

Graphical Abstract
To the Editor:
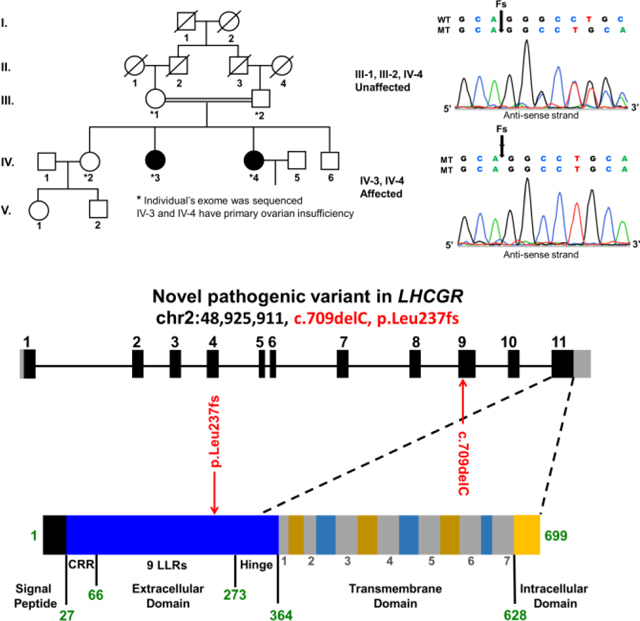
Primary Ovarian Insufficiency (POI) affects 1–2% of women and is a genetically heterogeneous condition characterized by hypergonadotropic hypogonadism, amenorrhea, and infertility. POI patients are evaluated for chromosomal, iatrogenic, endocrine, autoimmune, and infectious causes. Women with POI onset prior to age 20 are more likely to have a strong genetic component [1]. In this study, we investigated a consanguineous Pakistani family belonging to the district Hazara of Khyber Pakhtunkhwa. Parents were first cousins and had three daughters and one son. Two daughters presented with primary amenorrhea, hypergonadotropic hypogonadism, and normal 46, XX karyotypes. The Ethical Review Board of Khyber Medical University, approved this study and all participants provided informed consent.
The first proband presented at 20 years of age with primary amenorrhea and hormonal evaluation was consistent with hypergonadotropic hypogonadism. Luteinizing hormone (37.50 IU/L; normal follicular range is 1.8–11.78 IU/L) and follicle stimulating hormone (26.30 IU/L; normal range is 3.03–8.08 IU/L) levels were elevated, while estradiol (25 pg/mL; normal range is 21–251), anti-mullerian hormone (1.51 ng/mL; normal range is 1–4), and testosterone (17.52 ng/dL; normal range is 11–57) levels were normal. Ultrasonography showed an anteverted uterus measuring 5.5×2×3.3 cm with a thin endometrium of 0.2 cm and normal sized ovaries with an anechoic cyst (8 cm3) in the left ovary.
The second proband presented at 24 years of age with primary amenorrhea, hypergonadotropic hypogonadism, and had a history of hot flushes. Estradiol (48 pg/mL) and testosterone (31.83 ng/dL) levels were normal, anti-mullerian hormone (1.12 ng/mL) levels were relatively low, while follicle stimulating hormone (24.42 IU/L) and luteinizing hormone (32.16 IU/L) levels were high. Ultrasonography showed a slightly small uterus with a thin endometrium. The left ovary was of normal size with few follicles and two anechoic simple cysts (12 cm3; 18 cm3) were seen in the right.
We performed whole exome sequencing (WES) on five family members (parents and three daughters) with an average coverage of 150 reads. We assumed autosomal recessive inheritance, with a minor allele frequency of less than 0.01%, which identified a total of seven potential variants. Four were synonymous or intronic with unlikely splicing effects, while three were exonic and non-synonymous in LHCGR (MIM: 152790), PCLO (MIM: 604918), and OXER1 genes. The luteinizing hormone/choriogonadotropin receptor (LHCGR) was the only gene known to be associated with reproduction.
The LHCGR (chr2: 48,925,911, c.del709C, p.Leu237fs) variant was not present in dbSNP, HGMD, or general population variant databases (ExAC and gnomAD). Both patients were homozygous for the c.709delC variant, whereas the parents and unaffected sister were heterozygous, consistent with known recessive inheritance for LHCGR associated pathologies (Table 1). All individuals had high quality sequencing coverage (>35x) in this region and Sanger sequencing confirmed each genotype. The c.del709C variant is located in exon 9 and encodes a conserved leucine-rich repeat (LRR-8). The extracellular domain especially the LRR regions, is essential for LH/hCG binding affinity and specificity[2]. This variant (p.Leu237CysTer5) results in a frame-shift at amino acid 237 and is predicted to truncate the regularly sized 699 amino acid wild-type LHCGR protein. The truncated LHCGR protein lacks the highly conserved serpentine transmembrane, which is responsible for signal transduction[2]. Therefore, it is likely a loss of function variant, and co-segregation with the POI phenotype in the pedigree, further supports its likely pathogenicity.
Table 1:
Reported LHCGR inactivating variants associated with female infertility and relevant phenotypes.
| Variant | Inheritance pattern | Amenorrhea | Hormone analysis | Reproductive tract | Affected family members and associated diagnoses | Ethnic group |
|---|---|---|---|---|---|---|
| c.54_55insCTGCCG p.Gln18_Pro19insLeuPro |
Dominant | Secondary | Normal | Recurrent ovarian cysts and inverted uterus | No siblings of same genotype | Canadian |
| c.34_60del / c.1756_1758delTC p.Lys12_Pro20del / p.Ser586fs |
Compound Heterozygous | Primary | Normal | Normal uterus size Normal sized cystic ovaries |
Individual diagnosed with polycystic ovary syndrome; pseudo-hermaphroditism in one 46, XY sibling | Canadian |
| c.709delC p.Leu237fs |
Recessive | Primary | High LH/FSH levels | Normal sized cystic ovaries Small atrophic uterus |
Two sisters with POI | Pakistani |
| c.1060G>A p.Glu354Lys |
Recessive | Primary | High LH levels Low E2 levels |
Normal | Pseudo-hermaphroditism in two 46, XY siblings | Dominican |
| c.1199A>G p.Asn400Ser |
Recessive | Oligo | Normal | Normal | Study reported two sisters with empty follicle syndrome | Turkish |
| c.1345G>A p.Ala449Thr |
Recessive | Oligo | Normal | Normal uterus Ovaries of unequal sizes |
Individual diagnosed with empty follicle syndrome | Chinese |
| c.1660T>C p.Arg554ter |
Recessive | Primary | High LH levels Low E2 levels |
Small uterus Unequal sized cystic ovaries |
Pseudo-hermaphroditism in three 46, XY siblings | Brazilian |
| c.1777G>C p.Ala593Pr006F |
Recessive | Primary | High LH/FSH levels Low E2/progesterone levels |
Enlarged right ovary with cyst | Pseudo-hermaphroditism in two 46, XY siblings | Brazilian |
| c.1822_1827del p.Leu608_Val609del |
Recessive | Oligo | High LH levels | Enlarged right ovary with cyst and normal uterus | Pseudo- hermaphroditism in one 46, XY sibling | Brazilian |
Note: Phenotyping information was acquired from the following PMIDs: 22369774, 24849377, this study, 9626144, 21683950, 28175319, 8559204, 8923827, and 9514160, respectively.
LHCGR variant phenotype is confined to the ovaries and results in non-syndromic infertility. We report the tenth variant in LHCGR associated with female infertility and amenorrhea. Previous reports of women with LHCGR variants described phenotypes of infertility, cystic ovaries, empty follicles, and primary/secondary amenorrhea (Table 1) in Brazilian, Canadian, Chinese, Dominican, and Turkish individuals [3, 4]. To our knowledge, this novel variant in the LHCGR gene was first to be described in a Pakistani family, which further supports the importance of LHCGR function in female reproduction across diverse ethnic groups.
Acknowledgements:
The authors thank the family for their participation. Funding was provided by the Eunice Kennedy Shriver National Institute of Child Health and Human Development (HD070647 and HD074278 to A.R. and HD080755 to A.Y.), the Higher Education Commission of Pakistan (International Research Support Initiative Program to M.J.K), and the Magee-Womens Research Institute T32 training program on Reproductive Development from Gonads to Fetuses (HD087194 to A.C.Z)
Footnotes
Conflict of Interest: None declared.
References
- 1.Rebar RW, Premature ovarian “failure” in the adolescent. Annals of the New York Academy of Sciences, 2008. 1135(1): p. 138–145. [DOI] [PubMed] [Google Scholar]
- 2.Ascoli M, Fanelli F, and Segaloff DL, The lutropin/choriogonadotropin receptor, a 2002 perspective. Endocrine reviews, 2002. 23(2): p. 141–174. [DOI] [PubMed] [Google Scholar]
- 3.Latronico AC and Arnhold IJ. Inactivating mutations of the human luteinizing hormone receptor in both sexes. in Seminars in reproductive medicine 2012. Thieme Medical Publishers. [DOI] [PubMed] [Google Scholar]
- 4.Yuan P, et al. , Genetic evidence of ‘genuine’empty follicle syndrome: a novel effective mutation in the LHCGR gene and review of the literature. Human Reproduction, 2017. 32(4): p. 944–953. [DOI] [PubMed] [Google Scholar]