

Abstract
Class I ribonucleotide reductases (RNRs) share a common mechanism of nucleotide reduction in a catalytic α subunit. All RNRs initiate catalysis with a thiyl radical, generated in class I enzymes by a metallocofactor in a separate β subunit. Class Id RNRs use a simple mechanism of cofactor activation involving oxidation of a MnII2 cluster by free superoxide to yield a metal-based MnIIIMnIV oxidant. This simple cofactor assembly pathway suggests that class Id RNRs may be representative of the evolutionary precursors to more complex class Ia-c enzymes. X-ray crystal structures of two class Id α proteins from Flavobacterium johnsoniae (Fj) and Actinobacillus ureae (Au) reveal that this subunit is distinctly small. The enzyme completely lacks common N-terminal ATP cone allosteric motifs that regulate overall activity, a process that normally occurs by dATP-induced formation of inhibitory quaternary structures to prevent productive β subunit association. Class Id RNR activity is insensitive to dATP in the Fj and Au enzymes evaluated here, as expected. However, the class Id α protein from Fj adopts higher order structures, detected crystallographically and in solution. The Au enzyme does not exhibit these quaternary forms. Our study reveals structural similarity between bacterial class Id and eukaryotic class Ia α subunits in conservation of an internal auxiliary domain. Our findings with the Fj enzyme illustrate that nucleotide-independent higher-order quaternary structures can form in simple RNRs with truncated or missing allosteric motifs.
Graphical Abstract
INTRODUCTION
Ribonucleotide reductases (RNRs) convert ribonucleotides to 2′-deoxyribonucleotides, the building blocks required for DNA synthesis and repair.1 This essential transformation is initiated by a cysteine thiyl radical (Cys•) in the active site of the catalytic RNR subunit (α in class I RNR).2 The Cys• is transient, generated every turnover upon substrate binding by a stable oxidizing metallocofactor. RNRs are divided into three classes based on the cofactor used to generate the Cys•.3 Aerobic class I enzymes use a dinuclear metallocofactor and/or organic radical housed in a separate subunit, β.4–6 Class I RNRs can be further subdivided into subclasses Ia-e, differentiated by the identity of the cofactor utilized and the mechanism of cofactor activation and Cys• generation. In subclasses Ia/b, the metallocofactor is a stable tyrosine radical (Tyr•) coupled to an oxidized metal cluster (FeIII2 or MnIII2).7, 8 The Ic/d enzymes lack a stable Tyr• and instead use the oxidized metal cluster (MnIVFeIII or MnIVMnIII) directly.5, 9 Recently, a metal-free class Ie RNR was identified, which instead uses a dihydroxyphenylalanine radical (DOPA•) to generate Cys•.6, 10 In all class I RNRs, substrate binding triggers the formation of an active α/β complex (α2β2 in E. coli Ia RNR),11 and an oxidizing equivalent is transferred from the metallocofactor or DOPA•/Tyr• to the active site in α over a distance of >40 Å.12, 13 The radical translocation reaction requires a pathway of conserved redox-active amino acids (Tyr, Trp, etc.) in both subunits. Although diverse metallocofactors initiate the radical reaction in RNRs, the catalytic α subunits all utilize the same core structural motif, a 10-stranded α/β barrel. 14–17
Class I RNR activity can be tuned via allostery,18–20 all known mechanisms of which involve formation of specific inhibitory quaternary structures.21–26 These adaptations function to avoid errors during DNA replication and repair.27 Substrate specificity is tuned in response to intracellular levels of (d)NTPs, which bind to an allosteric specificity site (S-site).15 In class I RNR catalytic subunits, the S-site is found at an α2 dimer interface. Between the S-site and the active site, a flexible region facilitates communication between the two sites and becomes ordered upon effector binding.28 The identity of the effector nucleotide bound to the S-site directs the reduction of specific NDP substrates: ATP/dATP promotes pyrimidine (CDP and UDP) reduction, (d)TTP promotes GDP reduction, and dGTP promotes ADP reduction. The S-site and corresponding pairings of substrates/effectors are conserved amongst all RNRs characterized to date.20
The overall activity of class I RNRs can also be allosterically regulated, with significantly more diversity in mechanism.21–25, 29 All known strategies require an N-terminal activity (A) site, which is typically found in a separate discrete ATP-cone domain.30 ATP and dATP bind competitively to the A-site, and mediate the formation of active or inactive quaternary structures, respectively.21, 23, 25 In class Ia RNR from E. coli, the structural basis for this general phenomenon is well-characterized. In the absence of dATP, an active α2β2 complex can form, while addition of dATP promotes conversion to an α4β4 ring structure.11, 21 In the latter case, the α and β subunits are no longer aligned for radical translocation from β to the active site in α.21 Additional inhibited states have been observed in other class I enzymes (see Figure S1 for selected examples). In all known cases, dATP promotes higher-order oligomers via interactions involving the ATP-cone domain.22, 23, 25 Overall activity regulation mediated by the ATP-cone is a trait that has been gained, lost, or modified based on selective pressure.19 While some members of the Ia/Ic subclasses have been identified with up to three ATP-cones, the majority of cobalamin-dependent class II and all Mn-dependent class Ib/d enzymes lack a complete cone domain in the α subunit.29 Historically, the latter systems have been generally presumed to be insensitive to dATP inhibition.24, 31 However, the class Ib RNR from Bacillus subtilis is potently inactivated by dATP via a partial cone domain that co-purifies with tightly-bound dAMP.24 The dAMP ligand is essential for overall activity regulation. This mechanism of inhibition expands upon the ways in which the N-terminus of an RNR catalytic subunit can mediate inter-subunit interactions. It also provides the first example of overall allosteric regulation in an RNR that does not have a discrete ATP-cone.
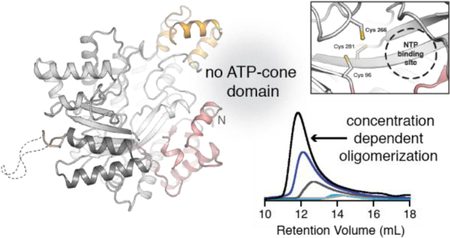
Class Id RNRs, including the previously-characterized enzyme from Flavobacterium johnsoniae (Fj), utilize a uniquely simple mode of metallocofactor assembly involving oxidation of the di-MnII cluster by direct scavenging of free superoxide.5 We propose that the Id activation mechanism may resemble evolutionarily primitive class I RNR β subunits with an ancestral relationship to the more complex Ia-c enzymes. Consistent with this hypothesis, bioinformatic analysis of the corresponding catalytic subunits indicates that the class Id α subunits are very small. The Id enzymes additionally lack even a partial N-terminal (or C-terminal) ATP-cone domain by sequence homology. This pattern suggests that the class Id α subunit could also be representative of a more primitive structure prior to the acquisition of the ATP-cone. Given the recent identification of activity regulation in a class Ib RNR by a mechanism that does not depend on a full ATP-cone, we investigated the structure and activity of the catalytic subunit of class Id RNRs in more detail to determine how, if at all, this new subset of class I RNRs is regulated. We establish that the class Id enzymes contain the expected catalytic residues and S-site effector binding motifs. Consistent with the lack of an ATP-cone, the enzyme is not inhibited by dATP. Surprisingly, the X-ray structure of Fj α (PDB ID: 6DQW) reveals an extended helical fibril with interfaces involving the canonical α2 dimer interface and a truncated N-terminal structural motif common to the class Id enzymes. The Fj enzyme can also form α4 structures in solution, detected in both size-exclusion chromatography (SEC) and small-angle X-ray scattering (SAXS) experiments. We also solved the structure of a second class Id α from the human opportunistic pathogen Actinobacillus ureae (Au) (PDB ID: 6DQX), and do not observe larger oligomers in this system. Nevertheless, our findings from the Fj enzyme suggest that novel quaternary structures can exist in class I RNRs that completely lack known overall activity regulatory domains.
MATERIALS AND METHODS
DNA vectors for overexpression of RNR α subunits and Fj TRR/Trx—
N-terminally His6-tagged Fj α and β were expressed from the plasmid pET28a(+) in E. coli BL21(DE3) cells (Stratagene) as described previously.5 The DNA sequences for Fj thioredoxin (Trx) and for the α subunit of the class Id RNR from Actinobacillus ureae (Au) were codon-optimized for expression in E. coli, synthesized, and cloned into the pET-28a(+) vector using NdeI and XhoI restriction sites (Invitrogen GeneArt Gene Synthesis, ThermoFisher). Fj thioredoxin reductase (TRR) was amplified by PCR using Fj genomic DNA as a template and the following primers: left (NdeI): 5’– GGATACCATATGATGGCATTAGCAATAACAGATGCTACTTTTGACG – 3’; right (XhoI): 5’ – AAGGATCTCGAGTTACAACAAAGCGTCTAAGCTGTCAG – 3’. The amplified DNA fragment was ligated into the pET28a(+) vector using NdeI and XhoI restriction sites. The DNA sequences for all genes were verified by Sanger sequencing (Genomics Core Facility, Penn State University). The expression vectors were used to transform E. coli BL21 (DE3) cells and successful transformants were selected on LB agar plates supplemented with 50 μg/ml kanamycin.
Overexpression and purification of Fj and Au α—
Fj α was overexpressed and purified by NiII-NTA affinity chromatography as previously described,5 with the following modifications: cells were lysed via sonication (QSonica Q500, 5 min total pulse time, 10 s on/30 s off, 60% amplitude) and the purified protein was exchanged into buffer A [50 mM HEPES pH 7.6, 150 mM NaCl, 5% (w/v) glycerol] containing 10 mM DTT by using a PD-10 pre-packed desalting column (GE Healthcare). Au α was expressed and purified following the same procedure except that overexpression was induced upon addition of 0.5 mM IPTG.
Additional purification steps were necessary to obtain α samples for X-ray crystallography and SAXS. Fj α was dialyzed into buffer B [50 mM HEPES pH 7.6, 50 mM NaCl, 5% (w/v) glycerol] with 10 mM DTT for an anion exchange chromatography step (DEAE Sepharose, GE Healthcare). Fj α elutes at 350 mM NaCl when a 50 mM – 2 M linear salt gradient is applied over 15 column volumes. Protein-containing fractions were concentrated in a 30 kDa MWCO filter device (Pall Corporation), injected onto a HiLoad 16/600 Superdex 200 (S200) gel filtration column connected to an ÄKTA Pure FPLC system (GE Healthcare), and eluted in an isocratic step into buffer A containing 10 mM DTT. Following the NiII-NTA affinity step, Au α was additionally purified on the S200 gel filtration column equilibrated in buffer C [50 mM HEPES pH 7.6, 300 mM NaCl, 5% (w/v) glycerol] with 5 mM DTT. Both proteins elute from the S200 column as primarily a single peak, with Fj α emerging at a volume of ~63 mL and Au eluting at ~78 mL. The difference in elution volume is consistent with the Fj enzyme forming larger oligomers in solution. Fractions containing α protein, as identified by SDS-PAGE, were concentrated in a 30 kDa MWCO centrifugal filter device. The yields of both the Fj and Au α proteins were ~1 mg of purified protein per gram of cell paste. Protein samples were flash-frozen in liquid N2 and stored at −80 °C until further use.
Overexpression and purification of Fj β—
N-terminally His6-tagged Fj β was expressed from the plasmid pET28a(+) and grown in LB-rich medium with 0.5 mM 1,10-phenanthroline added at the time of induction.5 Protein used for activity assays was purified by NiII-NTA affinity chromatography and exchanged into buffer A using a PD-10 pre-packed desalting column (GE Healthcare). Protein used for SAXS was further purified on a DEAE anion exchange column and an S200 gel filtration column.
Overexpression and purification of Fj Trx/TRR—
Single colonies selected on LB agar plates supplemented with 50 μg/mL kanamycin were used to inoculate starter cultures. Cells were grown in 1 L flasks of LB rich medium supplemented with 50 μg/mL kanamycin at 37 °C with shaking at 180 rpm. When the cultures reached an OD600 of ~0.8, IPTG was added to a concentration of 1 mM. Cultures overexpressing Fj Trx were incubated at 37 °C for an additional 3 h with shaking at 180 rpm, and cultures overexpressing Fj TRR were incubated at 18 °C for ~20 h with shaking at 180 rpm before harvesting. Cells were harvested by centrifugation at 6,000g for 15 min at 4 °C. Cells were lysed and purified using NiII-NTA affinity chromatography following the same protocol as Fj α, using buffer C with 10 mM imidazole as the lysis buffer and buffer C with 250 mM imidazole as the elution buffer. Fj Trx was further purified by gel filtration on an S200 column equilibrated in buffer A. Following the NiII-NTA chromatography step, Fj TRR was exchanged into buffer B for an anion exchange chromatography step (Q Sepharose, GE Healthcare). Fractions containing Fj TRR were exchanged into buffer A and concentrated in a 3 kDa MWCO centrifugal device. Protein was flash-frozen in liquid N2 and stored at −80 °C until further use.
Determination of protein concentration—
Concentrations of all proteins were determined spectrophotometrically using the computationally derived molar absorption coefficients (ε280) of 82,320 M−1 cm−1 for the Fj α monomer, 46,870 M−1 cm−1 for the Fj β monomer, 11,300 M−1 cm−1 for Fj Trx, 26,740 M−1 cm−1 for Fj TRR, and 82,740 M−1cm−1 for the Au α monomer (ExPASY).
General crystallographic methods—
All crystals of Fj and Au α were grown anaerobically in an 97% N2/3% H2 anoxic chamber (Coy Laboratory Products). Crystallographic datasets were collected at the Life Sciences Collaborative Access Team (LS-CAT) and the National Institute of General Medical Science and National Cancer Institute Collaborative Access Team (GM/CA-CAT) beamlines at the Advanced Photon Source (Argonne National Laboratory, Argonne, IL). All diffraction datasets were processed with the HKL2000 package.32 Phases were obtained by molecular replacement (MR) with the program PHASER33 implemented within the CCP4 software package.34 The coordinates of NrdA from Homo sapiens (PDB ID: 2WGH) were identified as the search model by the online version of BALBES35 for the initial Fj α structure solution. All subsequent structures of Fj α and Au α were phased by MR with the finalized coordinates of Fj α (PDB ID: 6DQW). Model building and refinement were carried out using Coot36 and Refmac5,37 respectively. Structures were validated and analyzed for Ramachandran outliers with the Molprobity server.38 Figures were prepared with the PyMOL molecular graphics software package (Schrödinger, LLC).
X-ray structure determination of Fj α—
Fj α was exchanged into buffer B containing 10 mM DTT. To ensure reduction of redox active cysteines, the protein was equilibrated in a 97% N2/3% H2 anoxic chamber (Coy Laboratory Products) prior to crystallization trials. Crystals were obtained by the hanging drop vapor diffusion method at room temperature with 0.25 M magnesium chloride, 0.1 M Tris pH 7.0, and 10% (w/v) PEG 8000 as the precipitating solution mixed in a 1:1 μL ratio with a protein solution of 32.5 μM Fj α, 32.5 μM Fj β, and 260 μM MnCl2 (prepared anaerobically). Samples used for data collection were harvested from drops seeded with microcrystals that were obtained from identical crystallization conditions. Oblong, hexagonal-shaped crystals appeared after 4–6 months. These crystals were soaked briefly in a cryoprotectant solution (freshly made well solution plus 30% (v/v) glycerol in place of water), harvested in rayon loops, and flash-frozen in liquid N2 for data collection.
Fj α crystallized in the P61 space group with four molecules in the asymmetric unit. The final model consists of residues 19–547 for chain A, 19–546 for chain B, 20–545 for chain C, 22–543 for chain D, and 134 water molecules. We note that while Fj α subunit was crystallized in the presence of the β subunit, we did not observe electron density in the final structure for the β component. Electron density was not observed for any of the residues associated with the N-terminal His6-tag in any of the chains. In each chain, short disordered regions could not be modeled (residues ~136–145, 163–169, and 431–441). These disordered loops correspond to the specificity site, consistent with the lack of bound effector molecules, and part of the auxiliary domain. In chain D, an additional large disordered region (residues 428–487) could not be confidently modeled. Of the residues modeled, 97% are in allowed or preferred regions indicated by the Ramachandran statistical analysis. The surface area of the interface between monomers in the helical fibril was calculated using PISA (http://www.ebi.ac.uk/pdbe/prot_int/pistart.html).39
X-ray structure determination of Au α—
Au α was exchanged into slightly modified buffer B, containing 75 mM NaCl, with 5 mM DTT. To ensure reduction of the redox active cysteines, Au α frozen protein solution was thawed and equilibrated in an anoxic chamber (Coy Laboratory Products) for at least one hour prior to crystallization trials. All dilutions were performed with argon saturated buffer B, containing 75 mM NaCl and 5 mM DTT. Crystals were generated via the hanging drop vapor diffusion method at room temperature by mixing 1 μL of the well solution, containing 16% (w/v) PEG 4000, 0.2 M magnesium chloride, and 0.1 M Tris pH 8.5, in a 1:1 μL ratio with a protein solution of 10 mg/mL Au α (160 μM). Large, diamond-shaped crystals appeared within one week. These crystals were soaked briefly in a cryoprotectant solution [freshly made well solution plus 10% (v/v) ethylene glycol in place of water], harvested in rayon loops, and flash-frozen in liquid N2.
Au α crystallizes in the P 42 21 2 space group with one monomer in the asymmetric unit. The final model consists of residues 6–551 in chain A, 2 Cl- ions, 2 Mg+ ions, 4 ethylene glycol molecules, 1 glycerol molecule, and 259 water molecules. Electron density was not visible for any of the residues associated with the N-terminal His6-tag or linker. Some electron density is visible for a portion of the typically disordered C-terminal tail (Figure S5). Due to a SCSSC sequence motif that interacts with a symmetry mate, Ser 552 and Cys 553 could not be modeled with confidence. Also excluded from the model are the residues 129–132, 154–158, 420–429 and 539–545, which are located either on the surface or at the specificity site. Of the residues modeled, 100% are in allowed or preferred regions, as indicated by the Ramachandran statistical analysis.
Analytical SEC—
Gel filtration analysis was performed on a Superdex 200 10/300 GL column (10 × 300 mm, ~24 mL column volume) connected to an ÄKTA Pure FPLC system (GE Healthcare) and equilibrated in buffer D [50 mM HEPES pH 7.6, 150 mM NaCl, 5% (w/v) glyvcerol, 12.5 mM MgSO4, and 5 mM DTT]. Samples of Fj and Au α were diluted to the desired concentration (5–300 μM), spun for 10 min at 10,000g to remove any precipitation, and a 100 μL sample was injected onto the column. Elutions were performed at a flow rate of 0.5 ml min-1. Apparent molecular weights were calculated based on the SEC retention times. The column was calibrated using a Low Molecular Weight and High Molecular Weight Gel Filtration kit (GE Healthcare) containing the following standards: aprotinin (6.5 kDa), RNAse A (13.7 kDa), carbonic anhydrase (29 kDa), ovalbumin (44 kDa), conalbumin (75 kDa), aldolase (158 kDa), and ferritin (440 kDa). For each standard, the gel phase distribution coefficient (Kav) was calculated from the elution volume, and a calibration curve was generated by plotting Kav vs. log MW) [Kav = 1.8269–0.29187(logMW)]. This curve was used to estimate the molecular weights of Fj and Au α oligomers.
Small-angle X-ray scattering (SAXS) data collection—
X-ray scattering experiments were performed at the Cornell High Energy Synchrotron Source (CHESS) G1 station40 using a 250 μm × 250 μm X-ray beam with an energy of ~9.85 keV and flux of ~1012 photons s−1 at the sample position. SAXS scattering images were collected on a Pilatus 100K photon-counting detector, covering a collected q-range of ~0.01–0.3 Å−1 in which q = 4π/λ sin θ; θ is the scattering angle, and λ is the X-ray wavelength. Samples were oscillated at 4 °C in the beam to minimize X-ray induced damage. 15–80 × 0.5 s exposures were taken for each protein and buffer sample.
Datasets were collected for SAXS by either manually loading the sample into the flow cell or by in-line size-exclusion chromatography. For the flow cell samples, aliquots of Fj α were centrifuged for 10 min at 10,000g and 4 °C to minimize aggregation before loading 30 μL by hand into the quartz capillary tube. Because Fj α tends to aggregate in buffer with low salt concentration during purification, we tested for aggregation in the SAXS setup by preparing samples in buffer containing 50 mM HEPES pH 7.6, 5 mM DTT and 5% glycerol with 75, 100, and 150 mM NaCl. No aggregation or repulsion effects were seen at 150 mM NaCl, so this concentration was used for all subsequent SAXS data collection (buffer A). For the concentration-dependence experiments, the protein was exchanged into buffer A with 5 mM DTT using a Bio-spin P-30 column (BioRad) and diluted to the desired concentration (10, 20, 35, 50, or 100 μM). All other SAXS data on Fj α were collected in buffer D. For experiments with nucleotides, each sample was prepared by incubating 20 μM α with CDP and variable amounts of ATP/dATP for ~5 min at room temperature before data collection. Background subtraction was performed using buffers prepared with the corresponding concentrations of CDP and ATP/dATP used for each sample respectively.
The in-line SEC setup was used for SEC-SAXS data collected on Fj α in buffer D at 4 °C. 100 μL of ~300 μΜ (~2 mg) Fj α was injected onto a Superdex 200 Increase 10/300 GL column (10 × 300 mm, ~24 mL column volume) connected to an ÄKTA Pure FPLC system (GE Healthcare) using a flow rate of 0.5 mL min-1. The samples are diluted ~10x on the column. Each frame was collected using a 0.5 s exposure, and the protein elution was monitored by UV-vis detection at 280 nm.
SAXS data analysis—
Data processing and analysis were performed using BioXTAS RAW41 and ATSAS.42 Scattering images were integrated around the beam center and data were normalized by the transmitted intensity. Images that did not exhibit radiation damage were averaged. Scattering from a matching buffer was subtracted from the protein scattering curve to yield the resultant one-dimensional protein scattering profile, I(q). The quality of the buffer subtraction was evaluated as described in Skou, et al.43 Radii of gyration (Rg) and molecular weights were estimated using Guinier analysis in RAW. The pair distance distribution function, P(r), and the maximum particle dimension (Dmax) were calculated using the program GNOM within the ATSAS software package.44
Theoretical scattering curves were calculated from structural models and fit to experimental curves using the FoXS server.45, 46 Alternative tetramer models of α were constructed using symmetry mates from the crystal structure generated in PyMOL, and by docking together two crystallographic dimers using the FoXSDock server.46 Ab initio reconstructions were generated using the programs DAMMIF47 and DAMAVER.48 For both α and β, the data from SEC-SAXS were used for the ab initio shape reconstructions. The generated envelopes were aligned with the Fj α tetramer models and the Fj β crystal structure (PDB ID: 6CWP) using SUPCOMB.42
Sequence similarity network (SSN) and phylogenetic analysis—
All searches were carried out and sequences were retrieved from the National Center for Biotechnology Information (NCBI) and Uniprot databases. The sequences used to generate the SSNs were retrieved from the InterPro Database. The SSNs were generated using the internet-based Enzyme Function Initiative Enzyme Similarity Tool (EFI-EST) (http://efi.igb.illinois.edu/efi-est/).49 Two protein families were used as input in the construction of the SSNs: The NrdA/NrdE family (IPR013346: ribonucleotide reductase, Class I, alpha subunit, contains 19,398 sequences as of August 2018) and a separate RNR_alpha family (IPR013350, contains 611 proteins as of August 2018) that represents a distinct clade of RNR alpha subunits that are divergent from the NrdA/NrdE sequences. Figures that illustrate SSN analyses, including the sequence length coloring, were created in Cytoscape (v3.4.0). Sequences were aligned with the MUSCLE (multiple sequence comparison by log-expectation) software package (http://www.ebi.ac.uk/Tools/msa/muscle/). Maximum-likelihood unrooted trees were computed with RaxML50 using the WAG amino acid replacement model. The bootstrap values, which reflect the confidence levels for the nodes, are reported for the major branches. The scale bar represents the number of amino acid substitutions per site. Trees were rooted using a set of class II RNR sequences as the outgroup. The branching between the class Ib and Id sequences remains the same when the class II sequences are omitted from the analysis, supporting the choice of the class II sequences as a valid outgroup.
Preparation of activated (MnIIIMnIV) Fj β—
Apo Fj β was activated by addition of 1.5 eq. MnII/β and 1 eq. naphthoquinone (NQ)/β.5 Fj β used in activity assays was verified to have a turnover number of ~ 1.3 s−1/β.
RNR activity assays—
The activities of His6-tagged α and β were determined by measuring the reduction of CDP in the presence of 5–10 fold excess of the other subunit. Assays were carried out aerobically at room temperature (21 ± 2 °C). Reaction mixtures contained 2–100 μM β, 2–100 μM α, 2 mM CDP, ATP or dATP at varying concentrations, 10 mM DTT (or 40 µM Fj Trx, 0.4 µM TRR and 1 mM NADPH), and 12.5 mM MgSO4 in buffer A. All reactions were initiated by addition of the desired quantity of activated β and terminated at selected time points by treatment of a 50 µL aliquot with 5 µL of 2 M formic acid. The acid-quenched samples were prepared for LC-MS analysis to quantify the relative amounts of CDP and dCDP at each time point.5, 51
RESULTS
Class Id α is universally small and lacks an ATP cone.
Previous bioinformatics analyses determined that class Id α subunits comprise a separate phylogenetic clade and cluster distinctly in a sequence similarity network (SSN).5 We repeated this analysis with updated database searches for a more accurate estimate of the number of sequences in the Id subclass. When using the Fj α sequence to query InterPro (IPR), the IPR013350 family was identified and described as sufficiently divergent from the other class I NrdA/E sequences to warrant a distinct grouping. Members are found in a variety of bacteria, archaea, and bacteriophage viruses. All of the members of class Id that had been previously identified by their β subunit sequence patterns are included in this family of α subunits. The Id α sequences have ≥ 36% sequence similarity, as calculated by BLASTp analysis. To compare these sequences to the rest of the class I RNR α subunits, IPR013350 was combined with IPR013346 (annotated as all class I NrdE/NrdA sequences) to generate a SSN of the entire class I α superfamily (Figure 1A). At a minimum alignment score of 165, both the Ib and Id sequences cluster distinctly, although we could not distinguish between the Ia and Ic α subunits.
Figure 1. Class Id RNR alpha subunits are universally small and lack regulatory ATP cone motifs.
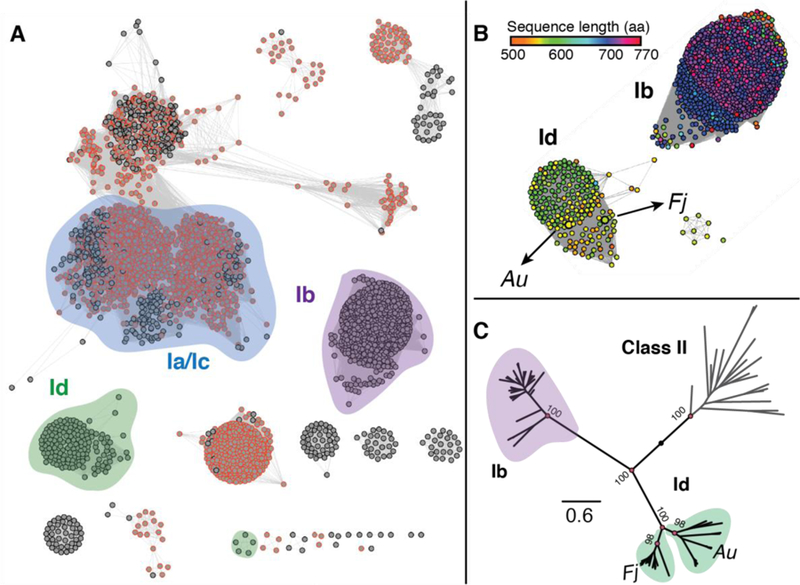
(A) Sequence similarity network (SSN) for the IPR013350 and IPR013346 superfamilies that contain the class I α sequences. The network was constructed using a minimum alignment score of 165. Each node represents a set of sequences with ≥80% identity. The nodes have been arranged in the organic layout. Nodes outlined in orange correspond to sequences that are annotated as containing an ATP-cone domain (IPR005144). (B) Daughter network of the Ib and Id sequences from panel A. The nodes are colored by amino acid sequence length. (C) Phylogenetic tree showing the Id sequences branching into two distinct groupings, one of Fj-like sequences and one of Au-like sequences. The numbers shown at the nodes are bootstrap values that correspond to the level of confidence in a given node and the scale bar indicates the distance in units of substitutions per site. The tree was rooted using the class II RNRs as the outgroup.
To visualize the distribution of the canonical ATP cone domain across the class I RNRs, we searched the entire network for sequences tagged with the IPR code for an intact ATP-cone domain (IPR005144, orange outline in Figure 1A). As reported previously, annotated ATP cones are widespread amongst class Ia and Ic RNRs, but completely absent from both the Ib and Id clusters.29 A daughter network containing the Id and Ib sequences and colored by sequence length shows that, even though both lack an ATP cone domain, the Id sequences are uniformly shorter. The median length of a Ib sequence is 709 amino acids, and the median length of a Id sequence is 565 (Figure 1B). Specifically, the NrdE (Ib) sequences retain approximately half of the full ATP cone that is present in the NrdA sequences, whereas the Id sequences are missing the entire domain.24 Maximum likelihood phylogenetic trees support this distinction and further show that the Id sequences branch into two distinct groupings (Figure 1C). The branch that contains Fj α is comprised of sequences from organisms similar to Fj that are exclusively from the Bacteroidetes phylum. The branch containing Au α is more diverse, and it contains both archaeal and bacterial species. The functional significance of this branching is not known, although a similar feature was observed in phylogenetic trees of the corresponding class Id β subunits.5
X-ray crystal structures (Table S1) of the α subunit from two different class Id organisms (one from each grouping represented in Figure 1C), Fj and Au, were solved to 2.6 Å resolution and 1.76 Å resolution, respectively (Figures 2A, S2–4). The Au structure is the highest resolution class I catalytic subunit structure determined to date. In the Fj α crystal structure, the asymmetric unit (ASU) contains two non-crystallographic symmetry-related copies of the canonical α2 dimer (Figure S2A). In the Au structure, the ASU contains a single α subunit with the α2 functional homodimer formed by crystallographic symmetry (Figure S2B). All components of the RNR catalytic subunit required for nucleotide reduction are conserved in both class Id enzymes (Figures 2B, 3, and S3), including the active site catalytic Cys• (Fj C266 and Au C255) and disulfide-forming Cys side chains (Fj C96/C281 and Au C85/C270). All visible Cys side chains are reduced in both the Fj and Au structures, likely because the proteins were purified in the presence of DTT and crystallized anaerobically. The class Id α subunits also contain key conserved proton donor/acceptor side chains (Fj E268 and Au E257) and the two C-terminal Tyr residues on the radical translocation pathway (Y542/Y543 in Fj and Y531/532 in Au) (Figure 3). In the Id structures, the C-terminal Y/Y pair is not stacked (Figures 3 and S3). Aromatic Tyr stacking is likely essential for radical translocation to and from the active site Cys. Consequently, a conformational change must occur in this region as part of the RT pathway alignment.52 Other class I enzymes exhibit similar unstacking (Figure S3).14, 24, 52
Figure 2. The class Id RNR α subunit uses a minimal domain architecture for ribonucleotide reduction.
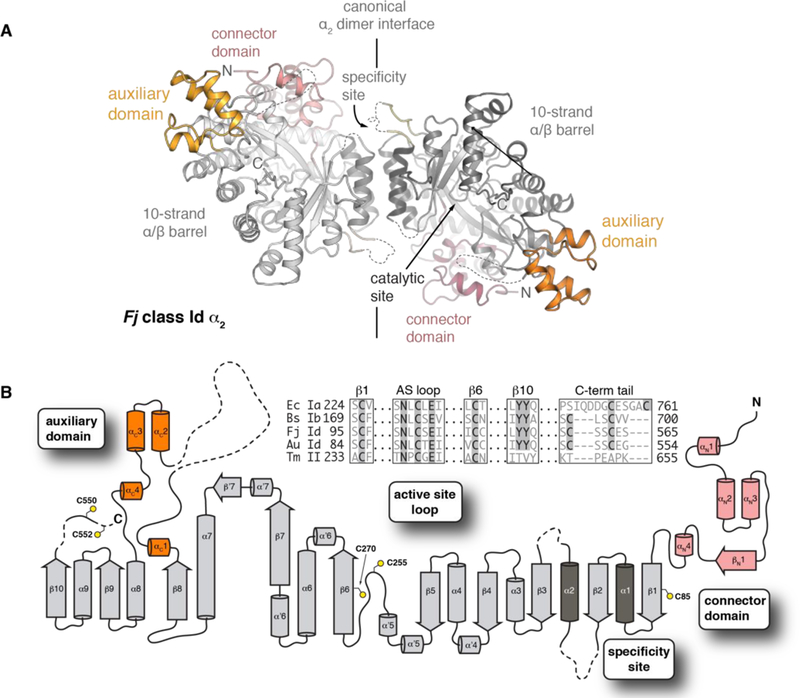
(A) The X-ray crystal structure of the Fj class Id α2 homodimer (PDB ID: 6DQW) reveals three distinct domains (pink, white, and orange). Disordered regions are indicated as dashed lines. These regions include the specificity site (residues 136–145, 164–169) and auxiliary domain (residues 431–441) loops. (B) Topology diagram showing N-terminal connector domain (pink), 10-strand α/β catalytic core (light gray), α2 dimer interface (dark gray), and intervening helical auxiliary domain (orange). Sequence alignment of conserved catalytic motifs is shown in inset.
Figure 3. The class Id RNR active site contains all key catalytic components.
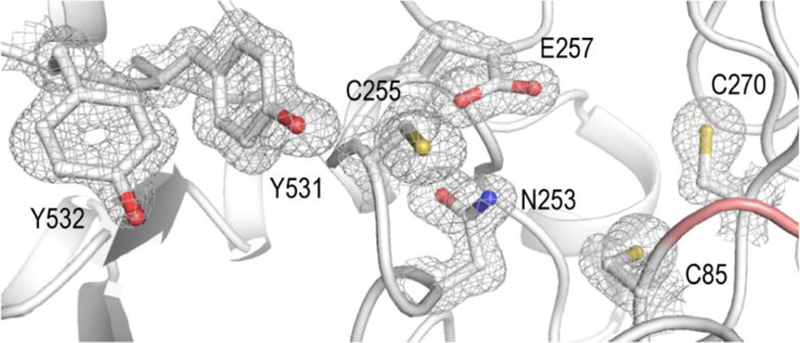
A 1.8 Å resolution X-ray crystal structure of the Au class Id α subunit (PDB ID: 6DQX) shows that the enzyme conserves residues responsible for radical translocation (Y532, Y531), thiyl radical formation/reaction initiation (C255), proton transfer (E257, N253), and redox chemistry/thiol-disulfide exchange (C85, C270). Selected side chains are shown in stick format with a corresponding 2Fo-Fc electron density map (gray mesh) contoured at 1.5σ.
In both Fj and Au α, a ten-stranded α/β barrel core domain houses the conserved catalytic residues described above (Figure 2B). Our structures show that the Id enzymes also retain the secondary structures that form the specificity site, namely the loops flanking the dimer interface. In the absence of substrate and effector nucleotides, these residues (136–145 and 164–169 in Fj α, 129−132 and 154–158 in Au α) are disordered. Also missing from the model are 11 residues in an auxiliary domain and, in Fj α, ~30 amino acids in the C-terminal tail, which contains a redox-cycling cysteine pair (C561 and C564) that is necessary for active site disulfide reduction in all class I RNRs. Although the overall secondary structure is conserved between Fj and Au α (Figure S4), the higher resolution of the Au structure provides information about the location of this cysteine pair (C550 and C553), not previously observed in an X-ray crystal structure. In the Au enzyme, C550 is visible and wrapped around the back of the protein, away from the active site. Clear difference density exists for amino acids beyond S551 (the last modeled residue). However, S552 and C553 could not be modeled with confidence, due to their location near a symmetry mate (Figure S5). The location of the tail does not represent its position during reduction of the active site disulfide bond that forms at the end of the cycle. However, the model could be representative of the tail binding site when interacting with the reducing system, before and after folding back inwards towards the active site to reduce the catalytic cysteine residues (Figure S6). At this stage, we also cannot rule out the possibility of a lattice artifact influencing the position of the tail.
As predicted from the SSN analysis, the overall size of the class Id α subunit is considerably diminished relative to all other class I α structures solved to date. In comparison to E. coli class Ia α, the small size of the Id subunit results from differences in seven external loops/domains (ranging in size from 7–120 amino acids) that are not present in the class Id enzymes (Figure S7). The core structure aligns closely with that of the eukaryotic class Ia RNRs with an rmsd of 1.45 Å over 481 amino acids when compared to Au α on the basis of secondary-structure matching superposition.53 The close match is consistent with the automated search model selection of the human α protein in our initial search for a molecular replacement model. The most significant distinction between the class Id and class Ia RNR α structures is the complete absence of the ATP-cone domain and truncation of a subsequent connector domain at the N-terminus of the protein (Figure S8). Interestingly, Fj and Au α append an additional small auxiliary domain (residues 447–482 in Fj, 436–471 in Au) in the middle of the protein, located near the N-terminus in 3D space (Figure 2B). This additional domain is not present in any of the bacterial class Ia or class Ib catalytic RNR subunits crystallized previously, but it does resemble a three-helix insert identified in eukaryotic class Ia RNRs and certain class II enzymes54 (Figures S8B and S9). Even with this auxiliary domain extension, the α subunits of Fj and Au are strikingly small. At ~ 150 amino acids shorter than a typical class Ib α subunit, the Id α subunits represent the simplest known structure of a class I RNR α subunit and appear to be the minimal functional domain that can support nucleotide reduction.
The Fj Id α subunit forms distinct higher order structures in the crystal lattice and in solution.
Inspection of the symmetry-related molecules in the Fj α crystal structure reveals an extended helix consisting of a repeating chain of canonical α2 dimers (Figure 4A, left). Interestingly, the dimers are connected by interactions between Id-specific structural motifs, including both the N-terminal and the auxiliary domain (Figure 4A, right). Of these, the N-terminal interface is the most extensive and involves ~500 Å2 of protein surface area (approximately 50% of the canonical α2 dimer interface). It is symmetric and linked by both H-bonding interactions and hydrophobic packing. All three interfaces bury a total surface area of ~1757 Å2 per monomer to yield a repeating fibril with a pitch of 185 Å and a width of 150 Å. The center of the helix is open with an inner pore ~50 Å wide.
Figure 4. Fj α forms an extended right-handed helical fibril in the crystal lattice.
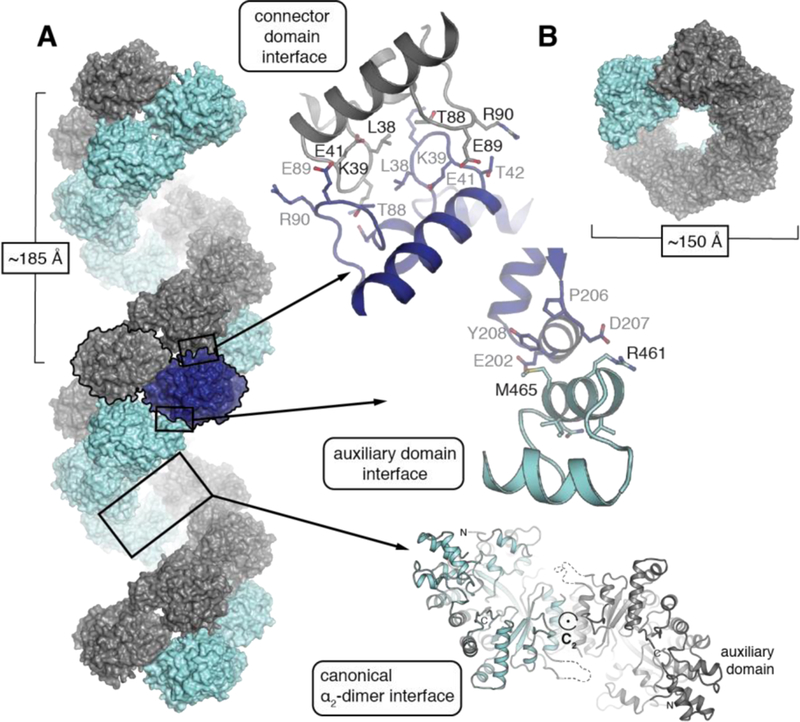
(A) Analysis of crystal packing interactions in the Fj α structure reveals a double-helical fibril (left) formed by symmetrical interactions at two interfaces involving the canonical α2 dimer (bottom right) and the connector domain (top right). The auxiliary domain (middle right) mediates a third asymmetric interface. (B) A top-down view reveals a central pore in the helix.
The canonical dimer itself contains a C2 symmetry axis that is roughly perpendicular to the 61 axis of the extended helix, but with an offset of ~10 degrees with respect to the unit cell (Figure S10). This phenomenon lowers the overall crystal symmetry and would be consistent with a hypothesis in which helical fibril formation occurs in solution prior to crystallization rather than solely as an artifact of lattice packing. In the Fj α X-ray structure, the predicted β2 binding site interface (Figure S11) is located on the interior of the helix, the inner dimensions of which are similar to the inner diameter of recent atomic resolution cryo-EM structures of the human α6 dATP-inhibited structure.55 The human hexamers prevent active β2 association by hiding the productive metallocofactor subunit binding site on the interior of a constricted ring. Interestingly, the auxiliary domain domain that is shared among the eukaryotic Ia and class Id enzymes is an integral part of the proposed inhibition mechanism of human RNR. The analogous structure in human α protrudes into the center of the ring to prevent β2 association with the inhibited α hexamer. In the Fj α helix, we propose a similar scenario in which the interior of the helix is too small to accommodate a β2 dimer, eliminating the possibility for a functional α2β2 complex when α is constrained in the fibril. Although we see no evidence of extended fibrils in the Au crystal lattice, it is possible that we have not yet identified the proper conditions for fibril formation in this system. Alternatively, the capacity to form extended quaternary structures may be a species-specific adaptation in class Id RNRs. In support of the latter conclusion, conservation analysis (Figure S12) of the residues that contribute to the N-terminal and auxiliary domain fibril interfaces observed in the Fj x-ray structure are more highly conserved within the Fj sequence clade than within the Au clade (see Figure 1 for phylogenetic analysis). This observation is consistent with a functional role for this interface in the Fj-like organisms but not for the Au grouping.
To understand whether higher-order fibrils can form in solution under more physiologically relevant conditions, we performed chromatography and X-ray scattering experiments under a variety of conditions. Surprisingly, although we found little evidence of filaments in solution for either Id enzyme, the Fj α subunit does undergo concentration-dependent changes in quaternary structure, forming tetramers under certain conditions. We initially used analytical size-exclusion chromatography (SEC) to assess the ability of both Fj and Au α to form higher-order oligomeric structures in solution at concentrations lower than those used for X-ray crystallography (~150 μM in the crystallization solution, and >1 mM in the crystal). For Fj α, chromatograms were analyzed in the absence of any substrate or effector molecules, at a range of concentrations from ~5 μM to ~300 μM, in order to probe the effect of protein concentration on oligomeric state (Figure 5A). Note that these values are the concentrations at which the protein mixture was loaded onto the column. We estimate that when the protein elutes off the column, it is diluted by a factor of 5–10. At the lowest concentration evaluated (~5 μM), Fj α elutes as a broad peak centered at an elution volume of 14.4 mL, corresponding to an apparent molecular weight (MW) of 76 kDa, which is in between the calculated MW of His6- α monomer (65 kDa) and His6- α2 dimer (130 kDa) (Table 1). Both the elution volume and the increased breadth of the peak are suggestive of interconverting monomer and dimer species, although at this concentration the equilibrium is likely shifted towards the monomer. As the protein concentration is increased to concentrations ≥100 μM, the peak gradually shifts to an elution volume that is consistent with the formation of a tetramer (260 kDa). At the highest concentrations tested, a small amount of aggregate is visible eluting with the void volume of the column at 8.23 mL, which could be attributed to the presence of fibrils or to approach of the solubility limit of many RNR αs. The tetrameric (non-aggregate) oligomer visible at the higher concentrations is consistent with our observation from crystallographic experiments that Fj α is capable of forming higher order quaternary structures in the absence of stabilizing nucleotide effectors. Our results further suggest that this phenomenon is influenced by protein concentration. By contrast, Au α does not form higher order oligomers at elevated protein concentrations. In ~ 10 μM solutions, Au α elutes as a mixture of monomer and dimer species with an average apparent MW of ~97 kDa (Figure 5B). At 100 μM concentration, the protein elutes at an apparent MW of ~115 kDa, which is close to the anticipated MW of the His6-α2 dimer (130 kDa).
Figure 5. Class Id RNR α subunits exhibit concentration-dependent changes in quaternary structure.
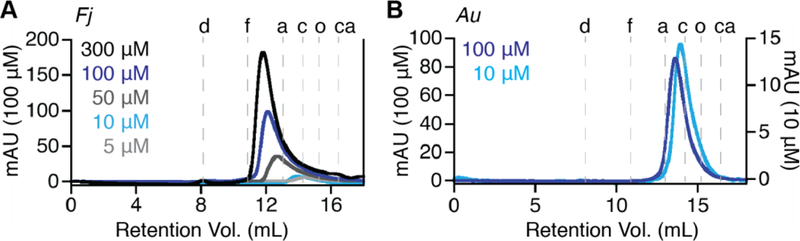
Size-exclusion chromatography traces of (A) Fj α, loaded onto the column over a range of concentrations from 5 μM to 300 μM, and (B) Au α, at 10 μM and 100 μM. Dashed lines indicate the elution volumes of globular standards [d: blue dextran; f: ferritin (440 kDa); a: aldolase (158 kDa); c: conalbumin (75 kDa); o: ovalbumin (44 kDa); ca: carbonic anhydrase (29 kDa)].
Table 1.
Predicted molecular weights of Fj α, β, and αxβx complexes
| Complex | MW (kDa) |
|---|---|
| α | 65.8 |
| α2 | 131.6 |
| α4 | 263.2 |
| β2 | 77.6 |
| α2β2 | 209.2 |
| α4β2 | 340.8 |
To further characterize the Fj α oligomers detected in solution, we used small-angle X-ray scattering (SAXS), a technique often employed in the study of active and inhibited quaternary forms of class I RNRs because the analysis can deconvolute mixtures of oligomeric states.56 Datasets were collected at 4 °C using Fj α concentrations ranging from 10 μM to 100 μM (0.66–6.6 mg/mL) and processed to generate scattering profiles (I, vs. q) in which I is the scattering intensity and q is a function of scattering angle. At low q, the scattering profile behaves linearly, suggesting minimal concentration-dependent aggregation or repulsion effects over the range of concentrations tested (Figure 6A, Table S2). All subsequent experiments were conducted at 20 μM α, in buffer D, which contains 12.5 mM MgSO4 to best mimic the conditions under which the protein is active. Subsequent Guinier fits yield consistent radii of gyration, Rg, of 49.35 ± 0.68 Å with an estimated MW of 211 kDa (Figure 6A, inset, Table S3). The estimated Rg value for the canonical α2 dimer as predicted by CRYSOL57 is 37.14 Å. Our observation of a larger Rg value than that predicted for a dimer suggests that Fj α is forming a higher-order structure. Given a monomer size of 65 kDa, the estimated MW could be indicative of a trimer. However, this assignment is inconsistent with all other RNR α subunits that have been studied to date.14, 23–25 We instead propose a dynamic mixture of dimer and tetramer species, which would yield an average consistent with the estimated molecular weight.
Figure 6. Flow-cell SAXS analysis of Fj α indicates the formation of a tetramer in solution.
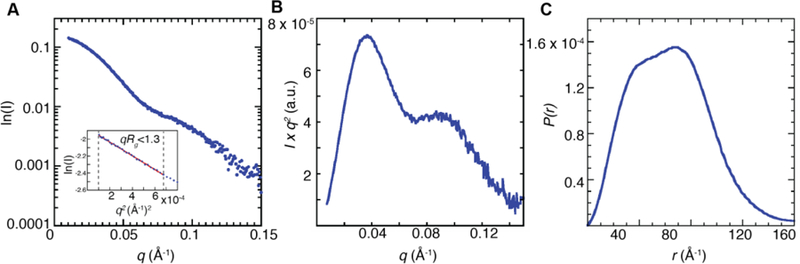
(A) Representative small-angle X-ray scattering data of apo Fj α at 20 μM. Inset, Guinier fit demonstrating that Fj α does not aggregate at these concentrations. The calculated Rg value (49.35 ± 0.68 Å) and estimated molecular weight (211.3 ± 8 Å) are consistent with formation of α4 tetramers. (B) Kratky representation and (C) P(r) plot of SAXS data collected on 20 μM Fj α samples. The bimodal curve suggests formation of a structure with two distinct length scales.
To resolve this mixture, we used in-line SEC-SAXS, in which the eluent from a Superdex200 size exclusion column was directed to the BioSAXS flow cell. At an Fj α concentration of ~300 μM (concentration loaded onto the column, estimated 30–60 μM in the X-ray beam), both the absorbance- and scattering-detected SEC traces show primarily a single peak corresponding to the α4 tetramer (Figures S13A, B). The estimated molecular weight obtained from averaging the scattering data across the sharpest point of the SEC peak is within 10% of the predicted 260 kDa of the α4 tetramer [271 kDa using the volume of correlation (Vc) method58 and 242 kDa using the Corrected Porod Volume (Vp) method59], and much closer than the estimates from the oscillating flow cell measurements (Table S3). However, changes to the overall scattering profile are minimal (Figure S13C). We predict that the flow cell datasets are dominated by the tetrameric form, with a small amount of dimer present in a dynamic equilibrium that artificially lowers the MW estimate.
Plotting both the flow-cell and SEC-SAXS data in a Kratky representation (Iq2 vs. q) produces a bimodal shape at each tested condition within this concentration regime, with distinct peaks at q ~ 0.035 and q ~ 0.092 Å−1 (Figure 6B, C and Figure S13D, E). Such a profile is typical of less compact, multi-domain species with two distinct length scales,60 such as a ring-shaped or other non-globular structure. Similar behavior has been observed in various ring-shaped dATP-inhibited forms of other class I RNRs, including the α4β4 complex of E. coli class Ia RNR,21 and the human RNR α6 hexamer.23 Intriguingly, the most similar RNR SAXS scattering profile to Fj α is that of the dATP-inhibited tetramer of Pseudomonas aeruginosa (Pa) NrdA.25 Pa NrdA forms an elongated ring-shaped tetramer via interactions of two dimers in the dATP-inhibited state (Figure S1). Due to the similarities between the scattering profiles, we anticipate that the Fj α4 tetramer might adopt a similarly non-globular structure.
Given the overwhelming evidence for a dominant tetramer population in solution, we evaluated possible models of this quaternary form. Two models were generated from crystallography data, both of which contain at least one canonical α2 dimer interface, with the tetramer formed by appending additional monomers using orientations observed in the helix in the crystal structure (Tetramers 1 and 2, Figure S14A, B). A third model was generated by rigid-body modeling of two crystallographic dimers, restrained by the SAXS data using the FoXSDock server46, 61 (Tetramer 3, Figure S14C). Theoretical scattering curves were then calculated for all tetramer models and compared to the SAXS data using the FoXS server (Figure S14).45 The χ2 values suggest that Tetramer 3, in which two α2 dimers associate asymmetrically, represents the best fit to the experimental data. The observed deviations between the theoretical and experimental SAXS curves may be due to scattering contributions from disordered loops/termini that are not modeled in the crystal structure. Attempts to model the disordered regions using the ensemble optimization method (EOM), as was done for Pa NrdA,25 did not improve the fit to the experimental scattering data. To further evaluate the different tetramer models, ab initio shape reconstructions using DAMMIF and DAMAVER were also generated from the SEC-SAXS data. Values for the excluded volume determined by DAMMIF and the alignment program SUPCOMB suggest that the best model can be generated by enforcing P2 symmetry (Table S4). This result is further supported by the normalized spatial discrepancy (NSD) obtained by the latter program and, once again, favors the asymmetric Tetramer 3 (Figure S14, Table S5). The program OLIGOMER62 (Table S6) was also used to confirm that the volume fractions predicted for tetramer 3 were ~98%. Given these findings, we propose that the tetramer found in solution is likely formed by contacts between two α2 dimers. However, the interactions between the dimers may be distinct from those observed in the crystal structure, as found in rigid-body docking model tetramer 3. Although this model is the current best fit to the SAXS data, we caution that deduction of the true interactions between α subunits will require additional validation by other methods. However, the general features of tetramer 3 – an open-core and asymmetric interaction between two α2 dimers – may be correct. We conclude from these experiments that four distinct quaternary forms of Fj α are possible - α, α2, α4, and an extended fibril structure (Figure S15).
We also attempted to use SEC-SAXS to obtain information about the stoichiometry of the active α/β holoenzyme complex. First, we collected datasets for the Fj β subunit by itself, previously shown, both crystallographically and via SEC, to be a globular dimer.5 Our datasets were consistent with this quaternary structure (Figure S16, Table S4) and we can fully model the SAXS envelope with the β2 crystal structure (PDB ID: 6CWP). In a 1:1 mixture of C96S α and β, two major peaks identified in the UV-vis SEC trace give rise to scattering intensity (Figure S17A, B). We assign the smaller of these to free β2. The larger peak, by contrast, is likely a mixture of species, because we were unable to obtain a consistent MW or Rg estimate across the scattering peak (Figure S17C, D). Preliminary SVD analysis identified three components: free α4, free β2, and a larger oligomer at the leading edge of the peak. However, deconvolution of the individual SAXS curves via EFA was inconclusive. The estimated molecular weight of this largest size oligomer is ~280 kDa, significantly larger than the calculated molecular weight of the α2β2 complex. Although this value is very close to the proposed MW of α4, the peak exhibits an accompanying increase in the Rg value (from ~48 Å to ~55 Å). We propose that these metrics indicate formation of an oligomer larger than α4, such as α4β2. Other complexes, including α4, α4β4 and/or α2β2, are likely present in this mixture, given that not all the β2 is associated with the complex. Although the stoichiometry of the active form(s) remains inconclusive, we propose that both α2 and α4 are capable of assembling into active holoenzyme complexes.
Class Id RNR α subunits lacking the ATP cone are not inhibited by dATP.
Due to the weak interactions between the α and β subunits in most RNR systems, the activity of each subunit is typically measured in the presence of a 5–10-fold excess of the other subunit. We have previously shown that MnIII/IV2-Fj β displays activity of ~1.3 s−1/β when assayed in the presence of a ten-fold excess of α.5 However, these assays were performed with the chemical reductant DTT, and it has been shown that use of the endogenous reducing system is necessary for obtaining high activity of the catalytic subunit when that component is limiting.63 In Fj RNR, when we used a candidate reducing system (Trx/TRR/NADPH) from Flavobacterium johnsoniae, we observed an ~six-fold increase in measured activity relative to assays using DTT. In both cases, the enzyme was analyzed at room temperature (21 ± 2 °C). The rate with DTT was 0.21 ± 0.05 s−1/ α, and the rate with Trx/TRR/NADPH was 1.24 ± 0.25 s−1/α. Importantly, the measured rate in the limiting α experiments with the native Trx/TRR reducing system approaches the maximum rate observed when using limiting β and DTT.5 In these previously published experiments, in which α concentrations were as high as 500 μM, concentration-dependent inhibition was not observed.5 For all subsequent experiments, in which the effects of ATP/dATP on regulation of overall RNR activity were measured, the native reducing system was used in place of DTT.
Assays performed in the presence of each of the four NDP substrates and corresponding effectors show that Fj RNR exhibits identical substrate specificity preferences as other characterized class I RNRs: both ATP and dATP stimulate CDP reduction (and UDP to a lesser extent), dGTP stimulates ADP reduction, and (d)TTP stimulates GDP reduction (Figure S18). These patterns are consistent with structural conservation of the specificity site. The observation that dATP is inhibitory in a B. subtilis Ib system lacking a full ATP cone led us to investigate whether the class Id system might also display nucleotide-dependent overall activity regulation. To test this idea, the activity of Fj α was measured in the presence of both ATP and dATP (Figure 7). In the absence of any effectors, the turnover number was quite low (~0.17 s−1/α), due to the lack of the appropriate specificity effector required to enhance CDP reduction. However, activity increased in the presence of ATP, with the maximal rate achieved at 1–2 mM ATP (~1.2 s−1/α). We observed a similar enhancement in Fj RNR activity in the presence of dATP, even at 1 mM (the highest concentration measured). The Au enzyme was also active in the presence of 1 mM dATP (Figure S19). From this observation, we conclude that dATP can act as a specificity effector for CDP reduction but cannot inhibit overall RNR activity in Fj (or Au) α, consistent with lack of an ATP-cone to bind this effector. Class Id enzymes exhibit relatively high activity even in the presence of dATP at levels that would inhibit the class Ia and B. subtilis Ib enzymes. To further support this conclusion, we compared the SAXS curves for Fj α in the presence and absence of nucleotides. Resultant Kratky plots are essentially identical, indicating that there are no significant changes in quaternary structure due to the presence of ATP or dATP (Figure S20).
Figure 7. Class Id α is not inhibited by dATP, consistent with lack of an ATP cone.
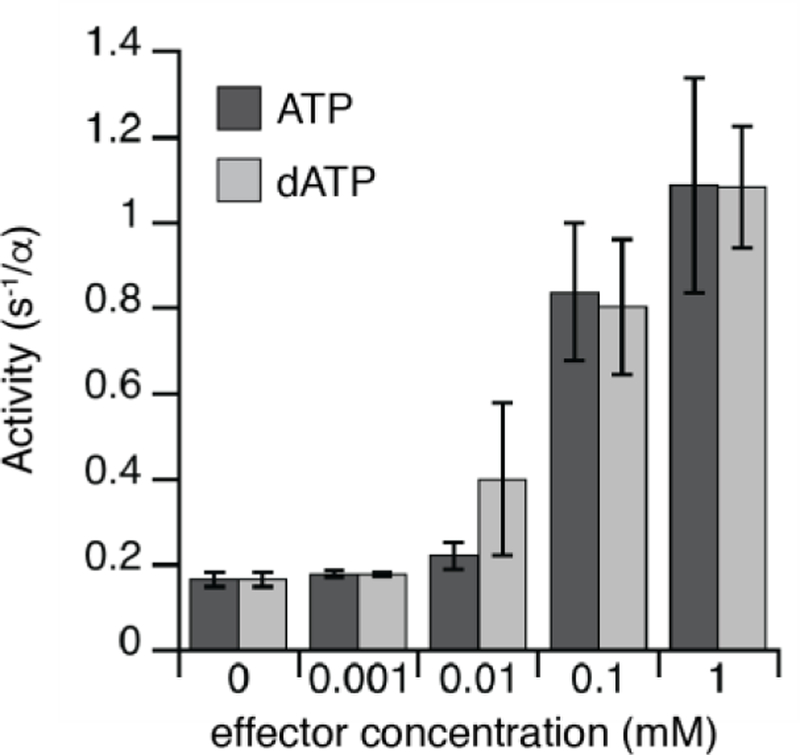
Effect of the addition of overall activity regulators ATP and dATP (0 – 1 mM) on CDP reduction by 5 μM Fj α, measured in the presence of 25 μM MnIVMnIII Fj β. Error bars represent one standard deviation from the mean of three independent trials.
DISCUSSION
Class Id RNR α forms complex and dynamic mixtures of oligomers that are similar to other class I RNRs.
Known mechanisms of overall activity inhibition in bacterial class I RNRs involve disruption of intersubunit radical translocation via formation of higher-order oligomers.11, 21, 64, 65 Characterized inhibited states of bacterial class I RNRs include non-canonical α2, α4, or α4β4 arrangements, all of which are mediated by the binding of dATP to a full or partial N-terminal ATP-cone domain. In E. coli class Ia RNR, the ATP-cone modulates the conversion between the active α2β2 and inhibited α4β4 ring structures. The class Ia RNR from Pseudomonas aeruginosa has two consecutive N-terminal ATP-cone domains, and the enzyme uses a distinct regulatory mechanism. Two molecules of dATP bind to a single N-terminal cone and, unexpectedly, the subsequent cone does not contain an effector-binding site. The dATP-inhibited state is a flat ring-shaped α4 tetramer (Figure S1) composed of two α2 dimers with a second interface involving the ATP-cone.25 Our analysis of Fj class Id RNR suggests that it is possible to form similar non-globular α4 structures in a nucleotide-independent manner in the absence of an ATP cone. However, we do not know if the resulting structures are inhibitory because we have not gathered any in vitro evidence of concentration-dependent α inhibition to date. Although we note that activity has not been measured at concentrations in which we would see fibrils (> 1 mM), and we cannot rule out the involvement of other cellular factors in stabilizing or altering these motifs.
The eukaryotic class I RNRs characterized to date all contain an N-terminal ATP cone and use this motif to form closed ring-shaped α6 quaternary structures (Figure S1).23, 55, 64, 65 Hexameric forms persist in the presence of either dATP or ATP, and the relative stability of each complex controls transition to the active form. With ATP, the α hexamers readily disassemble to bind productively with the β2 subunit, among other effects.64 By contrast, in the dATP-bound state, the α6 form remains intact to prevent RT between the subunits.23 Observation of persistent oligomeric α structures in the eukaryotic enzymes is a shared trait with Fj class Id RNR. Filamentous forms of human RNR α have also been observed, similar to the structures formed in our Fj α crystals. However, the human α fibril structures form upon addition of ATP.23 Higher-order α oligomers are present in active holoenzyme forms of the eukaryotic RNR with stoichiometries of α2nβ2. Our observation of class Id oligomers larger than α2β2 in SEC-SAXS analysis of the holoenzyme complex suggests that, as in the eukaryotic systems, Fj RNR may also form active complexes larger than α2β2.
Protein concentration as a factor in regulating overall activity in vivo could be relevant to Fj class Id RNR. Overproduction of dNTP precursors is detrimental to cells, and prior studies show that enzyme concentration can affect the quaternary structure of RNRs in addition to nucleotide-dependent mechanisms.22, 27 For example, crowding agents cause dATP-inhibited E. coli class Ia α4β4 oligomers to adopt (α4β4)2 concatenated rings, forming an inhibited structure in which it would be more difficult to directly convert between active/inactive states. Similar nucleotide-independent reversible oligomerization exists in other systems involved in nucleotide metabolism, including inosine monophosphate dehydrogenase (IMPDH)66 and CTP synthase (CTPS).67, 68 As in RNR, these enzymes are subject to feedback inhibition, and may do so via transient assembly into filaments.
Challenges in studying class I RNR α oligomerization.
Our work on class Id RNR α subunits shows that they share in common with other class I enzymes a propensity to interconvert between different oligomeric forms.21, 23 This feature of class I enzymes presents challenges in structural study, often requiring a suite of characterization techniques to discern the composition of complex mixtures and probe quaternary structure within different concentration regimes.23, 24 For example, our current understanding of the properties of the aforementioned dATP-inhibited human α6 oligomer required x-ray crystallography, SAXS, and electron microscopy analysis.23, 55, 65 Importantly, few experimental approaches exist to probe the role of dynamics in RNR allostery, even though relative stability of quaternary forms is known to govern transition to the active holoenzyme complex in the human enzyme.23 Recently, a similar combined approach was used to delineate six distinct oligomeric forms of B. subtilis class Ib RNR.24, 69 Initial detection of filaments in SAXS studies of dATP-inhibited Bs α required subsequent cryo-EM analysis to determine the precise structure. Observation of multiple oligomeric forms in the Bs Ib enzyme, including filamentous helical structures, resembles our findings from SAXS and crystallographic analysis of Fj class Id α. Notably, the Id higher-order structures can form in the absence of nucleotides, whereas the Ib oligomers only form upon binding of dATP/dAMP effector nucleotides. Similar electron microscopy studies of Fj class Id RNR might provide corroboration of the filamentous inhibited states formed in x-ray structures or additional structural details of the tetramer detected by SAXS.
Class Id α subunits universally lack an ATP-cone.
Our work suggests a subclass-wide absence of dATP-dependent activity inhibition in the α subunit of class Id RNRs. This claim is further substantiated by the observation that a significant fraction of the Id RNRs have an ATP cone domain instead appended to the radical-generating β subunit.26 The recently characterized class Id β from L. blandensis (Lb) contains an N-terminal ATP-cone that, as in Pa NrdA, can bind two molecules of dATP per monomer. dATP binding promotes the formation of an inactive ring-shaped β4 tetramer that presumably binds to NrdA (α) dimers in a nonproductive orientation. However, the structural basis for RT pathway disruption remains unclear because the putative α subunit binding interfaces are still theoretically accessible in the dATP-bound β4 tetramer, in contrast to the Pa RNR tetramer or the α6 hexamers formed in eukaryotic enzymes. In class Id RNRs, the ATP cone was likely acquired by selected β subunits to compensate for lack of nucleotide-dependent activity regulation in the α subunit. It is notable that the Id βs with an N-terminal ATP-cone all belong to the Au-like clade (Figure 1C), in which we do not observe any α subunit oligomerization. The impetus, in this subset of Id βs, to append the ATP-cone domain to this subunit supports the idea that the function of the tetramers/helix we observe in the Fj enzyme is not to mediate dATP inhibition but perhaps to function instead as an alternative method of dATP-independent activity regulation or inhibition. RNR subunits that lack a full ATP cone (Figure 8) were historically believed to be insensitive to any nucleotide-dependent overall activity regulation and incapable of oligomerization beyond α2 formation. For example, the crystal structure of the class Ib enzyme from S. typhimurium (St) was characterized as lacking an ATP-cone and it does not display overall activity regulation in vitro when isolated heterologously from E. coli.31 However, subsequent work on a class Ib enzyme from B. subtilis revealed that new mechanisms of activity regulation could be imparted by use of small molecule effectors.24 Bs NrdE uses an endogenously bound dAMP in a binding site that involves a partial ATP cone and a region of an extended connector domain to form a non-canonical α2 dimer. Upon dATP binding, the enzyme is proposed to form extended higher-order inhibited oligomers, recently validated by cryo-EM analysis.69 There are several possible explanations for the regulatory differences within the class Ib subclass. St NrdE does not have a bound dAMP molecule, which could contribute to the lack of observed dATP-dependent inhibition. Or the difference could be due to the use of the class Ib RNR as an auxiliary enzyme in St, whereas in Bs the Ib enzyme is the sole aerobic source of dNTPs. The need to rely on the Ib system under all circumstances, not only in times of low Fe and high oxidative stress, could mean that Bs was forced to adopt a more sophisticated regulatory system that is not required in St. Both Fj and Au have an annotated class Ia RNR in addition to the Id enzyme, although it is not known which RNR is used during normal aerobic growth in these organisms. However, the presence of the Ia enzyme could explain the lack of nucleotide-dependent activity regulation in these particular Id systems. Although the class Id αs are missing the region of the connector domain that is involved in the inhibitory interactions in Bs (Figure S8A-C), the crystal structure of Fj describes a possible new mode of inhibitory oligomerization via the truncated connector and auxiliary domains. It is conceivable that these regions could also bind a small molecule effector, similar to the dAMP component of Bs α. They could also interact with another protein, like the Sml1 protein that binds to a C-terminal insert in yeast RNR α.70 This possibility remains an open question but genetic tractability of class Id hosts, including Fj, makes it possible to isolate the enzyme from its native environment to test this hypothesis directly, as well as to examine the overall role of the class Id RNR during aerobic growth.
Figure 8. Class Id α lacks all known N-terminal RNR allosteric nucleotide binding motifs.
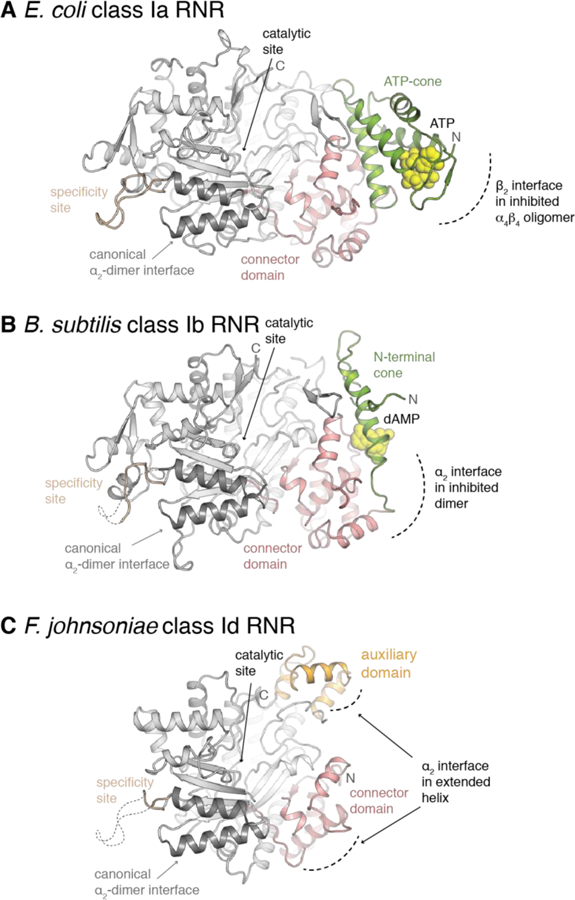
A structural comparison of the α subunit from class Ia (A) (PDB ID: 3R1R), class Ib (B) (PDB ID: 6CGL), and class Id RNRs (C) (this work) shows that N-terminus of the protein is truncated such that all known adenine nucleotide binding sites in the ATP-cone and connector domains are no longer present. An auxiliary domain shared with eukaryotic class Ia and class II RNRs occupies a nearby region.
CONCLUSIONS
The X-ray crystal structures of class Id RNR α subunits reveal a minimal domain architecture that completely lacks even the partial ATP cone structure present in other minimal class I RNRs, such as the Ib enzymes. The absence of this domain would imply an inability to form inhibitory oligomers but, surprisingly, the X-ray structure of Fj class Id α suggests a capacity for higher-order quaternary structure mediated by a truncated N-terminal structural motif. In yet another example of diversity within an RNR subclass, we do not see these larger oligomers in the Au system. This observation is supported by evidence of Fj tetramers in both size-exclusion chromatography (SEC) and small-angle X-ray scattering (SAXS) experiments. These higher-order structures change across different concentration regimes but do not respond to addition of nucleotides, consistent with observed lack of dATP-dependent enzyme inhibition in activity assays. Although the formation of oligomers may be an Fj-specific adaptation, Fj and Au come from different phylogenetic clades within the Id subclass, and it is possible that other Fj-like Id RNR αs exhibit similar behavior. The functional significance of these structures, as well as the potential involvement of other components, remains to be discovered.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by grants from the Searle Scholars Program (to A.K.B.) and the NIH (GM119707 to A.K.B.). Portions of this work were conducted at the Advanced Photon Source (APS), a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02–06CH11357. GM/CA at APS has been funded in whole or in part with Federal funds from the National Cancer Institute (ACB-12002) and the National Institute of General Medical Sciences (AGM-12006). The Eiger 16M detector was funded by the NIH-Office of Research Infrastructure Programs, High-End Instrumentation Grant (1S10OD012289–01A1). Use of LS-CAT Sector 21 was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor Grant (085P1000817). Portions of this work are based upon research conducted at the Cornell High Energy Synchrotron Source (CHESS), which is supported by the National Science Foundation and the National Institutes of Health/National Institute of General Medical Sciences under NSF award DMR-1332208, using the Macromolecular Diffraction at CHESS (MacCHESS) facility, which is supported by award GM-103485 from the National Institutes of Health, through its National Institute of General Medical Sciences. K.M.D. is grateful for support from the Arnold O. Beckman Postdoctoral Fellowship Program. We thank Richard Gillilan and Jesse Hopkins for help with small-angle X-ray scattering experiments, and Jens Kaiser (Beckman Institute Molecular Observatory, California Institute of Technology) for helpful discussion and critical analysis of the Fj crystal structure.
Footnotes
SUPPORTING INFORMATION
Additional experimental details on cloning Au α and β, Fj thioredoxin and thioredoxin reductase, and Fj α C96S variant; additional details for the overexpression and purification of Au β; SAXS data collection for Fj β and Fj α/β complex; supporting figures, and supplementary tables with SAXS parameters and crystallography data collection and refinement statistics.
UNIPROT ACCESSION IDs
Fj α: A5FCJ4
Fj β: A5FCJ5
Fj thioredoxin (Trx): A5FK98
Fj thioredoxin reductase (TRR): A5FNI6
Au α: E8KJ17
Au β: E8KJ18
PDB ACCESSION IDs:
Fj α: 6DQW
Au α: 6DQX
REFERENCES
- [1].Nordlund N, and Reichard P (2006) Ribonucleotide reductases, Annu. Rev. Biochem 75, 681–706. [DOI] [PubMed] [Google Scholar]
- [2].Licht S, Gerfen GJ, and Stubbe J (1996) Thiyl radicals in ribonucleotide reductases, Science 271, 477–481. [DOI] [PubMed] [Google Scholar]
- [3].Reichard P (1993) From RNA to DNA, why so many ribonucleotide reductases?, Science 260, 1773–1777. [DOI] [PubMed] [Google Scholar]
- [4].Cotruvo JA Jr., and Stubbe J (2011) Class I ribonucleotide reductases: metallocofactor assembly and repair in vitro and in vivo, Annu. Rev. Biochem 80, 733–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Rose HR, Ghosh MK, Maggiolo AO, Pollock CJ, Blaesi EJ, Hajj V, Wei Y, Rajakovich LJ, Chang W.-c., Han Y, Hajj M, Krebs C, Silakov A, Pandelia M-E, Bollinger JM Jr., and Boal AK (2018) Structural basis for superoxide activation of Flavobacterium johnsoniae class I ribonucleotide reductase and for radical initiation by its dimanganese cofactor, Biochemistry 57, 2679–2693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Blaesi EJ, Palowitch GM, Hu K, Kim AJ, Rose HR, Alapati R, Lougee MG, Kim HJ, Taguchi AT, Tan KO, Laremore TN, Griffin RG, Krebs C, Matthews ML, Silakov A, Bollinger JM Jr., Allen BD, and Boal AK (2018) Metal-free class Ie ribonucleotide reductase from pathogens initiates catalysis with a tyrosine-derived dihydroxyphenylalanine radical, Proc. Natl. Acad. Sci. U. S. A 115, 10022–10027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Bollinger JM Jr., Edmondson DE, Huynh BH, Filley J, Norton JR, and Stubbe J (1991) Mechanism of assembly of the tyrosyl radical-dinuclear iron cluster cofactor of ribonucleotide reductase, Science 253, 292–298. [DOI] [PubMed] [Google Scholar]
- [8].Cotruvo JA Jr., Stich TA, Britt RD, and Stubbe J (2013) Mechanism of assembly of the dimanganese-tyrosyl radical cofactor of class Ib ribonucleotide reductase: enzymatic generation of superoxide is required for tyrosine oxidation via a Mn(III)Mn(IV) intermediate, J. Am. Chem. Soc 135, 4027–4039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Dassama LMK, Boal AK, Krebs C, Rosenzweig AC, and Bollinger JM Jr. (2012) Evidence that the beta subunit of chlamydia trachomatis ribonucleotide reductase is active with the manganese ion of its manganese(IV)/iron(III) cofactor in site 1, J. Am. Chem. Soc 134, 2520–2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Srinivas V, Lebrette H, Lundin D, Kutin Y, Sahlin M, Lerche M, Eirich J, Branca RMM, Cox N, Sjöberg B-M, and Högbom M (2018) Metal-free ribonucleotide reduction powered by a DOPA radical in Mycoplasma pathogens, Nature 563, 416–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Rofougaran R, Crona M, Vodnala M, Sjöberg B-M, and Hofer A (2008) Oligomerization status directs overall activity regulation of the Escherichia coli class Ia ribonucleotide reductase, J. Biol. Chem 283, 35310–35318. [DOI] [PubMed] [Google Scholar]
- [12].Stubbe J, Nocera DG, Yee CS, and Chang MCY (2003) Radical initiation in the class I ribonucleotide reductase: long-range proton-coupled electron transfer?, Chem. Rev 103, 2167–2201. [DOI] [PubMed] [Google Scholar]
- [13].Minnihan EC, Nocera DG, and Stubbe J (2013) Reversible, long-range radical transfer in E. coli class la ribonucleotide reductase, Acc. Chem. Res 46, 2524–2535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Uhlin U, and Eklund H (1994) Structure of ribonucleotide reductase protein R1, Nature 370, 533–539. [DOI] [PubMed] [Google Scholar]
- [15].Eriksson M, Uhlin U, Ramaswamy S, Ekberg M, Regnström K, Sjöberg B-M, and Eklund H (1997) Binding of allosteric effectors to ribonucleotide reductase protein R1: reduction of active-site cysteines promotes substrate binding, Structure 5, 1077–1092. [DOI] [PubMed] [Google Scholar]
- [16].Logan DT, Andersson J, Sjöberg B-M, and Nordlund P (1999) A glycyl radical site in the crystal structure of a class III ribonucleotide reductase, Science 283, 1499–1504. [DOI] [PubMed] [Google Scholar]
- [17].Sintchak MD, Arjara G, Kellogg BA, Stubbe J, and Drennan CL (2002) The crystal structure of class II ribonucleotide reductase reveals how an allosterically regulated monomer mimics a dimer, Nat. Struct. Biol 9, 293–300. [DOI] [PubMed] [Google Scholar]
- [18].Reichard P (2010) Ribonucleotide reductases: substrate specificity by allostery, Biochem. Biophys. Res. Commun 396, 19–23. [DOI] [PubMed] [Google Scholar]
- [19].Lundin D, Berggren G, Logan DT, and Sjöberg B-M (2015) The origin and evolution of ribonucleotide reduction, Life (Basel) 5, 604–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Hofer A, Crona M, Logan DT, and Sjöberg B-M (2012) DNA building blocks: keeping control of manufacture, Crit. Rev. Biochem. Mol. Biol 47, 50–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Ando N, Brignole EJ, Zimanyi CM, Funk MA, Yokoyama K, Asturias FJ, Stubbe J, and Drennan CL (2011) Structural interconversions modulate activity of Escherichia coli ribonucleotide reductase, Proc. Natl. Acad. Sci. U. S. A 108, 21046–21051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Zimanyi CM, Ando N, Brignole EJ, Asturias FJ, Stubbe J, and Drennan CL (2012) Tangled up in knots: structures of inactivated forms of E. coli class Ia ribonucleotide reductase, Structure 20, 1374–1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Ando N, Li HR, Brignole EJ, Thompson S, McLaughlin MI, Page JE, Asturias FJ, Stubbe J, and Drennan CL (2016) Allosteric inhibition of human ribonucleotide reductase by dATP entails the stabilization of a hexamer, Biochemistry 55, 373–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Parker MJ, Maggiolo AO, Thomas WC, Kim A, Meisburger SP, Ando N, Boal AK, and Stubbe J (2018) An endogenous dAMP ligand in Bacillus subtilis class Ib RNR promotes assembly of a noncanonical dimer for regulation by dATP, Proc. Natl. Acad. Sci. U. S. A 115, E4594–E4603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Johansson R, Jonna VR, Kumar R, Nayeri N, Lundin D, Sjöberg B-M, Hofer A, and Logan DT (2016) Structural mechanism of allosteric activity regulation in a ribonucleotide reductase with double ATP cones, Structure 24, 906–917. [DOI] [PubMed] [Google Scholar]
- [26].Grinberg IR, Lundin D, Hasan M, Crona M, Jonna VR, Loderer C, Sahlin M, Markova N, Borovok I, Berggren G, Hofer A, Logan DT, and Sjöberg B-M (2018) Novel ATP-cone-driven allosteric regulation of ribonucleotide reductase via the radical-generating subunit, Elife 7, e31529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Wheeler LJ, Rajagopal I, and Mathews CK (2005) Stimulation of mutagenesis by proportional deoxyribonucleoside triphosphate accumulation in Escherichia coli, DNA Repair (Amst) 4, 1450–1456. [DOI] [PubMed] [Google Scholar]
- [28].Zimanyi CM, Chen PY, Kang G, Funk MA, and Drennan CL (2016) Molecular basis for allosteric specificity regulation in class Ia ribonucleotide reductase from Escherichia coli, Elife 5, e07141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Jonna VR, Crona M, Rofougaran R, Lundin D, Johansson S, Brännström K, Sjöberg B-M, and Hofer A (2015) Diversity in overall activity regulation of ribonucleotide reductase, J. Biol. Chem 290, 17339–17348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Aravind L, Wolf YI, and Koonin EV (2000) The ATP-Cone: An evolutionarily mobile, ATP-binding regulatory domain, J. Mol. Microbiol. Biotechnol 2, 191–194. [PubMed] [Google Scholar]
- [31].Uppsten M, Färnegårdh M, Jordan A, Eliasson R, Eklund H, and Uhlin U (2003) Structure of the large subunit of class Ib ribonucleotide reductase from Salmonella typhimurium and its complexes with allosteric effectors, J. Mol. Biol 330, 87–97. [DOI] [PubMed] [Google Scholar]
- [32].Otwinowski Z, and Minor W (1997) Processing of X-ray diffraction data collected in oscillation mode, Methods Enzymol 276, 307–326. [DOI] [PubMed] [Google Scholar]
- [33].McCoy AJ, Grosse-Kunstleve RW, Storoni LC, and Read RJ (2005) Likelihood-enhanced fast translation functions, Acta Crystallogr., Sect. D: Biol. Crystallogr 61, 458–464. [DOI] [PubMed] [Google Scholar]
- [34].Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AG, McCoy A, McNicholas SJ, Murshudov GN, Pannu NS, Potterton EA, Powell HR, Read RJ, Vagin A, and Wilson KS (2011) Overview of the CCP4 suite and current developments, Acta Crystallogr., Sect. D: Biol. Crystallogr 67, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Long F, Vagin AA, Young P, and Murshudov GN (2008) BALBES: a molecular-replacement pipeline, Acta Crystallogr., Sect. D: Biol. Crystallogr 64, 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Emsley P, and Cowtan K (2004) Coot: model-building tools for molecular graphics, Acta Crystallogr., Sect. D: Biol. Crystallogr 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- [37].Murshudov GN, Vagin AA, and Dodson EJ (1997) Refinement of macromolecular structures by the maximum-likelihood method, Acta Crystallogr., Sect. D: Biol. Crystallogr 53, 240–255. [DOI] [PubMed] [Google Scholar]
- [38].Chen VB, Arendall WB, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, and Richardson DC (2010) MolProbity: all-atom structure validation for macromolecular crystallography, Acta Crystallogr., Sect. D: Biol. Crystallogr 66, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Krissinel E, and Henrick K (2007) Inference of macromolecular assemblies from crystalline state, J. Mol. Biol 372, 774–797. [DOI] [PubMed] [Google Scholar]
- [40].Acerbo AS, Cook MJ, and Gillilan RE (2015) Upgrade of MacCHESS facility for X-ray scattering of biological macromolecules in solution, J. Synchrotron Radiat 22, 180–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Nielsen SS, Toft KN, Snakenborg D, Jeppesen MG, Jacobsen JK, Vestergaard B, Kutter JP, and Arleth L (2009) BioXTAS RAW, a software program for high-throughput automated small-angle X-ray scattering data reduction and preliminary analysis, J. Appl. Crystallogr 42, 959–964. [Google Scholar]
- [42].Petoukhov MV, Franke D, Shkumatov AV, Tria G, Kikhney AG, Gajda M, Gorba C, Mertens HDT, Konarev PV, and Svergun DI (2012) New developments in the ATSAS program package for small-angle scattering data analysis, J. Appl. Crystallogr 45, 342–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Skou S, Gillilan RE, and Ando N (2014) Synchrotron-based small-angle X-ray scattering of proteins in solution, Nat. Protoc 9, 1727–1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Svergun DI (1992) Determination of the regularization parameter in indirect-transform methods using perceptual criteria, J. Appl. Crystallogr 25, 495–503. [Google Scholar]
- [45].Schneidman-Duhovny D, Hammel M, Tainer JA, and Sali A (2013) Accurate SAXS profile computation and its assessment by contrast variation experiments, Biophys. J 105, 962–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Schneidman-Duhovny D, Hammel M, Tainer JA, and Sali A (2016) FoXS, FoXSDock and MultiFoXS: Single-state and multi-state structural modeling of proteins and their complexes based on SAXS profiles, Nucleic Acids Res 44, W424–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Franke D, and Svergun DI (2009) DAMMIF, a program for rapid ab-initio shape determination in small-angle scattering, J. Appl. Crystallogr 42, 342–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Volkov VV, and Svergun DI (2003) Uniqueness of ab initio shape determination in small-angle scattering, J. Appl. Crystallogr 36, 860–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Gerlt JA, Bouvier JT, Davidson DB, Imker HJ, Sadkhin B, Slater DR, and Whalen KL (2015) Enzyme Function Initiative-Enzyme Similarity Tool (EFI-EST): A web tool for generating protein sequence similarity networks, Biochim. Biophys. Acta 1854, 1019–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Stamatakis A (2014) RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies, Bioinformatics 30, 1312–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Jiang W, Saleh L, Barr EW, Xie J, Gardner MM, Krebs C, and Bollinger JM Jr. (2008) Branched activation- and catalysis-specific pathways for electron relay to the manganese/iron cofactor in ribonucleotide reductase from Chlamydia trachomatis, Biochemistry 47, 8477–8484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Greene BL, Taguchi AT, Stubbe J, and Nocera DG (2017) Conformationally Dynamic Radical Transfer within Ribonucleotide Reductase, J. Am. Chem. Soc 139, 16657–16665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Krissinel E, and Henrick K (2004) Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions, Acta Crystallogr., Sect. D: Biol. Crystallogr 60, 2256–2268. [DOI] [PubMed] [Google Scholar]
- [54].Xu H, Faber C, Uchiki T, Fairman JW, Racca J, and Dealwis C (2006) Structures of eukaryotic ribonucleotide reductase I provide insights into dNTP regulation, Proc. Natl. Acad. Sci. U. S. A 103, 4022–4027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Brignole EJ, Tsai KL, Chittuluru J, Li H, Aye Y, Penczek PA, Stubbe J, Drennan CL, and Asturias F (2018) 3.3-Å resolution cryo-EM structure of human ribonucleotide reductase with substrate and allosteric regulators bound, Elife 7, e31502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Meisburger SP, Thomas WC, Watkins MB, and Ando N (2017) X-ray scattering studies of protein structural dynamics, Chem. Rev 117, 7615–7672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Svergun DI, Barberato C, and Koch MHJ (1995) CRYSOL - A program to evaluate x-ray solution scattering of biological macromolecules from atomic coordinates, J. Appl. Crystallogr 28, 768–773. [Google Scholar]
- [58].Rambo RP, and Tainer JA (2013) Accurate assessment of mass, models and resolution by small-angle scattering, Nature 496, 477–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Fischer H, de Oliveira Neto M, Napolitano HB, Polikarpov I, and Craievich AF (2010) Determination of the molecular weight of proteins in solution from a single small-angle X-ray scattering measurement on a relative scale, J. Appl. Crystallogr 43, 101–109. [Google Scholar]
- [60].Mertens HD, and Svergun DI (2010) Structural characterization of proteins and complexes using small-angle X-ray solution scattering, J. Struct. Biol 172, 128–141. [DOI] [PubMed] [Google Scholar]
- [61].Schneidman-Duhovny D, Hammel M, and Sali A (2011) Macromolecular docking restrained by a small angle X-ray scattering profile, J. Struct. Biol 173, 461–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Konarev PV, Volkov VV, Sokolova AV, Koch MHJ, and Svergun DI (2003) PRIMUS - a Windows-PC based system for small-angle scattering data analysis, J Appl Cryst 36, 1277–1282. [Google Scholar]
- [63].Parker MJ, Zhu XL, and Stubbe J (2014) Bacillus subtilis class Ib ribonucleotide reductase: high activity and dynamic subunit interactions, Biochemistry 53, 766–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Rofougaran R, Vodnala M, and Hofer A (2006) Enzymatically active mammalian ribonucleotide reductase exists primarily as an alpha(6)beta(2) octamer, J. Biol. Chem 281, 27705–27711. [DOI] [PubMed] [Google Scholar]
- [65].Fairman JW, Wijerathna SR, Ahmad MF, Xu H, Nakano R, Jha S, Prendergast J, Welin RM, Flodin S, Roos A, Nordlund P, Li Z, Walz T, and Dealwis CG (2011) Structural basis for allosteric regulation of human ribonucleotide reductase by nucleotide-induced oligomerization, Nat. Struct. Mol. Biol 18, 316–U102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Anthony SA, Burrell AL, Johnson MC, Duong-Ly KC, Kuo YM, Simonet JC, Michener P, Andrews A, Kollman JM, and Peterson JR (2017) Reconstituted IMPDH polymers accommodate both catalytically active and inactive conformations, Mol. Biol. Cell 28, 2600–2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Barry RM, Bitbol AF, Lorestani A, Charles EJ, Habrian CH, Hansen JM, Li HJ, Baldwin EP, Wingreen NS, Kollman JM, and Gitai Z (2014) Large-scale filament formation inhibits the activity of CTP synthetase, Elife 3, e03638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Lynch EM, Hicks DR, Shepherd M, Endrizzi JA, Maker A, Hansen JM, Barry RM, Gitai Z, Baldwin EP, and Kollman JM (2017) Human CTP synthase filament structure reveals the active enzyme conformation, Nat. Struct. Mol. Biol 24, 507–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Thomas WC, Brooks FP III, Burnim AA, Bacik J-P, Stubbe J, Kaelber JT, CHen JZ, and Ando N (2018) Convergent Allostery in Ribonucleotide Reductase, bioRxiv 504290. [DOI] [PMC free article] [PubMed]
- [70].Zhang Z, Yang K, Chen CC, Feser J, and Huang M (2007) Role of the C terminus of the ribonucleotide reductase large subunit in enzyme regeneration and its inhibition by Sml1, Proc. Natl. Acad. Sci. U. S. A 104, 2217–2222. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.