

Abstract
Fusarium oxysporum is an economically important pathogen that widely exists in the environment and is capable of causing serious problems in crop production and animal/human health. One important step for characterization of a fungal protein with an unknown function is to determine its subcellular localization within the cell. To facilitate the study of important functional regulators or key virulence factors, we developed a CRISPR/Cas9-mediated endogenous gene tagging (EGT) system based on two different strategies, homology-independent targeted integration (HITI) and homology-dependent recombination integration (HDRI). The HITI strategy was able to facilitate integration of a large DNA fragment, ~8 kb in length, into the genome of F. oxysporum at the sgRNA cleavage site, and was used to insert a C-terminal 3×sGFP tag to the FoCHS5 gene and a N-terminal mCherry tag to the FoSSO2 gene. The HDRI strategy was used to tag the paralogous gene, FoSSO1, with a C-terminal mCherry marker. FoChs5–3×sGFP localized to conidia, some septa, and fungal tips. A majority of the FoSso1-mCherry was distributed in the conidia, septum, and hyphae that were distal from the fungal tips. While FoSso1-mCherry showed a very weak fluorescent signal at the fungal tips, mCherry-FoSso2 accumulated in the plasma membrane of conidia, germlings, fungal tips, hyphae, and phialides, suggesting FoSSO1 and FoSSO2 are regulated differently during fungal development. These results indicate this EGT system is efficient and can be another molecular tool to resolve the function(s) of proteins and infection strategies of F. oxysporum.
Keywords: CRISPR, endogenous gene tagging (EGT), homology-independent targeted integration (HITI), homology-dependent recombination integration (HDRI), SNARE
Graphical Abstract
1. Introduction
Members of the Fusarium oxysporum species complex (FOSC) are soil-borne pathogens which pose a serious threat to agricultural production and human health (Gordon, 2017). In agriculture, F. oxysporum is capable of causing several diseases, most notably Fusarium wilt resulting from colonized plant xylem tissue. Infected plants show a range of symptoms including chlorosis, necrosis, stunting, and wilting (Gordon, 2017). In addition, these fungi can be opportunistic human pathogens infecting immunocompromised patients with high mortality rates (Muhammed et al., 2013; Nucci and Anaissie, 2007).
Rapid improvement of high-throughput DNA and RNA sequencing techniques has facilitated large scale investigation of various biological processes, including fungal pathogenicity, utilizing whole genome and/or transcriptome analysis. Currently, several high-quality, well-annotated F. oxysporum genomes are publicly available, and enable further exploration of fungal pathogenesis and disease control (Ma et al., 2010; van Dam et al., 2017). Experimental evidence has shown that horizontal transfer could have contributed to the genetic diversity among FOSC isolates, as mobile chromosomes harboring virulence factor genes were able to expand the host range of the recipient and increase their environmental niche (Ma et al., 2010; Vlaardingerbroek et al., 2016).
Molecular manipulation of genes and proteins are essential to fully understand the role they have in an organism, and some of the more common techniques to elucidate cellular functions include gene deletion, RNAi knockdown, ectopic gene expression, and protein subcellular localization. The subcellular localization of a protein can greatly influence the function, as improperly localized proteins, including those that have lost their subcellular localization peptides, may result in decreased or abolished protein activity (Scott et al., 2005). Several studies have indicated that cellular location of a fungal protein is critical for proper function inside the cell. An example is the localization of the well characterized calcineurin-regulated transcription factor Crz1 which ultimately controls transcription of several stress response genes and cell wall integrity in most fungi (Lev et al., 2012; Schumacher et al., 2008; Spielvogel et al., 2008). Upon an increase in intracellular calcium levels, Crz1 is dephosphorylated resulting in rapid localization into the nucleus activing the expression of downstream genes.
Studies of fungal protein subcellular localization may rely on several methods, where the most commonly employed method is the use of fluorescent markers including GFP, eGFP, RFP, YFP, and mCherry. These fluorescent proteins are usually fused in frame with the coding sequence of the gene of interest and randomly inserted into the fungal genome under the control of a constitutive or native promoter (Gupta et al., 2015; Schuster et al., 2016). Based on different excited marker fluorescence, protein subcellular localization can be determined and protein translocation can be dynamically tracked (Schuster et al., 2016). While this method enables the protein subcellular localization to be detected or translation of the protein monitored, some limitations still exist including: 1) native promoters may not have been accurately predicted upstream of the coding regions, resulting in no or altered expression of the protein fused with tags; 2) protein expression that is driven by constitutive promoters might cause inaccurate protein subcellular localization, as constitutive expression may disrupt post-translational regulation; 3) ectopic insertion may disrupt other genes or important regions in the fungal genome, resulting in inaccurate protein subcellular localization or dynamic gene expression, necessitating multiple transformants to be assessed. While antibody-based immunofluorescence approaches to determine the subcellular localization of proteins exist (Rasmussen and Glass, 2005), the difficulty in obtaining an antibody with high specificity, compounded by the time required to generate the antibody, have limited the application of this approach.
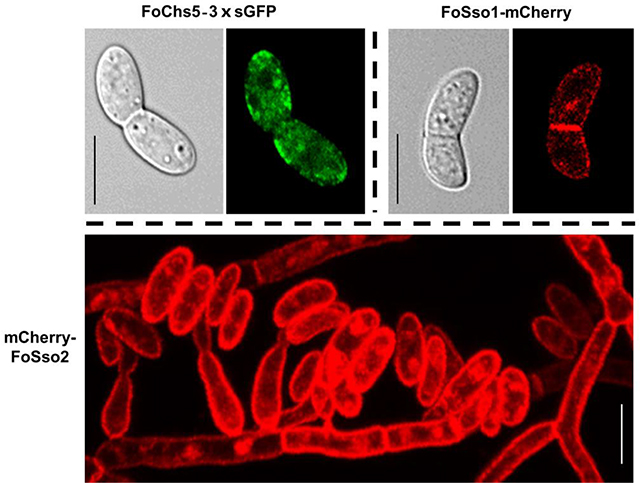
As molecular methods to generate gene fusions are an important component for functional studies, a rapid and efficient endogenous gene tagging system (EGT) in F. oxysporum was developed based on a CRISPR/Cas9 ribonucleoprotein complex transformation method (Wang et al., 2018). With this system, we could not only localize the protein encoded by the endogenous genes of interest, but also detect the relative protein abundance at various fungal cellular structures. Different fluorescent markers were integrated prior to the start/stop codon of the gene, allowing gene fusions to be generated with the gene under its native promoter, at the usual location within the genome, and containing the gene introns. Two strategies, a homology-independent targeted integration (HITI) and homology-dependent recombination integration (HDRI), were used in the development of the EGT system with fluorescent markers including sGFP (a variant of eGFP) and mCherry. The HITI strategy was initially developed based on the non-homologous end joining (NHEJ) DNA repair approach (Suzuki et al., 2016). We used this strategy to tag endogenous genes at either the N-terminus or the C-terminus, and generated FoChs5–3×sGFP (C-terminus tagging) and mCherry-FoSso2 (N-terminus) transformants. The protein subcellular localization showed FoChs5 in F. oxysporum was present in both conidia and hyphae, where a majority accumulated at the hyphal tips although some was present in the septum. FoChs5 was largely found on the surface of newly formed germ tubes, suggesting Chs5 plays an important role in cell wall modelling. The HDRI strategy was used to tag the plasma membrane syntaxin 1 SNARE protein FoSso1 with a mCherry marker at the C-terminus. FoSso1-mCherry was localized to the cell plasma membrane (PM) of hyphae and the septum of conidia while mCherry-FoSso2 showed strong PM localization in conidia, germlings, fungal tips, hyphae, and phialides. Overall, this CRISPR/Cas9 mediated EGT system was highly efficient and stable for use in protein subcellular localization studies and could provide a genetic engineering reference for gene tagging in other filamentous fungi.
2. Materials and methods
2.1. Strains and culture conditions.
The Fusarium oxysporum f.sp. vasinfectum strain (FGSC #10442) was used in this study (McCluskey et al., 2010), and was cultured in PDB (Potato Dextrose Broth), YG (1% yeast extract, 3% glucose) or M-100 (minimum medium). All transformants in this study were isolated on M-100 agar plates containing 75 μg/mL hygromycin B before observation by confocal microscopy. All transformed strains generated in this study are listed in Table A.1.
2.2. Homologous gene identification.
Two well-annotated CHS5 genes from Neurospora crassa (NCU04352) and Ustilago maydis (UM03204.1) were used as query sequences to identify the homolog of CHS5 in F. oxysporum with blastp in the EnsembleFungi database (https://fungi.ensembl.org/index.html). Several previously identified syntaxin 1 SNARE proteins from Saccharomyces cerevisiae (SSO1: YPL232W and SSO2: YMR183C), N. crassa (SSO1: XP_961235.1) and Zymoseptoria tritici (SSO1: XP_003857391.1) were used as the blastp queries to identify the homologous sequences in the above database. Since these genes perform similar functions during fungal development, their amino acid sequences were hypothesized to be conserved. To determine the sequence identity or splice variants, the potential homologous genes were compared with associated genes in other closely-related filamentous fungal pathogens which had well-annotated genomes including Fusarium oxysporum f. sp. lycopersici 4287, Magnaporthe oryzae 70–15, Fusarium graminearum PH1, and Fusarium solani (Nectria haematococca MPVI 77–13-4). The protein domains were predicted using PFAM (Finn et al., 2016), and multiple sequence alignments were conducted using the online bioinformatic program TCOFFEE (Notredame et al., 2000). The phylogenetic tree was generated using PhyML 3.1 with the maximum likelihood algorithm and 500 bootstraps (http://www.phylogeny.fr/one_task.cgi?task_type=phyml) (Dereeper et al., 2008). All accession numbers of predicted genes in this study are listed in Table A.1.
2.3. The evaluation of fusion protein topology and plasmid construction.
Since the integrated location of the different fluorescent protein markers may alter the original protein topology, the TMHMM v. 2.0 analysis software (http://www.cbs.dtu.dk/services/TMHMM/) was used to compare the differences of protein topology between the WT proteins and the fusion proteins. All plasmids were maintained, propagated, and stored in DH5α E. coli. All tagging plasmid constructs were based on a pUC19 plasmid, and cloned fragments were inserted between two enzyme sites, BamHI and HindIII, with the NEBuilder® HiFi DNA Assembly Master Mix kit (New England Biolabs). For the CHS5 gene tagging vector construction (HITI), the initial vector was first constructed by amplifying the following three fragments by PCR: 1) 1× sGFP from the plasmid pCT74 (Lorang et al., 2001), 2) the Tef-1 terminator from the plasmid pFFC332, and 3) the hygromycin cassette from the plasmid pCWHyg1. These three fragments were simultaneously inserted between the BamHI and HindIII sites in the pUC19 vector in the following order 1×sGFP-Tef-1-HYGB generating the primary vector pUC19–1×sGFP-Tef-1-HYGB. The final CHS5 HITI vector was constructed as follows: 1) a ~700 bp coding sequence prior to the stop codon of FoCHS5 was cloned as a bait sequence which contained a sgRNA cleavage site; 2) to construct a triple sGFP in the vector, the first copy of sGFP (1st sGFP) was cloned with the addition of a 3′-terminus marker sequence (nt: GACTACAAGGACGAC, aa: DYKDD) while the second copy of sGFP (2nd sGFP) was cloned with the addition of the 3′-terminus marker sequence (nt: AGCGCTGGCGCTTAC, aa: SAGAY); 3) the fragment of 1×sGFP-Tef-1-HYGB was cloned from the above primary plasmid; and 4) these four fragments were simultaneously inserted into the pUC19 plasmid to generate the final HITI plasmid containing the triple sGFP tag, which was named as pUC19-HITI-FoCHS5–3×sGFP.
The detailed N-terminal FoSSO2 gene tagging vector for HITI was constructed as follows: 1) a ~800 bp sequence in front of the FoSSO2 gene start codon was cloned with a sgRNA site; 2) 1 × mCherry was cloned from plasmid ss3591; 3) the FoSSO2 coding sequence including introns and a ~ 450 bp 3-UTR region were amplified from genomic DNA; 4) the hygromycin cassette was obtained from plasmid pCWHyg1; 5) the above four fragments were simultaneously inserted into the pUC19 plasmid, which was later labeled as pUC19-HITI-mCherry-FoSSO2.
The FoSSO1 C-terminal tagging plasmid contained a mCherry tag fused with a Tef-1 terminator and hygromycin resistance cassette. The detailed HDRI vector construction was carried out as follows: 1) a ~400 bp DNA template fragment prior to the gene stop codon was cloned from the genomic DNA (upstream sequence); 2) the other DNA fragment after the sgRNA cleavage site, ~400bp in length, was obtained by PCR (downstream sequence); 3) the mCherry was cloned from plasmid ss3591 and the Tef-1 terminator fused with the hygromycin cassette was cloned from pUC-HITI-FoCHS5–3×sGFP as constructed above; 4) these four fragments were assembled in the following order, upstream sequence, mCherry, Tef-1 fused with the hygromycin cassette, and downstream sequence, in pUC19 (pUC19-HDRI-FoSSO1-mCherry). All primers used in this study are listed in Table A.2.
2.4. sgRNA selection and in vitro Cas9 nuclease assay.
The sgRNA sites were located after the stop codon between 0 and 200 bp for the HDRI strategy (FoSSO1-mCherry), while those for C-terminal tagging of the FoCHS5 gene and N-terminal tagging of the FoSSO2 gene were located prior to the gene stop or start codon, respectively. All sgRNA sequences and PAM motifs are listed in Table A.1. A ~2kb fragment was cloned for use as a template for the in vitro Cas9 nuclease assay to confirm the Cas9 endonuclease cleavage activity for the synthesized sgRNA. The in vitro Cas9 nuclease assay was performed as previously described (Wang et al., 2018). In brief, a mixture of 200 ng Cas9, ~40 ng sgRNA, 1×Cas9 Nuclease Reaction Buffer, and ~ 75 ng DNA template in a 20 μL total volume was incubated at 37°C for 1 h. The cleaved products were evaluated on a 1% agarose gel. All sgRNA were able to effectively cleave PCR fragments with the Cas9 nuclease (Figs. S1C and S2B).
2.5. Fungal transformation and transformant identification.
Transformation of F. oxysporum was carried out as previously described (Wang et al., 2018). For the HDRI strategy, PCR fragments were amplified from the donor plasmids (pUC19-HDRI-FoSSO1-mCherry and pUC19-HDRI-FoSSO2-mCherry) with the primers, NA_FoSSO1upF/ NA_FoSSO1downR and NA_FoSSO2upF/ NA_FoSSO2downR, and used to conduct the transformations. The desired transformants will contain the gene of interest fused with the different marker tags under the native promoter, and were screened by three PCR reactions to determine the location of the integrated fragment in the fungal genome. Detailed illustrations of correct transformants are shown in Fig. S1A and S2A. The first pair of primers, Tf and Tr, were used to assess the full length near the sgRNA cleavage site after the fragments were integrated. Another two pairs of primers were used to determine the upstream and downstream sequences of the inserted fragments. All primers used are listed in Table A.2.
2.6. Confocal microscopy
Transformant hyphae were picked and inoculated into 50 mL YG media and cultured for 4 days at room temperature on a rotary shaker at 150 rpm. The 500 μL hyphae-conidia mixture of the F. oxysporum transformants were inoculated into 50 mL of fresh YG media and incubated on a shaker at 18°C at 220 rpm for 16 h. One mL of the fungal culture was centrifuged at 13,000 rpm for 1 min and the supernatant removed. The resulting pellet was suspended in 500 μL of 4% paraformaldehyde and the mixture was incubated at room temperature at least 15 min, followed by at least two washes of PBS, pH 7.4. Transformants were suspended in a final volume of 300 μL of PBS, pH 7.4 and imaged by confocal microscopy using an inverted agar method under 60 × oil magnification (Nikon A1 Confocal Microscopy) (Hickey and Read, 2009). For live cell imaging, the fungus was cultured in a four-chambered coverglass (Thermo Scientific™ Nunc™ Lab-Tek™ Chambered Coverglass) or the live fungal cells were dispensed on the surface of a 0.5 cm × 0.5 cm piece of agar on a slide and covered with the coverglass. The sGFP fluorescent marker was excited at 488 nm while the mCherry fluorescent marker was excited at 561 nm.
3. Results and Discussion
3.1. Identification of FoChs5, FoSso1, and FoSso2
Orthologs to several well-characterized genes in other filamentous fungi were selected in F. oxysporum to develop an EGT system to study subcellular localization. The well characterized localization of the transmembrane chitin synthases (CHSs) and their importance in production of the fungal cell wall component chitin, make this group of enzymes ideal to use in the development of an EGT system. Nine CHS genes reside in the F. oxysporum genome and the presence of multiple copies of CHSs suggest functional complexity exists in regards to chitin biosynthesis in these fungi (Kong et al., 2012). Among these CHSs, the class V CHS (CHS5) usually contains several protein domains including a Myosin head, Cyt-b5, Chitin_synth_2, and DEK C domains (Fig. 1A); importantly, this class of CHS enzymes have been shown to be involved in pathogenesis and vesicle transportation (Madrid et al., 2003; Treitschke et al., 2010). Among these domains, the myosin domain is involved in traversing on actin filaments and microtubules while the chitin synthase domain participates in chitin synthesis (Schuster et al., 2016; Treitschke et al., 2010). To characterize the subcellular localization of FoChs5 in different fungal structures and stages of development, the orthologous gene (FOTG_09933) in the F. oxysporum genome was identified based on the CHS5 genes from other fungi. FoChs5 shares 83.3% and 52.8% amino acid similarity to Chs5 in N. crassa and Mcs1 in U. maydis, respectively; supporting Chs5 performs similar functions among these fungi.
Fig. 1.

Identification of the FoChs5, FoSso1, and FoSso2 proteins in F. oxysporum. (A) The protein domain structures of the chitin synthase FoChs5, and the two Qa SNARE syntaxin proteins FoSso1 and FoSso2. (B) The maximum-likelihood phylogenetic tree of Sso1 and Sso2 amino acid sequences from select filamentous ascomycete fungi. The tree was generated with a bootstrap value of 500.
In addition to FoCHS5, we chose the duplicated genes SSO1 and SSO2, that encode homologues of syntaxin 1, as representative genes to develop this EGT system based on their known localization to membranes (Kienle et al., 2009). Syntaxins are Qa SNARE transmembrane proteins that play essential roles in membrane fusion events (Salaun et al., 2004). In the yeast S. cerevisiae, the SSO1 and SSO2 gene products are involved in the fusion between secretory vesicles and the plasma membrane. Deletion of SSO1 halted vesicle fusion and decreased sporulation efficiency, while mutation of SSO2 resulted in increased sensitivity to chemicals and temperature (Jantti et al., 2002; Nakanishi et al., 2006). Syntaxin 1 and its homologues contain, in their cytosolic region, an N-terminal syntaxin domain followed by a TMD-proximal SNARE domain (Fig. 1A), and the syntaxin Sso1 has been used for characterization of the plasma membrane in some fungi (Kilaru et al., 2017; Schuster et al., 2016; Taheri-Talesh et al., 2008; Valkonen et al., 2007). To identify the homologous genes of syntaxin 1, several previously described syntaxin 1 genes from S. cerevisiae, U. maydis, and Z. tritici were used as a query for the FoSSO1 and FoSSO2 gene sequences (Kilaru et al., 2017). FoSso1 and FoSso2 contained the two typical domains (syntaxin and SNARE) and shared ~44% amino acid sequence similarity between the two proteins. Although some strains of Aspergillus species only contain a single copy of the SSO genes (Kienle et al., 2009), phylogenetic analysis revealed that a majority of the ascomycete fungi encode two copies of the SSO genes, and can be resolved into two distinct phylogenetic clades suggesting that these two genes diverged some time ago and have since evolved to function differently (Fig. 1B). Both copies of the SSO genes were highly conserved among Fusarium spp. and were clustered in the phylogenetic tree supporting that the presence of two SSO genes is ancestral in this genus.
3.2. HITI-mediated endogenous gene tagging at the C-terminus of FoCHS5
A homology-independent targeted integration (HITI) strategy was developed utilizing CRISPR/Cas9 technology (Wang et al., 2018), enabling long DNA fragments to be inserted into the genome using the NHEJ repair mechanism. The main advantage of HITI is that it can occur in non-dividing cells, and has been validated in many cell types including human cell lines and zebrafish (Auer et al., 2014; Suzuki et al., 2016). The HITI strategy was used to insert a 3×sGFP (a variant of eGFP) tag before the FoCHS5 stop codon (C-terminal tagging), generating a fusion protein consisting of FoChs5 and a strong green fluorescent signal for protein subcellular localization. The donor plasmid DNA was composed of the following: a ~700 bp homologous fragment containing the sgRNA cleavage site before the FoCHS5 stop codon was fused with a 3×sGFP sequence, a Tef-1 terminator sequence, and the hygromycin resistance cassette sequence (Fig. 2A). During the transformation, both the genome and donor plasmid sequences can be simultaneously cleaved by the Cas9 RNPs. The activated DNA repair mechanism allows “cross-ligation” between the chromosome and the donor plasmid at some frequency (between C1—P2 and P1—C2, Fig. 2A). The desired transformants will contain the complete FoCHS5 endogenous gene sequence fused in frame with the 3×sGFP tag followed by the remaining portion of the donor plasmid containing the hygromycin resistance cassette for selection of the desired transformants (Fig. 2A).
Fig. 2.

FoCHS5 gene C-terminal tagging with a 3×sGFP fluorescent marker based on the HITI strategy. (A) The scheme for the homology-independent targeted integration (HITI) strategy of the C-terminal tagging of the FoCHS5 gene. A bait sequence, ~700 bp in length containing the sgRNA cleavage site, was cloned prior to gene stop codon and ligated to a 3×sGFP marker and the hygromycin resistance cassette. During transformation, two DNA cross–ligation events will occur between the donor plasmid and the fungal chromosome (C1-P2 and P1-C2). The whole plasmid will be inserted into the sgRNA cleavage site on the fungal chromosome in the desired transformants. The C1-C2 fragment is homologous to the P1-P2 fragments B) FoChs5–3×sGFP subcellular localization in different fungal structures. a and b: conidia, c and d: conidia with emerging germination tubes, e: fungal tips, f: hyphae, Scale bars, 10 μm.
Previous evidence indicated the HITI strategy allowed perfect repair of DNA at a higher frequency, and that error-prone DNA repair occurred infrequently (Auer et al., 2014; Suzuki et al., 2016). We hypothesized if the Cas9 RNP transformation of F. oxysporum protoplasts only allowed a miniscule amount of the Cas9 RNPs into the cell and the Cas9 RNPs carried out the cleavage a short time before their degradation, some proportion of the transformants would have perfect DNA repair occurring between the cleavage sites of the chromosome and donor plasmid. After transformation, two of the four generated transformants were used for PCR analysis to confirm that the whole plasmid (~ 8 kb) was inserted into the genome at the sgRNA cleavage site (Fig. S1D). In theory, the HITI-mediated gene editing may generate short losses or additions of nucleotides at the repair sites (Auer et al., 2014; Suzuki et al., 2016). However, sequencing of the sgRNA target sequence site of the two transformants showed identical nucleotide sequence with the wide-type strain (Fig. S1B), demonstrating that the NHEJ DNA repair mechanism for some F. oxysporum protoplast cells would be error free, consistent with previous publications (Auer et al., 2014; Suzuki et al., 2016).
The subcellular localization of FoChs5 revealed that the protein had variable distribution and accumulated in some areas (Fig. 2B). In conidia, FoChs5–3×sGFP accumulated at both ends of the spore (Fig. 2B a and b); however in F. oxysporum germlings FoChs5–3×sGFP was distributed in the cell membrane primarily at the newly formed hyphal tips emerging from the conidium showing that FoChs5, as a class V myosin chitin synthase, was strongly polarized (Fig. 2B c and d). In addition, confocal microscopy showed that FoChs5 localized to the hyphal tips and septa (Fig. 2B e and f, Fig. S3, Mov. S1). As FoChs5 is polarized and contains a myosin motor domain, it may have a similar function to Mcs1 of U. maydis and participate in the transportation of vesicles (Treitschke et al., 2010).
3.3. HDRI-mediated endogenous gene C-terminal tagging of FoSSO1
Our previous study showed Cas9 RNPs significantly improved the efficiency of homologous recombination (Wang et al., 2018). FoSSO1 was tagged with a mCherry peptide at the 3′-terminus of the genes by a homology-dependent recombination integration (HDRI) strategy. The sgRNA site was located after the gene stop codon (Fig. 3A). The donor DNA was constructed by the following: an upstream homologous fragment, ~400 bp in length, immediately prior to the gene stop codon was cloned and ligated with the mCherry-Tef-1-HYGB and a ~400 bp downstream homologous fragment after the sgRNA cleavage site. During the transformation with the donor DNA fragments and Cas9 RNPs, two homologous recombination events were expected to occur at the sgRNA cleavage site (Fig. 3A). The desired transformants will express the Sso-mCherry fusion protein and can be selected for by hygromycin resistance.
Fig. 3.

FoSSO1 gene C-terminal tagging with a mCherry fluorescent marker based on the HDRI strategy. (A) An illustration of the homology-dependent recombination integration (HDRI) strategy for the C-terminal tagging of the FoSSO1 gene. A homologous upstream fragment was amplified prior to the gene stop codon and fused with mCherry, the hygromycin resistance cassette, and a homologous downstream fragment that is immediately after the sgRNA cleavage site. During transformation, two homologous recombination events are necessary to occur and the mCherry-Tef-1-HYGB fragment will be integrated at the 3′-terminus of the endogenous gene. (B) the FoSso1-mCherry subcellular localization in different fungal structures. a and b: conidia, c and d: germlings, e and f: fungal tips and hyphae. Gradient red arrows weaken from strong to weak, indicating the different distribution of the FoSso1 protein in relation to the fungal hyphal tip. Scale bars, 10 μm.
Since the transformation efficiency was significantly increased with the CRISPR/Cas9 RNPs (Wang et al., 2018), three potential FoSSO1-mCherry transformants were randomly selected for further analysis. All of the transformants possessed the desired fusion (Fig. S2C) and produced a fluorescence signal when excited at a wavelength of 561 nm under confocal microscopy. In the previous study when the donor plasmid was directly used for the protoplast transformation, the maximum correct integrated frequency was ~50% (Wang et al., 2018). However, all our selected HDRI transformants contained the correctly integrated fragments at the expected sites, suggesting that PCR fragment transformation might have a higher integration efficiency in F. oxysporum than transformation with plasmid donors. In conidia, FoSso1-mCherry primarily accumulated at the septum and the cell plasma membrane (Fig. 3B a and b). FoSso1-mCherry had no specific accumulation except for the septum in the germinating conidia and showed a weak signal in the newly-formed hyphal tip (Fig. 3B c and d). Conversely, FoSso1-mCherry accumulated in well-formed, mature hyphae distal from the fungal tips, whereas in the newly formed hyphae and fungal tips there was very poor FoSso1-mCherry fluorescence (Fig. 3B e and f). To further confirm the FoSso1-mCherry subcellular localization, the Z-series scanning function on the confocal microscopy was used to observe the fungus at a fluorescent range of 3.2 μm with a step of 0.4 μm (Fig. S4, Mov. S2). The Z-series stack indicated that all three fungal tips emitted weak fluorescent signals while the septa and hyphae distal from the fungal tips had strong fluorescence accumulation.
3.4. HITI-mediated endogenous gene tagging at the N-terminus of FoSSO2
Previous studies have tagged SSO2 othologs at the N-terminus to avoid the fluorescent protein residing in the lumen of exocytic vesicles, which can result in improper folding of the protein tag (Taheri-Talesh et al., 2008; Valkonen et al., 2007). To further confirm the subcellular localization of FoSso2 proteins, we tagged the FoSSO2 gene at the N-terminus using the HITI strategy. The donor DNA was composed of a ~800 bp fragment prior to the FoSSO2 start codon containing a sgRNA cleavage site which was fused with the mCherry coding sequence fused in frame with the FoSSO2 coding sequence, including the introns and the 3′-UTR sequence, followed by the hygromycin resistance cassette (Fig. 4A). During the transformation the entire plasmid is intended to be integrated at the sgRNA cleavage site, and the desired transformants will contain an intact endogenous FoSSO2 gene fused with a mCherry marker gene at the N-terminus (Fig. 4A). The number of transformant colonies generated using the Cas9 RNPs was much greater than those without the Cas9 RNPs (Fig. 4B, Table A.3), indicating that the addition of the Cas9 RNPs significantly enhanced transformation efficiency. A total of 49 independent colonies were selected and screened for detection of mCherry fluorescence, and only five transformants failed to produce any detectable fluorescent signal under confocal microscopy (positive fluorescent efficiency was ~90% (44/49)). Nine randomly selected mCherry-FoSSO2 transformants were further assessed for precise integration at the expected site in the genome by PCR (data not shown) and sequencing the locus. Sequencing revealed eight of the transformants were correct, having the desired mCherry-FoSSO2 gene fusion, while a single transformant contained a 3-bp nucleotide deletion at the sgRNA cleavage site, although the resulting transformant was still fluorescent.
Fig. 4.

FoSSO2 gene tagging with a N-terminal mCherry fluorescent marker based on the HITI strategy. (A) The scheme for the HITI strategy of the N-terminal tagging of the FoSSO2 gene; a ~800 bp homologous sequence containing the sgRNA cleavage site in front of the FoSSO2 gene start codon was cloned and ligated to the mCherry marker coding sequence, the FoSSO2 endogenous gene, the 3′-UTR region of the FoSSO2 gene, and the hygromycin resistance cassette. During transformation, the entire plasmid will be inserted at the sgRNA cleavage site generating the N-terminal mCherry reporter for FoSSO2. (B) Comparison of the transformation efficiency with or without Cas9 RNPs for the FoSSO2 N-terminal tagging. (C) mCherry-FoSso2 subcellular localization in different fungal structures. a: conidia, b: fungal tips, c: a germling, d: fungal conidiation, *: phialides Scale bars, 10 μm.
The mCherry fluorescent marker for FoSso2 indicated that the Qa SNARE protein was localized to the fungal plasma membrane (Fig. 4C). To further investigate the mCherry-FoSso2 plasma membrane localization, confocal microscopy Z-series stacks of live cells were used to further observe key structures involved in F. oxysporum conidiation (Fig. 4C d, Fig. S5). mCherry-FoSso2 was localized to an extensive array of fungal structures including conidia, phialides, septa, germlings, hyphae, and fungal tips (Fig. 4C). The localization of FoSso2 to the plasma membrane of a broad range of fungal structures indicates it may serve as a better marker for the characterization of the plasma membrane than FoSso1.
Clearly the differences in subcellular localization between FoSso1 and FoSso2 indicate divergence during their evolution. While FoSso1-mCherry showed a weak fluorescent signal in germlings and at subapical regions of the hyphae, mCherry-FoSso2 showed strong plasma membrane localization in conidia, fungal germlings, and in hyphae at the apical fungal tips. Qa SNARE proteins encode a single type II transmembrane domain which allows it to be localized to the plasma membrane (Sharpe et al., 2010). Previously, the single Qa SNARE protein SSOA from Aspergillus nidulans was shown to localize to the plasma membrane of the hyphal tip with some accumulation at the apical plasma membrane (Taheri-Talesh et al., 2008). Alternatively, in the fungus Trichoderma reesei the site of SNARE complex formation between SNCI and SSOI or SSOII was spatially segregated (Valkonen et al., 2007). Both Qa SNARE proteins from T. reesei was localized to the plasma membrane; however the SNCI-SSOII complexes were mainly localized to the growing hyphal apical regions while SSOI and SNCI co-localized to subapical regions.
This EGT system is a rapid and efficient method for fungal protein subcellular localization studies. There are several advantages of this system over traditional methods including: 1) conventional methods for protein subcellular localization in fungi required cloning and fusing the promoter region with the gene coding region and fluorescent marker sequences (Kilaru et al., 2015). However, in this CRISPR/Cas9-based EGT system only one or two short homologous fragments around the sgRNA site are necessary to clone. This avoids the usually time-consuming process to clone large gene coding regions such as the entire FoCHS5 gene from cDNA; 2) this method does not require the promoter region of a gene to be predicted to generate a construct, which avoids inaccurate promoter prediction or incorrect expression; 3) the HITI strategy allows a large DNA fragment to be integrated into the fungal genome. This is a potential advantage whereby it may allow simultaneous integration and expression of multiple genes in fungal genomes and can be further evaluated in the future; 4) the EGT system could accurately insert DNA fragments at the desired site(s) in the genome, and has a clear advantage over traditional transformation methods since it limits the frequency of random insertion; 5) the desired transformant(s) can be obtained from a small quantity of transformants due to the high efficiency of the system. Despite these advantages, using this EGT system might be difficult for genes with low expression under the native promoter due to low detection of the fluorescent signal during microscopy.
In this study, we used two different strategies for tagging endogenous genes in situ with fluorescent markers in the plant pathogenic filamentous fungus F. oxysporum. Both methods allowed the gene of interest to be expressed with a fluorescent signal under the control of the native promoter. The HITI strategy allowed a long DNA fragment to be inserted into the fungal chromosome and was used for the C-terminal tagging of FoCHS5 and the N-terminal tagging of FoSSO2. In addition, the other Qa SNARE syntaxin 1 homologous gene, FoSSO1, was tagged by an HDRI strategy. All these tagging transformants carried the desired fluorescent signals. Overall, this CRISPR/Cas9 RNP-mediated transformation method for EGT may facilitate the study of genetic engineering and the elucidation of gene function(s) in F. oxysporum and other fungi.
Supplementary Material
Fig. S1. Screen of the correct HITI transformants for generation of FoCHS5-3×sGFP. (A) the illustration of the chromosomal region of a correct HITI transformant after the integration of the entire plasmid. Tf and Tr: a pair of primers located outside of the bait sequence and can be used to clone a ~2.1 kb fragment, from the wild-type strain DNA, which was used for the in vitro Cas9 nuclease assay. Three PCR products, Up, Down and Full, were used to confirm the location of the integrated plasmid at the correct target site. (B) The sequencing results of DNA fragments containing the sgRNA sites for the wild-type strain and two transformants. (C) In vitro Cas9 RNPs cleavage assay to assess sgRNA target sequence. (D) agarose gel electrophoresis for the three DNA fragments based on the three pairs of primers from (A).
Fig. S2. Screen of the correct HDRI transformants for generation of FoSSO1-mCherry. (A) the illustration of the chromosomal region for correct HDRI transformants after homologous recombination. Tf and Tr: a pair of primers located outside of the homologous arms and were used to amplify a fragment from the wild-type strain DNA for the in vitro Cas9 nuclease assay. Three fragments, Up, Down and Full, were used to determine the location of the integrated fragment at the desired positions. The two orange arrows represent different primers located in the region of the mCherry-Tef-1-HYGB. B) In vitro Cas9 RNP cleavage assay for the FoSSO1 gene. C) The PCR screening results, as resolved by agarose gel electrophoresis, for three FoSSO1-mCherry transformants displaying the three PCR products generating using the three pairs of primers depicted in A).
Fig. S3. Individual frames of a FoChs5-3×sGFP transformant observed in a confocal microscopy Z-series stack.
Fig. S4. Individual frames of a FoSso1-mCherry transformant observed in a confocal microscopy Z-series stack.
Fig. S5. Individual frames of a mCherry-FoSso1 transformant observed in a confocal microscopy Z-series stack.
Movie S1. Subcellular localization of FoChs5-3×sGFP in a single hyphae. The image is from a Z-series stack with a total scanning depth of 5 μm and a scanning step of 0.5 μm.
Movie S2. Subcellular localization of FoSso1-mCherry in a single hyphae. The image is from a Z-series stack with a total scanning depth of 3.2 μm and a scanning step of 0.4 μm.
Highlights.
Developed a CRISPR/Cas9-mediated EGT system for F. oxysporum
First instance of a HITI strategy to insert a large fragment into the fungal genome
FoChs5 showed polarized location at the hyphal tip
Used HDRI strategy to tag the paralogous genes, FoSSO1 and FoSSO2
FoSso1 and FoSso2 are localized to different fungal structures
Acknowledgements
This work was supported by the National Institute of Allergy and Infectious Diseases (K22AI100983), the Alabama Agricultural Experiment Station, and the Hatch program of the National Institute of Food and Agriculture, USDA. We would like to thank Lynda Ciuffetti for the pCT74 plasmid containing sGFP and Sang-Jin Suh for the ss3591 plasmid containing mCherry.
Footnotes
Appendix A. Supplementary material
Supplementary data associated with this article can be found in the online version, at https://doi.org/10.1016/j.fgb.2019.02.002
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Auer TO, et al. , 2014. Highly efficient CRISPR/Cas9-mediated knock-in in zebrafish by homology-independent DNA repair. Genome Res. 24, 142–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dereeper A, et al. , 2008. Phylogeny.fr: robust phylogenetic analysis for the non-specialist. Nucleic Acids Res. 36, W465–W469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn RD, et al. , 2016. The Pfam protein families database: towards a more sustainable future. Nucleic Acids Research. 44, D279–D285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon TR, 2017. Fusarium oxysporum and the Fusarium Wilt Syndrome. Annu Rev Phytopathol. 55, 23–39. [DOI] [PubMed] [Google Scholar]
- Gupta YK, et al. , 2015. Septin-Dependent Assembly of the Exocyst Is Essential for Plant Infection by Magnaporthe oryzae. Plant Cell. 27, 3277–3289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickey PC, Read ND, 2009. Imaging living cells of Aspergillus in vitro. Med Mycol. 47 Suppl 1, S110–9. [DOI] [PubMed] [Google Scholar]
- Jantti J, et al. , 2002. Characterization of temperature-sensitive mutations in the yeast syntaxin 1 homologues Sso1p and Sso2p, and evidence of a distinct function for Sso1p in sporulation. J Cell Sci. 115, 409–20. [DOI] [PubMed] [Google Scholar]
- Kienle N, et al. , 2009. Phylogeny of the SNARE vesicle fusion machinery yields insights into the conservation of the secretory pathway in fungi. BMC Evol Biol. 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilaru S, et al. , 2015. A gene locus for targeted ectopic gene integration in Zymoseptoria tritici. Fungal Genet Biol. 79, 118–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilaru S, et al. , 2017. Fluorescent markers of various organelles in the wheat pathogen Zymoseptoria tritici. Fungal Genet Biol. 105, 16–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong LA, et al. , 2012. Different chitin synthase genes are required for various developmental and plant infection processes in the rice blast fungus Magnaporthe oryzae. PLoS Pathog. 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lev S, et al. , 2012. The Crz1/Sp1 transcription factor of Cryptococcus neoformans is activated by calcineurin and regulates cell wall integrity. PLOS ONE. 7, e51403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorang JM, et al. , 2001. Green fluorescent protein is lighting up fungal biology. Applied And Environmental Microbiology. 67, 1987–1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma LJ, et al. , 2010. Comparative genomics reveals mobile pathogenicity chromosomes in Fusarium. Nature. 464, 367–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madrid MP, et al. , 2003. Class V chitin synthase determines pathogenesis in the vascular wilt fungus Fusarium oxysporum and mediates resistance to plant defence compounds. Mol Microbiol. 47, 257–66. [DOI] [PubMed] [Google Scholar]
- McCluskey K, et al. , 2010. The Fungal Genetics Stock Center: a repository for 50 years of fungal genetics research. J Biosci. 35, 119–26. [DOI] [PubMed] [Google Scholar]
- Muhammed M, et al. , 2013. Fusarium infection: report of 26 cases and review of 97 cases from the literature. Medicine (Baltimore). 92, 305–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakanishi H, et al. , 2006. Phospholipase D and the SNARE Sso1p are necessary for vesicle fusion during sporulation in yeast. J Cell Sci. 119, 1406–1415. [DOI] [PubMed] [Google Scholar]
- Notredame C, et al. , 2000. T-Coffee: A novel method for fast and accurate multiple sequence alignment. J Mol Biol. 302, 205–217. [DOI] [PubMed] [Google Scholar]
- Nucci M, Anaissie E, 2007. Fusarium infections in immunocompromised patients. Clin Microbiol Rev. 20, 695–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen CG, Glass NL, 2005. A rho-type GTPase, rho-4, is required for septation in Neurospora crassa. Eukaryot Cell. 4, 1913–1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salaun C, et al. , 2004. Plasma membrane targeting of exocytic SNARE proteins. Biochim Biophys Acta. 1693, 81–9. [DOI] [PubMed] [Google Scholar]
- Schumacher J, et al. , 2008. Calcineurin-responsive zinc finger transcription factor CRZ1 of Botrytis cinerea is required for growth, development, and full virulence on bean plants. Eukaryotic Cell. 7, 584–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuster M, et al. , 2016. Co-delivery of cell-wall-forming enzymes in the same vesicle for coordinated fungal cell wall formation. Nat Microbiol. 1, 16149. [DOI] [PubMed] [Google Scholar]
- Scott MS, et al. , 2005. Refining protein subcellular localization. PLoS Comput Biol. 1, e66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharpe HJ, et al. , 2010. A comprehensive comparison of transmembrane domains reveals organelle-specific properties. Cell. 142, 158–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spielvogel A, et al. , 2008. Two zinc finger transcription factors, CrzA and SltA, are involved in cation homoeostasis and detoxification in Aspergillus nidulans. Biochemical Journal. 414, 419–429. [DOI] [PubMed] [Google Scholar]
- Suzuki K, et al. , 2016. In vivo genome editing via CRISPR/Cas9 mediated homology-independent targeted integration. Nature. 540, 144–+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taheri-Talesh N, et al. , 2008. The tip growth apparatus of Aspergillus nidulans. Mol Biol Cell. 19, 1439–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treitschke S, et al. , 2010. The Myosin Motor Domain of Fungal Chitin Synthase V Is Dispensable for Vesicle Motility but Required for Virulence of the Maize Pathogen Ustilago maydis. Plant Cell. 22, 2476–2494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valkonen M, et al. , 2007. Spatially segregated SNARE protein interactions in living fungal cells. J Biol Chem. 282, 22775–85. [DOI] [PubMed] [Google Scholar]
- van Dam P, et al. , 2017. A mobile pathogenicity chromosome in Fusarium oxysporum for infection of multiple cucurbit species. Sci Rep. 7, 9042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlaardingerbroek I, et al. , 2016. Dispensable chromosomes in Fusarium oxysporum f. sp. lycopersici. Mol Plant Pathol. 17, 1455–1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, et al. , 2018. Efficient genome editing in Fusarium oxysporum based on CRISPR/Cas9 ribonucleoprotein complexes. Fungal Genet Biol. 117, 21–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Screen of the correct HITI transformants for generation of FoCHS5-3×sGFP. (A) the illustration of the chromosomal region of a correct HITI transformant after the integration of the entire plasmid. Tf and Tr: a pair of primers located outside of the bait sequence and can be used to clone a ~2.1 kb fragment, from the wild-type strain DNA, which was used for the in vitro Cas9 nuclease assay. Three PCR products, Up, Down and Full, were used to confirm the location of the integrated plasmid at the correct target site. (B) The sequencing results of DNA fragments containing the sgRNA sites for the wild-type strain and two transformants. (C) In vitro Cas9 RNPs cleavage assay to assess sgRNA target sequence. (D) agarose gel electrophoresis for the three DNA fragments based on the three pairs of primers from (A).
Fig. S2. Screen of the correct HDRI transformants for generation of FoSSO1-mCherry. (A) the illustration of the chromosomal region for correct HDRI transformants after homologous recombination. Tf and Tr: a pair of primers located outside of the homologous arms and were used to amplify a fragment from the wild-type strain DNA for the in vitro Cas9 nuclease assay. Three fragments, Up, Down and Full, were used to determine the location of the integrated fragment at the desired positions. The two orange arrows represent different primers located in the region of the mCherry-Tef-1-HYGB. B) In vitro Cas9 RNP cleavage assay for the FoSSO1 gene. C) The PCR screening results, as resolved by agarose gel electrophoresis, for three FoSSO1-mCherry transformants displaying the three PCR products generating using the three pairs of primers depicted in A).
Fig. S3. Individual frames of a FoChs5-3×sGFP transformant observed in a confocal microscopy Z-series stack.
Fig. S4. Individual frames of a FoSso1-mCherry transformant observed in a confocal microscopy Z-series stack.
Fig. S5. Individual frames of a mCherry-FoSso1 transformant observed in a confocal microscopy Z-series stack.
Movie S1. Subcellular localization of FoChs5-3×sGFP in a single hyphae. The image is from a Z-series stack with a total scanning depth of 5 μm and a scanning step of 0.5 μm.
Movie S2. Subcellular localization of FoSso1-mCherry in a single hyphae. The image is from a Z-series stack with a total scanning depth of 3.2 μm and a scanning step of 0.4 μm.