

Abstract
Aim: Lomitapide is an approved lipid-lowering agent indicated as adjunct to low-fat diet and standard lipid-lowering therapies (LLTs) including lipoprotein apheresis for the treatment of homozygous familial hypercholesterolemia (HoFH). Clinical data from Phase 3 studies have demonstrated the prolonged lipid-lowering capacity of lomitapide in patients with HoFH. We assessed the long-term lipid-lowering capacity of daily oral lomitapide in a cohort of Japanese patients with HoFH enrolled in a Phase 3 extension study.
Methods: Five of 8 Japanese HoFH patients completing a 56-week Phase 3 dose-escalation and safety study of lomitapide continued their maximum tolerated dose (MTD) until study drug was approved or commercially available or until treatment was discontinued. Lipid parameters were measured at Day 1 and at 12-week intervals through study end. Safety and tolerability were assessed.
Results: Daily lomitapide treatment with permitted LLTs maintained approximately 50% mean reductions in plasma low-density lipoprotein cholesterol (LDL-C) levels from baseline for > 60 weeks. Reductions in LDL-C levels varied across patients and were not associated with the HoFH genotype. Four patients achieved > 25% reductions and 1 patient achieved > 50% reduction in LDL-C; 2 patients achieved reduction in LDL-C to < 100 mg/dL. Lomitapide significantly reduced total cholesterol (−26.5%), triglycerides (−54.8%), and non-highdensity lipoprotein cholesterol (non-HDL-C) (−37.4%). All 5 patients continued their individual MTD of lomitapide throughout the extension study with acceptable safety and tolerability, and no deaths were reported.
Conclusions: Results from this extension study support the long-term safety and efficacy of lomitapide in significantly reducing plasma levels of atherosclerotic lipids in patients with HoFH.
Keywords: Homozygous familial hypercholesterolemia, Lipid-lowering therapy, Lomitapide, LDL-C
Introduction
Homozygous familial hypercholesterolemia (HoFH) is a rare and serious inherited disease originally characterized by levels of plasma cholesterol > 500 mg/dL (> 13 mmol/L), increased levels of low-density lipoprotein cholesterol (LDL-C), and physical manifestations of cholesterol deposition in the skin (cutaneous xanthomas), eyes (arcus corneae), and/or tendons (tendon xanthomas)1–4). Consequently, patients with HoFH have poor prognosis without intensive lipid-lowering treatment because of greatly increased risk of atherosclerotic cardiovascular disease such as myocardial infarction, supravalvular aortic stenosis1, 2, 5).
The binding, incorporation and intracellular transport of low-density lipoproteins (LDL) via the cell surface LDL-R on hepatocytes and intestinal epithelial cells are pivotal elements of circulatory and hepatic lipoprotein homeostasis6, 7). The absence of functional LDL-R derived from true homozygous or compound heterozygous mutations underlies the inability of patients with HoFH to uptake and metabolize LDL particles1). Consequently, patients with HoFH have limited interventional options beyond nutritional and lifestyle changes (e.g., reducing lipid intake, smoking cessation, limiting alcohol intake) and periodic lipoprotein apheresis to control elevated levels of circulating LDL-C2, 5). The same mutations in LDL-R also render patients with HoFH either refractory or poorly responsive to the therapeutic effects of standard LLTs including statins and ezetimibe because pharmacological effects of these agents depend on LDL-R activities5).
Lomitapide (Juxtapid [AEGR-733], Aegerion Pharmaceuticals Inc., Cambridge, MA; Lojuxta, Aegerion Pharmaceuticals Ltd, Uxbridge, UK, initial approval 2012, is an inhibitor of the microsomal triglyceride transport protein (MTP) involved in intracellular assembly and secretion of apoB-containing very low-density lipoprotein6). Results from 2 Phase 3 clinical studies and one extension study have demonstrated the capacity of lomitapide to significantly lower LDL-C by approximately 45% to 50% from baseline over 26 weeks of therapy when administered with standard LLTs and to maintain 38% to 45% reductions in LDL-C up to 78 to 126 weeks of therapy7–9).
This report describes the therapeutic effects of extended lomitapide treatment in combination with standard LLTs on plasma LDL-C levels and other circulating lipids in a subset of patients enrolled in a recently completed Phase 3 dose-escalation and safety study8) in Japanese patients with HoFH.
Materials and Methods
Study Design
The AEGR-733-301 extension study was conducted to confirm the safety of extended lomitapide therapy in Japanese patients with HoFH until commercial availability of lomitapide in Japan. Five of 8 patients who completed the 56-week Phase 3 study entered the single-arm, open-label, multicenter (n = 5 sites) extension study and continued their individual MTD of lomitapide established in the Phase 3 study throughout the extension study, with dosing of 10 mg/day (1 patient), 20 mg/day (3 patients), and 40 mg/day (1 patient). Lomitapide was administered as specified for the safety phase of the Phase 3 study, at the beginning of the extension study (Visit 1; Week 56 of Phase 3 study), and during visits at Weeks 12, 24, 36, and 48, and every 12 weeks thereafter until lomitapide was commercially available or until discontinuation of lomitapide treatment (Fig. 1). During each visit, patients underwent a physical examination, concomitant medication therapy, laboratory tests (hematology and serum chemistry), and assessments for adverse events and vital signs (body weight, heart rate, blood pressure).
Fig. 1:
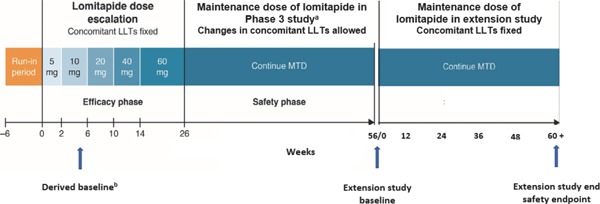
Study design
LLTs, lipid-lowering therapies; MTD, maximum tolerated dose
aLomitapide dose reduction permitted where dose modification rules applied. Subsequent re-escalation allowed if ≤ MTD (determined during efficacy phase)
bDerived baseline was calculated from average value of Visit 2 and Visit Visit 3 at Phase 3 study
All patients provided informed written consent to continue lomitapide treatment in the extension study. The study protocol and patient informed consent were reviewed and approved by an institutional review board before study initiation at 5 clinical sites in Japan from 1 July 2015 to 24 March 2017.
Patient Disposition
Four of the 5 patients entering the extension study continued the MTD dose of lomitapide through the time after drug approval and commercial availability until study completion at the last visit. Two of the 5 patients had true HoFH, 2 had compound heterozygous HoFH, and 1 patient had double mutations in the LDL-R and PCSK9 genes. This latter patient discontinued lomitapide (10 mg/day) after approximately 349 days of treatment and was permanently discontinued prior to the last study visit because of a change to evolocumab therapy. Prohibited concomitant medications and therapies (e.g., hepatotoxic medications, moderate/strong chromosome P450 3A4 (CYP3A4) inhibitors, or > 1 alcoholic drinks/day) were the same as those specified for the safety phase of the Phase 3 study8).
All 5 patients took at least 1 dose of study drug and completed at least 1 visit assessment. Therefore, 5 patients were included in the safety population and the full analysis set (FAS). Patients could not have a coexisting malignant tumor or use concomitant medications prohibited in the protocol or deemed inappropriate for the study by the investigator.
Patients continued their concomitant LLTs during the extension study, specifically: patient 1 (rosuvastatin 5 mg, ezetimibe 10 mg, colestilan 3.62 g), patient 2 (rosuvastatin 20 mg, ezetimibe 10 mg, colestilan 1 g), patient 3 (ezetimibe 10 mg, ethyl eicosapentaenoic acid 900mg), patient 4 (atorvastatin 20 mg, ezetimibe 10 mg) and patient 5 (rosuvastatin 7.5 mg, ezetimibe 10 mg). All patents with the exception of 1 received LDL apheresis (2 patients with weekly and 2 patients with biweekly frequency).
All patients were counseled by a registered dietitian in each center on adhering to a low-fat diet, and a low-fat diet (< 20% of food energy as fat) was prescribed with daily intake of supplements; vitamin E (400 IU) and fatty acids (200 mg linoleic acid), 210 mg α–linolenic acid, 110 mg eicosapentaenoic acid, and 80 mg docosahexaenoic acid. Dietary counseling sessions were held once every 5 weeks.
Study Endpoints
The primary efficacy endpoint of the study was change in LDL-C during the extension study period from a derived baseline at the end of the 26-week efficacy period of the Phase 3 study and from extension study start (i.e., end of the 56-week Phase 3 study). Secondary efficacy endpoints were changes in other plasma lipids including TC, apoB, apoA1, triglycerides, non-HDL-C, HDL-C, and lipoprotein(a) [Lp(a)]. The number of patients with reductions of > 15%, > 25%, and > 50% in LDL-C from derived baseline and number achieving target LDL-C levels of < 100 mg/dL and < 70 mg/dL were also measured. The safety endpoint was occurrence of adverse events and side effects from the start of lomitapide treatment in the Phase 3 study through end of the extension study and from extension study start.
Statistical Analyses
Descriptive statistics (number of samples, mean, median, minimum, and maximum) of measured parameters were calculated. For the efficacy endpoint, a statistical analysis was performed on changes in measurements. Adverse events and side effects were classified by system organ class and preferred term using the Medical Dictionary for Regulatory Activities (MedDRA; ver.17.1). The incidence of events, including incidence by severity, were recorded.
Results
Patient Demographics and Study Drug Exposure
The patient demographic characteristics, including frequency of apheresis at Day 1 for the 5 patients in the safety population/FAS, are summarized in Table 1. The mean and median cumulative dose, average daily dosage, and duration of exposure to lomitapide during the extension study are listed in Table 2. The mean lomitapide exposure was 441.8 days and the treatment duration ranged from 349 to 487 days. Total median cumulative dose of lomitapide generally reflected the product of daily dose and duration of exposure, except the single patient (10 mg dose) who discontinued due to a change in treatment at study day 421 with 349 days of drug exposure. Total median duration of exposure in patients receiving 20 mg or 40 mg lomitapide daily exceeded 60 weeks (420 days) of therapy from approximately 26 to 46 days.
Table 1. Baseline patient demographics and characteristics by the MTD of lomitapide at Day 1 and in the overall FAS populationa.
| Characteristic | Lomitapide MTD at Visit 1 |
FAS (N = 5) |
||
|---|---|---|---|---|
| 10 mg (n = 1) | 20 mg (n = 3) | 40 mg (n = 1) | ||
| Age (years) | ||||
| Mean (SD) | 47.0 | 50.7 (14.22) | 53.0 | 50.4 (10.29) |
| Median | 47.0 | 44.0 | 53.0 | 47.0 |
| (Min., Max.) | (41, 67) | (41, 67) | ||
| Gender, n (%) | ||||
| Male | 1 (100.0) | 2 (66.7) | 0 | 3 (60.0) |
| Female | 0 | 1 (33.3) | 1 (100.0) | 2 (40.0) |
| Ethnicity, n (%) | ||||
| Japanese | 1 (100.0) | 3 (100.0) | 1 (100.0) | 5 (100.0) |
| Weight (Day 1) (kg) | ||||
| Mean (SD) | 78.50 | 55.10 (10.213) | 41.20 | 57.00 (15.259) |
| Median | 78.50 | 57.20 | 41.20 | 57.20 |
| (Min., Max.) | (44.0, 64.1) | (41.2, 78.5) | ||
| BMI (Day 1) (kg/m2) | ||||
| Mean (SD) | 29.10 | 20.10 (2.615) | 17.10 | 21.30 (4.911) |
| Median | 29.10 | 18.90 | 17.10 | 18.90 |
| (Min., Max.) | (29.1, 29.1) | (18.3, 23.1) | (17.1, 17.1) | (17.1, 29.1) |
| Use of Apheresis at Day 1, n | 1 | 3 | 1 | 5 |
| Yes | 0 | 3 (100.0) | 1 (100.0) | 4 (80.0) |
| No | 1 (100.0) | 0 | 0 | 1 (20.0) |
| Frequency of Apheresis at Day 1, n | 0 | 3 | 1 | 4 |
| Every week | 0 | 2 (66.7) | 0 | 2 (50.0) |
| Every 2 weeks | 0 | 1 (33.3) | 1 (100.0) | 2 (50.0) |
BMI, body mass index; FAS, full analysis set; MTD, maximum tolerated dose; SD, standard deviation
Full analysis set and safety population
Table 2. Cumulative lomitapide exposure and duration of treatment.
| Variable | Lomitapide MTD at Visit 1 |
FAS (N = 5) |
||
|---|---|---|---|---|
| 10 mg (n = 1) | 20 mg (n = 3) | 40 mg (n = 1) | ||
| Cumulative Dose (mg)a | ||||
| Mean (SD) | 3490.0 | 9426.7 (331.26) | 17840.0 | 9922.0 (5123.98) |
| Median | 3490.0 | 9460.0 | 17840.0 | 9460.0 |
| (Min., Max.) | (9080, 9740) | (3490, 17840) | ||
| Study Drug Exposure (days)b | ||||
| Mean (SD) | 349.0 | 471.3 (16.56) | 446.0 | 441.8 (54.30) |
| Median | 349.0 | 473.0 | 446.0 | 454.0 |
| (Min., Max.) | (454, 487) | (349, 487) | ||
| Average Daily Dose (mg/day)c | ||||
| Mean (SD) | 10.00 | 20.00 | 40.00 | 22.00 (10.95) |
| Median | 10.00 | 20.00 | 40.00 | 20.00 |
| (Min., Max.) | (10.00, 40.00) | |||
| Treatment Duration (days)d | ||||
| Mean (SD) | 349.0 | 473.0 (15.72) | 446.0 | 442.8 (54.86) |
| Median | 349.0 | 476.0 | 446.0 | 456.0 |
| (Min., Max.) | (456, 487) | (349, 487) | ||
FAS, full analysis set; MTD, maximum tolerated dose; SD, standard deviation
Cumulative dose is the total dose taken during the study phase
Study medication exposure is calculated as the actual number of days on study medication (Last dose date − First dose date + 1 − Days study medication is interrupted)
Average daily dose is the cumulative dose/study medication exposure
Treatment duration is calculated as the total number of days on study medication (Last dose date − First dose date + 1)
Efficacy
Changes in LDL-C levels from the end of the Phase 3 MTD study (Visit 1) and the derived Phase 3 study baseline (primary efficacy endpoint) to the 60-week visit and 60 weeks last observation carried forward (LOCF) in the FAS population of 5 patients are illustrated graphically in Fig. 2.
Fig. 2.
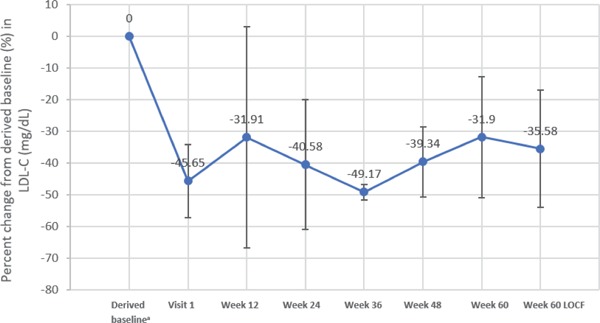
Percent change in LDL-C from end of MTD study and from derived baseline–FAS population
FAS, full analysis set; LDL-C, low-density lipoprotein cholesterol; LOCF, last observation carried forward; MTD, maximum tolerated dose. Values were obtained from all 5 patients for each measure except for week 60 where n = 4.
aDerived baseline was calculated from average value of Visit 2 and Visit 3 at Phase 3 study.
Across all 5 patients, the mean LDL-C at Visit 1 had already decreased to −88.3 mg/dL from the derived Phase 3 study baseline, representing a mean percent change from derived baseline to Visit 1 of −45.65%. The mean percent change in LDL-C from derived baseline to Week 60 LOCF was −35.58% (P = 0.0135 vs derived baseline), with mean values fluctuating between −49.17% and −31.9%.
Individual patient percent changes in LDL-C levels from the Phase 3 derived baseline to Week 60 LOCF by MTD at Visit 1 of the extension study are illustrated graphically in Fig. 3. These data illustrate variability in percent decreases in LDL-C observed across the study patients that did not appear to correlate with lomitapide dosage, although a > 30% reduction from derived baseline of Phase 3 study at final assessment was observed for all patients except the minimal decrease (−5%) observed in 1 patient receiving a 20 mg/day lomitapide dose.
Fig. 3.
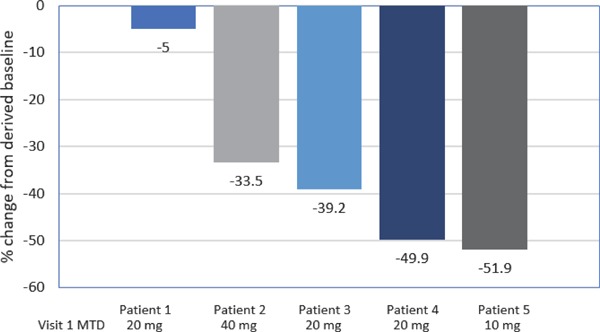
Waterfall plot of percent change from derived baseline of Phase 3 study in LDL-C at Week 60 LOCF by MTD at baseline of Phase 3 extension study (Visit 1)
LDL-C, low-density lipoprotein cholesterol; LOCF, last observation carried forward; MTD, maximum tolerated dose
Results of an LDL-C responder analysis to assess the number and percent of patients who experienced a > 15%, > 25%, or > 50% decrease in LDL-C levels and those with decreases in LDL-C to < 100 mg/dL and < 70 mg/dL from the Phase 3 study–derived baseline through final visit assessment are listed in Table 3. Four of 5 patients experienced a decrease in LDL-C of > 25% through Week 60 and 1 patient exhibited a > 50% decrease in LDL-C. Two patients experienced a decrease in LDL-C to < 100 mg/dL during the study, while no patients experienced decrease in LDL-C to < 70 mg/dL.
Table 3. Patient decreases in LDL-C from Phase 3 study–derived baseline to final visit assessment.
| Responder Parameter, N (%) | Lomitapide MTD at Visit 1 |
FAS (N = 5) |
||
|---|---|---|---|---|
| 10 mg (n = 1)a | 20 mg (n = 3) | 40 mg (n = 1) | ||
| > 15% Decrease in LDL-C | 1 | 2 | 1 | 4 |
| > 25% Decrease in LDL-C | 1 | 2 | 1 | 4 |
| > 50% Decrease in LDL-C | 1 | 0 | 0 | 1 |
| LDL-C < 100 mg/dL | 1 | 1 | 0 | 2 |
| LDL-C < 70 mg/dL | 0 | 0 | 0 | 0 |
FAS, full analysis set; LDL-C, low-density lipoprotein cholesterol; MTD, maximum tolerated dose
Final assessment was made at the termination of study at Day 349 for this patient
The mean and median percent changes in the study secondary lipid parameters from the Phase 3 study–derived baseline to Week 60 LOCF of the extension study are listed in Table 4. Treatment with the MTD of lomitapide significantly reduced the mean levels of TC, triglycerides, and non-HDL-C by 26.5%, 54.8%, and 37.5% from derived baseline, respectively, and near significantly in the mean Lp(a) level (p = 0.058). Conversely, lomitapide treatment significantly increased the mean level of HDL-C by 17.0%, with no changes observed in levels of either apoB or apoA1. Levels of these secondary lipids did not significantly change from the extension study baseline to Week 60 LOCF (data not shown).
Table 4. Mean and median percentage changes in secondary endpoint lipid parameters from Phase 3 study–derived baseline to Week 60 LOCF of extension study.
| Variable Statistic | Week 60 LOCF |
P-Valuea | ||
|---|---|---|---|---|
| Observed Value | Observed Change | Percent Change | ||
| Total Cholesterol (mg/dL), N | 5 | 5 | 5 | |
| Mean (SD) | 203.6 (58.46) | −71.2 (36.28) | −26.53 (13.967) | |
| Median | 178.0 | −77.5 | −33.18 | |
| (Min., Max.) | (142, 269) | (−102, −11) | (−38.1, −3.8) | |
| [95% CI] | [131.0, 276.2] | [−116.2, −26.2] | [−43.88, −9.19] | 0.012 |
| apoB (mg/dL) N | 2 | 2 | 2 | |
| Mean (SD) | 98.5 (38.89) | −39.5 (28.28) | −29.50 (22.763) | |
| Median | 98.5 | −39.5 | −29.50 | |
| (Min., Max.) | (71, 126) | (−60, −20) | (−45.6, −13.4) | |
| [95% CI] | [−250.9, 447.9] | [−293.6, 214.6] | [−234.02, 175.02] | NS |
| Triglycerides (mg/dL), N | 5 | 5 | 5 | |
| Mean (SD) | 41.2 (13.92) | −50.1 (18.48) | −54.75 (12.248) | |
| Median | 41.0 | −49.5 | −50.24 | |
| (Min., Max.) | (19, 54) | (−79, −28) | (−68.9, −40.6) | |
| [95% CI] | [23.9, 58.5] | [−73.1, −27.1] | [−69.96, −39.54] | 0.004 |
| Non-HDL-C (mg/dL), N | 5 | 5 | 5 | |
| Mean (SD) | 140.4 (53.98) | −80.6 (34.92) | −37.45 (17.114) | |
| Median | 112.0 | −85.0 | −40.83 | |
| (Min., Max.) | (92, 201) | (−112, −27) | (−54.9, −11.8) | |
| [95% CI] | [73.4, 207.4] | [−124.0, −37.2] | [−58.70, −16.20] | 0.007 |
| Lp(a) (nmol/L), N | 2 | 2 | 2 | |
| Mean (SD) | 25.2 (0.21) | −36.9 (4.74) | −59.32 (2.906) | |
| Median | 25.2 | −36.9 | −59.32 | |
| (Min., Max.) | (25, 25) | (−40, −34) | (−61.4, −57.3) | |
| [95% CI] | [23.2, 27.1] | [−79.4, 5.7] | [−85.42, −33.21] | NS |
| HDL-C (mg/dL), N | 5 | 5 | 5 | |
| Mean (SD) | 63.2 (12.30) | 9.4 (7.06) | 17.04 (14.036) | |
| Median | 66.0 | 10.0 | 15.63 | |
| (Min., Max.) | (42, 74) | (−2, 17) | (−3.4, 32.0) | |
| [95% CI] | [47.9, 78.5] | [0.6, 18.2] | [−0.39, 34.47] | 0.041 |
| apoA1 (mg/dL), N | 2 | 2 | 2 | |
| Mean (SD) | 131.0 (26.87) | −1.5 (32.53) | −0.61 (24.523) | |
| Median | 131.0 | −1.5 | −0.61 | |
| (Min., Max.) | (112, 150) | (−25, 22) | (−17.9, 16.7) | |
| [95% CI] | [−110.4, 372.4] | [−293.7, 290.7] | [−220.94, 219.72] | NS |
apoA1, apolipoprotein A1; apoB, apolipoprotein B; CI, confidence interval; HDL-C, high-density lipoprotein cholesterol; LOCF, last observation carried forward; Lp(a), lipoprotein(a); non-HDL-C, non-high-density lipoprotein cholesterol; SD, standard deviation; NS, not significant
P-value from paired t-test
Safety
TEAEs
A summary of treatment-emergent adverse events (TEAEs) by lomitapide MTD at Day 1 during the extension study is provided in Table 5. All 5 patients experienced at least 1 TEAE during the extension study, and drug-related TEAEs were reported in 3 (60.0%) of the 5 patients. There were no severe TEAEs or deaths during the extension study. A serious TEAE (angina pectoris) was reported in 1 (20.0%) of the 5 patients (a 20 mg/day patient); this serious TEAE was not considered related to study drug. Discontinuation due to TEAEs or the dose of study drug interrupted or reduced due to a TEAE were not reported during the study.
Table 5. Summary of treatment-emergent adverse eventsa.
| Number of Patients with (%) | Lomitapide MTD at Day 1 |
FAS (N= 5) |
||
|---|---|---|---|---|
| 10 mg (n = 1) | 20 mg (n = 3) | 40 mg (n = 1) | ||
| Adverse events (AEs) | 1 (100.0) | 3 (100.0) | 1 (100.0) | 5 (100.0) |
| Drug-related AEsb | 1 (100.0) | 2 (66.7) | 0 | 3 (60.0) |
| Severe AEs | 0 | 0 | 0 | 0 |
| Serious AEs | 0 | 1 (33.3) | 0 | 1 (20.0) |
| AEs leading to study discontinuation | 0 | 0 | 0 | 0 |
| Dose held or reduced due to an AE | 0 | 0 | 0 | 0 |
| Deaths due to an AE | 0 | 0 | 0 | 0 |
FAS, full analysis set; MTD, maximum tolerated dose
Events with onset date that occurred during this study phase are included. The FAS column includes all adverse events reported, including events where a dose was not indicated or study drug was interrupted/stopped. A subject's AE was counted according to the MTD at Day 1. Only 1 occurrence of that event was counted within each dose. Subjects may have been counted more than once for AEs occurring during multiple dose values.
Related is defined as definite by the investigator on the CRF
The most common TEAEs by system organ class were gastrointestinal disorders, general disorders, and administration site condition. The most common TEAEs by preferred term were upper abdominal pain (2 patients, 40.0%) and chest pain (2 patients, 40.0%). All reported TEAEs were assessed by the investigators as mild to moderate in intensity. The moderate TEAEs were reported as iron-deficiency anemia and eczema in 1 of the 5 patients (20.0%), which were considered related to the study drug by the investigators. The single patient receiving 10 mg/day of lomitapide experienced an abnormal liver function test during the extension study.
Clinical Laboratory Tests
Four of the 5 patients exceeded the upper limit of normal range (ULN) at each site for aspartate aminotransferase (AST) and or alanine aminotransferase (ALT) at any of assessment visits, but none of these patients had AST or ALT levels ≥ 3 × ULN. One patient receiving the 10 mg/day dose of lomitapide had liver function tests (LFT) increased as a related TEAE throughout the study. None of the other patients reported TEAEs related to liver function tests. Alkaline phosphatase and total bilirubin were within normal range throughout the study for all patients. There were no clinically meaningful changes in vital signs or physical findings at each study visit.
Discussion
Patients with HoFH remain at very high risk for life-threatening cardiovascular events. Even in the setting of treatment with current standard of care LLTs, these patients do not exhibit reduction in their plasma LDL-C to levels at or near normal10). In addition, patients with HoFH generally respond poorly or do not respond to newer treatment agents that increase LDL-R activity, such as the PCSK9 inhibitors, because of the inherited lack of or greatly reduced level of LDL-R activity that affects individual treatment responses to these inhibitors11).
Lomitapide inhibits the activity of endoplasmic microsomal triglyceride transfer protein (MTP) that functions to load triglycerides onto apoB lipoprotein in intestinal epithelial cells and hepatocytes, thereby blocking the production and secretion of VLDL, the precursor of LDL, and VLDL-mediated secretion of triglycerides10–12). In this manner, lomitapide reduces plasma levels of LDL-C in patients with HoFH, independent of cell surface expression of LDL-R, and those with loss-of-function mutations in the MTTP gene encoding the MTP protein.
Data supporting the approval of lomitapide come from a single-arm, open-label, dose-escalation study in 29 patients with HoFH or compound heterozygous FH7). In that study, a median maximum tolerated dose (MTD) of 40 mg/day oral lomitapide (range, 5 to 60 mg/day) significantly reduced mean LDL-C levels by 50% from baseline at 26 weeks of therapy, along with significant reductions in total cholesterol, apoB, and triglycerides from 45% to 49% from baseline7). Notably, 3 patients in this study discontinued lipoprotein apheresis and 3 other patients extended their time intervals between apheresis during the study. The significant lipid-lowering activity of lomitapide in these patients was maintained to 78 weeks of continued treatment during the safety follow-up period of the study. Additional data supporting the therapeutic efficacy of lomitapide come from a recent Phase 3, open-label, single dose-escalation study in 9 Japanese HoFH patients, in which an MTD of lomitapide (up to 60 mg/day) significantly reduced LDL-C, non-HDL-C, VLCL-C, triglycerides, and apoB within 26 weeks of treatment and continuing to 56 weeks follow-up8).
Here the results of the long-term extension study in 5 Japanese patients demonstrate that prolonged lomitapide treatment produces controlled levels of plasma LDL-C to near or at < 100 mg/dL, while also sustaining reduced levels of triglycerides, non-HDL-C, and total cholesterol without deterioration in the profile of TEAEs. These results support the long-term lipid-lowering effects of lomitapide evaluated in 19 HoFH patients enrolled in a treatment extension study of the pivotal Phase 3 clinical trial9). In that extension study, individual MTDs of daily lomitapide maintained decreases in plasma LDL-C levels from extension study baseline by approximately 45% to 50% or lower over the course of 294 weeks of treatment. In that patient cohort, median percent increases of up to approximately 12% in hepatic fat were observed, with increases in ALT and AST above 5x ULN in a minority of patients (21%) that were attributed to use of CYP3A4 inhibitors or excess alcohol consumption and which were controlled by discontinuation of causative agents or dose reduction/cessation of lomitapide9). In the extension study reported here, hepatic fat was not measured; however, none of the 5 patients exhibited ≥ 3-fold increases in ALT or AST during treatment over 60 weeks.
The data from our study also demonstrated acceptable safety and tolerability of long-term lomitapide treatment, with no observed serious TEAEs or treatment discontinuations attributed to its use. The tolerability of lomitapide was also demonstrated by the observed sustained dosage in each patient for at least approximately 400 days of treatment or longer. One patient with combined PCSK9 and LDL-R mutations receiving 10 mg/day lomitapide discontinued treatment with this agent in favor of evolocumab following observed increases in hepatic fat and on the advice of the treating physician.
The results of this extension study and other clinical and real-world data demonstrate that long-term treatment with lomitapide, in addition to other LLTs and in the setting of a low-fat diet consistently reduces plasma LDL-C levels in patients with HoFH, while also producing beneficial profiles of other plasma lipids, including HDL-C. In conjunction with results of the aforementioned Phase 3 and extension studies7 and a separate report of long-term lomitapide treatment of 4 patients with HoFH in real-world settings13), the results of this study support the safety and efficacy of lomitapide as an effective LLT for patients with different genetic types of HoFH.
In conclusion, we have demonstrated that lomitapide therapy, in conjunction with other standard LLTs, is an effective lipid-lowering agent that significantly and consistently reduces circulating levels of LDL-C and other atherosclerotic lipids in patients with HoFH. For patients with inherited mutations adversely affecting LDL metabolism that do not respond to standard and newer LLTs including the PCSK9 inhibitors, lomitapide offers an effective treatment option that can be administered over extended periods of therapy. Careful monitoring of lipid responses to lomitapide treatment and its potential side effects on hepatic function may help to optimize its use in patients with HoFH over extended periods of time.
Acknowledgements and Study Support
This study was supported by Aegerion Pharmaceuticals Inc. All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this manuscript, take responsibility for the integrity of the work, and have given final approval to the version to be published. Aegerion provided funding for medical writing support in the development of this manuscript; THA, Inc. wrote the first draft of the manuscript based on input from authors, and copyedited and styled the manuscript per journal requirements. Aegerion reviewed and provided feedback on the manuscript to the authors. The authors had full editorial control of the manuscript and provided their final approval of all content.
COI
Dr. Nohara reports grants from Aegerion Pharmaceuticals, personal fees from Sanofi K.K., Amgen and Amgen Astellas Biopharma. Dr. Otsubo, Dr. Yanagi, Dr. Yoshida, Dr. Ikewaki, report no conflict of interest. Dr. Harada-Shiba reports honoraria from Astellas, Amgen Astellas Biopharma, and Sanofi; grants from Astellas, Amgen Astellas Biopharma, and Aegerion Pharmaceuticals. Dr. Jurecka has equity in, and is an employee of Aegerion Pharmaceuticals.
References
- 1). Cuchel M, Bruckert E, Ginsberg HN, Raal FJ, Santos RD, Hegele RA, Kuivenhoven JA, Nordestgaard BG, Descamps OS, Steinhagen-Thiessen E, Tybjßrg-Hansen A, Watts GF, Averna M, Boileau C, Borén J, Catapano AL, Defesche JC, Hovingh GK, Humphries SE, Kovanen PT, Masana L, Pajukanta P, Parhofer KG, Ray KK, Stalenhoef AF, Stroes E, Taskinen MR, Wiegman A, Wiklund O, Chapman MJ, European Atherosclerosis Society Consensus Panel on Familial Hypercholesterolaemia Homozygous familial hypercholesterolaemia: new insights and guidance for clinicians to improve detection and clinical management. A position paper from the Consensus Panel on Familial Hypercholesterolaemia of the European Atherosclerosis Society. Eur Heart J, 2014; 35: 2146-2157.40-46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2). France M, Rees A, Datta D, Thompson G, Capps N, Ferns G, Ramaswami U, Seed M, Neely D, Cramb R, Shoulders C, Barbir M, Pottle A, Eatough R, Martin S, Bayly G, Simpson B, Halcox J, Edwards R, Main L, Payne J, Soran H, for HEART UK Medical Scientific and Research Committee HEART UK statement on the management of homozygous familial hypercholesterolaemia in the United Kingdom. Atherosclerosis, 2016; 255: 128-139 [DOI] [PubMed] [Google Scholar]
- 3). Sjouke B, Kusters DM, Kindt I, Besseling J, Defesche JC, Sijbrands EJ, Roeters van Lennep JE, Stalenhoef AF, Wiegman A, de Graaf J, Fouchier SW, Kastelein JJ, Hovingh GK. Homozygous autosomal dominant hypercholesterolaemia in the Netherlands: prevalence, genotype-phenotype relationship, and clinical outcome. Eur Heart J, 2015; 36: 560-565 [DOI] [PubMed] [Google Scholar]
- 4). Defesche JC, Gidding SS, Harada-Shiba M, Hegele RA, Santos RD, Wierzbicki AS. Familial hypercholesterolemia. Nat Rev Dis Primers, 2017; 7: 17093. [DOI] [PubMed] [Google Scholar]
- 5). Ito MK, McGowan MP, Moriarty PM, National Lipid Association Expert Panel on Familial Hypercholesterolemia : Management of familial hypercholesterolemias in adult patients: recommendations from the National Lipid Association Expert Panel on Familial Hypercholesterolemia. J Clin Lipidol, 2011; 5: S38-S45 [DOI] [PubMed] [Google Scholar]
- 6). Juxtapid® (lomitapide), full prescribing information. Aegerion Pharmaceuticals. December 2012 [Google Scholar]
- 7). Cuchel M, Meagher EA, du Toit Theron H, Blom DJ, Marais AD, Hegele RA, Averna MR, Sirtori CR, Shah PK, Gaudet D, Stefanutti C, Vigna GB, Du Plessis AM, Propert KJ, Sasiela WJ, Bloedon LT, Rader DJ, and for the Phase 3 HoFH Lomitapide Study investigators Efficacy and safety of a microsomal triglyceride transfer protein inhibitor in patients with homozygous familial hypercholesterolaemia. Lancet, 2013; 381: 40-46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8). Harada-Shiba M, Ikewaki K, Nohara A, Otsubo Y, Koji Yanagi K, Yoshida M, Chang Q, Foulds P. Efficacy and safety of lomitapide in Japanese patients with homozygous familial hypercholesterolemia. J Atheroscler Thromb, 2017; 24: 402-411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9). Blom DJ, Averna MR, Meagher EA, du Toit Theron H, Sirtori CR, Hegele RA, Shah PK, Gaudet D, Stefanutti C, Vigna GB, Larrey D, Bloedon LT, Foulds P, Rader DJ, Cuchel M. Long-term efficacy and safety of the microsomal triglyceride transfer protein inhibitor lomitapide in patients with homozygous familial hypercholesterolemia. Circulation 2017; 136: 332-335 [DOI] [PubMed] [Google Scholar]
- 10). Rader DJ, Kastelein JJ. Lomitapide and mipomersen: two first-in-class drugs for reducing low-density lipoprotein cholesterol in patients with homozygous familial hypercholesterolemia. Circulation, 2014; 129: 1022-1032 [DOI] [PubMed] [Google Scholar]
- 11). Thedrez A, Blom DJ, Ramin-Mangata S, Blanchard V, Croyal M, Chemello K, Nativel B, Pichelin M, Cariou B, Bourane S, Tang L, Farnier M, Raal FJ, Lambert G. Homozygous Familial Hypercholesterolemia Patients With Identical Mutations Variably Express the LDLR (Low-Density Lipoprotein Receptor): Implications for the Efficacy of Evolocumab. Atherioscler Thromb Vasc Biol, 2018; 38: 592-598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12). Jamil H, Dickson JK, Jr, Chu CH, Lago MW, Rinehart JK, Biller SA, Gregg RE, Wetterau JR. Microsomal triglyceride transfer protein. Specificity of lipid binding and transport. J Biol Chem, 1995; 270: 6549-6554 [DOI] [PubMed] [Google Scholar]
- 13). Roeters van Lennep J, Averna M, Alonso R. Treating homozygous familial hypercholesterolemia in a real-world setting: Experiences with lomitapide. J Clin Lipidol, 2015; 9: 607-617 [DOI] [PubMed] [Google Scholar]