads mapping statistics of cDNA libraries; swimming motility of P. putida KT2440 on swimming medium in the presence of DMSO (control) or promysalin; swimming and swarming assays with promysalin treatment; Western blot of P. putida KT2440 cells when exposed to DMSO (control) or promysalin; volcano plots showing mean counts and fold change of genes involved in iron binding and/or iron homeostasis; and genes involved in iron homeostasis that were down-regulated in P. putida KT2440 with promysalin treatment (PDF)
Relative expression levels measured by qRT-PCR (XLSX)
The authors declare no competing financial interest.
REFERENCES
- (1).Fisher RA, Gollan B, and Helaine S (2017) Persistent bacterial infections and persister cells. Nat. Rev. Microbiol. 15 (8), 453–464. [DOI] [PubMed] [Google Scholar]
- (2).Fletcher J, Bender C, Budowle B, Cobb WT, Gold SE, Ishimaru CA, Luster D, Melcher U, Murch R, Scherm H, Seem RC, Sherwood JL, Sobral BW, and Tolin SA (2006) Plant pathogen forensics: capabilities, needs, and recommendations. Microbiol Mol. Biol. Rev. 70 (2), 450–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Holmes AH, Moore LS, Sundsfjord A, Steinbakk M, Regmi S, Karkey A, Guerin PJ, and Piddock LJ (2016) Understanding the mechanisms and drivers of antimicrobial resistance. Lancet 387 (10014), 176–87. [DOI] [PubMed] [Google Scholar]
- (4).Vidaver AK (2002) Uses of antimicrobials in plant agriculture. Clin. Infect. Dis. 34, S107–10. [DOI] [PubMed] [Google Scholar]
- (5).Gunatilaka AA (2006) Natural products from plant-associated microorganisms: distribution, structural diversity, bioactivity, and implications of their occurrence. J. Nat. Prod. 69 (3), 509–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Li W, Estrada-de los Santos P., Matthijs S, Xie GL, Busson R, Cornelis P, Rozenski J, and De Mot R (2011) Promysalin, a salicylate-containing Pseudomonas putida antibiotic, promotes surface colonization and selectively targets other Pseudomonas. Chem. Biol. 18 (10), 1320–30. [DOI] [PubMed] [Google Scholar]
- (7).Vlassak K, Vanholm L, Duchateau L, Vanderleyden J, and Demot R (1992) Isolation and Characterization of Fluorescent Pseudomonas Associated with the Roots of Rice and Banana Grown in Sri-Lanka. Plant Soil 145 (1), 51–63. [Google Scholar]
- (8).Knouse KW, and Wuest WM (2016) The enantioselective synthesis and biological evaluation of chimeric promysalin analogs facilitated by diverted total synthesis. J. Antibiot. 69 (4), 337–9. [DOI] [PubMed] [Google Scholar]
- (9).Steele AD, Keohane CE, Knouse KW, Rossiter SE, Williams SJ, and Wuest WM (2016) Diverted Total Synthesis of Promysalin Analogs Demonstrates That an Iron-Binding Motif Is Responsible for Its Narrow-Spectrum Antibacterial Activity. J. Am. Chem. Soc. 138 (18), 5833–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Steele AD, Knouse KW, Keohane CE, and Wuest WM (2015) Total synthesis and biological investigation of (–)-promysalin. J. Am. Chem. Soc. 137 (23), 7314–7. [DOI] [PubMed] [Google Scholar]
- (11).Keohane CE, Steele AD, Fetzer C, Khowsathit J, Van Tyne D, Moynie L, Gilmore MS, Karanicolas J, Sieber SA, and Wuest WM (2018) Promysalin Elicits Species-Selective Inhibition of Pseudomonas aeruginosa by Targeting Succinate Dehydrogenase. J. Am. Chem. Soc. 140 (5), 1774–1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Heo A, Jang HJ, Sung JS, and Park W (2014) Global transcriptome and physiological responses of Acinetobacter oleivorans DR1 exposed to distinct classes of antibiotics. PLoS One 9 (10), e110215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Slager J, Kjos M, Attaiech L, and Veening JW (2014) Antibiotic-induced replication stress triggers bacterial competence by increasing gene dosage near the origin. Cell 157 (2), 395–406. [DOI] [PubMed] [Google Scholar]
- (14).Rivera M (2017) Bacterioferritin: Structure, Dynamics, and Protein-Protein Interactions at Play in Iron Storage and Mobilization. Acc. Chem. Res. 50 (2), 331–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Gao T, and O’Brian MR (2005) Iron-dependent cytochrome c1 expression is mediated by the status of heme in Bradyrhizobium japonicum. J. Bacteriol. 187 (15), 5084–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Lim CK, Hassan KA, Tetu SG, Loper JE, and Paulsen IT (2012) The effect of iron limitation on the transcriptome and proteome of Pseudomonas fluorescens Pf-5. PLoS One 7 (6), e39139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Goddard AD, Stevens JM, Rao F, Mavridou DA, Chan W, Richardson DJ, Allen JW, and Ferguson SJ (2010) c-Type cytochrome biogenesis can occur via a natural Ccm system lacking CcmH, CcmG, and the heme-binding histidine of CcmE. J. Biol. Chem. 285 (30), 22882–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Stevens JM, Mavridou DA, Hamer R, Kritsiligkou P, Goddard AD, and Ferguson SJ (2011) Cytochrome c biogenesis System I. FEBS J. 278 (22), 4170–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Tsu BV, and Saier MH, Jr (2015) The LysE Superfamily of Transport Proteins Involved in Cell Physiology and Pathogenesis. PLoS One 10 (10), e0137184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Fluman N, and Bibi E (2009) Bacterial multidrug transport through the lens of the major facilitator superfamily. Biochim. Biophys. Acta, Proteins Proteomics 1794 (5), 738–47. [DOI] [PubMed] [Google Scholar]
- (21).Brocker M, Schaffer S, Mack C, and Bott M (2009) Citrate utilization by Corynebacterium glutamicum is controlled by the CitAB two-component system through positive regulation of the citrate transport genes citH and tctCBA J. Bacteriol. 191 (12), 3869–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Widenhorn KA, Somers JM, and Kay WW (1989) Genetic regulation of the tricarboxylate transport operon (tctI) of Salmonella typhimurium. J. Bacteriol. 171 (8), 4436–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Gallegos MT, Williams PA, and Ramos JL (1997) Transcriptional control of the multiple catabolic pathways encoded on the TOL plasmid pWW53 of Pseudomonas putida MT53. J. Bacteriol. 179 (16), 5024–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Fernandez M, Duque E, Pizarro-Tobias P, Van Dillewijn P, Wittich RM, and Ramos JL (2009) Microbial responses to xenobiotic compounds. Identification of genes that allow Pseudomonas putida KT2440 to cope with 2,4,6-trinitrotoluene. Microb. Biotechnol 2 (2), 287–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Frota CC, Papavinasasundaram KG, Davis EO, and Colston MJ (2004) The AraC family transcriptional regulator Rv1931c plays a role in the virulence of Mycobacterium tuberculosis. Infect. Immun. 72 (9), 5483–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Khoroshkin MS, Leyn SA, Van Sinderen D, and Rodionov DA (2016) Transcriptional Regulation of Carbohydrate Utilization Pathways in the Bifidobacterium Genus. Front. Microbiol. 7, 120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Rowe SE, Campbell C, Lowry C, O’Donnell ST, Olson ME, Lindgren JK, Waters EM, Fey PD, and O’Gara JP (2016) AraC-Type Regulator Rbf Controls the Staphylococcus epidermidis Biofilm Phenotype by Negatively Regulating the icaADBC Repressor SarR. J. Bacteriol. 198 (21), 2914–2924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Kaduskar RD, Scala GD, Al Jabri Z. J. H., Arioli S, Musso L, Oggioni MR, Dallavalle S, and Mora D (2017) Promysalin is a salicylate-containing antimicrobial with a cell-membrane-disrupting mechanism of action on Gram-positive bacteria. Sci. Rep. 7 (1), 8861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Aronesty E (2011) ea-utils: Command-line tools for processing biological sequencing data. Expression Analysis. Durham, NC. Available online at: http://code.google.com/p/ea-utils. [Google Scholar]
- (30).Aronesty E (2013) Comparison of sequencing utility programs. Open Bioinf. J. 7 (1), 1–8. [Google Scholar]
- (31).Langmead B, and Salzberg SL (2012) Fast gapped-read alignment with Bowtie 2. Nat. Methods 9 (4), 357–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Filiatrault MJ, Stodghill PV, Bronstein PA, Moll S, Lindeberg M, Grills G, Schweitzer P, Wang W, Schroth GP, Luo S, Khrebtukova I, Yang Y, Thannhauser T, Butcher BG, Cartinhour S, and Schneider DJ (2010) Transcriptome analysis of Pseudomonas syringae identifies new genes, noncoding RNAs, and antisense activity. J. Bacteriol. 192 (9), 2359–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Love MI, Huber W, and Anders S (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15 (12), 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.