

SUMMARY
The nature of cell state transitions during the transit-amplifying phases of many developmental processes - hematopoiesis in particular - is unclear. Here we use single cell RNA sequencing to demonstrate a continuum of transcriptomic states in committed transit-amplifying erythropoietic progenitors, which correlates with a continuum of proliferative potentials in these cells. We show glucocorticoids enhance erythrocyte production by slowing rate of progression through this developmental continuum of transit-amplifying progenitors, permitting more cell divisions prior to terminal erythroid differentiation. Mechanistically, glucocorticoids prolong expression of genes that antagonize, and slow induction of genes that drive terminal erythroid differentiation. Erythroid progenitor daughter cell pairs have similar transcriptomes with or without glucocorticoid stimulation, indicating largely symmetric cell division. Thus, the rate of progression along a developmental continuum dictates the absolute number of erythroid cells generated from each transit-amplifying progenitor, suggesting a paradigm for regulating total output of differentiated cells in numerous other developmental processes.
eTOC BLURB
Li and Natarajan et al. utilize single cell RNA-seq and functional assays to demonstrate erythropoiesis progresses through a continuum of both transcriptomic and phenotypic states. Perturbation of developmental progression through this continuum with glucocorticoid steroids reveals differentiation speed can be uncoupled from cell cycle progression, generating greater numbers of erythrocytes.
Graphical Abstract:
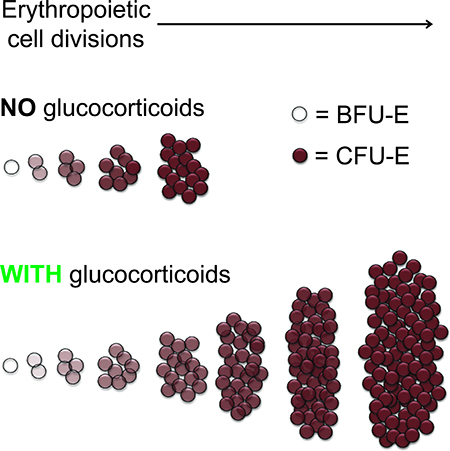
INTRODUCTION
Tissue development and regeneration represent fundamental biological processes with distinct relevance to health and disease. Blood is a continuously regenerating organ producing trillions of erythrocytes each day (Koury, 2016), requiring committed erythroid progenitors to exponentially expand in number during the transit-amplifying phase of erythropoiesis. The concept of erythroid progenitor self-renewal was proposed as an explanation for this biological phenomenon (Wendling et al., 1983, Koury, 2016), with subsequent extension of progenitor cell self-renewal models to numerous other developmental systems (Basta et al., 2014, Jin et al., 2013, Lui et al., 2011, Collins et al., 2005, McCulloch et al., 1991, Bonyadi et al., 2003). However, strict stem cell-like self-renewal, where either one or both daughter cells are identical to the parent cell, has yet to be demonstrated for committed erythroid progenitor cells. Indirect evidence for erythroid progenitor self-renewal was inferred from findings that extended ex vivo culture of unfractionated hematopoietic tissues results primarily in an erythroid cell population (Wendling et al., 1983, Hayman et al., 1993, England et al., 2011, von Lindern et al., 1999), and from studies suggesting that glucocorticoids increase the number of self-renewal divisions of early committed erythroid progenitor cells (Flygare et al., 2011, Zhang et al., 2013, von Lindern et al., 1999, Narla et al., 2011).
Distinct committed erythroid progenitor cell stages are currently defined based on colony morphology in methylcellulose colony-forming assays (Koury, 2016). The earliest committed erythroid progenitor cell, the transit-amplifying burst forming unit-erythroid (BFU-E), is thought to give rise to a number of colony forming unit-erythroid (CFU-E) progenitor cells after several cell divisions. In the presence of erythropoietin (EPO), CFU-E progenitor cells then undergo 4–5 terminal cell divisions contemporaneous with induction of ~400 erythrocyte-important genes, giving rise to erythroblasts and then enucleated reticulocytes (Hattangadi et al., 2011). BFU-E and CFU-E cell numbers are decreased in the bone marrow of patients with Diamond-Blackfan anemia (DBA) (Nathan et al., 1978, Chan et al., 1982, Iskander et al., 2015). Glucocorticoids are the only known effective medical treatment for EPO-resistant hypoplastic anemias such as DBA, and successfully treated DBA patients have increased numbers of bone marrow BFU-E and CFU-E cells (Iskander et al., 2015, Chan et al., 1982). In mice, the glucocorticoid receptor is required for stress erythropoiesis (Bauer et al., 1999, Reichardt et al., 1998), and BFU-E and CFU-E cell numbers increase in the spleen during stress erythropoiesis (Voorhees et al., 2013, Vignjevic et al., 2015, Harandi et al., 2010).
Early ex vivo culture studies of unfractionated hematopoietic tissues were equivocal in identifying the erythroid cell type upon which glucocorticoids act, but glucocorticoids unequivocally increase total erythroid cellular output in culture of committed erythroid progenitors (Ohene-Abuakwa et al., 2005, von Lindern et al., 1999, Golde et al., 1976). Later studies on populations enriched for BFU-E and CFU-E cells demonstrated that when both cell types are stimulated with glucocorticoids, the proliferative capacity of BFU-E enriched populations is increased by a much greater magnitude than the proliferative capacity of CFU-E enriched populations in both mouse (Flygare et al., 2011) and human systems (Narla et al., 2011).
Recent advances in single cell transcriptome profiling have suggested a continuum of progenitor cell states in differentiating hematopoietic stem and progenitor cells, as well as in other developmental pathways (Macaulay et al., 2016, Tusi et al., 2018, Karamitros et al., 2018, Velten et al., 2017, Zeng et al., 2017, Treutlein et al., 2016, Dulken et al., 2017, Lescroart et al., 2018, Nestorowa et al., 2016), but functional validation of developmental continuum models is currently lacking. Here, we demonstrate that the continuum of transcriptomic states in early erythropoiesis correlates with a continuum of functional states, and that ex vivo cultured BFU-E cells progressively advance through this developmental continuum without significant evidence for stem cell-like self-renewal. We identify the mechanism for glucocorticoid-stimulated increase in erythroid output as slowing progression through this developmental continuum without affecting the rate of cell division or the transcriptome symmetry of daughter cells following BFU-E cell divisions. Lastly, we demonstrate that glucocorticoid treatment increases the ratio of early relative to late erythropoietic progenitor cells in vivo.
RESULTS
BFU-E cells possess greater proliferative capacity and glucocorticoid-responsiveness than CFU-E cells
To experimentally assay erythroid progenitor cells, we isolated BFU-Es and CFU-Es from the fetal liver of embryonic day 14.5 (E14.5) mice. Previous studies showed that depletion of cells expressing Sca-1 and cell surface lineage markers from E14.5 mouse fetal liver, followed by isolation of cells positive for c-Kit, results in a population highly enriched for early erythroid progenitor cells (Lin-/c-Kit+ population). Co-staining for the transferrin receptor (CD71) and CD24 allows further enrichment, with the Lin-/c-Kit+ population expressing the lowest 10% CD71/CD24 being highly enriched for cells forming BFU-E colonies, and the cells with the highest 20% CD71/CD24 expression being highly enriched for cells forming CFU-E colonies (Figure 1B and Supplementary Figure S1A) (Flygare et al., 2011). Ex vivo culture of BFU-E and CFU-E enriched populations in progenitor culture media (PCM), a serum-free media containing stem cell factor, insulin-like growth factor 1, and EPO, demonstrated BFU-E enriched cells proliferate to reach a maximum cell number approximately 7.5-times greater than the maximum cell number reached by proliferating CFU-E enriched cells (Figure 1C). Similar to prior studies (Flygare et al., 2011), addition of the synthetic glucocorticoid dexamethasone (Dex) to PCM resulted in a 14-times greater maximum proliferation of BFU-E enriched cells, and only a 1.3-times greater maximum proliferation of CFU-E enriched cells (Figure 1C). Importantly, exposure to Dex did not alter the rate of BFU-E cell division, as evidenced by an unchanged rate of CFSE dilution (Supplementary Figure S1B). Furthermore, during the initial 72 hours of culture in which the majority of BFU-E cells transition into CFU-E cells and erythroblasts (Flygare et al., 2011), cell survival was greater than 96% for BFU-E cells cultured in both the presence and absence of Dex (Supplementary Figure S1C). Taken together, these results indicate glucocorticoid treatment increases BFU-E proliferative capacity and stimulates 3–4 additional rounds of cell division in culture to yield an overall increase of 14 times greater cellular output.
Figure 1 – Glucocorticoids slow progression of early erythroid progenitor cells through a continuum of transcriptomic states.

(A) Existing model for distinct cell stages of erythropoiesis based on colony forming assays and cellular morphology. (B) FACS-sorting strategy for isolation of BFU-E and CFU-E enriched populations from mouse fetal liver, and experimental schema for serial single cell transcriptome profiling of ex vivo cultured BFU-E cells. (C) Proliferative capacities of BFU-E and CFU-E enriched populations with and without 100nM Dex when cultured in progenitor culture media (PCM). Cultures initiated with 10,000 cells in 1mL PCM, with cells split every 2–3 days. Population expansion normalized to a single cell. Error bars indicate standard deviation. P-values comparing BFU-E + Dex and BFU-E no Dex calculated with Student’s T-Test. N = 8 biological replicates. (D-J) PCA of all single cell transcriptomes in this study, displayed as: (D) single BFU-E and CFU-E cell transcriptomes with proposed trajectory of developmental progression. (E-J) BFU-E cells cultured for 1, 2, and 3 days without Dex (H-J), or with Dex (E-G). Blue dots indicate transcriptomes for individual cells on each day with indicated Dex treatment status, background gray dots indicate transcriptomes of all single cells analyzed in this study. N denotes number of single cell transcriptomes that passed all sequence quality control filters. P-values calculated with multivariate Pillai’s statistic comparing the likelihood that the transcriptomes of the two cell populations indicated by the double arrow occupy the same distributions in two-dimensional PC1 vs PC2 space. See also Supplementary Figures S1-S3.
Single cell transcriptome profiling determines glucocorticoids slow progression through a continuum of early erythroid progenitor states
To characterize the global gene expression states of erythroid progenitor cells, we performed single cell transcriptome profiling using SmartSeq2 (Picelli et al., 2013) on 96 individual BFU-E cells and 96 individual CFU-E cells. On average, 7,500 genes with measurable expression were detected per cell (Supplementary Figure S2A), and aggregated single cell BFU-E and single cell CFU-E transcriptome data correlated well with previously published (Flygare et al., 2011) bulk RNA-seq BFU-E and CFU-E cell transcriptome data (Supplementary Figure S2B). Principal component analysis (PCA) demonstrated a relatively more homogenous CFU-E population than the BFU-E population, and also suggested a developmental trajectory for progression of BFU-E cells to CFU-E cells (Supplementary Figure S2C).
To interrogate whether erythropoiesis followed this proposed developmental trajectory, we cultured BFU-E enriched cells in PCM with and without Dex, isolated 96 cells per culture condition each day for three days, and performed SmartSeq2 on these cells. BFU-E enriched cells cultured in PCM were previously shown to result in generation of CFU-E progenitors, and ultimately erythroblasts, within three days (Flygare et al., 2011). We confirmed that BFU-E enriched cells cultured three days in PCM acquire high surface expression of CD71 and the terminal erythroid marker Ter119 (Supplementary Figure S1A). PCA of transcriptomes of BFU-E and CFU-E cells (Figure 1D), and BFU-E cells cultured in PCM with Dex (Figure 1E-G) or without Dex (Figure 1H-J) indicated daily progression along the same proposed developmental trajectory through a continuum of transcriptomic states. Additionally, multivariate analysis of single cell transcriptomes demonstrated Dex treated BFU-E cells occupied a significantly earlier distribution along the developmental trajectory than untreated BFU-E cells at days 1, 2, and 3 of culture (Figure 1E-J). Similar results were obtained when performing PCA on all transcriptomes using only the genes driving variability among BFU-Es, CFU-Es, and BFU-Es cultured without Dex (Supplementary Figure S2D,F). Similar results were also obtained when performing Diffusion Mapping (Haghverdi et al., 2015, Butler et al., 2018) of the first 50 principal components of transcriptomes of BFU-Es, CFU-Es, and BFU-Es cultured in PCM with and without Dex (Supplementary Figure S3B). Lastly, we employed the Monocle 2 (Qiu et al., 2017) algorithm, which maps developmental trajectories in multiple dimensions taking into account all principal components that contribute to variability between cells, and also obtained similar results (Supplementary Figure S3C). Overall, we determined that 100% of transcriptome variability among cells was captured with the first 20 principal components (Figure S3G).
The continuum of early erythroid transcriptomic states reflects a continuum of phenotypic states
We next sought to evaluate if the developmental trajectory revealed by ex vivo BFU-E cell culture faithfully represented the trajectory of early erythroid development in vivo. Given the presence of a continuum of CD71/CD24 expression levels in the Lin-/c-Kit+ population of E14.5 mouse fetal liver cells, we reasoned there were likely developmental intermediates between the BFU-E enriched (10% lowest CD71/CD24) and the CFU-E enriched (20% highest CD71/CD24) populations (Figure 2A). To this end we isolated 23 Lin-/c-Kit+ cells with 25–35th percentile (25–35%) and 24 cells with 50–60th percentile (50–60%) CD71/CD24 expression (Figure 2B), as well as 48 BFU-E and 24 CFU-E cells during the same isolation, and performed SmartSeq2 on single cells from these four populations. Consistent with our hypothesis, when performing PCA of all cells in this study, 25–35% and 50–60% cell transcriptomes were both located within our previously defined developmental trajectory, occupying the region linking BFU-E cells to CFU-E cells (Figure 2C). Additionally, we used Diffusion Mapping to compare all cells from our study to an independent study profiling all c-Kit+ cells in fetal liver (Tusi et al., 2018); we found that all 848 cells from our study were located within the same single cell transcriptome distribution defined by the 6,054 fetal liver erythroid progenitor cells profiled by Tusi and colleagues (Supplementary Figure S4).
Figure 2 – The continuum of transcriptomic states in early erythropoiesis reflects a continuum of phenotypic states.

(A) Region within existing erythropoiesis model for proposed developmental intermediates between BFU-Es and CFU-Es. (B) FACS-sorting strategy for enriching for 25–35% and 50–60% CD71/24 developmental intermediates between BFU-Es and CFU-Es. (C) Display of BFU-E, 25–35%, 50–60%, and CFU-E cells isolated from the same sample when performing PCA on all cells in this study. (D) Maximum expansion of cell numbers of CFU-E, 50–60%, 25–35%, and BFU-E populations cultured in PCM in the absence of Dex. Results normalized relative to maximum expansion of CFU-Es. N=10 biological replicates. (E) Maximum expansion of CFU-E, 50–60%, 25–35%, and BFU-E populations cultured in PCM with and without Dex, with magnitude of increase in population expansion (glucocorticoid-responsiveness) induced by Dex indicated. Results normalized to maximum expansion of CFU-Es without Dex. N=8 biological replicates. (F) Experimental schema for serial measurement of proliferative capacity (maximum expansion without Dex) for freshly isolated BFU-Es, or BFU-Es cultured for 1–3 days in PCM with or without Dex prior to proliferative capacity assay. (G) Maximum expansion in cultures without Dex of freshly isolated BFU-Es, or BFU-Es that have been cultured for 1, 2, or 3 days in PCM without (black bars) or with (orange bars) Dex. Results normalized to maximum expansion of freshly isolated BFU-Es. N=4 biological replicates. All p-values calculated with Student’s T-Test. All error bars indicate standard deviation. See also Supplementary Figures S4 & S5.
Using a previously developed algorithm for predicting cell cycle state based on gene expression pattern (Butler et al., 2018), we confirmed that developmental progression from the BFU-E to CFU-E stage was associated with decreased residence in the G1 cell cycle phase (Supplementary Figure S5), as previously demonstrated (Hwang et al., 2017, Tusi et al., 2018). We additionally found that PCM-cultured BFU-E cells treated with Dex for 24 hours demonstrated increased residence in G1 compared to BFU-E cells cultured without Dex, consistent with an earlier developmental state for Dex-treated cells (Supplementary Figure S5).
We then sought to evaluate whether the continuum of transcriptomic states in early erythroid development truly reflected a continuum of cell states at the functional level. We assayed BFU-E, 25–35%, 50–60%, and CFU-E cells with two different functional assays: proliferative capacity and glucocorticoid-responsiveness. Consistent with loss of proliferative capacity while progressing through a developmental continuum, the proliferative capacity decreased significantly from BFU-Es to 25–35% to 50–60% to CFU-Es (Figure 2D). Also consistent with a developmental continuum, glucocorticoid-responsiveness, as assessed by the magnitude of increase in maximum proliferation with Dex treatment, decreased from BFU-Es to 25–35% to 50–60% to CFU-Es (Figure 2E).
Our findings that BFU-E cells cultured with Dex do not persist in an identical transcriptomic state are seemingly in contrast to our previous work claiming glucocorticoid treatment induces self-renewal of BFU-E cells. These early studies found that BFU-E enriched cells cultured in PCM with Dex led to an increase in the number of BFU-E colony-forming cells over the course of 3 days in culture, before declining as the cells generated CFU-Es and then terminally differentiated erythroid cells (Flygare et al., 2011). These results were interpreted as glucocorticoid stimulation of a limited number of BFU-E self-renewal cell divisions. However, a well-known limitation of colony forming assays is the heterogeneity of BFU-E colonies – some very large colonies (presumed to be generated from early BFU-Es with higher proliferative potential) and others small (presumed from later BFU-Es). Here we improved upon these limitations through the use of quantitative proliferative capacity assays. To this end we cultured BFU-E enriched cells in PCM with and without Dex, measuring maximum proliferative capacity of the cultured population each day for three days (Figure 2F). Compared to BFU-E enriched cells assayed immediately after isolation, BFU-E enriched cells cultured in PCM without Dex demonstrated decreased proliferative capacity with each day of culture. At each time point, BFU-E enriched cells cultured with Dex demonstrated a greater proliferative capacity compared to cells cultured without Dex. However, BFU-E enriched cells cultured with Dex also demonstrated decreasing proliferative capacity with each day in culture, consistent with progressive advancement through a developmental continuum from “early” to “late” BFU-Es to CFU-Es.
Pseudotemporal ordering confirms that glucocorticoids slow the progression of erythroid transit amplifying cells through a developmental continuum.
Because of our findings that erythropoiesis progresses along a single developmental trajectory, regardless of treatment with Dex, we applied pseudotemporal ordering to the PCA of all transcriptomes in this study. We inferred a linear developmental progression from BFU-E cells through CFU-E cells and terminal erythroid differentiation by calculating a regression function fitting the single developmental path of cells in PC1 vs PC2 (Figure 3A, Supplementary Figure S3A,D). At each time point, the linear developmental progression of BFU-E cells cultured with Dex lagged behind those cultured without Dex. A similar pseudotemporal order was calculated by inferring a linear developmental progression based on the developmental trajectory of cells with Diffusion Mapping (Supplementary Figure S3B,E,H), as well as by using the Monocle 2 (Qiu et al., 2017) pseudotime algorithm (Supplementary Figures S3C,F,I). To understand early erythroid development gene expression patterns, we quantified the expression changes during development for the 2,880 most variable genes identified during the process of performing PCA (Figure 3B). Our analysis confirmed that genes known to decrease or increase in expression during erythroid development did indeed follow these expression patterns (Figure 3C-N).
Figure 3 – Pseudotemporal ordering identifies continuous gene expression changes in erythroid development and confirms that glucocorticoids slow the rate of differentiation of cultured BFU-E cells.

(A) Pseudotemporal order of cell position within the erythroid developmental continuum (earliest pseudotime point of 0, latest pseudotime point of 125 in arbitrary units), as calculated in Supplementary Figure S3 for BFU-Es, CFU-Es, and BFU-Es cultured with and without 100nM Dex for 1–3 days. P-values indicate Student’s T-test comparing distribution of pseudotemporal position each day for BFU-E’s cultured with or without Dex. (B) Relative expression kinetics over the course of development for the 2,880 most variable genes. (C-N) Examples of expression kinetics of individual genes known to decrease in expression after the BFU-E stage (C,D), genes known to antagonize erythroid terminal differentiation (E-H), and genes known to increase in expression during erythroid terminal differentiation (I-N). Each cell is plotted as log-transformed read counts for that gene versus the cell’s position in the developmental continuum, with the blue line depicting the LOESS fit for gene expression over the course of development. See also Supplementary Figure S3
Glucocorticoids alter expression profiles of key developmental regulatory proteins
Given the transcription factor functions of the glucocorticoid receptor (Reichardt et al., 1998, Flygare et al., 2011), we reasoned that gene regulation mechanistically contributes to glucocorticoid-induced slowing of progression of BFU-E cells through the erythroid developmental continuum. As Dex treatment results in slower global transcriptome progression, we asked whether expression of genes known to antagonize terminal differentiation (Flygare et al., 2011, Zhang et al., 2013, Back et al., 2004, Briegel et al., 1993) is higher in Dex treated cells than untreated cells. Indeed, at each of the three days of culture, Gata2, Zfp36l2, Hif1a, and Spi1 all demonstrated higher expression levels (Figure 4A-D) in Dex treated BFU-E cells compared to untreated BFU-E cells. We then asked whether expression of Gata1 and Klf1, master regulators driving erythroid terminal differentiation (Orkin and Zon, 2008), is delayed following Dex treatment. Over three days of culture, Klf1 and Gata1 expression was lower in BFU-E cells treated with Dex compared to untreated cells (Figure 4E,F). We additionally assessed the pseudotemporal expression kinetics of these six genes, and consistent with our global transcriptome findings of Dex-induced developmental slowing, Gata2, Zfp36l2, Hif1a, and Spi1 all demonstrated prolonged expression kinetics with Dex treatment (Figure 4G-J), and Klf1 and Gata1 demonstrated slower onset of expression with Dex treatment (Figure 4K,L).
Figure 4 – Expression kinetics of key developmental regulatory proteins are altered by glucocorticoids.

(A-F) Gene expression levels in individual freshly isolated BFU-Es, and BFU-Es cultured with or without 100nM Dex for 1, 2, or 3 days, of four genes that antagonize (A-D) and two genes that promote terminal erythroid differentiation (E,F). All p-values calculated by the Mann-Whitney U-Test compare gene expression level in no Dex (black) versus +Dex (orange) cells at each day of culture. (G-L) Individual cell log-transformed read counts of all 6 genes from panels A-F, plotted versus cell position within the erythroid developmental continuum to illustrate alterations in expression kinetics over the course of development associated with Dex treatment. Individual no Dex cells are plotted in black, and individual +Dex cells are plotted in orange. Blue line denoting LOESS fit for gene expression over the course of development reproduced from Figure 3.
Glucocorticoids increase the ratio of early to late erythroid progenitors in vivo
Our studies using cultured BFU-E cells support the notion that glucocorticoids increase BFU-E proliferative capacity by slowing progression through the erythroid developmental trajectory, without altering cell survival or the rate of proliferation during the BFU-E to CFU-E developmental transition. To test whether these principles reflect erythroid development in vivo, we treated pregnant mice at 13.5 days gestation with vehicle control or Dex at a dose equivalent to the glucocorticoid dosing used in DBA patients (0.3 mg dexamethasone per kg of body weight), then isolated fetal liver erythroid progenitor cells at 14.5 days gestation to assess cell survival, cell proliferation, and relative distribution of early and late erythroid progenitor cells (Figure 5A). To confirm that administration of Dex results in a biological effect in vivo, we isolated lineage-negative fetal liver cells at E14.5 following vehicle or Dex treatment at E13.5. Western blot analysis of glucocorticoid receptors in nuclear and cytoplasmic cell fractions of these fetal liver erythroid progenitors demonstrated the expected increase in nuclear accumulation of glucocorticoid receptor in Dex treated mice (Figure 5B).
Figure 5 – Glucocorticoid treatment increases relative numbers of early fetal liver erythroid progenitors in vivo without affecting cell survival or cell proliferation rate.

(A) Experimental schema for in vivo Dex (0.3mg/kg) administration followed by analysis of cell survival, cell proliferation, and developmental stage. (B) Western blot for glucocorticoid receptor in nuclear and cytoplasmic protein fractions from lineage-depleted fetal liver cells isolated at E14.5 from pregnant mice treated 24 hours prior with vehicle control or Dex. Lamin B serves as a control nuclear protein marker and GAPDH serves as a cytoplasmic marker protein. (C) Propidium iodide staining to assay cell viability and survival of BFU-E, 25–35%, 50–60%, and CFU-E cells isolated from E14.5 fetal liver from pregnant mice treated 24 hours prior with vehicle or Dex. N=6 biological replicates. (D) Schema indicating S phase transit status after in vivo dual-labeling with EdU and BrdU. (E) Representative flow cytometry gating for fetal liver erythroid progenitor cells following in vivo dual-labeling with EdU and BrdU, denoting BrdU+/EdU- cells as having entered S phase in the 2 hour period between EdU and BrdU labeling, and EdU+/BrdU- cells as having been in S phase during EdU labeling, but exited during the 2 hours prior to BrdU labeling. (F,G) Rate of S phase entrance (F) and exit (G) for BFU-E, 25–35%, 50–60%, and CFU-E fetal liver cells at E14.5 after treatment of pregnant mice 24 hours prior with vehicle or Dex. N=4 biological replicates. (H) Representative flow cytometry histogram of CD71/CD24 expression of Lin-/c-Kit+ cells from E14.5 fetal livers isolated from paired pregnant mice treated 24 hours prior with vehicle or Dex. The demarcation between “Early” and “Late” progenitors occurs at the point where the vehicle and Dex histograms intersect. (I) Ratio of “Early” divided by “Late” progenitors, displayed relative to the ratio obtained from vehicle treated mice. N=6 paired biological replicates. All error bars indicate standard deviation, and all p-values calculated with Student’s T-Test. Due to shifts in CD71/CD24 expression patterns induced by Dex treatment, CD71/CD24 gates for BFU-E, 25–35%, 50–60%, and CFU-E populations in panels C-G were drawn based on no Dex cells, and these fluorescence intensity gates were then applied to + Dex cells in order to match CD71/CD24 expression levels.
To assess cell survival, we performed propidium iodide staining on BFU-E, 25–35%, 50–60%, and CFU-E fetal liver populations isolated from vehicle or Dex-treated mice. For all populations, we did not find significant differences in rate of cell death between vehicle and Dex-treated cells, with the minor exception of the 25–35% population (Figure 5C). Importantly, for all four populations, greater than 97% of cells were viable (negative for propidium iodide staining) regardless of vehicle or Dex treatment, indicating cell death contributes minimally to in vivo erythroid progenitor biology during transit amplification.
To assess rates of cell proliferation, we adapted a previously developed technique (Hwang et al., 2017, Akinduro et al., 2018) for in vivo dual pulse labeling with ethynyl-deoxyuridine (EdU) followed by bromo-deoxyuridine (BrdU) to measure rates of S phase entrance and exit as a marker of cell cycling and proliferation. 24 hours after treatment of pregnant mice at E13.5 with vehicle or Dex, on E14.5 we injected EdU, and then after 2 hours injected BrdU. At 25 minutes following BrdU administration we isolated fetal liver erythroid progenitor cells. Cells entering S phase during the 2 hour period between EdU and BrdU injections incorporate BrdU into their DNA, but will not have incorporated EdU. Cells that were in S phase at the start of the 2 hour interval, but exited S phase prior to BrdU administration, will have incorporated EdU into their DNA, but will not have incorporated BrdU (Figure 5D,E). Through flow cytometric detection of EdU and BrdU, we found no significant difference in rate of S phase entrance and exit between vehicle and Dex-treated BFU-E, 25–35%, 50–60%, or CFU-E populations (Figure 5F,G). Thus cell cycle speed, and by extension cell proliferation rate, are unchanged by in vivo Dex treatment.
To determine the relative distribution of progenitor cells along the BFU-E to CFU-E developmental continuum in vivo, we assayed combined surface expression of the developmental stage markers CD71 and CD24 on fetal liver erythroid progenitors from vehicle and Dex-treated mice. For each biological replicate, a vehicle treated pregnant mouse was paired with a Dex treated pregnant mouse, and fetal liver cells were isolated and analyzed in parallel. When comparing CD71/CD24 expression on Lin-/c-Kit+ fetal liver cells from pairs of vehicle and Dex-treated mice, it was readily visualized that the Dex-treated population was characterized by a greater number of “early” erythroid progenitors with low expression of CD71/CD24, and fewer numbers of “late” progenitors with high expression of CD71/CD24 (Figure 5H). For each vehicle and Dex-treated pair, we quantified the ratio of “early” to “late” progenitors. Dex treatment increased the ratio of “early” to “late” progenitors on average by 35% (Figure 5I). Thus, Dex increases the proportion of “early” erythroid progenitors, consistent with a slowing of developmental progression without altering the rate of cell division.
Early erythroid progenitor cell division is symmetric
Because of our results demonstrating Dex increases the proliferative capacity of BFU-E cells more than other erythroid progenitor populations, we sought to determine whether glucocorticoid stimulation affected the degree of daughter cell symmetry following BFU-E cell division. We chose whole transcriptome profiling as a measure of cell state to assess symmetry of cell division. To this end, we deposited a single BFU-E cell per well into PCM with or without Dex. Four to six hours after the first cell division of each cell, we manually separated the two daughter cells and performed SmartSeq2 on both. In total, we collected 44 daughter cells. Daughter cells from a BFU-E cell division without Dex treatment demonstrated remarkable transcriptome symmetry, with a median separation between daughter cells less than 5% of the entire developmental transcriptome continuum (Figure 6B). Importantly, we compared the average transcriptional separation between 10 daughter pairs cultured without Dex, to the average separation between 10 random pairs of BFU-E cells cultured for 24 hours without Dex, and determined that the average separation of daughter pairs was more symmetric than by chance (average separation of random pairs) in 99.88% of random trials (Figure 6C). Daughter cells from a BFU-E cell division with Dex treatment also demonstrated comparable symmetry of transcriptomes (Figure 6D). When comparing the average separation between 12 daughter pairs cultured with Dex, to the average separation between 12 random pairs of BFU-E cells cultured for 24 hours with Dex, again the average separation of daughter pairs was more symmetric than by chance (average separation of random pairs) in greater than 99.99% of random trials (Figure 6E). Thus Dex treatment did not significantly alter the degree of daughter cell symmetry following BFU-E cell division (Figure 6F).
Figure 6 – Early erythroid progenitor cell divisions are symmetric both in the presence and absence of glucocorticoids.

(A) PCA of BFU-E cells redisplayed for relative comparison. (B,D) PCA of daughter cells resulting from the first cell division of a freshly isolated BFU-E cell cultured without Dex (B), or with 100nM Dex (D). (C,E) Probability that the average daughter pair pseudotemporal separation distance for 10 no Dex daughter pairs (C), and 12 +Dex daughter pairs (E) is much less than obtained by chance. As detailed in STAR Methods, all possible distances between BFU-Es cultured for 24 hours without Dex and all possible distances between BFU-Es cultured for 24 hours with Dex were calculated, then 1 million replicates of randomly sampling 10 distances for no Dex cells, and 1 million replicates of randomly sampling 12 distances for +Dex cells were performed, with the distribution of these randomly sampled averages displayed as histograms. Red dashed lines indicate the observed average distance for no Dex and +Dex daughter pairs. P-values are the calculated empiric probability that the randomly sampled average distances are less than or equal to the observed average distances for the paired daughter cells. (F) Degree of BFU-E daughter cell symmetry with and without Dex, as measured by distance between both daughter cells along the developmental continuum, and displayed as percent of the total developmental continuum. P-value from Student’s T-Test.
DISCUSSION
Principles of erythroid progenitor cell biology are broadly relevant to numerous developmental systems, with studies of terminal erythroid differentiation long serving as models for other differentiation processes. However, despite past advances in elucidating erythroid biology, the nature of the erythroid BFU-E progenitor population and its expansion during the transit-amplifying phase of erythropoiesis was elusive. Coincident with this was a lack of molecular understanding for how glucocorticoids mediate stress erythropoiesis in mice, and improve anemia in DBA patients. Our study addresses these decades-old questions in erythroid biology, and provides a paradigm for modulating total cellular output in many developmental and regenerative contexts.
Our study profiling single cell transcriptomes demonstrates that the widely used erythroid progenitor cell culture system we employed faithfully mimics in vivo development. Prior studies performed single cell transcriptome profiling during ex vivo culture (Trapnell et al., 2014, Shalek et al., 2014), but our work establishes that these techniques in fact can benchmark how well these culture systems mimic in vivo development. Indeed this culture system allows a molecular dissection of progenitor cell expansion. One such example is our finding that a BFU-E enriched erythroid progenitor population advances through the developmental continuum to less of an extent per cell cycle with glucocorticoid exposure than without. In effect, this indicates that glucocorticoid treatment uncouples differentiation speed from cell division. Additionally, we used this culture system to demonstrate that the proliferative capacity of a BFU-E progenitor cell along the developmental continuum correlates with its transcriptomic state. Although our ex vivo culture experiments utilized a fetal liver erythroid progenitor source, as opposed to a bone marrow source which may potentially behave differently, Tusi and colleagues recently demonstrated using single cell transcriptomics that erythroid transit amplification in both fetal liver and bone marrow progress through similar developmental states (Tusi et al., 2018). We also demonstrated that in vivo glucocorticoid administration yielded findings consistent with our ex vivo culture experiments – that glucocorticoids slow the rate of progression through a developmental continuum.
We additionally discovered that glucocorticoid-induced slowing of progenitor cell progression through the erythroid developmental continuum occurs through repression of genes driving terminal differentiation, and maintenance of expression of genes antagonizing terminal differentiation. We also demonstrated the symmetry of daughter cell transcriptomes following BFU-E cell division, and that this is not significantly affected by glucocorticoid stimulation. This result was in the context of a cell culture system where cytokine receptor ligands are in solution and not supplied by stroma, but our results indicate single cell transcriptome profiling can be used as a measure to quantify global cell state of daughter cell pairs.
Our results clarify the true nature of erythroid progenitor transit-amplification, a poorly understood process since Axelrad and colleagues proposed the concept of progenitor self-renewal in 1983 (Wendling et al., 1983). The new paradigm we uncovered indicates that the majority of BFU-E progenitor cells do not undergo true stem cell-like self-renewal when treated with glucocorticoids, but rather progressively advance through the developmental continuum with each cell division, and importantly, at a slower rate than untreated BFU-E cells. Furthermore, these findings indicate that the rate of progression through the erythroid developmental continuum dictates number of progenitor cell divisions, and thus total number of erythroid cells generated prior to terminal differentiation. This mechanism for quantitatively modulating cellular output has relevance for a number of developmental and regenerative biology systems, as well as disease states where a progenitor cell pool cannot generate sufficient numbers of differentiated cells, as is the case with Diamond-Blackfan Anemia.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Harvey Lodish (lodish@wi.mit.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice.
C57bl/6J mice were housed in the Whitehead Institute for Biomedical Research animal facility, and all experiments were performed in accordance with guidelines from the Massachusetts Institute of Technology Division of Comparative Medicine’s Committee on Animal Care. Fetal livers of male and female fetuses were isolated on day E14.5 and combined together so that erythroid progenitor cells were a mix of male and female.
METHOD DETAILS
Erythroid progenitor cell isolation.
Isolation of BFU-E and CFU-E enriched populations was performed as previously described (Zhang et al., 2013, Flygare et al., 2011). In detail, fetal liver was dissected from embryonic day 14.5 mice and disrupted and homogenized with manual pipetting, followed by lysis of mature red blood cells with ammonium chloride solution (Stem Cell Technologies). Remaining fetal liver cells were stained with biotinylated antibodies against the following surface proteins: CD3e, CD45R, Ly-6G, CD11b, Ter119 (all 5 from the BD-Pharmingen biotin mouse lineage depletion panel), Ly-6A/E (i.e. Sca-1, clone D7, E-Biosciences), CD16/32 (clone 93, E-Biosciences), CD41 (clone EbioMWReg30, E-Biosciences), and Ter119 (E-biosciences). Cells were then incubated with streptavidin-labeled magnetic beads (BD) to deplete cells labeled with biotinylated antibodies. Remaining unlabeled cells were then purified by FACS (staining procedure described below) on a FACS Aria sorter (BD).
In vivo Dexamethasone treatment and labeling with EdU and BrdU.
13.5 day of gestation pregnant female were injected subcutaneously with 200 microliters of vehicle control or 10 micrograms of Dexamethasone (USP-grade, Sigma) dissolved in 200 microliters of 1% DMSO (Sigma) in sterile phosphate-buffered saline (PBS) (Sigma). For those mice undergoing cell cycle analysis, 24 hours following Dexamethasone injection, 5 mg of EdU (Click Chemistry Tools) dissolved in 500 microliters of sterile PBS was injected intraperitoneal into pregnant mice. 2 hours following EdU injection, 5 mg of BrdU (Sigma) dissolved in 500 microliters of sterile PBS was injected intraperitoneal into the same pregnant mice. 25 minutes after BrdU injection, pregnant mice were euthanized and fetal livers were isolated for erythroid progenitor cell isolation.
Antibody staining, FACS, and flow cytometry.
For FACS purification of erythroid progenitors, lineage-depleted fetal liver cells were stained with the following: Propidium Iodide (Sigma), APC anti-CD117 (clone ACK2, E-Biosciences), PE anti-CD71 (clone R127217, E-Biosciences), PE anti-CD24 (clone M1/69, E-biosciences), FITC streptavidin (E-Biosciences). For isolation of erythroid progenitor subpopulations, lineage-depleted fetal liver cells first had cell fragments and large aggregates excluded with the FSC-A and SSC-A channels, and doublets excluded by the FSC-H and FSC-W channels followed by the SSC-H and SSC-W channels. Dead cells were then excluded by gating on the PI-negative population, and remaining lineage-marked cells were excluded by gating on the FITC-negative population. The remaining cells were then viewed gated on the CD117-high population to obtain c-Kit+ cells, and of the c-Kit+ cells, BFU-Es were gated as expressing the 0–10%ile of PE fluorescence, 25–35% cells were gated as expressing the 25–35%ile of PE fluorescence, 50–60% cells were gated as expressing the 50–60%ile of PE fluorescence, and CFU-Es were gated as expressing the 80–100%ile of PE fluorescence. For quantification of erythroid differentiation markers, cultured BFU-E cells were stained with PE anti-CD71 (clone R127217, E-Biosciences) and APC anti-Ter119 (E-Biosciences). For quantification of cell death/viability of cultured BFU-E and CFU-E cells, cells were stained with DAPI (ThermoFisher). For cells undergoing in vivo cell death/viability and CD71/CD24 distribution, lineage-depleted fetal liver cells were stained in the same manner as FACS isolation. For in vivo cell death/viability, PI-positive cell exclusion was not performed following doublet exclusion, and was performed after BFU-E, 25–35%, 50–60%, and CFU-E populations had been gated. For in vivo CD71/CD24 distribution, paired samples of c-Kit+ lineage-depleted cells for vehicle and Dex treated mice were overlayed as PE-expression histograms. The intersection point between Dex and vehicle curves in the middle of the histogram was identified and used as the bisecting gating point to delineate between “early” and “late” progenitors. For cells undergoing cell cycle analysis, lineage-depleted fetal liver cells stained with the Live/Dead-Near IR kit (ThermoFisher) and then fixed and permeabilized with BD Fix/Perm solution (BD) for 20 minutes at 4 degrees C. All washes between steps were performed with BD Perm/Wash solution (BD). Cells were then incubated with DNase (EMD Millipore) I at 37 degrees C for 1 hour. EdU DNA content was then labeled with the Click-It EdU AlexaFluor647 flow cytometry kit (ThermoFisher) and then samples were then stained with AlexaFluor488 anti-BrdU (Biolegend). Cells were then stained with PE anti-CD71 (clone R127217, E-Biosciences), PE anti-CD24 (clone M1/69, E-biosciences), BrilliantViolet421 anti-CD117 (Biolegend), and BrilliantViolet711 streptavidin (Biolegend). Flow cytometry was performed on a FACS Fortessa cytometer (BD)
Methylcellulose colony assay.
Lineage-depleted fetal liver cells were resuspended in MethoCult M3234 with cytokines supplemented at the following concentrations: erythropoietin (Amgen) 10 U/mL, murine stem cell factor (Peprotech) 50 ng/mL, murine IL-3 (Peprotech) 20 ng/mL, and murine IL-6 (Peprotech) 20 ng/mL. Cells were cultured at 37 degrees Celsius and colonies were counted at 3 and 7 days of culture. Colonies were visualized with a Nikon Eclipse TS100 microscope.
Proliferation assay.
FACS-isolated BFU-E enriched cells were cultured in progenitor culture media (PCM) consisting of serum free expansion media (SFEM II, Stem Cell Technologies) supplemented with 100 ng/mL murine SCF (Peprotech), 40 ng/mL murine insulin-like growth factor 1 (Peprotech), 2 U/mL human erythropoietin (Amgen), 1% penicillin/streptomycin (Gibco), and for indicated cultures 100 nM Dexamethasone (Sigma). Cells were seeded at known quantities and cultured at 37 degrees Celsius. Live cell numbers were counted at indicated days by both hemocytometer using Trypan Blue (Gibco) for dead cell exclusion, and by FACS on a FACS Fortessa cytometer (BD) normalizing to Precision Count Beads (Biolegend) and using Propidium Iodide (Sigma) for dead cell exclusion.
CFSE staining.
FACS-isolated BFU-E enriched cells were incubated in 5 uM CFSE (Invitrogen) in SFEM2 for 15 minutes at 37 deg C, then washed twice with 2% FBS (Gibco) in phosphate-buffered saline. Cells analyzed immediately after CFSE labeling were first incubated for 10 minutes in PCM before fixation with 4% paraformaldehyde (Electron Microscopy Sciences). The remainder of CFSE labeled cells were cultured in PCM at 37 degrees Celsius, and at indicated time points were fixed with 4% paraformaldehyde. All samples were analyzed at the same on a FACS Fortessa cytometer (BD).
Western blot and nuclear/cytoplasmic fractionation.
Lineage-depleted E14.5 fetal liver erythroid progenitor cells were isolated as described above. Nuclear and cytoplasmic cell fractions were then separated using the nuclei EZ lysis buffer (Sigma). Nuclei were then lysed with RIPA buffer. Protein sample concentrations were then quantified using the DC protein assay (Bio-Rad). 10 micrograms of protein from each cell fraction were then loaded onto a Bis-Tris 4–12% gel (Invitrogen), followed by polyacrylamide gel electrophoresis. Protein was then transferred to a PVDF membrane, blocked with 3% BSA, and the membrane was then cut along SeeBlue Plus 2 (Invitrogen) protein markers to isolate membrane portions corresponding to appropriate migration patterns of glucocorticoid receptor, lamin B, GAPDH, and beta-actin. Appropriate membrane portions were then incubated overnight with rabbit polyclonal anti-glucocorticoid receptor (ThermoFisher, PAI-511A), mouse monoclonal (clone A-11) anti-Lamin B (Santa Cruz Biotechnology, sc-377000), rabbit monoclonal (clone 13E5) anti-beta actin (Cell Signaling Technologies, 4970), or mouse monoclonal (clone 6C5) anti-GAPDH (Santa Cruz Biotechnology, sc-32233) overnight. Appropriate membranes were then incubated overnight with either HRP-conjugated anti-rabbit IgG (ThermoFisher) or HRP-conjugated anti-mouse IgG (Santa Cruz Biotechnology). Membranes were then incubated with ECL reagent (Perkin Elmer) and developed with CL-XPosure film (ThermoFisher).
Single cell transcriptome library preparation.
SmartSeq2 library prep was performed as previously described (Picelli et al., 2013). In detail, single cells were lysed in either 0.2% Triton X-100 (Sigma), or TCL buffer (Qiagen), containing RNase inhibitor (Clontech), Oligo-dT (5′-AAGCAGTGGTATCAACGCAGAGTACT30VN-3′, IDT), and dNTPs (Invitrogen). Lysates were then reverse transcribed at 42 deg C with SuperScript II reverse transcriptase (Invitrogen) in the presence of First-Strand Buffer (Invitrogen) containing DTT (Invitrogen), betaine (Sigma), magnesium chloride (Sigma), and template switching oligonucleotide (5′ -AAGCAGTGGTATCAACGCAGAGTACATrGrG+G-3′ where rG = riboguanosine, and +G = LNA-modified guanosine, Exiqon/Qiagen). Reverse-transcribed products were then amplified using Kapa HiFi Hotstart PCR master mix (Kapa) and an IS PCR oligo primer (5′ -AAGCAGTGGTATCAACGCAGAGT-3′, IDT). Amplified double-stranded cDNA libraries were then purified using Agencourt Ampure XP beads (Beckman Coulter), followed by DNA concentration quantification using the Qubit dsDNA HS kit (Life Technologies). 0.2 ng of double-stranded cDNA was used in a tagmentation reaction using the Nextera XT DNA sample preparation kit (Illumina) per manufacturers protocol. Tagmented transcriptomes were then indexed using the Nextera XT 96-index kit for 384 samples (Illumina) per manufacturer’s instructions, and samples were then sequenced on a HiSeq 2500 sequencer (Illumina).
Read mapping and counting.
The sample preparation was done in two locations with different lysis buffer protocols (generic buffer versus buffer TCL). We corrected for this batch effect during the Seurat analysis as described below.
Samples prepared using buffer TCL: Single BFU-Es and CFU-Es were isolated and processed on a 96 well plate for each cell type. We obtained between 35 thousand and 4 million 125 nt long paired end reads per cell. Reads were mapped against the GRCm38 mouse genome using STAR version 2.4, gene annotations from ENSEMBL version 87 (using only canonical chromosomes) and the overhang parameter from STAR set to 100. BFU-Es cultured for 24, 48 or 72 hours in the presence or absence of DEX were processed on six 96 well plates (one plate per condition) and sequenced. We obtained between 30 thousand and 6 million 40 nt long paired end reads. Reads were mapped against the GRCm38 mouse genome using STAR version 2.4, gene annotations from ENSEMBL version 87 (using only canonical chromosomes) and the overhang parameter from STAR set to 39.
Samples prepared using the generic lysis buffer: BFU-Es, CFU-Es and cells with intermediate levels of expression of CD71/CD24 (25–35% and 50–60%) were processed on a 96 well plate, 24 cells per cell type. We obtained between 1.6 and 6.4 million, 75 nt long, paired end reads. We mapped the reads against the GRCm38 mouse genome using STAR version 2.4, gene annotations from ENSEMBL version 87 (using only canonical chromosomes), and the overhang parameter from STAR set to 74. We sequenced an additional 24 BFU-Es, obtaining between 1–3 million, 50 nt long, paired end reads that were mapped as described above but with overhang parameter from STAR set to 49. For the daughter experiment we obtained between 500 thousand and 6 million, 40 nt, long paired end reads and mapped as described above but with the overhang parameter from STAR set to 39.
Read counts per gene were obtained with featureCounts (Liao et al., 2014) using the option –p and the same GTF file used to map the reads. We processed all 1040 sequences and kept only cells that had between 4000 and 11000 genes detected and less than 0.06 % of the reads mapping to mitochondrial genes, see Seurat analysis below.
Single cell vs bulk RNAseq correlation, Seurat analysis, heatmap drawing, and monocle analysis were done in R:
Correlation of single cell RNAseq and bulk RNAseq.
Bulk RNA-seq data for CFU-Es and BFU-Es was downloaded from the GEO accession number GSE26086 (Flygare et al., 2011). We mapped the reads against the GRCm38 mouse genome using STAR version 2.5.4b, gene annotations from ENSEMBL version 87 (using only canonical chromosomes), and the overhang parameter from STAR set to 35. (Read counts per gene were obtained with featureCounts using the option –p and the same GTF file used to map the reads.). To compare single cell RNA-seq to the bulk RNA-seq experiment, all the cells from the single cell experiment were summed to obtained one total count per gene. The gene counts from the single cell experiment and the bulk RNA-seq were normalized with DEseq (Anders and Huber, 2010) to account for the difference on sequencing depth. Scatter plots of the log2 of the counts plus 1 pseudocount were done in R. Correlation was calculated with the R function cor.test.
Seurat analysis.
Seurat v2.2 (Butler et al., 2018) was used to analyze the single cell RNA-seq data, including filtering out cells with too few reads or low quality cells. We followed the Seurat Guided Clustering Tutorial (https://satijalab.org/seurat/pbmc3k_tutorial.html) changing the following cut offs: 1) “FilterCells” step, “nGene” thresholds 4000–11000, “percent.mito” thresholds -Inf - 0.06. We kept 848 cells in total after the filtering step. 2) “NormalizeData” step, normalization method “LogNormalize”, scale.factor 1e4. 3) Since our samples’ libraries were prepared with two different lysis buffers, on the “ScaleData” step we regressed out the variation driven by the batch effect of library preparation. To get variable genes we used the “FindVariableGenes” function with cutoffs “x.low.cutoff = 0.0125, x.high.cutoff = 7, y.cutoff = 0.7” which gave us 2880 genes. We also selected variable genes with two other cutoffs: “x.low.cutoff = 0.0125, x.high.cutoff = 7, y.cutoff = 0.5” gave us 4864 genes and “x.low.cutoff = 0.0125, x.high.cutoff = 7, y.cutoff = 1” gave us 1347 variable genes. The PCA obtained was similar using any of the three variable genes sets selected.
PCA using only the cells not treated with Dex.
The data counts were used to make a Seurat object. We filtered out cells keeping cells with a number of genes detected between 4000 and 11000, and percentage of mitochondrial <= 0.06, keeping 848 cells. Counts were log normalized and scaled to remove the batch effect introduced by the lysis buffer. We made a new object taking the subset of cells that had not been treated with DEX, and selected 2732 variable genes using “x.low.cutoff = 0.0125, x.high.cutoff = 7, and y.cutoff = 0.7”. We used the no Dex cells variable genes to make a PCA plot with all the cells included on the study. Additionally, we performed the normalization, scale with batch removal, selection of variable genes, and PCA with only no treated cells, and obtained the same type of trajectory on the PCA plot.
Diffusion mapping.
Since the diffusion map function requires log normalized data as input, we removed the batch effect from the log normalized data with ComBat. To use the ComBat parameter “par.prior=TRUE”, we only kept genes that had a log normalized value of 1 in at least 50 cells. This gave us 4003 genes (848 cells). We selected variable genes with the same cut offs as before (“x.low.cutoff = 0.0125, x.high.cutoff = 7, and y.cutoff = 0.7”) and obtained 645 variable genes that we used run the PCA and diffusion (RunDiffusion function) using either 15 or 50 principal components. The diffusion maps obtained with 15 or 50 components were the same.
Comparison to Tusi et. al. transcriptome distribution.
Using the visualization tool from the Tusi et. al. paper, <https://kleintools.hms.harvard.edu/tools/springViewer_1_6_dev.html?datasets/mouse_HPCs/fetal_liver/full>, we selected 6054 cells on the erythroid branch (probability of erythroid > ~0.6). Of those, we used the 4753 cells that had 1 on column 5, “pass_filter”, of the counts file “GSM2388074_fetal_liver.raw_umifm_counts.csv” from the GSE89754 series of the GEO repository. To combine this data with ours we used the 26729 gene symbols present in both count matrices. We made Seurat objects with the Tusi et. al count matrix (4753 cells) and our count matrix (888 cells), and combined them in one Seurat object. We filtered cells on number of genes detected, between 1000 and 10000, and percentage of mitochondrial genes, between –Inf and 0.1. After this filtering we kept 5412 cells. We normalized the data using Seurat log normalization and made diffusion maps for only the 4527 erythroid cells from the Tusi et. al study and for all 5412 cells as described below.
We first subset 4527 erythroid cells from the Seurat object, scaled the data, and selected variable genes using the following cut offs: “x.low.cutoff = 0.03, x.high.cutoff = 7, y.cutoff = 0.8”. We obtained 3208 variable genes that we used to run PCA and diffusion maps using Seurat functions “RunPCA” and “RunDiffusion” using principal components 1–10, respectively. Next we analyzed both datasets together using the variable genes selected during the analysis of erythroid FL cells only. To combine both datasets and remove batch effects we first filtered genes, keeping only genes with a minimum log normalized expression value of 1 in at least 100 cells. After this filtering we kept 7915 genes across the 5412 samples. Of these genes, 2129 were variable genes selected when analyzing only the Tusi et. al data. We adjusted for batch effects with the function ComBat(), from the R package sva, using Seurat log normalized data as input, default parameters, and defining 3 batches, one for the cells from Tusi et. al. paper, and one from each of the 2 batches present in our cells due to the lysis buffer used. We replaced the normalized data with the ComBat corrected data on the Seurat object, scaled the data, and run PCA and diffusion maps using Seurat as described above (using Seurat functions “RunPCA” and “RunDiffusion” using principal components 1–10) using the 2129 variable genes that had passed the expression filter. The diffusion map of the combined data had the same shape than the DM of the FL only and showed that our cells/data follow the same trajectory than the FL erythroid cells.
Regression of PCA developmental trajectory and pseudotime calculation.
PC1 and PC2 values, or DM1 and DM2 values, for all 848 cells after filtering were input into Prism 7 (Graphpad) and subjected to a 6th order regression that resulted in the y(x) function (where y = PC2 or DM2 value and x = PC1 or DM1 value) displayed in Supplementary Figure S4. (x0,y0) represent the position of each cell in PC1 vs PC2, or DM1 vs DM2, two-dimensional space. We used the regression function, y(x), and the coordinates for each cell to define the distance function for each cell.
To determine the x-mapped coordinate, xmapn, along y(x) or in other words the position along the curve in which a given cell, celln, travels the shortest distance to reach, we optimized the distance function, D(x), using the optimize function from the stats package in R, and obtained the minimum of this optimization using the optimize(D(x))$min operation.
With the x-mapped coordinates for each cell, we can then use arc length as a metric for “pseudotime,” with x-mapped coordinate of the earliest cell in development, xmap1, and xmapn serving as the bounds for integration.
We define pseduotime for celln as the result of the integration of the arc-length formula from the mapped position of the earliest cell in development, xmap1, to the mapped position of celln, xmapn, with the first cell defined as pseudotime 0.
Gene expression plots for individual genes along pseudotime (LOESS fit line).
The gene plots showing the expression in all cells, or in BFU-Es at day 1, 2 or 3, either treated or not treated with DEX, along pseudotime for individual genes were done with ggplot. The blue line was drawn using LOESS (span = 0.75) to fit the expression of all 848 cells used in the Seurat analysis and heatmap.
Heatmap for expression of genes used to build the PCA over pseudotime.
Cells were ordered based on the distance from their projected point in the curve to the beginning of the curve as described above. We used the logged normalized expression of the 2880 variable genes selected with Seurat as described above. To draw the heatmap we followed a strategy similar to the Monocle 2 (Qiu et al., 2017) “pheatmap” function. We selected 100 time points equally distributed between 0 and the furthest (highest) pseudotime value. For each gene we calculated the expected expression at those 100 pseudotime points using the R function “LOESS” to fit the real values, with the span parameter set to 1. We centered and scaled the expression values of each gene across pseudotime using the R function scale(). We ordered the genes based on the pseudotime point when they peak and we broke ties by displaying first the genes with higher scaled values, from zero to time point 50, and the opposite way from time point 51 to 100. We used the Monocle 2 color scheme.
Monocle 2 analysis (Qiu et al., 2017).
We analyzed the single BFU-Es and CFU-Es, sequenced with 125 nt long paired reads, and the BFU-Es cultured for 24, 48 or 72 hours in the presence or absence of DEX, with Monocle2.2. The libraries for all of these cells were prepared in the same way, so we didn’t have to correct for batch effects. We followed the standard analysis proposed on “http://cole-trapnell-lab.github.io/monocle-release/docs/ “. When creating a new monocle object with the “newCellDataSet” function we used the following parameters: “lowerDetectionLimit = 0.5, expressionFamily=negbinomial.size”. As ordering genes in the monocle function “setOrderingFilter” we used the 1347 most variable genes selected with Seurat “FindVariableGenes” function and cut offs “x.low.cutoff = 0.0125, x.high.cutoff = 7, y.cutoff = 1”, as described above.
Transcriptome-based cell cycle state predictions.
To score and classify cells based on cell cycle gene expression, we used the Seurat function “CellCycleScoring” following the cell cycle vignette from Seurat “https://satijalab.org/seurat/cell_cycle_vignette.html”. The vignette uses the human cell cycle markers genes from (Kowalczyk MS1, Tirosh I, et. al 2015). We obtained the mouse orthologs for those genes using ESEMBL Biomart (https://academic.oup.com/nar/article/46/D1/D754/4634002) and use those instead.
Comparison of PCA pseudotime ordering to Diffusion Mapping and Monocle 2 pseudotime ordering.
For each of the 3 methods of calculating pseudotime, BFU-E and CFU-E cells, and cultured BFU-E cells were ordered based on pseudotime coordinate. To compare pseudotime order from PCA to pseudotime order from Diffusion Mapping, we plotted pseudotime order position for PCA vs pseudotime order position for Diffusion Mapping and calculated the correlation coefficient with the identity line. To compare pseudotime order from PCA to pseudotime order from Monocle 2, we plotted pseudotime order position for PCA vs pseudotime order position for Monocle 2 and calculated the correlation coefficient with the identity line.
Daughter cell transcriptome analysis.
Single BFU-E cells were FACS-deposited into individual wells of a 96-well plate containing PCM either with or without 100 nM dexamethasone. Plates were briefly spun down and visualized by light microscopy using a Zeiss Primovert microscope to confirm the wells with only a single cell. Those wells were further assessed 16 hours later to identify the cells that completed a single cell division. Cells from wells that contained a single pair of daughter cells were removed from the 96-well plate by mouth pipetting, using hand-drawn glass capillary pipettes (Drummond Scientific) and a Nikon SMZ 1500 microscope. Both daughter cells were deposited in a 30 uL drop of 2% FBS (Gibco) in PBS and the two cells were manually separated by mouth pipetting. Each individual daughter was then mouth pipetted into SmartSeq2 lysis buffer to perform single cell transcriptome profiling with SmartSeq2.
QUANTIFICATION AND STATISTICAL ANALYSIS
Student’s T-Test, Mann-Whitney U-Test, Pillai’s multivariate statistic, and correlation coefficient for regression functions were utilized as indicated. Student’s T-Test, the Mann-Whitney U-Test, and correlation coefficients were calculated in Prism 7 (Graphpad), and Pillai’s multivariate statistic was calculated in R. For calculating the empiric probability of daughter cell symmetry, we performed the following: We measured the pseudotime for 10 pairs of daughter BFU-E cells cultured without Dex and 96 non-paired control BFU-E cells cultured without Dex for a total of 116 paired and unpaired BFU-E cells cultured without Dex. We calculated the average observed difference between the 10 pairs of daughter BFU-E cells cultured without Dex. To determine whether this average observed difference is smaller than expected by chance, we generated a distribution of average differences from 10 randomly generated pairs of cells. To do so, we first generated all possible pairs of 116 BFU-E cells (116 “choose” 2 = 6,670 total combinations). We calculated the absolute difference in pseudotime for each possible pair. We randomly sampled 10 pairs of cells, repeated 1 million times to generate a distribution of average differences. Finally using this distribution, we calculated the empirical probability of observing an average difference less than or equal to the averaged observed difference for the 10 paired daughter cells. We repeated this analysis for 12 pairs of daughter cells from BFU-Es cultured with Dex and 96 non-paired control BFU-E cells cultured with Dex (total = 120 paired and unpaired DEX cells).
DATA AND SOFTWARE AVAILABILITY
Single cell RNA sequencing data has been deposited in NCBI GEO under accession GSE117233.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT OR RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-mouse CD3e biotin (clone 145–2C11) | BD-Pharmingen | 559971; RRID:AB_394592 |
| Anti-mouse CD45R biotin (clone RA3–6B2) | BD-Pharmingen | 559971; RRID:AB_394616 |
| Anti-mouse Ly-6G biotin (clone RB6–8C5) | BD-Pharmingen | 559971; RRID:AB_394640 |
| Anti-mouse CD11b biotin (clone M1/70) | BD-Pharmingen | 559971; RRID:AB_394773 |
| Anti-mouse Ter119 biotin (clone Ter-119) | BD-Pharmingen | 559971; RRID:AB_394985 |
| Anti-mouse Ly-6A/E biotin (clone D7) | E-Biosciences | 13–5981–85 |
| Anti-mouse CD16/32 biotin (clone 93) | E-Biosciences | 13–0161–86 |
| Anti-mouse CD41 biotin (clone EbioMWReg30) | E-Biosciences | 13–0411–85 |
| Anti-mouse Ter119 biotin (clone Ter-119) | E-Biosciences | 13–5921–81; RRID:AB_469756 |
| Anti-mouse CD117 APC (clone ACK2) | E-Biosciences | 17–1172–83; RRID:AB_469429 |
| Anti-mouse CD71 PE (clone R127217) | E-Biosciences | 12–0711–81; RRID:AB_465739 |
| Anti-mouse CD24 PE (clone M1/69) | E-Biosciences | 12–0242–82; RRID:AB_465598 |
| Streptavidin FITC | E-Biosciences | 11–4317–87 |
| Anti-BrdU AlexaFluor488 | Biolegend | 364106 |
| Anti-mouse CD117 BrilliantViolet 421 (clone ACK2) | Biolegend | 135123 |
| Streptavidin BrilliantViolet 711 | Biolegend | 405241 |
| Anti-glucocorticoid receptor polyclonal | ThermoFisher | PAI-511A |
| Anti-lamin B (clone A-11) | Santa Cruz Biotechnologies | sc-377000 |
| Anti-beta actin (clone 13E5) | Cell Signaling Technologies | 4970 |
| Anti-GAPDH (clone 6C5) | Santa Cruz Biotechnologies | sc-32233 |
| Goat anti-mouse IgG HRP | Santa Cruz Biotechnologies | sc-2005 |
| anti-rabbit IgG HRP | ThermoFisher | NC0322305 |
| Chemicals, Peptides, and Recombinant proteins | ||
| Dexamethasone, USP | Sigma | D9184 |
| 5-ethynyl deoxyuridine (EdU) | Click-Chemistry Tools | 1149–100 |
| 5-bromo deoxyuridine (BrdU) | Sigma | B5002 |
| Dimethyl sulfoxide (DMSO) | Sigma | D2650 |
| Phosphate buffered saline (PBS) | Sigma | P4417 |
| Propidium Iodide | Sigma | P4864 |
| DAPI | ThermoFisher | D1306 |
| DNase I | EMD Millipore | 260913 |
| Erythropoietin | Amgen | CAS # 113427–24–0 |
| Murine stem cell factor | Peprotech | 250–03C |
| Murine insulin-like growth factor 1 | Peprotech | 250–19 |
| Murine interleukin-3 | Peprotech | 213–13 |
| Murine interleukin-6 | Peprotech | 216–16 |
| Penicillin/Streptomycin | Gibco | 15140122 |
| Trypan blue | Gibco | 15250061 |
| Precision count beads | Biolegend | 424902 |
| CFSE | Invitrogen | C34554 |
| Paraformaldehyde | Electron Microscopy Sciences | 100504–858 |
| Bovine serum albumin | Sigma | A7906 |
| Triton X-100 | Sigma | T9284 |
| RNase inhibitor | Clontech | 2313B |
| dNTPs | Invitrogen | 10297018 |
| DTT | Invitrogen | D1532 |
| Betaine | Sigma | 61962 |
| Magnesium chloride | Sigma | M8266 |
| Critical Commercial Assays | ||
| Click-It EdU AlexaFluor 647 flow cytometry kit | ThermoFisher | C10424 |
| Cytofix/Cytoperm buffer | BD | BDB554714 |
| Live/Dead Fixable Near-IR Dead Cell Stain Kit | ThermoFisher | L34975 |
| Nuclei EZ lysis buffer | Sigma | NUC101–1KT |
| DC protein assay | Bio-Rad | 5000111 |
| Bis-Tris 4–12% gel | Invitrogen | NP0322 |
| See Blue plus 2 ladder | Invitrogen | LC5925 |
| ECL reagent | Perkin Elmer | NEL103001 |
| TCL buffer | Qiagen | 1070498 |
| Superscript II | Invitrogen | 18064014 |
| First-strand buffer | Invitrogen | 18064014 |
| Kapa HiFi hotstart PCR master mix | Kapa | KK2602 |
| Qubit dsDNA HS kit | Life Technologies | Q32854 |
| Nextera XT DNA sample preparation kit | Illumina | FC-131–1096 |
| Nextera XT 96-index kit for 384 samples | Illumina | FC-121–1012 |
| Deposited Data | ||
| BFU-E and CFU-E bulk RNA-seq | Flygare, J. et. al., 2011 | GEO: GSE26086 |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6J | Jackson Labs | 000664; RRID:IMSR_JAX:000664 |
| Oligonucleotides | ||
| Oligo-dT (5’-AAGCAGTGGTATCAACGCAGAGTACT30VN-3’) | Integrated DNA Technologies | |
| Template switching oligonucleotide (5’-AAGCAGTGGTATCAACGCAGAGTACATrGrG+G-3’) | Exiqon/Qiagen | |
| IS PCR (5’-AAGCAGTGGTATCAACGCAGAGT-3’) | Integrated DNA Technologies | |
| Software and Algorithms | ||
| Prism 7 | Graphpad | www.graphpad.com |
| FlowJo v10 | FlowJo | www.flowjo.com |
| R studio | RStudio | www.rstudio.com |
| Other | ||
| streptavidin-labeled magnetic bead particles | BD Biosciences | BDB557812 |
| MethoCult M3234 | Stem Cell Technologies | 03234 |
| Serum-free expansion media II | Stem Cell Technologies | 09655 |
| Fetal bovine serum | Gibco | 10438026 |
| CL-XPosure film | ThermoFisher | 34091 |
| Agencourt Ampure XP beads | Beckman Coulter | 102492–758 |
HIGHLIGHTS.
Transcriptomic continuums correlate with phenotypic continuums in erythropoiesis
Degree of developmental progression per cell cycle governs proliferative capacity
Glucocorticoids uncouple cell division and differentiation speed in erythropoiesis
Erythroid progenitor cell divisions are symmetric
ACKNOWLEDGEMENTS
The authors would like to thank George Bell, Stuart Orkin, Peter Reddien, Vijay Sankaran, and Akiko Shimamura for helpful discussion, Yung Hwang and Merav Socolovsky for sharing experimental protocols, and the Whitehead Flow Cytometry core, the Whitehead Genome Technology Core, and the Broad Technology Labs for technical assistance. This work was supported by NIH grants to H.F.L. (DK06834813, HL032262-25), and an ASH RTAF award to H.L.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing financial interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- AKINDURO O, WEBER TS, ANG H, HALTALLI MLR, RUIVO N, DUARTE D, RASHIDI NM, HAWKINS ED, DUFFY KR & LO CELSO C 2018. Proliferation dynamics of acute myeloid leukaemia and haematopoietic progenitors competing for bone marrow space. Nat Commun, 9, 519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ANDERS S & HUBER W 2010. Differential expression analysis for sequence count data. Genome Biol, 11, R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BACK J, DIERICH A, BRONN C, KASTNER P & CHAN S 2004. PU.1 determines the self-renewal capacity of erythroid progenitor cells. Blood, 103, 3615–23. [DOI] [PubMed] [Google Scholar]
- BASTA JM, ROBBINS L, KIEFER SM, DORSETT D & RAUCHMAN M 2014. Sall1 balances self-renewal and differentiation of renal progenitor cells. Development, 141, 1047–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BAUER A, TRONCHE F, WESSELY O, KELLENDONK C, REICHARDT HM, STEINLEIN P, SCHUTZ G & BEUG H 1999. The glucocorticoid receptor is required for stress erythropoiesis. Genes Dev, 13, 2996–3002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BONYADI M, WALDMAN SD, LIU D, AUBIN JE, GRYNPAS MD & STANFORD WL 2003. Mesenchymal progenitor self-renewal deficiency leads to age-dependent osteoporosis in Sca-1/Ly-6A null mice. Proc Natl Acad Sci U S A, 100, 5840–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BRIEGEL K, LIM KC, PLANK C, BEUG H, ENGEL JD & ZENKE M 1993. Ectopic expression of a conditional GATA-2/estrogen receptor chimera arrests erythroid differentiation in a hormone-dependent manner. Genes Dev, 7, 1097–109. [DOI] [PubMed] [Google Scholar]
- BUTLER A, HOFFMAN P, SMIBERT P, PAPALEXI E & SATIJA R 2018. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol, 36, 411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHAN HS, SAUNDERS EF & FREEDMAN MH 1982. Diamond-Blackfan syndrome. I. Erythropoiesis in prednisone responsive and resistant disease. Pediatr Res, 16, 474–6. [DOI] [PubMed] [Google Scholar]
- COLLINS CA, OLSEN I, ZAMMIT PS, HESLOP L, PETRIE A, PARTRIDGE TA & MORGAN JE 2005. Stem cell function, self-renewal, and behavioral heterogeneity of cells from the adult muscle satellite cell niche. Cell, 122, 289–301. [DOI] [PubMed] [Google Scholar]
- DULKEN BW, LEEMAN DS, BOUTET SC, HEBESTREIT K & BRUNET A 2017. Single-Cell Transcriptomic Analysis Defines Heterogeneity and Transcriptional Dynamics in the Adult Neural Stem Cell Lineage. Cell Rep, 18, 777–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ENGLAND SJ, MCGRATH KE, FRAME JM & PALIS J 2011. Immature erythroblasts with extensive ex vivo self-renewal capacity emerge from the early mammalian fetus. Blood, 117, 2708–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FLYGARE J, RAYON ESTRADA V, SHIN C, GUPTA S & LODISH HF 2011. HIF1alpha synergizes with glucocorticoids to promote BFU-E progenitor self-renewal. Blood, 117, 3435–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GOLDE DW, BERSCH N & CLINE MJ 1976. Potentiation of erythropoiesis in vitro by dexamethasone. J Clin Invest, 57, 57–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HAGHVERDI L, BUETTNER F & THEIS FJ 2015. Diffusion maps for high-dimensional single-cell analysis of differentiation data. Bioinformatics, 31, 2989–98. [DOI] [PubMed] [Google Scholar]
- HARANDI OF, HEDGE S, WU DC, MCKEONE D & PAULSON RF 2010. Murine erythroid short-term radioprotection requires a BMP4-dependent, self-renewing population of stress erythroid progenitors. J Clin Invest, 120, 4507–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HATTANGADI SM, WONG P, ZHANG L, FLYGARE J & LODISH HF 2011. From stem cell to red cell: regulation of erythropoiesis at multiple levels by multiple proteins, RNAs, and chromatin modifications. Blood, 118, 6258–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HAYMAN MJ, MEYER S, MARTIN F, STEINLEIN P & BEUG H 1993. Self-renewal and differentiation of normal avian erythroid progenitor cells: regulatory roles of the TGF alpha/c-ErbB and SCF/c-kit receptors. Cell, 74, 157–69. [DOI] [PubMed] [Google Scholar]
- HWANG Y, FUTRAN M, HIDALGO D, POP R, IYER DR, SCULLY R, RHIND N & SOCOLOVSKY M 2017. Global increase in replication fork speed during a p57(KIP2)-regulated erythroid cell fate switch. Sci Adv, 3, e1700298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ISKANDER D, PSAILA B, GERRARD G, CHAIDOS A, EN FOONG H, HARRINGTON Y, KARNIK LC, ROBERTS I, DE LA FUENTE J & KARADIMITRIS A 2015. Elucidation of the EP defect in Diamond-Blackfan anemia by characterization and prospective isolation of human EPs. Blood, 125, 2553–7. [DOI] [PubMed] [Google Scholar]
- JIN L, FENG T, SHIH HP, ZERDA R, LUO A, HSU J, MAHDAVI A, SANDER M, TIRRELL DA, RIGGS AD & KU HT 2013. Colony-forming cells in the adult mouse pancreas are expandable in Matrigel and form endocrine/acinar colonies in laminin hydrogel. Proc Natl Acad Sci U S A, 110, 3907–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KARAMITROS D, STOILOVA B, ABOUKHALIL Z, HAMEY F, REINISCH A, SAMITSCH M, QUEK L, OTTO G, REPAPI E, DOONDEEA J, USUKHBAYAR B, CALVO J, TAYLOR S, GOARDON N, SIX E, PFLUMIO F, PORCHER C, MAJETI R, GOTTGENS B & VYAS P 2018. Single-cell analysis reveals the continuum of human lympho-myeloid progenitor cells. Nat Immunol, 19, 85–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KLEIN AM, MAZUTIS L, AKARTUNA I, TALLAPRAGADA N, VERES A, LI V, PESHKIN L, WEITZ DA & KIRSCHNER MW 2015. Droplet barcoding for single-cell transcriptomics applied to embryonic stem cells. Cell, 161, 1187–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KOURY MJ 2016. Tracking erythroid progenitor cells in times of need and times of plenty. Exp Hematol, 44, 653–63. [DOI] [PubMed] [Google Scholar]
- LESCROART F, WANG X, LIN X, SWEDLUND B, GARGOURI S, SANCHEZ-DANES A, MOIGNARD V, DUBOIS C, PAULISSEN C, KINSTON S, GOTTGENS B & BLANPAIN C 2018. Defining the earliest step of cardiovascular lineage segregation by single-cell RNA-seq. Science, 359, 1177–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LIAO Y, SMYTH GK & SHI W 2014. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics, 30, 923–30. [DOI] [PubMed] [Google Scholar]
- LUI JH, HANSEN DV & KRIEGSTEIN AR 2011. Development and evolution of the human neocortex. Cell, 146, 18–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MACAULAY IC, SVENSSON V, LABALETTE C, FERREIRA L, HAMEY F, VOET T, TEICHMANN SA & CVEJIC A 2016. Single-Cell RNA-Sequencing Reveals a Continuous Spectrum of Differentiation in Hematopoietic Cells. Cell Rep, 14, 966–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MCCULLOCH CA, STRUGURESCU M, HUGHES F, MELCHER AH & AUBIN JE 1991. Osteogenic progenitor cells in rat bone marrow stromal populations exhibit self-renewal in culture. Blood, 77, 1906–11. [PubMed] [Google Scholar]
- NARLA A, DUTT S, MCAULEY JR, AL-SHAHROUR F, HURST S, MCCONKEY M, NEUBERG D & EBERT BL 2011. Dexamethasone and lenalidomide have distinct functional effects on erythropoiesis. Blood, 118, 2296–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NATHAN DG, CLARKE BJ, HILLMAN DG, ALTER BP & HOUSMAN DE 1978. Erythroid precursors in congenital hypoplastic (Diamond-Blackfan) anemia. J Clin Invest, 61, 489–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NESTOROWA S, HAMEY FK, PIJUAN SALA B, DIAMANTI E, SHEPHERD M, LAURENTI E, WILSON NK, KENT DG & GOTTGENS B 2016. A single-cell resolution map of mouse hematopoietic stem and progenitor cell differentiation. Blood, 128, e20–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OHENE-ABUAKWA Y, ORFALI KA, MARIUS C & BALL SE 2005. Two-phase culture in Diamond Blackfan anemia: localization of erythroid defect. Blood, 105, 838–46. [DOI] [PubMed] [Google Scholar]
- ORKIN SH & ZON LI 2008. Hematopoiesis: an evolving paradigm for stem cell biology. Cell, 132, 631–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PICELLI S, BJORKLUND AK, FARIDANI OR, SAGASSER S, WINBERG G & SANDBERG R 2013. Smart-seq2 for sensitive full-length transcriptome profiling in single cells. Nat Methods, 10, 1096–8. [DOI] [PubMed] [Google Scholar]
- QIU X, MAO Q, TANG Y, WANG L, CHAWLA R, PLINER HA & TRAPNELL C 2017. Reversed graph embedding resolves complex single-cell trajectories. Nat Methods, 14, 979–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- REICHARDT HM, KAESTNER KH, TUCKERMANN J, KRETZ O, WESSELY O, BOCK R, GASS P, SCHMID W, HERRLICH P, ANGEL P & SCHUTZ G 1998. DNA binding of the glucocorticoid receptor is not essential for survival. Cell, 93, 531–41. [DOI] [PubMed] [Google Scholar]
- SHALEK AK, SATIJA R, SHUGA J, TROMBETTA JJ, GENNERT D, LU D, CHEN P, GERTNER RS, GAUBLOMME JT, YOSEF N, SCHWARTZ S, FOWLER B, WEAVER S, WANG J, WANG X, DING R, RAYCHOWDHURY R, FRIEDMAN N, HACOHEN N, PARK H, MAY AP & REGEV A 2014. Single-cell RNA-seq reveals dynamic paracrine control of cellular variation. Nature, 510, 363–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TRAPNELL C, CACCHIARELLI D, GRIMSBY J, POKHAREL P, LI S, MORSE M, LENNON NJ, LIVAK KJ, MIKKELSEN TS & RINN JL 2014. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat Biotechnol, 32, 381–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TREUTLEIN B, LEE QY, CAMP JG, MALL M, KOH W, SHARIATI SA, SIM S, NEFF NF, SKOTHEIM JM, WERNIG M & QUAKE SR 2016. Dissecting direct reprogramming from fibroblast to neuron using single-cell RNA-seq. Nature, 534, 391–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TUSI BK, WOLOCK SL, WEINREB C, HWANG Y, HIDALGO D, ZILIONIS R, WAISMAN A, HUH JR, KLEIN AM & SOCOLOVSKY M 2018. Population snapshots predict early haematopoietic and erythroid hierarchies. Nature, 555, 54–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VELTEN L, HAAS SF, RAFFEL S, BLASZKIEWICZ S, ISLAM S, HENNIG BP, HIRCHE C, LUTZ C, BUSS EC, NOWAK D, BOCH T, HOFMANN WK, HO AD, HUBER W, TRUMPP A, ESSERS MA & STEINMETZ LM 2017. Human haematopoietic stem cell lineage commitment is a continuous process. Nat Cell Biol, 19, 271–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VIGNJEVIC S, BUDEC M, MARKOVIC D, DIKIC D, MITROVIC O, DIKLIC M, SUBOTICKI T, COKIC V & JOVCIC G 2015. Glucocorticoid receptor mediates the expansion of splenic late erythroid progenitors during chronic psychological stress. J Physiol Pharmacol, 66, 91–100. [PubMed] [Google Scholar]
- VON LINDERN M, ZAUNER W, MELLITZER G, STEINLEIN P, FRITSCH G, HUBER K, LOWENBERG B & BEUG H 1999. The glucocorticoid receptor cooperates with the erythropoietin receptor and c-Kit to enhance and sustain proliferation of erythroid progenitors in vitro. Blood, 94, 550–9. [PubMed] [Google Scholar]
- VOORHEES JL, POWELL ND, MOLDOVAN L, MO X, EUBANK TD & MARSH CB 2013. Chronic restraint stress upregulates erythropoiesis through glucocorticoid stimulation. PLoS One, 8, e77935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WENDLING F, SHREEVE MM, MCLEOD DL & AXELRAD AA 1983. Long-term culture of murine erythropoietic progenitor cells in the absence of an adherent layer. Nature, 305, 625–7. [DOI] [PubMed] [Google Scholar]
- ZENG C, MULAS F, SUI Y, GUAN T, MILLER N, TAN Y, LIU F, JIN W, CARRANO AC, HUISING MO, SHIRIHAI OS, YEO GW & SANDER M 2017. Pseudotemporal Ordering of Single Cells Reveals Metabolic Control of Postnatal beta Cell Proliferation. Cell Metab, 25, 1160–1175 e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHANG L, PRAK L, RAYON-ESTRADA V, THIRU P, FLYGARE J, LIM B & LODISH HF 2013. ZFP36L2 is required for self-renewal of early burst-forming unit erythroid progenitors. Nature, 499, 92–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Single cell RNA sequencing data has been deposited in NCBI GEO under accession GSE117233.