

Abstract
The blood glycoprotein von Willebrand factor (VWF) plays an important role in hemostasis and thrombosis. VWF is produced and secreted as large multimers by endothelial cells and megakaryocytes. It is then cleaved in a sheer-stress dependent manner by a specific protease, ADAMTS13, into multimers consisting of 2–80 subunits. Among VWF multimers, high molecular weight (HMW) multimers play important roles in platelet aggregation. Therefore, their loss induces a hemostatic disorder known as von Willebrand disease (VWD) type 2A. Various cardiovascular diseases, such as aortic stenosis, hypertrophic obstructive cardiomyopathy (HOCM), and several congenital structural diseases, as well as mechanical circulatory support systems, generate excessive high shear stress in the bloodstream. These cause excessive cleavage of VWF multimers resulting in a loss of HMW multimers, known as acquired von Willebrand syndrome (AVWS), a hemostatic disorder similar to VWD type 2A. Bleeding often occurs in the gastrointestinal tract since a fragile angiodysplasia develops associated with these diseases. Radical treatment for AVWS is to remove the pathological high shear causing AVWS.
Keywords: Aortic stenosis, Heyde's syndrome, von Willebrand factor, Acquired von Willebrand's syndrome, ECMO, Left ventricular assist device
The Function of von Willebrand Factor and its Regulation by a Specific Protease ADAMTS13
VWF is produced and secreted from endothelial cells and megakaryocytes as large multimers1). It is constitutively secreted from endothelial cells and also stored in Weibel-Palade bodies2) that undergo regulated secretion by various stimuli such as exercise3, 4), inflammation5, 6), infection5), pregnancy7), and mental stress8, 9). Therefore, the plasma concentration of VWF is increased by these conditions.
The level of plasma VWF antigen (VWF:Ag) is relatively expressed using that of standard plasma at 100 IU/dL (1 IU/mL). In some cases, this is expressed as a percentage of the standard (100% as a control). It is noted that VWF:Ag increases in an age-dependent manner10). The VWF:Ag of 80-year-old people is 1.5–2.0 times the VWF:Ag of 40-year-old people on average11).
VWF:Ag also varies among individuals. In middle-aged people, VWF:Ag is widely distributed in 60–180%12). Furthermore, VWF is modified by ABO-type glycosylation as found in the erythrocyte plasma membrane. The plasma VWF:Ag level of persons with O-type is approximately 70% of that with other blood types, on average11). VWF is present not only in plasma, but also partially in the sub-endothelial tissues of blood vessels1). While VWF in such tissues plays an important role13–15), plasma VWF is also important for the regulation of thrombosis and hemostasis since excessive cleavage of VWF causes acquired von Willebrand syndrome (AVWS)16) as described in this review. VWF is also stored in the α-granules of platelets and secreted upon their activation17).
VWF has several functional domains (Fig. 1A). When endothelial cells are lost because of vascular damage, VWF binds to collagen fibers in the subendothelial tissues through its A3 domain18). Attached VWF changes its conformation in response to the shear stress of blood flow to interact with the glycoprotein Ib (GPIb) complex on the platelet surface through its A1 domain19). By this interaction, platelets start rolling along the vessel wall and finally attach firmly through their collagen receptors, GPIa/IIa (integrin α2β1) and GPVI. During the process, platelets are activated and release self-agonists such as thromboxane A2 produced by arachidonic acid breakdown, and ADP stored in dense-core granule by regulated exocytosis, resulting in the local enhancement of platelet activation. On the surface of activated platelets, GPIIb/IIIa (integrin αIIbβ3) that is abundantly present on platelets changes its conformation to allow it to bind its ligands, VWF and fibrinogen20, 21). A part (∼30%) of GPIIb/IIIa is also stored on the α-granule membrane and transported to the surface upon platelet activation22, 23). VWF is a multimer and fibrinogen is a heterohexamer consisting of two sets of α, β, and γ subunits24), both of which contain multiple binding sites for activated GPIIb/IIIa. Activated platelets are bridged with these ligands by protein-protein interaction, resulting in the formation of platelet aggregates. Thus, VWF plays a critical role in platelet thrombosis at multiple steps. Furthermore, VWF forms a complex with coagulation factor VIII (FVIII) through its D3 domain25). This interaction is critical for the stable existence of FVIII. Therefore, severe von Willebrand disease (VWD) such as VWD type 3 (see below) causes a loss of FVIII and concomitantly causes hemophilia-like symptoms26).
Fig. 1.
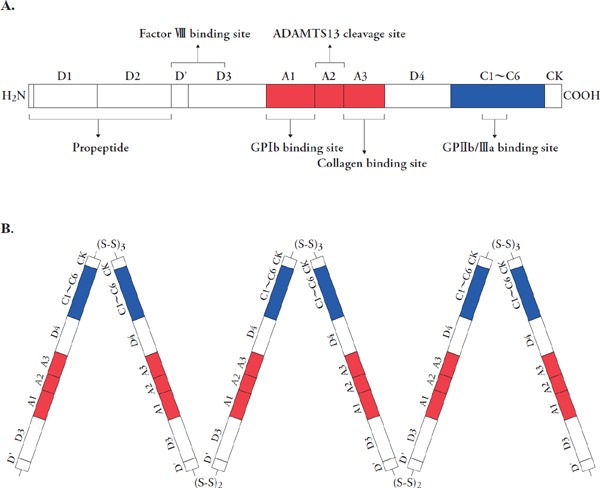
The structure of von Willebrand factor (VWF)
A) Schematic structure of VWF monomer with its functional domains. Binding sites are indicated for FVIII (D' and D3 domains), GPIb (A1 domain), collagen (A3 domain) and GPIIb/IIIa (C1 domain). An ADAMTS13 cleavage site is located in the A2 domain. B) The VWF multimer structure. The subunits of pro-VWF is initially dimerized by disulfide bonds between C-terminal cysteine knot (CK) domains of each subunit in endoplasmic reticulum and then multimerized by at least two disulfide bonds in D'D3 domains concomitantly with the removal of short peptide in the C-terminal region in the Golgi apparatus.
VWF monomers form multimers by covalent disulfide binding between C-terminal regions in the endoplasmic reticulum and between N-terminal regions in the Golgi apparatus, followed by removal of the propeptide domain (Fig. 1B)27). Subsequently, VWF is secreted as so-called unusually large VWF (UL-VWF) multimers28). Extracellularly, they are cleaved in a shear stress-dependent manner in the A2 domain by a specific zinc-containing metalloprotease, ADAMTS13,29) which is secreted mainly from hepatic stellate cells (Ito cells) in the liver30). The trigger for the cleavage is the change in VWF conformation. Thus, in healthy plasma, VWF is present as multimers consisting of 2–80 subunits. Notably, larger multimers show stronger activity for platelet aggregation, namely hemostatic activity31).
A genetic deficiency of ADAMTS13 causes the emergence of non-physiological UL-VWF multimers and induces thrombotic thrombocytopenic purpura (TTP) due to excessive thrombus formation in the microcirculation32). In comparison, genetic mutations in the VWF A2 domain, which increase susceptibility to cleavage by ADAMTS13, cause reduced HMW multimers and induce a hemostatic disorder classified as VWD type 2A (see below). A similar reduction in HMW multimers develops in various conditions such as cardiovascular diseases with excessive high shear stress, and autoimmune diseases that develop a hemostatic disorder, AVWS.
Mechanism of Development of AVWS
VWD is a hereditary disease caused by deficiency or dysfunction of VWF33). VWD is classified into several types: type 1 characterized by reduced VWF:Ag, type 2 by a qualitative change in VWF and type 3 by a complete loss of VWF33). Type 2 is divided into four subtypes:33) Type 2A is characterized by a selective deficiency of HMW multimers, type 2B shows increased affinity to GPIb on the surface of platelets, type 2M shows a decreased binding affinity of VWF to platelets without selective reduction of HMW multimers, and type 2N is characterized by a decreased binding affinity for FVIII.
Similar VWF dysfunctions occur even without genetic mutations, which are designated as AVWS34). Lymphoproliferative diseases such as multiple myeloma and malignant lymphoma, myeloproliferative diseases such as polycythemia vera, malignant tumors, autoimmune diseases such as systemic lupus erythematosus, and some drugs such as griseofulvin and ciprofloxacin cause AVWS type 2 due to various causes such as autoimmunity and the absorption of VWF multimers35–42). It is, however, noted that the incidence of these is quite low. Hypothyroidism is sometimes accompanied by AVWS due to low protein production according to the nature of the disease43). The reduction of VWF:Ag is mild and hypothyroidism is not thought to be related to a bleeding disorder. The hypothyroidism-induced low expression of VWF is recovered when thyroid function becomes normal43).
However, some cardiovascular diseases, such as aortic stenosis, cause AVWS16). Non-physiological high shear stress in blood excessively cleaves VWF multimers, resulting in AVWS with decreased HMW multimers. This pathophysiology of AVWS is similar to that of hereditary VWD type 2A.
Diagnosis of Shear Stress–Induced AVWS
A diagnosis of shear stress–induced AVWS includes the demonstration of the loss of HMW multimers. The current standard method is VWF multimer analysis. The multimers are separated by sodium dodecyl sulfate–containing agarose (not polyacrylamide) gel electrophoresis in a non-reduced condition and detected by western blot analysis with anti-VWF antibody to analyze giant molecules 500,000∼20,000,000 Daltons in size. VWF multimer ladders from the lowest to the fifth, from the sixth to the tenth, and higher than the eleventh, are classified as low molecular weight (LMW), medium molecular weight (MMW), and HMW multimers, respectively (Fig. 2). The loss of HMW multimers is evaluated by comparison with the plasma of a healthy subject analyzed in the same gel44).
Fig. 2.
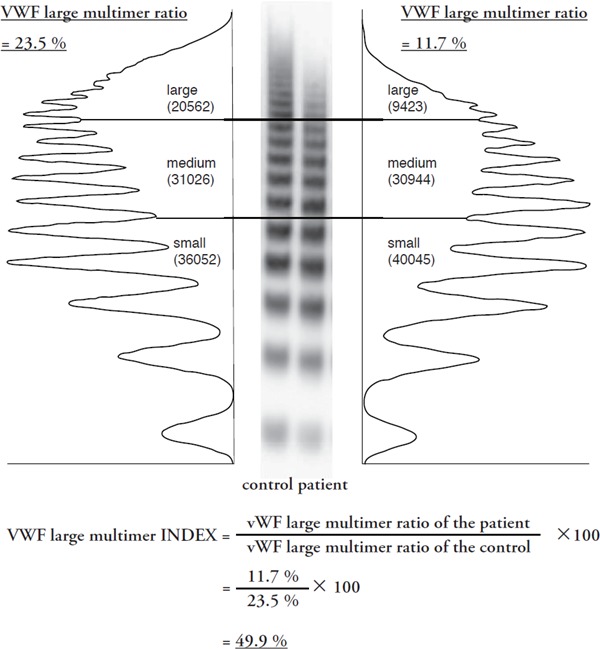
Quantification method for the calculation of the von Willebrand factor (VWF) large multimer index
This is defined as the ratio of the patient's HMW multimer ratio to the healthy subject's ratio analyzed in the same gel. With the index, a patient's high molecular weight (HMW) multimers are expressed as a percentage of those of a healthy control. The patient's plasma in this figure was from the patient with severe aortic stenosis with mean aortic pressure of 68 mmHg and peak blood flow of 5.24 m/sec.
VWF multimer analysis has not usually been evaluated quantitatively except for several studies, where ‘VWF HMW multimer ratios’ have been measured16, 44, 45). The ratio is calculated based on the densitometric analysis of a western blot of patients' plasma, defined as HMW multimer area per total VWF area44). Patients with aortic stenosis exhibited ratios ranging from 4–15% in a previous report16) and 15–25% in our study44), while an aortic stenosis severity–dependent decrease in the ratios was observed in both studies. Furthermore, the ratios of patients treated with LVAD have ranged from 20–60%45). Thus, the ‘ratios’ vary from study to study, probably due to the nature of the methods used. It seems difficult to use HMW multimer ratios for a severity classification of AVWS or as a comparison of severity across studies. To overcome the issue, we have recently proposed a value, named the VWF large multimer index, that is defined as the ratio of the patient's HMW multimer ratio to the healthy subject's ratio that is analyzed in the same gel (Fig. 2)44). With the index, a patient's HMW multimers are expressed as a percentage of those of a healthy control. The plasma of the patient with severe aortic stenosis with mean aortic pressure of 68 mmHg and peak blood flow of 5.24 m/sec exhibits the VWF large multimer ratio 23.7% and the index 49.9% (Fig. 2).
Ristocetin induces platelet agglutination in the presence of HMW multimers46). With pre-fixed platelets, VWF ristocetin co-factor activity (VWF:RCo) can be measured automatically by laboratory analyzer in hospitals and expressed as a percentage of that of standard plasma. Usually, VWF:RCo and VWF:Ag are well correlated to each other, with VWF:RCo/VWF:Ag ratios normally 1.047, 48). For the diagnosis of VWD type 2, a VWF:RCo/VWF:Ag ratio less than 0.649) or 0.750) is used as a reference for its diagnosis. It would be feasible if this value can be used for a diagnosis. However, it is not usually used for the diagnosis of cardiovascular disease-associated AVWS at the moment. In our preliminary analysis, the VWF:RCo/VWF:Ag ratio may be less sensitive than VWF multimer analysis, although they are moderately correlated. We are conducting further analysis to elucidate the precise reasons and improve the method.
Studies to evaluate AVWS severity with suitable quantitative values for HMW multimers and simultaneous bleeding events are definitely required. This would provide us with an AVWS severity classification useful for a clinical setting.
Heyde's Syndrome; Aortic Stenosis with Gastrointestinal Bleeding
Since aortic stenosis is an age-related disease, the number of patients is drastically increasing in many countries. Generally, severe aortic stenosis is defined as a mean aortic pressure higher than 40 mmHg, with peak blood flow faster than 4 m/sec, or an aortic valve area less than 1.0 cm2 51). When severe aortic stenosis is accompanied by heart failure, syncope, or angina, invasive treatment such as a surgical aortic valve replacement and a transcatheter aortic valve implantation (TAVI) is considered52, 53).
Aortic stenosis with gastrointestinal bleeding is designated as Heyde's syndrome since Dr. Edward Heyde first described such patients54). The mechanism of this syndrome has recently been uncovered: the gastrointestinal bleeding is caused by a hemostatic disorder AVWS (aortic stenosis–AVWS) and gastrointestinal angiodysplasia55). It has been reported that 7–24% of patients with gastrointestinal bleeding of unknown origin are associated with aortic stenosis56).
The severity of AVWS is dependent on the severity of the aortic stenosis. HMW multimer ratios in patients with severe aortic stenosis negatively correlated with the mean transvalvular gradient16). We have also demonstrated that VWF large multimer indices in 31 severe aortic stenosis patients are decreased depending on the severity of the aortic stenosis44). Importantly, in these studies, the values of HMW multimers start to be affected at around 40 mmHg of mean pressure gradient, namely 50–60 mmHg of the maximal pressure gradient, through the aortic valve. Thus, severe aortic stenosis in most patients may be associated with AVWS. It is speculated that no less than 100,000 patients with AVWS associated with aortic stenosis may exist in Japan.
The prevalence of Heyde's syndrome, namely the development of gastrointestinal bleeding in patients with aortic stenosis, is rather high. Among patients with severe aortic stenosis, 27.5–38.7% develop the syndrome, while the definition of the gastrointestinal bleeding varies from study to study44, 57). It is noted that the exclusion of Heyde's syndrome is difficult even when the origin of the bleeding cannot be identified by upper and lower endoscopic examinations since approximately 20% of angiodysplasia develop in the small intestine58).
For treatment of the acute phase of bleeding, supplementation of VWF by VWF-containing blood products, such as fresh frozen plasma and cryoprecipitate containing much VWF, could transiently be effective in addition to endoscopic hemostasis59). For radical therapy, the stenotic aortic valve itself should be treated by surgery16, 60) or, more recently, TAVI61, 62). Without proper treatment of the valve, bleeding events would become repetitive. For example, a case who initially refused surgery experienced 10 bleeding events over a period of 2 years until he was treated with a surgical aortic valve replacement44). AVWS, namely the loss of HMW multimers, is rapidly recovered in a couple of days after treatment of the valve63, 64). It is noted that the HMW multimer is not recovered when perivalvular leaks occur associated with TAVI65). During surgery, strong anti-coagulation is required for the use of the cardiopulmonary bypass apparatus. Since a case with a subdural hematoma associated a surgical aortic valve operation has been reported66), supplementation of VWF may be beneficial during treatment. Further evidence is required to clarify the operation-related bleeding risk and possible prevention by supplementation.
AVWS Caused by Other Cardiovascular Diseases
In addition to aortic stenosis, several cardiovascular diseases causing excessive high shear stress are associated with AVWS.
Hypertrophic cardiomyopathy sometimes causes an intraventricular obstruction, known as hypertrophic obstructive cardiomyopathy (HOCM). HOCM is sometimes associated with an intraventricular pressure gradient of more than 50 mmHg67). With a mechanism similar to that of aortic stenosis, HOCM can cause AVWS68–70). Several congenital structural heart diseases, such as a ventricular septal defect and tetralogy of Fallot, contribute to jet flow in the cardiovascular system. In these diseases, the development of AVWS and bleeding have been reported71–73).
Pulmonary hypertension is a critical and progressive disease, sometimes resulting in fatal outcomes. AVWS has been reported to occur in patients with pulmonary hypertension74–76). Some patients with pulmonary hypertension are treated with anti-coagulant therapy to prevent the progression of the disease itself77). Since hemoptysis is a complication of pulmonary hypertension78), careful attention should be paid to anti-coagulation therapy for such patients although only a proportion of patients with pulmonary hypertension may suffer from AVWS. Obstructive sleep apnea is associated with AVWS79). This could be caused by pulmonary hypertension during apnea periods.
Mitral regurgitation also causes AVWS80, 81). One study reported that 13.2% of patients with mitral regurgitation developed gastrointestinal bleeding and 41.5% of the patients had at least one positive bleeding according to a questionnaire80). In a clinical setting, however, mitral regurgitation has hardly been considered a hemostatic disorder. Thus, the association between mitral regurgitation and AVWS needs to be further evaluated in future.
AVWS Caused by Mechanical Circulatory Support
Mechanical circulatory supports play an important role in the treatment of life-threatening cardiopulmonary failure. Extracorporeal membrane oxygenation (ECMO; another name is V-V ECMO) is used for the treatment of acute respiratory failure. Under ECMO therapy, blood is collected through the outflow cannula inserted into the jugular vein and returned through the inflow cannula in the femoral vein after oxygenation by a centrifugal pump at 2–4 L/min. Bleeding is one of the major complications and has been thought to be exclusively due to antithrombotic therapy for the prevention of thrombosis. However, it has been demonstrated that ECMO is associated with AVWS, probably due to extraordinarily high shear stress inside the pump82). More recently, AVWS is thought to contribute to ECMO-associated bleeding complications82–84). Percutaneous cardiopulmonary support (PCPS), otherwise known as V-A ECMO or extracorporeal life support (ECLS), is used for the treatment of acute circulatory failure. PCPS is also associated with bleeding and AVWS is noted as one of its major cause85, 86). AVWS caused by PCPS is far more severe, with much lower VWF large multimer indces, compared to that caused by aortic stenosis86).
A left ventricular assist device (LVAD) is a powerful tool for the treatment of end-stage heart failure87, 88). By taking continuous flow instead of pulsatile flow, the pump has become smaller and implantable in the thoracic cavity. Since patients can be discharged from hospital after successful LVAD implantation, the number of patients is drastically increasing. Up to 2016, 18,987 patients were treated with an implantable LVAD in the world89). LVAD-implantation is only permitted for bridging to cardiac implantation in Japan in 2011. While heart transplantation is performed in approximately 50 cases in Japan, 174 patients were treated with an LVAD from October 2015 to September 2016 90).
Three-year survival rates after implantable LVAD are 59% in the world and 81.6% in Japan89, 91). The major complications of an implantable LVAD are infection of the drive-line that connects the implanted pump to the battery and computer outside of the body, intra-pump thrombosis and its related embolism, and bleeding. Bleeding most often occurs in the gastrointestinal tract in 10–33% of patients. To prevent pump thrombosis, antiplatelet and anticoagulation therapies are usually employed. It had been thought that LVAD-associated bleeding was due to the strong anti-thrombotic therapy. Nevertheless, LVAD-caused AVWS has been recently thought to contribute much to the bleeding92, 93). Bleeding occurs in not only an immediate or early phase but also even after years of LVAD implantation92, 94). The origin of the bleeding at an early phase could be from gastrointestinal mucosal damage, as observed in our patient who developed massive gastrointestinal bleeding at approximately 1 month after LVAD implantation from multiple ulcers and erosions of the colon95). In comparison, the gastrointestinal bleeding that develops at a later phase is considered to be from gastrointestinal angiodysplasia, since it would take some time, possibly several months, for the angiodysplasia to form after LVAD implantation. We have recently demonstrated that all 41 examined LVAD-treated patients exhibited far more severe AVWS compared to that in patients with aortic stenosis by quantitative analysis of AVWS96). Twelve patients (approximately 30%) developed gastrointestinal bleeding, where bleeding frequently occurred in patients with a VWF large multimer index below 40%96), suggesting a critical role of AVWS in gastrointestinal bleeding in patients treated with LVAD. It is noted that four cases developed bleeding within 40 days after LVAD implantation, with 8 cases after 142 days96). One case developed bleeding on the 1,106th day after LVAD implantation96).
Two kinds of rotary pumps, centrifugal and axial types, are utilized for LVAD. Sufficient blood flow is provided at ∼4,000 rpm by centrifugal-type pumps and at ∼8,000 rpm by axial pumps. Accordingly, axial type LVAD causes more severe AVWS than the centrifugal type97). Recently, it is found that platelet function is also impaired in patients treated with LVAD. This may also contribute to the bleeding associated with LVAD98). Thus, antiplatelet therapy, platelet dysfunction, AVWS, angiodysplasia and/or mucosal damage such erosion and ulcer are implicated in the pathogenesis of the LVAD-associated gastrointestinal bleeding (Fig. 3).
Fig. 3.
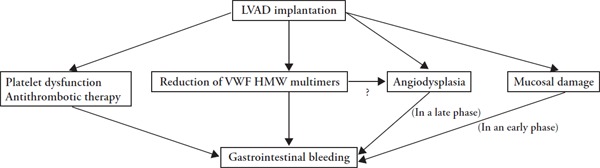
Pathogenesis of gastrointestinal bleeding associated in patients treated with an left ventricular assist device (LVAD). The gastrointestinal bleeding is caused by mucosal damage, platelet dysfunction, antithrombotic therapy, a reduction of von Willebrand factor (VWF) high molecular weight (HMW) multimers and angiodysplasia as possible causes. In an early phase, bleeding could be caused mainly by mucosal damage. In a late phase, bleeding could be caused mainly by angiodysplasia formed after LVAD implantation.
Much effort has been made to develop LVAD with reduced shear stress in the pump. HEART-MATE3 has achieved this by using a magnetically levitated centrifugal-flow pump99). However, HEARTMATE3 unexpectedly did not reduce the rate of gastrointestinal bleeding compared to the axial-flow pumps in the MOMENTUM3 trial100). Although the degree of reduction of HMW multimers was not shown, and therefore it is difficult to evaluate the contribution of the reduction in shear stress, it is possible that other factors may also contribute to LVAD-associated bleeding.
AVWS-Associated Gastrointestinal Angiodysplasia
Gastrointestinal angiodysplasia is frequently the origin of the gastrointestinal bleeding associated with cardiovascular disease-induced AVWS55). Angiodysplasia is an accumulation of dilated vessels in the subepithelial tissue susceptible to easy bleeding (Fig. 4). This lesion develops through the gastrointestinal tract from the stomach to rectum, more usually in the right colon in Western patients, while in the left colon in Japanese patients101). Furthermore, approximately 20% of the angiodysplasia develops in the small intestine58). Angiodysplasia increases in an age-dependent manner102). However, it develops in younger patients after LVAD implantation103, 104). Therefore, it is conceivable that the disease per se causes angiodysplasia, possibly through impaired perfusion of blood, reduced pulse pressure and/or a decrease of HMW multimers as well as for other reasons.
Fig. 4.
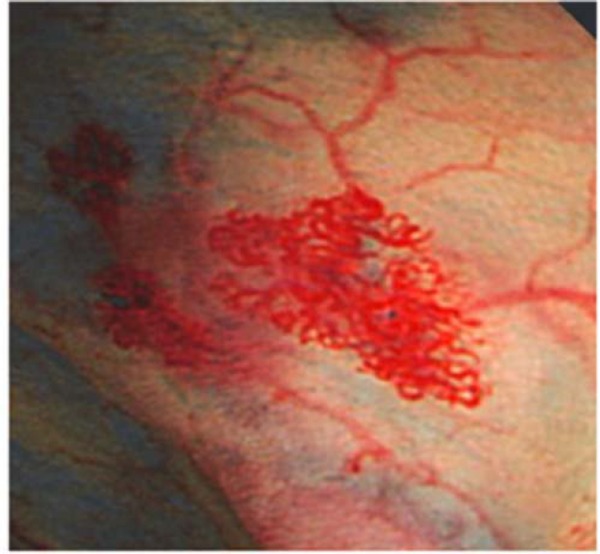
Typical angiodysplasia in the small intestine. The photo is adopted from Tamura et al.44).
Angiodysplasia may be generated by the acceleration of immature angiogenesis105). Angiogenesis is initiated by endothelial migration and proliferation followed by maturation with surrounding pericytes and/or vascular smooth muscle cells106). Vascular endothelial growth factor (VEGF) induces endothelial migration and proliferation and angiopoietin-1 (Ang-1) plays a critical role in maturation through activation of its receptor Tie-2, where Ang-1 function is antagonized by another factor, Ang-2107–109). VEGF and Ang-2 are generated in endothelial cells in ischemia, which may contribute to the enhancement of angiogenesis in ischemic tissue110, 111). Such angiogenesisregulating factors may be involved in the formation of angiodysplasia since their expression is increased in the affected tissue112,113). Plasmas of LVAD-treated patients are known to strongly enhance endothelial proliferation and migration114), suggesting the involvement of humoral factors. These may be VEGF and/or Ang2 in the tissue, the expression of which may be induced by the abnormal blood flow. Further, since VWF has an inhibitory function for angiogenesis115), decreased HMW multimers per se may contribute to the formation of angiodysplasia116–118). Thus, much is still to be elucidated for a full understanding of the mechanism of angiodysplasia formation.
Conclusion and Perspectives
Non-physiological high shear stress generated in various cardiovascular diseases causes the hemostatic disorder AVWS by the excessive cleavage of VWF multimers.
Many issues remain to be elucidated: (1) how often and how severely does AVWS occur in each cardiovascular disease, (2) how often do bleeding events occur in each disease, (3) what severity of AVWS is a risk for bleeding, and (4) what treatment is suitable for hemostasis upon bleeding and in an operation. A severity classification, namely bleeding risk stratification, of cardiovascular disease-associated AVWS needs to be established.
Acknowledgements
This work was supported by a Health and Labor Sciences Research Grant for Research on rare and intractable diseases from the Ministry of Health, Labor and Welfare, Japan, Grants from AMED under Gr a n t Nu m b e r J P 1 8 e k 0 1 0 9 3 7 0 h 0 0 0 1 , JP18ek0109256h0002 and JP18ek0109246h0002, and Grants from the SENSHIN Medical Research Foundation and the Suzuken Memorial Foundation. This work was also supported by the Cooperative Research Project Program of Joint Usage/Research Center at the Institute of Development, Aging and Cancer, Tohoku University, and the Research grant from the Japanese Society of Thrombosis and Hemostasis, in part.
Disclosures
The AVeC Study and the LVAD-AVWS Study are supported, in part, by Sysmex Corp.
References
- 1). Sadler JE: Biochemistry and genetics of von Willebrand factor. Annu Rev Biochem, 1998; 67: 395-424 [DOI] [PubMed] [Google Scholar]
- 2). Rondaij MG, Bierings R, Kragt A, van Mourik JA, Voorberg J: Dynamics and plasticity of Weibel-Palade bodies in endothelial cells. Arterioscler Thromb Vasc Biol, 2006; 26: 1002-1007 [DOI] [PubMed] [Google Scholar]
- 3). El-Sayed MS, Sale C, Jones PG, Chester M: Blood hemostasis in exercise and training. Med Sci Sports Exerc, 2000; 32: 918-925 [DOI] [PubMed] [Google Scholar]
- 4). van Loon JE, Sonneveld MA, Praet SF, de Maat MP, Leebeek FW: Performance related factors are the main determinants of the von Willebrand factor response to exhaustive physical exercise. PloS One, 2014; 9: e91687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5). Pottinger BE, Read RC, Paleolog EM, Higgins PG, Pearson JD: von Willebrand factor is an acute phase reactant in man. Throm Res, 1989; 53: 387-394 [DOI] [PubMed] [Google Scholar]
- 6). Chen J, Chung DW: Inflammation, von Willebrand factor, and ADAMTS13. Blood, 2018; 132: 141-147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7). Bremme KA: Haemostatic changes in pregnancy. Best Pract Res Clin Haematol, 2003; 16: 153-168 [DOI] [PubMed] [Google Scholar]
- 8). Musumeci V, Baroni S, Cardillo C, Zappacosta B, Zuppi C, Tutinelli F, Folli G: Cardiovascular reactivity, plasma markers of endothelial and platelet activity and plasma renin activity after mental stress in normals and hypertensives. J Hypertens Suppl, 1987; 5: S1-4 [PubMed] [Google Scholar]
- 9). Jern C, Eriksson E, Tengborn L, Risberg B, Wadenvik H, Jern S: Changes of plasma coagulation and fibrinolysis in response to mental stress. Thromb Haemost, 1989; 62: 767-771 [PubMed] [Google Scholar]
- 10). Konkle BA: Von Willebrand factor and aging. Semin Thromb Hemost, 2014; 40: 640-644 [DOI] [PubMed] [Google Scholar]
- 11). Gill JC, Endres-Brooks J, Bauer PJ, Marks WJ, Jr, Montgomery RR: The effect of ABO blood group on the diagnosis of von Willebrand disease. Blood, 1987; 69: 1691-1695 [PubMed] [Google Scholar]
- 12). Wang Z, Dou M, Du X, Ma L, Sun P, Cao H, Ye S, Jiang P, Liu F, Lin F, Zhang R, Li C: Influences of ABO blood group, age and gender on plasma coagulation factor VIII, fibrinogen, von Willebrand factor and ADAMTS13 levels in a Chinese population. Peer J, 2017; 5: e3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13). Nichols TC, Bellinger DA, Tate DA, Reddick RL, Read MS, Koch GG, Brinkhous KM, Griggs TR: von Willebrand factor and occlusive arterial thrombosis. A study in normal and von Willebrand's disease pigs with diet-induced hypercholesterolemia and atherosclerosis. Arteriosclerosis, 1990; 10: 449-461 [DOI] [PubMed] [Google Scholar]
- 14). Nichols TC, Bellinger DA, Reddick RL, Smith SV, Koch GG, Davis K, Sigman J, Brinkhous KM, Griggs TR, Read MS: The roles of von Willebrand factor and factor VIII in arterial thrombosis: studies in canine von Willebrand disease and hemophilia A. Blood, 1993; 81: 2644-2651 [PubMed] [Google Scholar]
- 15). Nichols TC, Samama CM, Bellinger DA, Roussi J, Reddick RL, Bonneau M, Read MS, Bailliart O, Koch GG, Vaiman M, et al. : Function of von Willebrand factor after crossed bone marrow transplantation between normal and von Willebrand disease pigs: effect on arterial thrombosis in chimeras. Proc Natl Acad Sci USA, 1995; 92: 2455-2459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16). Vincentelli A, Susen S, Le Tourneau T, Six I, Fabre O, Juthier F, Bauters A, Decoene C, Goudemand J, Prat A, Jude B: Acquired von Willebrand syndrome in aortic stenosis. N Engl J Med, 2003; 349: 343-349 [DOI] [PubMed] [Google Scholar]
- 17). Cramer EM, Meyer D, le Menn R, Breton-Gorius J: Eccentric localization of von Willebrand factor in an internal structure of platelet alpha-granule resembling that of Weibel-Palade bodies. Blood, 1985; 66: 710-713 [PubMed] [Google Scholar]
- 18). Lankhof H, van Hoeij M, Schiphorst ME, Bracke M, Wu YP, Ijsseldijk MJ, Vink T, de Groot PG, Sixma JJ: A3 domain is essential for interaction of von Willebrand factor with collagen type III. Thromb Haemost, 1996; 75: 950-958 [PubMed] [Google Scholar]
- 19). Ruggeri ZM: von Willebrand factor. J Clin Invest, 1997; 99: 559-564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20). Lombardo VT, Hodson E, Roberts JR, Kunicki TJ, Zimmerman TS, Ruggeri ZM: Independent modulation of von Willebrand factor and fibrinogen binding to the platelet membrane glycoprotein IIb/IIIa complex as demonstrated by monoclonal antibody. J Clin Invest, 1985; 76: 1950-1958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21). Litjens PE, Van Willigen G, Weeterings C, Ijsseldijk MJ, Van Lier M, Koivunen E, Gahmberg CG, Akkerman JW: A tripeptide mimetic of von Willebrand factor residues 981–983 enhances platelet adhesion to fibrinogen by signaling through integrin alpha(IIb)beta3. J Thromb Haemost, 2005; 3: 1274-1283 [DOI] [PubMed] [Google Scholar]
- 22). Phillips DR, Charo IF, Parise LV, Fitzgerald LA: The platelet membrane glycoprotein IIb-IIIa complex. Blood, 1988; 71: 831-843 [PubMed] [Google Scholar]
- 23). Bennett JS: Structure and function of the platelet integrin alphaIIbbeta3. J Clin Invest, 2005; 115: 3363-3369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24). Kolodziejczyk J, Ponczek MB: The role of fibrinogen, fibrin and fibrin(ogen) degradation products (FDPs) in tumor progression. Contemp Oncol, 2013; 17: 113-119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25). Saenko EL, Scandella D: A mechanism for inhibition of factor VIII binding to phospholipid by von Willebrand factor. J Biol Chem, 1995; 270: 13826-13833 [DOI] [PubMed] [Google Scholar]
- 26). Nichols WL, Hultin MB, James AH, Manco-Johnson MJ, Montgomery RR, Ortel TL, Rick ME, Sadler JE, Weinstein M, Yawn BP: von Willebrand disease (VWD): evidence-based diagnosis and management guidelines, the National Heart, Lung, and Blood Institute (NHLBI) Expert Panel report (USA). Haemophilia, 2008; 14: 171-232 [DOI] [PubMed] [Google Scholar]
- 27). Wagner DD: Cell biology of von Willebrand factor. Annu Rev Cell Biol, 1990; 6: 217-246 [DOI] [PubMed] [Google Scholar]
- 28). Furlan M: Von Willebrand factor: molecular size and functional activity. Ann Hematol, 1996; 72: 341-348 [DOI] [PubMed] [Google Scholar]
- 29). Zheng X, Chung D, Takayama TK, Majerus EM, Sadler JE, Fujikawa K: Structure of von Willebrand factor-cleaving protease (ADAMTS13), a metalloprotease involved in thrombotic thrombocytopenic purpura. J Biol Chem, 2001; 276: 41059-41063 [DOI] [PubMed] [Google Scholar]
- 30). Uemura M, Tatsumi K, Matsumoto M, Fujimoto M, Matsuyama T, Ishikawa M, Iwamoto TA, Mori T, Wanaka A, Fukui H, Fujimura Y: Localization of ADAMTS13 to the stellate cells of human liver. Blood, 2005; 106: 922-924 [DOI] [PubMed] [Google Scholar]
- 31). Matsumoto M, Kawaguchi S, Ishizashi H, Yagi H, Iida J, Sakaki T, Fujimura Y: Platelets treated with ticlopidine are less reactive to unusually large von Willebrand factor multimers than are those treated with aspirin under high shear stress. Pathophysiol Haemost Thromb, 2005; 34: 35-40 [DOI] [PubMed] [Google Scholar]
- 32). Furlan M, Robles R, Solenthaler M, Wassmer M, Sandoz P, Lammle B: Deficient activity of von Willebrand factor-cleaving protease in chronic relapsing thrombotic thrombocytopenic purpura. Blood, 1997; 89: 3097-3103 [PubMed] [Google Scholar]
- 33). Sadler JE, Budde U, Eikenboom JC, Favaloro EJ, Hill FG, Holmberg L, Ingerslev J, Lee CA, Lillicrap D, Mannucci PM, Mazurier C, Meyer D, Nichols WL, Nishino M, Peake IR, Rodeghiero F, Schneppenheim R, Ruggeri ZM, Srivastava A, Montgomery RR, Federici AB, Working Party on von Willebrand Disease C : Update on the pathophysiology and classification of von Willebrand disease: a report of the Subcommittee on von Willebrand Factor. J Thromb Haemost, 2006; 4: 2103-2114 [DOI] [PubMed] [Google Scholar]
- 34). Federici AB, Rand JH, Bucciarelli P, Budde U, van Genderen PJ, Mohri H, Meyer D, Rodeghiero F, Sadler JE, Subcommittee on von Willebrand F : Acquired von Willebrand syndrome: data from an international registry. Thromb Haemost, 2000; 84: 345-349 [PubMed] [Google Scholar]
- 35). Castaman G, Rodeghiero F: Acquired transitory von Willebrand syndrome with ciprofloxacin. Lancet, 1994; 343: 492. [DOI] [PubMed] [Google Scholar]
- 36). Maas D, Laros-van Gorkom B, Gianotten S, Cruijsen M, van Heerde W, Nijziel M: Acquired von Willebrand Disease Associated with Mantle Cell Lymphoma. Case Reports in Hematology, 2018; 2018: 7973297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37). Mohri H: Acquired von Willebrand disease in patients with polycythemia rubra vera. Am J Hematol, 1987; 26: 135-146 [DOI] [PubMed] [Google Scholar]
- 38). Michiels JJ, Budde U, van der Planken M, van Vliet HH, Schroyens W, Berneman Z: Acquired von Willebrand syndromes: clinical features, aetiology, pathophysiology, classification and management. Best Pract Res Clin Haematol, 2001; 14: 401-436 [DOI] [PubMed] [Google Scholar]
- 39). Scott JP, Montgomery RR, Tubergen DG, Hays T: Acquired von Willebrand's disease in association with Wilm's tumor: regression following treatment. Blood, 1981; 58: 665-669 [PubMed] [Google Scholar]
- 40). Sampson BM, Greaves M, Malia RG, Preston FE: Acquired von Willebrand's disease: demonstration of a circulating inhibitor to the factor VIII complex in four cases. Br J Haematol, 1983; 54: 233-244 [DOI] [PubMed] [Google Scholar]
- 41). Rao KP, Kizer J, Jones TJ, Anunciado A, Pepkowitz SH, Lazarchick J: Acquired von Willebrand's syndrome associated with an extranodal pulmonary lymphoma. Arch Pathol Lab Med, 1988; 112: 47-50 [PubMed] [Google Scholar]
- 42). Soff GA, Green D: Autoantibody to von Willebrand factor in systemic lupus erythematosus. J Lab Clin Med, 1993; 121: 424-430 [PubMed] [Google Scholar]
- 43). Federici AB: Acquired von Willebrand syndrome associated with hypothyroidism: a mild bleeding disorder to be further investigated. Semin Thromb Hemost, 2011; 37: 35-40 [DOI] [PubMed] [Google Scholar]
- 44). Tamura T, Horiuchi H, Imai M, Tada T, Shiomi H, Kuroda M, Nishimura S, Takahashi Y, Yoshikawa Y, Tsujimura A, Amano M, Hayama Y, Imamura S, Onishi N, Tamaki Y, Enomoto S, Miyake M, Kondo H, Kaitani K, Izumi C, Kimura T, Nakagawa Y: Unexpectedly High Prevalence of Acquired von Willebrand Syndrome in Patients with Severe Aortic Stenosis as Evaluated with a Novel Large Multimer Index. J Atheroscler Thromb, 2015; 22: 1115-1123 [DOI] [PubMed] [Google Scholar]
- 45). Heilmann C, Geisen U, Beyersdorf F, Nakamura L, Benk C, Berchtold-Herz M, Trummer G, Schlensak C, Zieger B: Acquired von Willebrand syndrome in patients with ventricular assist device or total artificial heart. Thromb Haemost, 2010; 103: 962-967 [DOI] [PubMed] [Google Scholar]
- 46). Jenkins CS, Clemetson KJ, Luscher EF: Studies on the mechanism of ristocetin-induced platelet agglutination: binding of ristocetin to platelets. J Lab Clin Med, 1979; 93: 220-231 [PubMed] [Google Scholar]
- 47). Trossaert M, Ternisien C, Lefrancois A, Llopis L, Goudemand J, Sigaud M, Fouassier M, Caron C: Evaluation of an automated von Willebrand factor activity assay in von Willebrand disease. Clinical and applied thrombosis/hemostasis: Clin Appl Thromb Hemost, 2011; 17: E25-29 [DOI] [PubMed] [Google Scholar]
- 48). Geisen U, Zieger B, Nakamura L, Weis A, Heinz J, Michiels JJ, Heilmann C: Comparison of Von Willebrand factor (VWF) activity VWF:Ac with VWF ristocetin cofactor activity VWF:RCo. Thromb Res, 2014; 134: 246-250 [DOI] [PubMed] [Google Scholar]
- 49). James PD, Paterson AD, Notley C, Cameron C, Hegadorn C, Tinlin S, Brown C, O'Brien L, Leggo J, Lillicrap D, Association Of Hemophilia Clinic Directors Of C : Genetic linkage and association analysis in type 1 von Willebrand disease: results from the Canadian type 1 VWD study. J Thromb Haemost, 2006; 4: 783-792 [DOI] [PubMed] [Google Scholar]
- 50). Hillery CA, Mancuso DJ, Evan Sadler J, Ponder JW, Jozwiak MA, Christopherson PA, Cox Gill J, Paul Scott J, Montgomery RR: Type 2M von Willebrand disease: F606I and I662F mutations in the glycoprotein Ib binding domain selectively impair ristocetin- but not botrocetin-mediated binding of von Willebrand factor to platelets. Blood, 1998; 91: 1572-1581 [PubMed] [Google Scholar]
- 51). Nishimura RA, Otto CM, Bonow RO, Carabello BA, Erwin JP, 3rd, Guyton RA, O'Gara PT, Ruiz CE, Skubas NJ, Sorajja P, Sundt TM, 3rd, Thomas JD, American College of Cardiology/American Heart Association Task Force on Practice G : 2014 AHA/ACC guideline for the management of patients with valvular heart disease: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol, 2014; 63: e57-185 [DOI] [PubMed] [Google Scholar]
- 52). Baumgartner H, Falk V, Bax JJ, De Bonis M, Hamm C, Holm PJ, Iung B, Lancellotti P, Lansac E, Munoz DR, Rosenhek R, Sjogren J, Mas PT, Vahanian A, Walther T, Wendler O, Windecker S, Zamorano JL: 2017 ESC/EACTS Guidelines for the Management of Valvular Heart Disease. Rev Esp Cardiol (English ed), 2018; 71: 110. [DOI] [PubMed] [Google Scholar]
- 53). Bonow RO, Brown AS, Gillam LD, Kapadia SR, Kavinsky CJ, Lindman BR, Mack MJ, Thourani VH, Dehmer GJ, Bonow RO, Lindman BR, Beaver TM, Bradley SM, Carabello BA, Desai MY, George I, Green P, Holmes DR, Jr., Johnston D, Leipsic J, Mick SL, Passeri JJ, Piana RN, Reichek N, Ruiz CE, Taub CC, Thomas JD, Turi ZG, Doherty JU, Dehmer GJ, Bailey SR, Bhave NM, Brown AS, Daugherty SL, Dean LS, Desai MY, Duvernoy CS, Gillam LD, Hendel RC, Kramer CM, Lindsay BD, Manning WJ, Mehrotra P, Patel MR, Sachdeva R, Wann LS, Winchester DE, Allen JM, ACC/AATS/AHA/ASE/EACTS/HVS/SCA/SCAI/SCCT/SCMR/STS 2017 Appropriate Use Criteria for the Treatment of Patients With Severe Aortic Stenosis : A Report of the American College of Cardiology Appropriate Use Criteria Task Force, American Association for Thoracic Surgery, American Heart Association, American Society of Echocardiography, European Association for Cardio-Thoracic Surgery, Heart Valve Society, Society of Cardiovascular Anesthesiologists, Society for Cardiovascular Angiography and Interventions, Society of Cardiovascular Computed Tomography, Society for Cardiovascular Magnetic Resonance, and Society of Thoracic Surgeons. J Am Soc Echocardiogr, 2018; 31: 117-147 [DOI] [PubMed] [Google Scholar]
- 54). Heyde EC: Gastrointestinal bleeding in aortic stenosis. N Engl J Med, 1958; 259: 196 [Google Scholar]
- 55). Warkentin TE, Moore JC, Morgan DG: Aortic stenosis and bleeding gastrointestinal angiodysplasia: is acquired von Willebrand's disease the link? Lancet, 1992; 340: 35-37 [DOI] [PubMed] [Google Scholar]
- 56). Shoenfeld Y, Eldar M, Bedazovsky B, Levy MJ, Pinkhas J: Aortic stenosis associated with gastrointestinal bleeding. A survey of 612 patients. Am Heart J, 1980; 100: 179-182 [DOI] [PubMed] [Google Scholar]
- 57). Yoshida K, Tobe S, Kawata M, Yamaguchi M: Acquired and reversible von Willebrand disease with high shear stress aortic valve stenosis. Ann Thorac Surg, 2006; 81: 490-494 [DOI] [PubMed] [Google Scholar]
- 58). Steger AC, Galland RB, Hemingway A, Wood CB, Spencer J: Gastrointestinal haemorrhage from a second source in patients with colonic angiodysplasia. Br J Surg, 1987; 74: 726-727 [DOI] [PubMed] [Google Scholar]
- 59). Perkins HA: Correction of the hemostatic defects in Von Willebrand's disease. Blood, 1967; 30: 375-380 [PubMed] [Google Scholar]
- 60). Spangenberg T, Budde U, Schewel D, Frerker C, Thielsen T, Kuck KH, Schafer U: Treatment of acquired von Willebrand syndrome in aortic stenosis with transcatheter aortic valve replacement. JACC Cardiovasc Interv, 2015; 8: 692-700 [DOI] [PubMed] [Google Scholar]
- 61). Marggraf O, Schneppenheim S, Daubmann A, Budde U, Seiffert M, Reichenspurner H, Treede H, Blankenberg S, Diemert P: Correction of acquired von Willebrand syndrome by transcatheter aortic valve implantation. J Invasive Cardiol, 2014; 26:654-658 [PubMed] [Google Scholar]
- 62). Caspar T, Jesel L, Desprez D, Grunebaum L, Samet H, Trinh A, Petit-Eisenmann H, Kindo M, Ohlmann P, Morel O: Effects of transcutaneous aortic valve implantation on aortic valve disease-related hemostatic disorders involving von Willebrand factor. Can J Cardiol, 2015; 31: 738-743 [DOI] [PubMed] [Google Scholar]
- 63). Yamashita K, Yagi H, Hayakawa M, Abe T, Hayata Y, Yamaguchi N, Sugimoto M, Fujimura Y, Matsumoto M, Taniguchi S: Rapid Restoration of Thrombus Formation and High-Molecular-Weight von Willebrand Factor Multimers in Patients with Severe Aortic Stenosis After Valve Replacement. J Atheroscler Thromb, 2016; 23: 1150-1158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64). Kokame K: Subsequent Response of VWF and ADAMTS13 to Aortic Valve Replacement. J Atheroscler Thromb, 2016; 23: 1141-1143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65). Blackshear JL: Gastrointestinal Bleeding in Native and Prosthetic Valve Disease. Curr Treat Options Cardiovasc Med, 2018; 20: 6. [DOI] [PubMed] [Google Scholar]
- 66). Uchida T, Hamasaki A, Ohba E, Yamashita A, Hayashi J, Sadahiro M: Life-threatening subdural hematoma after aortic valve replacement in a patient with Heyde syndrome: a case report. J Cardiothorac Surg, 2017; 12: 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67). Panza JA, Petrone RK, Fananapazir L, Maron BJ: Utility of continuous wave Doppler echocardiography in the noninvasive assessment of left ventricular outflow tract pressure gradient in patients with hypertrophic cardiomyopathy. J Am Coll Cardiol, 1992; 19: 91-99 [DOI] [PubMed] [Google Scholar]
- 68). Pruthi RK: Hypertrophic obstructive cardiomyopathy, acquired von Willebrand syndrome, and gastrointestinal bleeding. Mayo Clin Proc, 2011; 86: 181-182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69). Blackshear JL, Schaff HV, Ommen SR, Chen D, Nichols WL: Hypertrophic obstructive cardiomyopathy, bleeding history, and acquired von Willebrand syndrome: response to septal myectomy. Mayo Clin Proc, 2011; 86: 219-224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70). Wake M, Takahashi N, Yoshitomi H, Tanabe K: A case of hypertrophic obstructive cardiomyopathy and acquired von Willebrand syndrome: response to medical therapy. J Echocardiogr, 2014; 12: 112-114 [DOI] [PubMed] [Google Scholar]
- 71). Onimoe G, Grooms L, Perdue K, Ruymann F: Acquired von Willebrand Syndrome in congenital heart disease: does it promote an increased bleeding risk? Br J Haematol, 2011; 155: 622-624 [DOI] [PubMed] [Google Scholar]
- 72). Waldow HC, Westhoff-Bleck M, Widera C, Templin C, von Depka M: Acquired von Willebrand syndrome in adult patients with congenital heart disease. Int J Cardiol, 2014; 176: 739-745 [DOI] [PubMed] [Google Scholar]
- 73). Loeffelbein F, Funk D, Nakamura L, Zieger B, Grohmann J, Siepe M, Kroll J, Stiller B: Shear-stress induced acquired von Willebrand syndrome in children with congenital heart disease. Interact Cardiovasc Thorac Surg, 2014; 19: 926-932 [DOI] [PubMed] [Google Scholar]
- 74). Geggel RL, Carvalho AC, Hoyer LW, Reid LM: von Willebrand factor abnormalities in primary pulmonary hypertension. Am Rev Respir Dis, 1987; 135: 294-299 [DOI] [PubMed] [Google Scholar]
- 75). Lopes AA, Maeda NY, Aiello VD, Ebaid M, Bydlowski SP: Abnormal multimeric and oligomeric composition is associated with enhanced endothelial expression of von Willebrand factor in pulmonary hypertension. Chest, 1993; 104: 1455-1460 [DOI] [PubMed] [Google Scholar]
- 76). Veyradier A, Nishikubo T, Humbert M, Wolf M, Sitbon O, Simonneau G, Girma JP, Meyer D: Improvement of von Willebrand factor proteolysis after prostacyclin infusion in severe pulmonary arterial hypertension. Circulation, 2000; 102: 2460-2462 [DOI] [PubMed] [Google Scholar]
- 77). Robinson JC, Pugliese SC, Fox DL, Badesch DB: Anticoagulation in Pulmonary Arterial Hypertension. Curr Hypertens Rep, 2016; 18: 47. [DOI] [PubMed] [Google Scholar]
- 78). Yuceoglu YZ, Dresdale DT, Valensi QJ, Narvas RM, Gottlieb NT: Primary pulmonary hypertension with hoarseness and massive (fatal) hemoptysis. Review of the literature and report of a case. Vascular Diseases, 1967; 4: 290-304 [PubMed] [Google Scholar]
- 79). Koyama N, Matsumoto M, Tamaki S, Yoshikawa M, Fujimura Y, Kimura H: Reduced larger von Willebrand factor multimers at dawn in OSA plasmas reflect severity of apnoeic episodes. Eur Respir J, 2012; 40: 657-664 [DOI] [PubMed] [Google Scholar]
- 80). Blackshear JL, Wysokinska EM, Safford RE, Thomas CS, Shapiro BP, Ung S, Stark ME, Parikh P, Johns GS, Chen D: Shear stress-associated acquired von Willebrand syndrome in patients with mitral regurgitation. J Thromb Haemost, 2014; 12: 1966-1974 [DOI] [PubMed] [Google Scholar]
- 81). Wan SH, Liang JJ, Vaidya R, Blackshear JL, Chen D: Acquired Von Willebrand syndrome secondary to mitral and aortic regurgitation. Can J Cardiol, 2014; 30: 1108.e1109-1108.e1110 [DOI] [PubMed] [Google Scholar]
- 82). Kalbhenn J, Schmidt R, Nakamura L, Schelling J, Rosenfelder S, Zieger B: Early diagnosis of acquired von Willebrand Syndrome (AVWS) is elementary for clinical practice in patients treated with ECMO therapy. J Atheroscler Thromb, 2015; 22: 265-271 [DOI] [PubMed] [Google Scholar]
- 83). Goto S: Acquired von Willebrand Syndrome (AVWS) as an important diagnostic category of disease. J Atheroscler Thromb, 2015; 22: 231-232 [DOI] [PubMed] [Google Scholar]
- 84). Kalbhenn J, Schlagenhauf A, Rosenfelder S, Schmutz A, Zieger B: Acquired von Willebrand syndrome and impaired platelet function during venovenous extracorporeal membrane oxygenation: Rapid onset and fast recovery. J Heart Lung Transplant, 2018; 37: 985-991 [DOI] [PubMed] [Google Scholar]
- 85). Heilmann C, Geisen U, Beyersdorf F, Nakamura L, Benk C, Trummer G, Berchtold-Herz M, Schlensak C, Zieger B: Acquired von Willebrand syndrome in patients with extracorporeal life support (ECLS). Intensive Care Med, 2012; 38: 62-68 [DOI] [PubMed] [Google Scholar]
- 86). Tamura T, Horiuchi H, Obayashi Y, Fuki M, Imanaka M, Kuroda M, Nishimura S, Amano M, Sakamoto J, Tamaki Y, Enomoto S, Miyake M, Kondo H, Izumi C, Nakagawa Y: Acquired von Willebrand syndrome in patients treated with veno-arterial extracorporeal membrane oxygenation. Cardiovasc Interv Ther, 2019; in press [DOI] [PubMed] [Google Scholar]
- 87). Rose EA, Gelijns AC, Moskowitz AJ, Heitjan DF, Stevenson LW, Dembitsky W, Long JW, Ascheim DD, Tierney AR, Levitan RG, Watson JT, Meier P, Ronan NS, Shapiro PA, Lazar RM, Miller LW, Gupta L, Frazier OH, Desvigne-Nickens P, Oz MC, Poirier VL: Long-term use of a left ventricular assist device for endstage heart failure. N Engl J Med, 2001; 345: 1435-1443 [DOI] [PubMed] [Google Scholar]
- 88). Hata H, Fujita T, Shimahara Y, Sato S, Yanase M, Seguchi O, Sato T, Nakatani T, Kobayashi J: Early and mid-term outcomes of left ventricular assist device implantation and future prospects. Gen Thorac Cardiovasc Surg, 2015; 63: 557-564 [DOI] [PubMed] [Google Scholar]
- 89). Kirklin JK, Pagani FD, Kormos RL, Stevenson LW, Blume ED, Myers SL, Miller MA, Baldwin JT, Young JB, Naftel DC: Eighth annual INTERMACS report: Special focus on framing the impact of adverse events. J Heart Lung Transplant, 2017; 36: 1080-1086 [DOI] [PubMed] [Google Scholar]
- 90). J-MACS : Japanese registry for Mechanically Assisted Circulatory Support. (Japanese) http://www.pmda.go.jp/safety/surveillance-analysis/0009.html Accessed October 10, 2018. [Google Scholar]
- 91). Nakatani T, Sase K, Oshiyama H, Akiyama M, Horie M, Nawata K, Nishinaka T, Tanoue Y, Toda K, Tozawa M, Yamazaki S, Yanase M, Ohtsu H, Ishida M, Hiramatsu A, Ishii K, Kitamura S: Japanese registry for Mechanically Assisted Circulatory Support: First report. J Heart Lung Transplant, 2017; 36: 1087-1096 [DOI] [PubMed] [Google Scholar]
- 92). Draper KV, Huang RJ, Gerson LB: GI bleeding in patients with continuous-flow left ventricular assist devices: a systematic review and meta-analysis. Gastrointest Endosc, 2014; 80: 435-446.e431 [DOI] [PubMed] [Google Scholar]
- 93). Slaughter MS: Hematologic effects of continuous flow left ventricular assist devices. J Cardiovasc Transl Res, 2010; 3: 618-624 [DOI] [PubMed] [Google Scholar]
- 94). Demirozu ZT, Radovancevic R, Hochman LF, Gregoric ID, Letsou GV, Kar B, Bogaev RC, Frazier OH: Arteriovenous malformation and gastrointestinal bleeding in patients with the HeartMate II left ventricular assist device. J Heart Lung Transplant, 2011; 30: 849-853 [DOI] [PubMed] [Google Scholar]
- 95). Sakatsume K, Akiyama M, Saito K, Kawamoto S, Horiuchi H, Saiki Y: Intractable bleeding tendency due to acquired von Willebrand syndrome after Jarvik 2000 implant. J Artif Organs, 2016; 19: 289-292 [DOI] [PubMed] [Google Scholar]
- 96). Sakatsume K, Saito K, Akiyama M, Sasaki K, Kawatsu S, Takahashi G, Adachi O, Kawamoto S, Horiuchi H, Saiki Y: Association between the severity of acquired von Willebrand syndrome and gastrointestinal bleeding after continuous-flow left ventricular assist device implantation. Eur J Cardiothorac Surg, 2018; 54: 841-846 [DOI] [PubMed] [Google Scholar]
- 97). Meyer AL, Malehsa D, Budde U, Bara C, Haverich A, Strueber M: Acquired von Willebrand syndrome in patients with a centrifugal or axial continuous flow left ventricular assist device. JACC Heart Fail, 2014; 2: 141-145 [DOI] [PubMed] [Google Scholar]
- 98). Muslem R, Caliskan K, Leebeek FWG: Acquired coagulopathy in patients with left ventricular assist devices. J Thromb Haemost, 2018; 16: 429-440 [DOI] [PubMed] [Google Scholar]
- 99). Bourque K, Cotter C, Dague C, Harjes D, Dur O, Duhamel J, Spink K, Walsh K, Burke E: Design Rationale and Preclinical Evaluation of the HeartMate 3 Left Ventricular Assist System for Hemocompatibility. ASAIO J, 2016; 62: 375-383 [DOI] [PubMed] [Google Scholar]
- 100). Mehra MR, Goldstein DJ, Uriel N, Cleveland JC, Jr., Yuzefpolskaya M, Salerno C, Walsh MN, Milano CA, Patel CB, Ewald GA, Itoh A, Dean D, Krishnamoorthy A, Cotts WG, Tatooles AJ, Jorde UP, Bruckner BA, Estep JD, Jeevanandam V, Sayer G, Horstmanshof D, Long JW, Gulati S, Skipper ER, O'Connell JB, Heatley G, Sood P, Naka Y: Two-Year Outcomes with a Magnetically Levitated Cardiac Pump in Heart Failure. N Engl J Med, 2018; 378: 1386-1395 [DOI] [PubMed] [Google Scholar]
- 101). Ueno S, Nakase H, Kasahara K, Uza N, Kitamura H, Inoue S, Mikami S, Matsuura M, Chiba T: Clinical features of Japanese patients with colonic angiodysplasia. J Gastroenterol Hepatol, 2008; 23: e363-366 [DOI] [PubMed] [Google Scholar]
- 102). Sharma R, Gorbien MJ: Angiodysplasia and lower gastrointestinal tract bleeding in elderly patients. Arch Intern Med, 1995; 155: 807-812 [PubMed] [Google Scholar]
- 103). Nubret K, Mauriat P, Roubertie F, James C, Tafer N, Ouattara A: Acquired von Willebrand disease in a child with a ventricular assist device. J Thorac Cardiovasc Surg, 2013; 146: e30-32 [DOI] [PubMed] [Google Scholar]
- 104). Costello JP, Diab YA, Philippe-Auguste M, Jones MB, Shankar V, Friedman KD, Nath DS: Acquired von Willebrand syndrome in a child following Berlin Heart EXCOR Pediatric Ventricular Assist Device implantation: case report and concise literature review. World J Pediatr Congenit Heart Surg, 2014; 5: 592-598 [DOI] [PubMed] [Google Scholar]
- 105). Bauditz J, Lochs H: Angiogenesis and vascular malformations: antiangiogenic drugs for treatment of gastrointestinal bleeding. World J Gastroenterol, 2007; 13: 5979-5984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106). Carmeliet P: Mechanisms of angiogenesis and arteriogenesis. Nat Med, 2000; 6: 389-395 [DOI] [PubMed] [Google Scholar]
- 107). Suri C, Jones PF, Patan S, Bartunkova S, Maisonpierre PC, Davis S, Sato TN, Yancopoulos GD: Requisite role of angiopoietin-1, a ligand for the TIE2 receptor, during embryonic angiogenesis. Cell, 1996; 87: 1171-1180 [DOI] [PubMed] [Google Scholar]
- 108). Davis S, Aldrich TH, Jones PF, Acheson A, Compton DL, Jain V, Ryan TE, Bruno J, Radziejewski C, Maisonpierre PC, Yancopoulos GD: Isolation of angiopoietin-1, a ligand for the TIE2 receptor, by secretion-trap expression cloning. Cell, 1996; 87: 1161-1169 [DOI] [PubMed] [Google Scholar]
- 109). Maisonpierre PC, Suri C, Jones PF, Bartunkova S, Wiegand SJ, Radziejewski C, Compton D, McClain J, Aldrich TH, Papadopoulos N, Daly TJ, Davis S, Sato TN, Yancopoulos GD: Angiopoietin-2, a natural antagonist for Tie2 that disrupts in vivo angiogenesis. Science, 1997; 277: 55-60 [DOI] [PubMed] [Google Scholar]
- 110). Zhang ZG, Zhang L, Jiang Q, Chopp M: Bone marrow-derived endothelial progenitor cells participate in cerebral neovascularization after focal cerebral ischemia in the adult mouse. Circ Res, 2002; 90: 284-288 [DOI] [PubMed] [Google Scholar]
- 111). Brandao D, Costa C, Canedo A, Vaz G, Pignatelli D: Endogenous vascular endothelial growth factor and angiopoietin-2 expression in critical limb ischemia. International angiology: Int Angiol, 2011; 30: 25-34 [PubMed] [Google Scholar]
- 112). Junquera F, Saperas E, de Torres I, Vidal MT, Malagelada JR: Increased expression of angiogenic factors in human colonic angiodysplasia. Am J Gastroenterol, 1999; 94: 1070-1076 [DOI] [PubMed] [Google Scholar]
- 113). Holleran G, Hall B, O'Regan M, Smith S, McNamara D: Expression of Angiogenic Factors in Patients With Sporadic Small Bowel Angiodysplasia. J Clin Gastroenterol, 2015; 49: 831-836 [DOI] [PubMed] [Google Scholar]
- 114). Kang J, Hennessy-Strahs S, Kwiatkowski P, Bermudez CA, Acker MA, Atluri P, McConnell PI, Bartoli CR: Continuous-Flow LVAD Support Causes a Distinct Form of Intestinal Angiodysplasia. Circ Res, 2017; 121: 963-969 [DOI] [PubMed] [Google Scholar]
- 115). Starke RD, Ferraro F, Paschalaki KE, Dryden NH, McKinnon TA, Sutton RE, Payne EM, Haskard DO, Hughes AD, Cutler DF, Laffan MA, Randi AM: Endothelial von Willebrand factor regulates angiogenesis. Blood, 2011; 117: 1071-1080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116). Fressinaud E, Meyer D: International survey of patients with von Willebrand disease and angiodysplasia. Thromb Haemost, 1993; 70: 546. [PubMed] [Google Scholar]
- 117). Satoh Y, Kita H, Kihira K, Mutoh H, Osawa H, Satoh K, Ido K, Sakata Y, Sugano K: Gastrointestinal angiodysplasia in a patient with type 2 von Willebrand's disease and analysis of exon 28 of the von Willebrand factor gene. Am J Gastroenterol, 2004; 99: 2495-2498 [DOI] [PubMed] [Google Scholar]
- 118). Makris M: Gastrointestinal bleeding in von Willebrand disease. Thromb Res, 2006; 118 Suppl 1: S13-17 [DOI] [PubMed] [Google Scholar]