

Abstract
Duchenne muscular dystrophy (DMD) is an X-linked recessive fatal neuromuscular disorder characterized by progressive muscle degeneration which affects one in 3500-5000 males born worldwide. DMD is caused by loss-of-function mutations in the dystrophin (DMD) gene encoding for dystrophin, a cytoskeletal protein that supports the structural integrity of myofibers during cycles of muscle contraction and relaxation. DMD patients do not only experience skeletal muscle deterioration but also severe cardiomyopathy, which is recognized as the current leading cause of death for the disease. Among the therapies being developed, exon skipping using antisense oligonucleotides (AOs) is one of the most promising approaches. AOs effectively restore dystrophin expression in skeletal muscles; however, they are highly inefficient in the heart due to endosomal entrapment. Improving skeletal muscle function without restoring dystrophin expression in cardiac tissue may exacerbate cardiomyopathy due to increased voluntary activity. This review consolidates the preclinical antisense approaches to improve dystrophin restoration, with a special focus on the heart.
Keywords: Duchenne muscular dystrophy, dystrophin, cardiomyopathy, antisense therapy, morpholinos, drug delivery
Introduction
Duchenne muscular dystrophy (DMD) is an X-linked recessive disorder characterized by progressive muscle degeneration [1]. The prevalence of DMD is estimated to be 1 in 3500-5000 live male births, making it the most common lethal neuromuscular disorder [2,3]. Patients with DMD generally stay asymptomatic at birth although they could show signs of delayed gross motor development compared to their peers [4-6]. The disease progresses rapidly, with muscle weakness and wasting observed first in the proximal muscles then extending to the more distal muscles [7]. Affected boys often lose ambulation by the age of 12 and experience multiple organ system dysfunction such as neuromuscular scoliosis, joint contracture, osteoporosis, restrictive lung disease, obstructive sleep apnea, cardiomyopathy, and psychological problems [8-10]. The age of onset and the rate of decline are highly variable among patients. Due to continuous muscle damage, patients with DMD have markedly elevated serum levels of creatine kinase (CK), even at birth [11]. Death usually occurs in their 20s due to respiratory or cardiac complications [11-14].
DMD is caused by mutations in the DMD gene, which encodes a membrane-associated protein called dystrophin. Dystrophin has 4 domains: an actin-binding N-terminal domain, a rod domain consisting of 24 spectrin-like repeat motifs for structural flexibility, a cysteine-rich domain for facilitating protein-protein interaction, and a C-terminal domain for binding sarcolemmal proteins [15,16]. As a member of the dystrophin-glycoprotein complex (DGC), dystrophin functions to link the actin cytoskeleton of muscle cells to the extracellular matrix, providing mechanical support and membrane stabilization during muscle contraction and relaxation cycles [15,17-20]. In the absence of dystrophin, muscle fibers experience mechanical stress and become susceptible to tearing and fragmentation, resulting in degeneration [21,22]. Other studies have suggested that dystrophin also serves non-mechanical roles [23].
The DMD gene is located on the short arm of the X chromosome (Xp21.3-p21.2) spanning 2.4 Mb with 79 exons which produces a 14 kb transcript [24-26]. The DMD gene has 3 main promoters that produce full-length dystrophin (M, B, and P), with other promoters responsible for the transcription of different, smaller isoforms [27,28]. The M promoter produces the Dp427m isoform which is expressed in cardiac and skeletal muscle, the B promoter produces the Dp427c isoform which is expressed in neurons of the cortex and the Cornu Ammonis (CA) region of the hippocampus, and the P promoter produces the Dp427p isoform which is expressed in the Purkinje cells of the brain. Other shorter isoforms include Dp260 expressed in the retina, Dp140 expressed in the kidney and brain, Dp116 expressed in the peripheral nerves and Dp71 which is ubiquitously expressed [29-33].
DMD is considered one of the largest genes in the human genome. Furthermore, it is located in a genomic region with high rates of recombination [34-36]. Due to its length and location, DMD is highly susceptible to mutation. Approximately 60% of DMD cases are due to deletions of one or more exons, ~6% due to duplications, and the rest due to small insertions/deletions or point mutations [11,16,37,38]. These mutations usually disrupt the reading frame or introduce a premature stop codon, both of which lead to the absence of dystrophin. There is no significant correlation between mutation size and disease severity [39-41]. Most mutations that maintain the DMD reading frame produce truncated yet partly functional dystrophin, and often result in a milder form of the disease known as Becker muscular dystrophy (BMD) [42-44]. Although the skeletal muscle symptoms are less severe, the majority of BMD patients also develop cardiomyopathy, and it is the leading cause of death in patients with BMD [45].
Cardiomyopathy in DMD patients
The life expectancy for patients with DMD used to be less than 20 years of age just two decades ago. Due to advancements in ventilator support and spinal surgery, the average age of mortality has been pushed into the late 20s [12,13]. Since respiratory dysfunction is now better managed, cardiomyopathy is presently the leading cause of death among DMD patients. Cardiomyopathy is estimated to present in 25% of patients at the age of 6, and 59% of patients at the age of 10 [27,46]. The majority of patients will have developed cardiomyopathy by 18 years of age. Recognition of signs and symptoms of cardiac dysfunction can be challenging since DMD patients are usually wheelchair-bound and do not perform increased cardiac workload [47]. The correlation between a patient’s genotype and the severity of their cardiac phenotype remains uncertain [48-52].
The earliest clinical signs of cardiomyopathy are either decreased systolic function or sinus tachycardia, with the latter commonly seen in teenage DMD patients [53,54]. Sinus tachycardia results in elevated heart rate, which increases the workload of the already deteriorating dystrophic myocardium [55-57]. Arrhythmias are a common cardiac involvement in patients with DMD and are significantly correlated with decreased heart function. Approximately 44% of DMD patients show signs of arrhythmias [58]. Supraventricular tachycardia (SVT) and ventricular tachycardia (VT) are observed in 10% of individuals with DMD [58]. Although DMD cardiomyopathy is routinely described as dilated cardiomyopathy (DCM), patients with decreased systolic or diastolic function do not necessarily show ventricular dilation [49,59,60]. Additionally, myocardial fibrosis usually precedes left ventricular dysfunction (LVD) [61,62]. The age of onset and the rate of progression are variable for LVD. However, patients with onset of LVD before 18 years of age were found to have a significantly shorter lifespan compared to those who had LVD after 18 years old [48]. Other features of DMD cardiomyopathy include abnormal cardiac conduction (as caused by, for example, vacuole degeneration in the Purkinje fibers), congestive heart failure and sudden cardiac arrest [27,28,47,63-66].
Myocardial issues in DMD patients are unavoidable since dystrophin serves the same functions in cardiomyocytes as in skeletal muscle cells [67]. In the absence of dystrophin, cardiomyocytes experience increased structural vulnerability and are susceptible to membrane instability and tearing [22]. Cardiomyocyte remodeling occurs secondary to myocardial wasting, which leads to ventricular enlargement and fibrosis [68]. As previously mentioned, affected hearts do not always show signs of ventricular dilation. Another consequence of myocardial tearing and fragmentation is the dysregulation of ion influx/efflux in the heart [69,70]. Notably, damages to the myocardial membrane disrupt Ca2+ homeostasis. Since Ca2+ acts as a second messenger in many cellular pathways, Ca2+ dysregulation results in multiple downstream abnormalities within myocardial tissue. One significant consequence of Ca2+ dysregulation includes increased reactive oxygen species (ROS) production and mitochondrial dysfunction. Elevated Ca2+ levels within cardiomyocytes activate the mitochondria to produce ROS which leads to apoptosis and further muscle wasting [69-79]. An important second messenger affected by Ca2+ dysregulation is nitric oxide (NO) which has roles in blood flow and vasoregulation [80,81]. Increased intracellular Ca2+ concentrations also enhance the risk of Ca-dependent arrhythmias and cellular Ca2+ overload in DMD patients [69].
Cardiomyopathy in DMD patients is currently not curative. Therapies commonly used include: corticosteroids, angiotensin-converting enzyme inhibitors (ACEIs), angiotensin II receptor blockers (ARBs), beta-adrenergic receptor blockers, mineralocorticoid receptor antagonists and other interventions such as ventricular assist devices (VADs) or cardiac transplantation [9,47,82-92]. Treatment with corticosteroids has been shown to improve cardiac function. DMD patients who received steroids such as prednisolone/prednisone or deflazacort had significantly delayed onset of cardiomyopathy, less ventricular dilation and systolic dysfunction than those who did not. However, patients using corticosteroids can experience some long-term complications such as weight gain, bone fractures, cataracts, etc. [9,10,93-96], which limits their use for therapy. ACEIs and ARBs have proven effective in reducing left ventricular hypertrophy and fibrosis. Furthermore, initiation of beta-adrenergic receptor blocker treatment along with ACEIs has been demonstrated to improve left ventricular systolic function in DMD patients [88,91,97]. However, most of these studies have their inherent limitations, which makes it challenging for data interpretation as well as treatment application in clinical settings.
Antisense oligonucleotides for the treatment of DMD
At present, there is no cure for DMD. Current treatments are mostly palliative, aiming at alleviating the symptoms of the disease [11,14]. Several promising approaches have been investigated such as utrophin up-regulation, viral gene therapy, cell-based therapy, antisense oligonucleotides (AOs) for exon skipping and, most recently, CRISPR-Cas9-mediated gene editing [69]. In this review, we will discuss the use of AOs in the treatment of DMD with a specific focus on treating the heart.
AOs are short, synthetic nucleic acid sequences which bind to complementary target mRNA sequences and lead to either endonuclease-mediated transcript knockdown or splice modulation [98,99]. AO-mediated exon skipping can correct the reading frame by removing an out-of-frame exon or exons from the DMD pre-mRNA, producing a truncated but partly functional dystrophin protein [100-102] (Figure 1). The first generation of AOs has the unmodified phosphoribose backbone, making them highly susceptible to degradation by nucleases [103,104] (Figure 2). Due to their short half-life, first generation AOs rarely achieve sufficient intracellular concentrations to have a therapeutic effect [105,106]. Second and third generation AOs contain chemically modified structures. These modifications not only increase AO resistance to nuclease degradation, but also enhance their pharmacological properties. Alterations at the 2’ position of the ribose sugar have yielded a class of AOs with improved safety and efficacy profiles. 2’-O-methyl (2’-OMe) and 2’-O-methoxyethyl (2’-MOE) alkyl substituents are the most studied members of this group. In addition, the use of a phosphorothioate backbone which contains a sulfur in a non-bridging location on the phosphate backbone significantly improves AO nuclease resistance and binding affinity to serum proteins [98,107-109]. Antisense chemistries that contain both a phosphorothioate backbone and a 2’-alkyl substituent, such as 2’-OMe-phosphorothioate (2’OMePS) AOs, have shown several favorable properties compared to their unmodified counterparts [105,110-113]. The phosphodiester backbone can also be replaced with a polyamide structure made up of repeating N-(2-aminoethyl) glycine units, which has given rise to another class of AOs termed peptide nucleic acids (PNAs) [114]. This new class of AO can sterically block splicing factors, inhibit ribosome recruitment and bind non-coding RNAs, thereby further expanding therapeutic potential [69].
Figure 1.
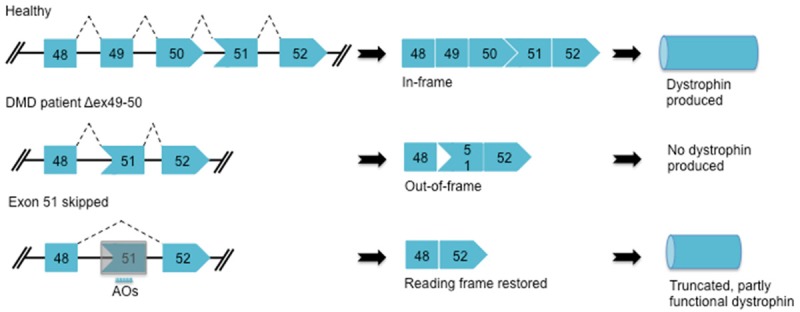
Antisense-induced exon skipping in DMD. Deletion of exon 49 and 50 in the DMD gene creates a frameshift mutation which results in a premature stop codon and no dystrophin produced. In the case shown, AOs can specifically bind to exon 51 and interfere with the splicing machinery to exclude exon 51 from the mature mRNA, thereby restoring the reading frame and producing truncated but partly functional dystrophin.
Figure 2.

Chemical structures commonly used in antisense therapy. (A) RNA, (B) 2’-OMe-phosphorothioate, (C) Tricyclo-DNA, (D) Phosphorodiamidate morpholino oligomers. X or Y can be a peptide; Y can be an octaguanidine moiety (E) as seen in Vivo-morpholino.
Among these next-generation chemistries, phosphorodiamidate morpholino oligomers (PMOs) represent the most advanced use of antisense therapy for DMD. PMOs have the deoxyribose/ribose moiety replaced by a morpholine ring, and the charged phosphodiester inter-subunit linkage replaced by an uncharged phosphorodiamidate linkage, making PMOs nuclease-resistant and charge-neutral [115,116]. One of the challenges with using nucleic acid-based molecules as therapeutics is their stability and potential toxicity in cells. The advantage of being charge-neutral, as in the case of PMOs, is that this imparts an even greater resistance to nucleases, which typically target charged molecules. Additionally, due to the lack of charge, PMOs are safer since they are unlikely to activate Toll-like receptors, a class of receptors involved in producing innate immune responses against pathogenic material [117]. In fact, a PMO targeting DMD exon 51, called eteplirsen or Exondys 51 (Sarepta Therapeutics, Cambridge, MA, USA), was conditionally approved by the U.S. Food and Drug Administration (FDA) in 2016, and several PMOs targeting other DMD exons, including golodirsen or SRP-4053 (Sarepta Therapeutics) and NS-065/NCNP-01 (NS Pharma, Paramus, NJ, USA) are currently undergoing clinical trials [118-121].
Although AOs effectively rescue dystrophin expression in skeletal muscles, they are highly inefficient in cardiac muscles [122,123]. Failure to restore dystrophin expression in the heart in the presence of rescued dystrophin in skeletal muscles can exacerbate cardiac issues due to increased patient voluntary activity [124,125]. The major barrier to effective use of AOs in therapy is the delivery of antisense drugs to their intracellular targets. Upon reaching the cell surface, AOs are internalized by endocytosis. A variety of cell-surface receptors have been suggested to bind AOs and facilitate their entry into the cell including integrins, scavenger receptors and Toll-like receptors [126-128]. Regardless of the endocytosis route taken, AOs entering a cell will encounter an intricate network of membrane compartments that include early and recycled endosomes, late endosomes and multi-vesicular bodies (MVBs), and lysosomes. Most AOs are initially delivered to early endosomes. Subsequently, they can be trafficked to lysosomes for degradation or sequestered in late endosomes/MVBs. Antisense drugs trapped within endomembrane compartments are pharmacologically inactive as they remain separated from their target sites by membrane barriers [106,129]. Nevertheless, a small portion of AOs escapes endosomes and only then can they reach their targets and become pharmacologically active. The concept of endosomal escape has been widely accepted as the most important barrier to the effective use of antisense drugs [127,130]. Importantly for developing heart-effective AOs, the research found that AOs (particularly PMOs) tend to be trapped in endosomes in cardiomyocytes more than in skeletal muscle cells [131]. Since cardiomyopathy is the leading cause of death in DMD patients, researchers have looked into different approaches to enhance delivery of AOs to cardiac tissues. The next section will discuss some of the most promising approaches to improving AO efficacy for treating the heart.
Strategies to improve the efficacy of antisense drugs for the treatment of cardiomyopathy in DMD patients
Tricyclo-DNA
In the late nineties, a novel class of AOs known as tricyclo-DNAs (tcDNAs) was shown to display unique pharmacological properties. tcDNA is a conformationally-constrained DNA analog that deviates from the natural DNA structure by the presence of an ethylene bridge connecting C3’ and C5’ [132-134]. tcDNA shows enhanced RNA affinity and nuclease resistance, as well as the inability to elicit RNase H activity [135-138]. Furthermore, tcDNAs can spontaneously form defined nanoparticles which potentially improves delivery and cellular uptake compared to other antisense chemistries [139-141].
In a study by Goyenvalle and colleagues, a mouse model carrying a nonsense mutation in exon 23 of the Dmd gene (hereafter referred to as mdx mice) was injected intravenously with a 15 nucleotide long (15-mer) tcDNA-AO that targets the skipping of Dmd exon 23 [139]. Treatment with 200 mg/kg/week of the drug for 12 weeks rescued up to 40% of wild type (WT) dystrophin expression in the heart. Additionally, tcDNA-treated mice showed significantly higher levels of dystrophin rescue, especially in the diaphragm and the heart, compared to those treated with PMO and 2’-OMe AOs at an equimolar dosage. Similar results were observed in a dystrophin/utrophin double knockout mouse model where treatment with weekly intravenous (IV) injections of 200 mg/kg of tcDNA over a period of 5-20 weeks restored 53% of WT dystrophin levels in the heart. Treatment with tcDNA also ameliorated cardiac systolic function as demonstrated by improved ventricular ejection fractions and shortening fractions in echocardiography.
One of the advantages of tcDNA is its increased RNA binding affinity. Due to this feature, shorter tcDNAs can be designed, reducing the mass of AOs administered and potential AO-induced toxicity while retaining their therapeutic effect. To investigate the efficacy of shorter tcDNAs, mdx mice were injected intravenously with a 13-mer tcDNA targeting Dmd exon 23 at 200 mg/kg/week for 12 weeks. The 13-mer tcDNA induced particularly high levels of exon skipping and restored dystrophin in the heart as shown by RT-PCR, Western blotting and immunostaining. The safety profile of the treatment also showed that tcDNAs were well-tolerated, with only minimal renal toxicity and liver inflammation observed [138]. Although tcDNAs have the potential to rescue dystrophin in cardiac tissue, treatment with these AOs requires high dosage and multiple dose administrations.
Peptides
Among the various delivery systems studied, cell-penetrating peptides (CPPs) have gained the most considerable interest. CPPs are composed of cationic and/or amphipathic amino acids that are capable of delivering a wide range of nucleic acid-based molecules both in vitro and in vivo [142-144]. These peptides either possess natural translocating properties or were engineered by combining different protein domains to have these properties [145]. Many studies have investigated peptide-conjugated AOs for the potential treatment of DMD. This review will discuss some of the most promising approaches, namely: arginine-rich peptides, B peptides, PNA/PMO internalization peptides (Pips), and phage peptides. Not all experiments are mentioned in this paper, but a selected few will be discussed for each approach.
The (RXR)4 peptide (with R standing for arginine and X standing for aminohexanoic acid) was the first arginine-rich CPP tested in the mdx model of DMD [146]. In a study by Yin and colleagues, (RXR)4 peptides were conjugated to PMOs targeting Dmd exon 23. A single IV injection at 25 mg/kg via tail vein resulted in ~50% exon 23 skipping in the heart of treated mice. A widespread, uniform distribution of dystrophin-positive fibers was also observed in cardiac tissue as shown by immunostaining. Western blot analysis demonstrated dystrophin restoration at levels between 10 and 20% of that found in the WT mouse heart. Even at lower doses administered (weekly injections at 6 mg/kg for 3 weeks), dystrophin restoration was still observed in cardiac tissue as shown by immunostaining and Western blotting.
Subsequently, another arginine-rich peptide, named the B-peptide, demonstrated high levels of exon skipping in various muscles including the heart. The B-peptide contains arginine, aminohexanoic acid and beta alanine (abbreviated B) in an (RXRRBR)2 sequence [147-149]. A single IV injection at 30 mg/kg of B-peptide-conjugated PMO (PPMO) restored 94% of dystrophin expression in cardiac muscle fibers of treated mdx mice by immunohistochemistry, 50% of WT dystrophin levels by Western blot, and 63% exon 23 skipping by RT-PCR. Repeated treatment for 3 months at the same dose at biweekly intervals further improved dystrophin restoration in cardiac muscle. Skipped transcript levels increased to 72%. Immunohistochemistry and Western blot demonstrated dystrophin rescue at levels comparable to that in WT hearts. Dystrophin restoration was also observed in the muscles of the atria and large vessels such as the aorta or vena cava and the pulmonary arteries. PPMO treatment significantly improved cardiac function and prevented cardiac failure under dobutamine stress [150]. A PPMO cocktail was also shown to restore dystrophin expression in the myocardium and cardiac Purkinje fibers, and improve cardiac conduction abnormalities in a canine model of DMD [151].
Another promising class of CPPs is the Pips. Pips are a novel series of transduction peptides developed from mutagenesis and functional studies of a derivative of Penetratin, with the six arginine residues added to the N terminus of the CPP (R6Pen) [152]. Penetratin is originally derived from the homeobox peptide of Drosophila Antennapedia. The CPPs in the Pip series are characterized by a central hydrophobic motif anchored on each side by arginine-rich sequences similar to that of the B-peptide which contains arginine, aminohexanoic acid, and beta-alanine residues. Among the highly efficient members of this series are Pip5 and Pip6, both of which have demonstrated high levels of dystrophin restoration in various muscles including the heart [153,154]. A single IV injection of 25 mg/kg of Pip5e-PMO (a member of the Pip5 family) induced almost complete exon skipping in cardiac tissue where full length uncorrected transcript was barely detectable. Immunohistochemistry data revealed more than 90% of dystrophin-positive fibers in cardiac muscle. Dystrophin restoration was further confirmed by Western blot which revealed more than 50% of WT dystrophin levels in the heart [153]. Modifications to the central hydrophobic core of the Pip5 peptide gave rise to a novel derivative of the Pip series, consecutively named Pip6. When directly compared to Pip5e-PMO treatment, a member of the Pip6 family exhibited significantly higher dystrophin restoration in the heart of treated mice at very low doses (12.5 mg/kg). The Pip6 series have also been demonstrated to prevent exercise-induced cardiomyopathy following Pip-PMO treatment in dystrophic mice [154].
While the above-mentioned CPPs work well for neutral AOs like PMOs, it is challenging to conjugate positively charged peptides to negatively-charged AOs such as 2’OMePS due to the presence of strong electrostatic interactions. In an effort to find uncharged CPPs, Jirka and colleagues screened a phage display peptide library and identified a 7-mer peptide (P4) that significantly enhanced AO uptake in cardiac tissue [155]. Dystrophic mdx mice were injected subcutaneously with 200 mg/kg/week doses of 2’OMePS-P4 conjugate targeting mouse Dmd exon 23 for 6 weeks. Treatment with peptide-conjugated AOs resulted in enhanced exon skipping efficacy with a significant difference in cardiac tissue compared to naked AOs, possibly due to improved uptake in cardiac muscle. The safety profile of the peptide conjugate was also evaluated and no significant changes were detected for liver and kidney damage. This result is encouraging since the phage peptides could be used as an alternative to arginine-rich peptides, which have shown toxicity in higher animals [156-158].
Octaguanidine morpholino
In an effort to improve the efficacy of AO systemic delivery and to reduce the immunogenicity of cell penetrating peptides, dendrimeric octaguanidine moieties have been conjugated to PMOs to form a new class of AOs named Vivo-Morpholinos or vPMOs [150,159].
The guanidine groups in vPMOs work similarly to the arginine-rich CPPs found conjugated to PMOs. As such, vPMOs have been shown to induce high levels of dystrophin expression especially in the heart [149]. Additionally, the combination of a synthetic scaffold and multiple unnatural side chains makes vPMOs unlikely to elicit an immune response, thus allowing for multiple administrations to maintain adequate levels of dystrophin in the body [160].
vPMOs have been shown to rescue cardiac dystrophin expression in vivo [161]. A single IV injection of a vPMO targeting mouse Dmd exon 23 (Vivo-ME23) at 6 mg/kg resulted in a strong dystrophin signal in a significant number of cardiomyocytes in mdx mice. Repeated treatment at the same dose for five times biweekly rescued dystrophin expression in more than 40% of cardiomyocytes as observed by immunostaining. Western blot showed dystrophin rescue to approximately 10% of normal levels in cardiac muscles. vPMOs have also been shown to restore dystrophin expression in a canine model of DMD, however, no systemic injection was performed so whether vPMOs can rescue dystrophin in canine cardiac tissues remains to be determined [162]. Regardless, these encouraging results represent an important step forward in our effort to overcome the challenges associated with the delivery of AOs to the heart.
Ultrasound and microbubbles
Another promising approach for improving AO delivery to cardiac tissue is the use of ultrasound in combination with contrast-enhancing microbubbles [163]. Microbubbles are used to carry the drugs to an area of interest, then ultrasound is applied to burst the microbubbles, resulting in tissue-specific delivery of the therapeutic materials [164-167]. Ultrasound exposure improves the efficacy of intracellular delivery due to a phenomenon known as sonoporation or cellular sonification. This effect creates transient pores in the cellular plasma membrane by a process known as inertial cavitation, thus allowing bioactive materials to enter the cells. Inertial cavitation is enhanced by using microbubbles of contrast agents [164,167-170]. AOs can be mixed with or incorporated into microbubbles in a number of ways such as binding to the microbubble shell or using site-specific ligands. The combination of ultrasound and microbubbles not only improves intracellular delivery but also reduces the amount of drugs used systemically thereby lessening the potential toxicity and side effects of treatment.
In order to investigate the microbubble properties that are important for influencing drug delivery efficacy in vivo, PMOs targeting Dmd exon 23 of the mdx mouse were injected systemically at 16 mg/kg via the tail vein [171]. PMOs were incorporated into three different commercially available microbubbles: Sonazoid, Optison, and SonoVue. Ultrasound was then applied to the heart for two minutes after injections. Treated hearts were collected 24 hours or one-week post injection. Of the three microbubbles tested, Sonazoid and Optison induced significant amounts of dystrophin-positive fibers in cardiac tissues. Treatments with PMOs only (no ultrasound or microbubbles) were ineffective in terms of restoring dystrophin in cardiomyocytes. Microbubbles were also found to increase AO delivery to the heart in a dose-dependent manner. Histological analysis showed no signs of tissue damage or observable morphological differences between samples treated with ultrasound and microbubbles and non-treated controls. Additionally, microbubble stability was determined to be the major factor affecting the efficiency of therapeutic drug delivery.
Nanoparticles
In order to improve AO stability and enable the use of lower AOs concentrations in vivo, AO delivery using nanoparticles was developed and demonstrated to be another promising approach. Two classes of nanoparticles, T1 and ZM2, have been shown to restore dystrophin expression in the heart of mdx mice [172,173].
T1 nanoparticles are a type of cationic core-shell nanospheres, made up of a core of polymethylmethacrylate (PMMA) and surrounded by a shell of cationic groups, which facilitates the binding of charged AOs. In a study by Rimessi and colleagues, T1 nanoparticles were demonstrated to deliver 2’OMePS AOs and induce dystrophin rescue in body-wide striated muscles. AOs targeting the boundary sequences of exon and intron 23 of mouse Dmd (M23D) were conjugated with T1 nanoparticles, and delivered by weekly intraperitoneal (IP) injections at 2.7 mg/kg to mdx mice [172]. T1 nanoparticles showed a wide distribution in the body, including the heart. At one week post-injection, immunohistochemical analysis of treated mice showed the presence of dystrophin-positive cardiomyocytes in various areas of the heart. Dystrophin was however undetectable in cardiac tissues 6 weeks after the last injection. Total and skipped dystrophin transcript levels in the hearts of treated mice were also significantly increased with 80% and 16% more transcripts, respectively. Although dystrophin was undetected in the heart by Western blot, the results were positive considering a significantly reduced dose compared to previous studies. From a safety aspect, T1 nanoparticles may cause adverse effects due to their slow biodegradability, indicating a possibility of accumulative toxicity for long-term treatments [172]. Also, T1 nanoparticles can form small aggregates and are therefore not suitable for IV injections.
To improve the efficacy of T1 nanoparticles, a more efficient class of nanoparticles termed ZM2 was designed [173]. ZM2 nanoparticles are composed of a PMMA core and a random copolymer shell consisting of units derived from N-isopropylacrylamide+ (NIPAM) and reactive methacrylate-bearing cationic groups. mdx mice were injected IP weekly for 7 weeks at 7.5 mg/kg with M23D AOs loaded onto ZM2 nanoparticles. Immunohistochemical findings showed 3% by manual count and 6% by semi-automated count of dystrophin-positive signal in the heart. Dystrophin was also detectable, though faintly, in the heart of ZM2-M23D treated mice via Western blot. However, dystrophin was not detectable in the heart at 3 months post-injection as demonstrated in a follow-up study.
Polymers
Different series of polymers were investigated for their ability to improve the delivery of AOs in vitro and in vivo. The first class is a series of low molecular weight polyethylenimine (LPEI) conjugated pluronic copolymers (PCMs) [174]. Amphiphilic pluronic, poly (ethylene oxide)-b-poly (propylene oxide)-b-poly (ethylene oxide) (PEO-PPO-PEO triblock copolymer) has been used widely as a pharmaceutical adjuvant and a free adjuvant for gene delivery. Depending on the molecular weight, hydrophilic-lipophilic balance (HLB), and other characteristics of the pluronics, PCMs can have different AO delivery efficiencies and toxicities. In a study by Wang and colleagues, mdx mice were injected intravenously with 0.4 mg of PCMs and 2 mg of PMOs targeting the boundary sequences of exon and intron 23 of mouse Dmd gene (PMOE23), or 2 mg of PMOE23 only as the control. Two weeks after injection, dystrophin was not detectable in cardiac muscle of control mice, while immunohistochemistry showed membrane-localized dystrophin in more than 5% of cardiomyocytes in some areas of the heart from treated mice. Treatment also demonstrated no detectable toxicity in muscles, liver, and kidneys after systemic administration.
Another class of amphiphilic polymers, termed Z polymers, is constructed from Tween 85 and LPEI [175]. Based on preliminary results, two Z polymers Z7 and Z8 were selected for systemic injection. Dystrophic mdx mice were treated by IV injection with 1 mg Z7/1 mg PMO complex, or 0.5 mg Z8/1 mg PMO conjugate, or 1 mg PMO as a control. Dystrophin expression was not detected in the PMO-only group, however, Z8-PMO treated mice demonstrated membrane-localized dystrophin-positive fibers in some areas of the heart. No adverse effects were observed in systemic delivery under the experimental conditions. The mechanism/s by which amphiphilic polymers improve gene transfer is/are not fully understood. Their hydrophobic interaction with cell membranes could reduce membrane viscosity and facilitate the entry of AOs [173]. Amphiphilic polymers could also prevent nonspecific binding of AOs to charged extracellular components, thus increasing the effective concentrations of therapeutic drugs.
Conclusion
To summarize, with the exception of tricyclo-DNAs, naked AOs are generally ineffective in treating the heart. Conjugation to CPPs, especially arginine-rich peptides and the Pip series, appears to be a promising approach to improve AO uptake and dystrophin restoration in cardiac tissue. However, the practicality of CPPs in the clinical setting remains to be determined largely due to concerns over our complex immune system. Phage peptides and octaguanidine moieties were developed in an effort to reduce the immunogenicity of arginine-rich peptides, however, they have been less successful in rescuing dystrophin in systemic delivery of antisense drugs to the heart. Other non-covalent conjugation approaches such as the use of nanoparticles, polymers, or ultrasound and microbubbles are promising strategies to enhance the efficacy of AOs in cardiac tissue, especially for charged antisense chemistries. With a growing number of AO-based approaches being investigated, antisense therapy holds the potential to improve the treatments for DMD patients, with special focus on cardiac aspects of the disease.
Tricyclo-DNA clearly appears as a promising chemistry for the antisense therapy of DMD-associated cardiomyopathy due to its unique pharmacological properties and high therapeutic potential. Besides their ability to improve delivery and uptake into cardiac tissue, tcDNAs can also cross the blood-brain barrier at low levels after systemic administration [139]. Along with other distinctive properties, this outstanding feature opens up more therapeutic options for other neuromuscular and neurodegenerative disorders such as spinal muscular atrophy and Huntington’s disease [135]. Although the safety profile of tcDNAs has been evaluated, observed toxicities were ascribed more to the phosphorothioate linkages rather than the tcDNA chemistry itself [138]. More extensive toxicological studies are recommended to assess the full safety profile of this promising chemistry. Given its positive performance in the preclinical phase, the evaluation of tcDNAs in human clinical trials seems to be the most logical next step. Similar to other chemistries, the outcome essentially depends on how well tcDNAs will be tolerated in patients.
An active body of research on DMD has significantly broadened our knowledge on the pathology as well as potential therapeutic treatments for the disease. To this day, DMD is still an invariably fatal disease that affects the whole body. It is, therefore, essential to focus on treatments for individual systems such as the heart. Advances in the understanding of antisense therapy have provided a strong foundation for the transition of AOs drugs from the lab bench to the clinic. However, delivery and uptake in cardiac tissue remain the key limiting factor in the bioavailability of AO-based therapies for the treatment of DMD. Perhaps more basic research into understanding the underlying mechanism of these processes is needed to overcome this major obstacle. Up to this point, clinical trials have focused mostly on the efficacy of AOs treatments on skeletal muscle, without much emphasis on the heart. Given how cardiomyopathy is currently the leading cause of death in patients with DMD, an increased focus on treatments for the heart is very much needed. In 2017, a clinical trial investigating the efficacy of a peptide-conjugated version of eteplirsen was initiated. The trial is expected to be completed in January 2019. With the promising results shown by peptide conjugated AOs in pre-clinical studies, it would be exciting to see the therapeutic outcome of the trial, particularly the cardiac aspects of it. Hopefully, these efforts will facilitate the progress of therapeutic development so that one day DMD will no longer be as devastating a disease as it is now.
Disclosure of conflict of interest
None.
References
- 1.Nakamura A, Takeda S. Mammalian models of Duchenne muscular dystrophy: pathological characteristics and therapeutic applications. J Biomed Biotechnol. 2011;2011:184393. doi: 10.1155/2011/184393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Emery AE. Population frequencies of inherited neuromuscular diseases-A world survey. Neuromuscul Disord. 1991;1:19–29. doi: 10.1016/0960-8966(91)90039-u. [DOI] [PubMed] [Google Scholar]
- 3.Mendell JR, Shilling C, Leslie ND, Flanigan KM, Al-Dahhak R, Gastier-Foster J, Kneile K, Dunn DM, Duval B, Aoyagi A, Hamil C, Mahmoud M, Roush K, Bird L, Rankin C, Lilly H, Street N, Chandrasekar R, Weiss RB. Evidence-based path to newborn screening for Duchenne muscular dystrophy. Ann Neurol. 2012;71:304–313. doi: 10.1002/ana.23528. [DOI] [PubMed] [Google Scholar]
- 4.Moser H. Duchenne muscular dystrophy: pathogenetic aspects and genetic prevention. Hum Genet. 1984;66:17–40. doi: 10.1007/BF00275183. [DOI] [PubMed] [Google Scholar]
- 5.Duchenne The pathology of paralysis with muscular degeneration (paralysie myosclerotique), or paralysis with apparent hypertrophy. Br Med J. 1867;2:541–542. doi: 10.1136/bmj.2.363.541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hoffman EF, Kunkel LM. Dystrophin abnormalities in Duchenne/becker muscular review dystrophy genes. Cell. 1989;2:1019–1029. doi: 10.1016/0896-6273(89)90226-2. [DOI] [PubMed] [Google Scholar]
- 7.Mercuri E, Muntoni F. Muscular dystrophies. Lancet. 2013;381:845–860. doi: 10.1016/S0140-6736(12)61897-2. [DOI] [PubMed] [Google Scholar]
- 8.Tsuda T. Clinical manifestations and overall management strategies for Duchenne muscular dystrophy. Methods Mol Biol. 2018;1687:19–28. doi: 10.1007/978-1-4939-7374-3_2. [DOI] [PubMed] [Google Scholar]
- 9.Bushby K, Finkel R, Birnkrant DJ, Case LE, Clemens PR, Cripe L, Kaul A, Kinnett K, McDonald C, Pandya S, Poysky J, Shapiro F, Tomezsko J, Constantin C. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and pharmacological and psychosocial management. Lancet Neurol. 2010;9:77–93. doi: 10.1016/S1474-4422(09)70271-6. [DOI] [PubMed] [Google Scholar]
- 10.Strehle EM, Straub V. Recent advances in the management of Duchenne muscular dystrophy. Arch Dis Child. 2015;100:1173–1177. doi: 10.1136/archdischild-2014-307962. [DOI] [PubMed] [Google Scholar]
- 11.Manzur AY, Kinali M, Muntoni F. Update on the management of Duchenne muscular dystrophy. Arch Dis Child. 2008;93:986–990. doi: 10.1136/adc.2007.118141. [DOI] [PubMed] [Google Scholar]
- 12.Eagle M, Baudouin SV, Chandler C, Giddings DR, Bullock R, Bushby K. Survival in Duchenne muscular dystrophy: improvements in life expectancy since 1967 and the impact of home nocturnal ventilation. Neuromuscul Disord. 2002;12:926–929. doi: 10.1016/s0960-8966(02)00140-2. [DOI] [PubMed] [Google Scholar]
- 13.Passamano L, Taglia A, Palladino A, Viggiano E, D’Ambrosio P, Scutifero M, Cecio MR, Torre V, De Luca F, Picillo E, Paciello O, Piluso G, Nigro G, Politano L. Improvement of survival in Duchenne muscular dystrophy: retrospective analysis of 835 patients. Acta Myol. 2012;31:121–125. [PMC free article] [PubMed] [Google Scholar]
- 14.Mah JK. Current and emerging treatment strategies for Duchenne muscular dystrophy. Neuropsychiatr Dis Treat. 2016;12:1795–1807. doi: 10.2147/NDT.S93873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ervasti JM. Dystrophin, its interactions with other proteins, and implications for muscular dystrophy. Biochim Biophys Acta. 2007;1772:108–117. doi: 10.1016/j.bbadis.2006.05.010. [DOI] [PubMed] [Google Scholar]
- 16.van Deutekom JC, van Ommen GJ. Advances in Duchenne muscular dystrophy gene therapy. Nat Rev Genet. 2003;4:774–783. doi: 10.1038/nrg1180. [DOI] [PubMed] [Google Scholar]
- 17.Campbell KP. Three muscular dystrophies: loss of cytoskeleton-extracellular matrix linkage. Cell. 1995;80:675–679. doi: 10.1016/0092-8674(95)90344-5. [DOI] [PubMed] [Google Scholar]
- 18.Whitmore C, Morgan J. What do mouse models of muscular dystrophy tell us about the DAPC and its components? Int J Exp Pathol. 2014;95:365–377. doi: 10.1111/iep.12095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ervasti JM, Campbell KP. A role for the dystrophin-glycoprotein complex as a transmembrane linker between laminin and actin. J Cell Biol. 1993;122:809–823. doi: 10.1083/jcb.122.4.809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ervasti JM, Campbell KP. Membrane organization of the dystrophin-glycoprotein complex. Cell. 1991;66:1121–1131. doi: 10.1016/0092-8674(91)90035-w. [DOI] [PubMed] [Google Scholar]
- 21.Lapidos KA, Kakkar R, McNally EM. The dystrophin glycoprotein complex: signaling strength and integrity for the sarcolemma. Circ Res. 2004;94:1023–1031. doi: 10.1161/01.RES.0000126574.61061.25. [DOI] [PubMed] [Google Scholar]
- 22.Petrof BJ, Shrager JB, Stedman HH, Kelly AM, Sweeney HL. Dystrophin protects the sarcolemma from stresses developed during muscle contraction. Proc Natl Acad Sci U S A. 1993;90:3710–3714. doi: 10.1073/pnas.90.8.3710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nichols B, Takeda S, Yokota T. Nonmechanical roles of dystrophin and associated proteins in exercise, neuromuscular junctions, and brains. Brain Sci. 2015;5:275–98. doi: 10.3390/brainsci5030275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hoffman EP, Brown RH, Kunkel LM. Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell. 1987;51:919–928. doi: 10.1016/0092-8674(87)90579-4. [DOI] [PubMed] [Google Scholar]
- 25.Koenig M, Hoffman EP, Bertelson CJ, Monaco AP, Feener C, Kunkel LM. Complete cloning of the Duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individuals. Cell. 1987;50:509–517. doi: 10.1016/0092-8674(87)90504-6. [DOI] [PubMed] [Google Scholar]
- 26.Roberts RG, Coffey AJ, Bobrow M, Bentley DR. Exon structure of the human dystrophin gene. Genomics. 1993;16:536–538. doi: 10.1006/geno.1993.1225. [DOI] [PubMed] [Google Scholar]
- 27.Tsuda T, Fitzgerald K. Dystrophic cardiomyopathy: complex pathobiological processes to generate clinical phenotype. J Cardiovasc Dev Dis. 2017;4:14. doi: 10.3390/jcdd4030014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kamdar F, Garry DJ. Dystrophin-deficient cardiomyopathy. J Am Coll Cardiol. 2016;67:2533–2546. doi: 10.1016/j.jacc.2016.02.081. [DOI] [PubMed] [Google Scholar]
- 29.Ahn AH, Kunkel LM. The structural and functional diversity of dystrophin. Nat Genet. 1993;3:283–291. doi: 10.1038/ng0493-283. [DOI] [PubMed] [Google Scholar]
- 30.Mehler MF. Brain dystrophin, neurogenetics and mental retardation. Brain Res Rev. 2000;32:277–307. doi: 10.1016/s0165-0173(99)00090-9. [DOI] [PubMed] [Google Scholar]
- 31.Chelly J, Hamard G, Koulakoff A, Kaplan JC, Kahn A, Berwald-Netter Y. Dystrophin gene transcribed from different promoters in neuronal and glial cells. Nature. 1990;344:64–65. doi: 10.1038/344064a0. [DOI] [PubMed] [Google Scholar]
- 32.Lidov HG, Selig S, Kunkel LM. Dp140: a novel 140 kDa CNS transcript from the dystrophin locus. Hum Mol Genet. 1995;4:329–335. doi: 10.1093/hmg/4.3.329. [DOI] [PubMed] [Google Scholar]
- 33.Pillers DA, Bulman DE, Weleber RG, Sigesmund DA, Musarella MA, Powell BR, Murphey WH, Westall C, Panton C, Becker LE, Worton RG, Ray PN. Dystrophin expression in the human retina is required for normal function as defined by electroretinography. Nat Genet. 1993;4:82–86. doi: 10.1038/ng0593-82. [DOI] [PubMed] [Google Scholar]
- 34.Takeshima Y, Yagi M, Okizuka Y, Awano H, Zhang Z, Yamauchi Y, Nishio H, Matsuo M. Mutation spectrum of the dystrophin gene in 442 Duchenne/becker muscular dystrophy cases from one Japanese referral center. J Hum Genet. 2010;55:379–388. doi: 10.1038/jhg.2010.49. [DOI] [PubMed] [Google Scholar]
- 35.Buzin CH, Feng J, Yan J, Scaringe W, Liu Q, Den Dunnen J, Mendell JR, Sommer SS. Mutation rates in the dystrophin gene: a hotspot of mutation at a CpG dinucleotide. Hum Mutat. 2005;25:177–188. doi: 10.1002/humu.20132. [DOI] [PubMed] [Google Scholar]
- 36.Nachman MW, Crowell SL. Contrasting evolutionary histories of two introns of the Duchenne muscular dystrophy gene, Dmd, in humans. Genetics. 2000;155:1855–1864. doi: 10.1093/genetics/155.4.1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yokota T, Pistilli E, Duddy W, Nagaraju K. Potential of oligonucleotide-mediated exon-skipping therapy for Duchenne muscular dystrophy. Expert Opin Biol Ther. 2007;7:831–842. doi: 10.1517/14712598.7.6.831. [DOI] [PubMed] [Google Scholar]
- 38.White S, Kalf M, Liu Q, Villerius M, Engelsma D, Kriek M, Vollebregt E, Bakker B, van Ommen GJ, Breuning MH, den Dunnen JT. Comprehensive detection of genomic duplications and deletions in the DMD gene, by use of multiplex amplifiable probe hybridization. Am J Hum Genet. 2002;71:365–374. doi: 10.1086/341942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ferlini A, Neri M, Gualandi F. The medical genetics of dystrophinopathies: molecular genetic diagnosis and its impact on clinical practice. Neuromuscul Disord. 2013;23:4–14. doi: 10.1016/j.nmd.2012.09.002. [DOI] [PubMed] [Google Scholar]
- 40.Flanigan KM, Dunn DM, Von Niederhausern A, Soltanzadeh P, Gappmaier E, Howard MT, Sampson JB, Mendell JR, Wall C, King WM, Pestronk A, Florence JM, Connolly AM, Mathews KD, Stephan CM, Laubenthal KS, Wong BL, Morehart PJ, Meyer A, Finkel RS, Bonnemann CG, Medne L, Day JW, Dalton JC, Margolis MK, Hinton VJ, Weiss RB, Bromberg M, Swoboda K, Kerr L, Gurvich O, Tuohy T, Taylor L, Zhao L, Hart K, Moural C, Hak K, Duval B, Hamil C, Mahmoud M, Aoyagi A, Viollet L, Gailey S, Lopate G, Golumbek P, Schierbecker J, Malkus B, Siener C, Baldwin K, Glanzman AM, Flickinger J, Naughton CE. Mutational spectrum of DMD mutations in dystrophinopathy patients: application of modern diagnostic techniques to a large cohort. Hum Mutat. 2009;30:1657–1666. doi: 10.1002/humu.21114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tuffery-Giraud S, Béroud C, Leturcq F, Yaou RB, Hamroun D, Michel-Calemard L, Moizard MP, Bernard R, Cossée M, Boisseau P, Blayau M, Creveaux I, Guiochon-Mantel A, De Martinville B, Philippe C, Monnier N, Bieth E, Van Kien PK, Desmet FO, Humbertclaude V, Kaplan JC, Chelly J, Claustres M. Genotype-phenotype analysis in 2,405 patients with a dystrophinopathy using the UMD-DMD database: a model of nationwide knowledgebase. Hum Mutat. 2009;30:934–945. doi: 10.1002/humu.20976. [DOI] [PubMed] [Google Scholar]
- 42.Hoffman EP, Kunkel LM, Angelini C, Clarke A, Johnson M, Harris JB. Improved diagnosis of Becker muscular dystrophy by dystrophin testing. Neurology. 1989;39:1011–1017. doi: 10.1212/wnl.39.8.1011. [DOI] [PubMed] [Google Scholar]
- 43.Arahata K, Hoffman EP, Kunkel LM, Ishiura S, Tsukahara T, Ishihara T, Sunohara N, Nonaka I, Ozawa E, Sugita H. Dystrophin diagnosis: comparison of dystrophin abnormalities by immunofluorescence and immunoblot analyses. Proc Natl Acad Sci U S A. 1989;86:7154–7158. doi: 10.1073/pnas.86.18.7154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Neri M, Torelli S, Brown S, Ugo I, Sabatelli P, Merlini L, Spitali P, Rimessi P, Gualandi F, Sewry C, Ferlini A, Muntoni F. Dystrophin levels as low as 30% are sufficient to avoid muscular dystrophy in the human. Neuromuscul Disord. 2007;17:913–918. doi: 10.1016/j.nmd.2007.07.005. [DOI] [PubMed] [Google Scholar]
- 45.Melacini P, Fanin M, Danieli GA, Villanova C, Martinello F, Miorin M, Freda MP, Miorelli M, Mostacciuolo ML, Fasoli G, Angelini C, Dalla Volta S. Myocardial involvement is very frequent among patients affected with subclinical Becker’s muscular dystrophy. Circulation. 1996;94:3168–3175. doi: 10.1161/01.cir.94.12.3168. [DOI] [PubMed] [Google Scholar]
- 46.Nigro G, Comi LI, Politano L, Bain RJ. The incidence and evolution of cardiomyopathy in Duchenne muscular dystrophy. Int J Cardiol. 1990;26:271–277. doi: 10.1016/0167-5273(90)90082-g. [DOI] [PubMed] [Google Scholar]
- 47.McNally EM, Kaltman JR, Benson D, Canter CE, Cripe LH, Duan D, Finder JD, Hoffman EP, Judge DP, Kertesz N, Kinnett K, Kirsch R, Metzger JM, Pearson GD, Rafael-Fortney JA, Raman SV, Spurney CF, Targum SL, Wagner KR, Markham LW. Contemporary cardiac issues in Duchenne muscular dystrophy. Working group of the national heart, lung, and blood institute in collaboration with parent project muscular dystrophy. Circulation. 2015;131:1590–1598. doi: 10.1161/CIRCULATIONAHA.114.015151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang M, Birnkrant DJ, Super DM, Jacobs IB, Bahler RC. Progressive left ventricular dysfunction and long-term outcomes in patients with Duchenne muscular dystrophy receiving cardiopulmonary therapies. Open Hear. 2018;5:e000783. doi: 10.1136/openhrt-2018-000783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jefferies JL, Eidem BW, Belmont JW, Craigen WJ, Ware SM, Fernbach SD, Neish SR, Smith EOB, Towbin JA. Genetic predictors and remodeling of dilated cardiomyopathy in muscular dystrophy. Circulation. 2005;112:2799–2804. doi: 10.1161/CIRCULATIONAHA.104.528281. [DOI] [PubMed] [Google Scholar]
- 50.Nigro G, Politano L, Nigro V, Petretta VR, Comi LI. Mutation of dystrophin gene and cardiomyopathy. Neuromuscul Disord. 1994;4:371–379. doi: 10.1016/0960-8966(94)90073-6. [DOI] [PubMed] [Google Scholar]
- 51.Ashwath ML, Jacobs IB, Crowe CA, Ashwath RC, Super DM, Bahler RC. Left ventricular dysfunction in Duchenne muscular dystrophy and genotype. Am J Cardiol. 2014;114:284–289. doi: 10.1016/j.amjcard.2014.04.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Magri F, Govoni A, D’Angelo MG, Del Bo R, Ghezzi S, Sandra G, Turconi AC, Sciacco M, Ciscato P, Bordoni A, Tedeschi S, Fortunato F, Lucchini V, Bonato S, Lamperti C, Coviello D, Torrente Y, Corti S, Moggio M, Bresolin N, Comi GP. Genotype and phenotype characterization in a large dystrophinopathic cohort with extended follow-up. J Neurol. 2011;258:1610–1623. doi: 10.1007/s00415-011-5979-z. [DOI] [PubMed] [Google Scholar]
- 53.Spurney CF. Cardiomyopathy of Duchenne muscular dystrophy: current understanding and future directions. Muscle Nerve. 2011;44:8–19. doi: 10.1002/mus.22097. [DOI] [PubMed] [Google Scholar]
- 54.Thomas TO, Morgan TM, Burnette WB, Markham LW. Correlation of heart rate and cardiac dysfunction in Duchenne muscular dystrophy. Pediatr Cardiol. 2012;33:1175–1179. doi: 10.1007/s00246-012-0281-0. [DOI] [PubMed] [Google Scholar]
- 55.Miller G, D’Orsogna L, O’Shea JP. Autonomic function and the sinus tachycardia of Duchenne muscular dystrophy. Brain Dev. 1989;11:247–250. doi: 10.1016/s0387-7604(89)80044-0. [DOI] [PubMed] [Google Scholar]
- 56.Lanza GA, Dello Russo A, Giglio V, De Luca L, Messano L, Santini C, Ricci E, Damiani A, Fumagalli G, De Martino G, Mangiola F, Bellocci F. Impairment of cardiac autonomic function in patients with Duchenne muscular dystrophy: relationship to myocardial and respiratory function. Am Heart J. 2001;141:808–812. doi: 10.1067/mhj.2001.114804. [DOI] [PubMed] [Google Scholar]
- 57.Kedem J, Sonn J, Scheinowitz M, Weiss HR. Relationship between local oxygen consumption and local and external cardiac work: effect of tachycardia. Cardiovasc Res. 1989;23:1043–1052. doi: 10.1093/cvr/23.12.1043. [DOI] [PubMed] [Google Scholar]
- 58.Chiang DY, Allen HD, Kim JJ, Valdes SO, Wang Y, Pignatelli RH, Lotze TE, Miyake CY. Relation of cardiac dysfunction to rhythm abnormalities in patients with Duchenne or becker muscular dystrophies. Am J Cardiol. 2016;117:1349–1354. doi: 10.1016/j.amjcard.2016.01.031. [DOI] [PubMed] [Google Scholar]
- 59.Ramaciotti C, Heistein LC, Coursey M, Lemler MS, Eapen RS, Iannaccone ST, Scott WA. Left ventricular function and response to enalapril in patients with Duchenne muscular dystrophy during the second decade of life. Am J Cardiol. 2006;98:825–827. doi: 10.1016/j.amjcard.2006.04.020. [DOI] [PubMed] [Google Scholar]
- 60.Su JA, Ramos-Platt L, Menteer JD. Left ventricular tonic contraction as a novel biomarker of cardiomyopathy in Duchenne muscular dystrophy. Pediatr Cardiol. 2016;37:678–685. doi: 10.1007/s00246-015-1331-1. [DOI] [PubMed] [Google Scholar]
- 61.Silva MC, Meira ZM, Gurgel Giannetti J, da Silva MM, Campos AF, Barbosa Mde M, Starling Filho GM, Ferreira Rde A, Zatz M, Rochitte CE. Myocardial delayed enhancement by magnetic resonance imaging in patients with muscular dystrophy. J Am Coll Cardiol. 2007;49:1874–1879. doi: 10.1016/j.jacc.2006.10.078. [DOI] [PubMed] [Google Scholar]
- 62.Silva MC, Magalhães TA, Meira ZM, Rassi CH, Andrade AC, Gutierrez PS, Azevedo CF, Gurgel-Giannetti J, Vainzof M, Zatz M, Kalil-Filho R, Rochitte CE. Myocardial fibrosis progression in Duchenne and becker muscular dystrophy: a randomized clinical trial. JAMA Cardiol. 2017;2:190–199. doi: 10.1001/jamacardio.2016.4801. [DOI] [PubMed] [Google Scholar]
- 63.Mavrogeni SI, Markousis-Mavrogenis G, Papavasiliou A, Papadopoulos G, Kolovou G. Cardiac involvement in Duchenne muscular dystrophy and related dystrophinopathies. Methods Mol Biol. 2018;1687:31–42. doi: 10.1007/978-1-4939-7374-3_3. [DOI] [PubMed] [Google Scholar]
- 64.Urasawa N, Wada MR, Machida N, Yuasa K, Shimatsu Y, Wakao Y, Yuasa S, Sano T, Nonaka I, Nakamura A, Takeda S. Selective vacuolar degeneration in dystrophin-deficient canine Purkinje fibers despite preservation of dystrophin-associated proteins with overexpression of Dp71. Circulation. 2008;117:2437–2448. doi: 10.1161/CIRCULATIONAHA.107.739326. [DOI] [PubMed] [Google Scholar]
- 65.Nomura H, Hizawa K. Histopathological study of the conduction system of the heart in Duchenne progressive muscular dystrophy. Acta Pathol Jpn. 1982;32:1027–33. doi: 10.1111/j.1440-1827.1982.tb02082.x. [DOI] [PubMed] [Google Scholar]
- 66.Yanagisawa A, Miyagawa M, Yotsukura M, Tsuya T, Shirato C, Ishihara T, Aoyagi T, Ishikawa K. The prevalence and prognostic significance of arrhythmias in Duchenne type muscular dystrophy. Am Heart J. 1992;124:1244–50. doi: 10.1016/0002-8703(92)90407-m. [DOI] [PubMed] [Google Scholar]
- 67.Wallace GQ, McNally EM. Mechanisms of muscle degeneration, regeneration, and repair in the muscular dystrophies. Annu Rev Physiol. 2009;71:37–57. doi: 10.1146/annurev.physiol.010908.163216. [DOI] [PubMed] [Google Scholar]
- 68.Konstam MA, Kramer DG, Patel AR, Maron MS, Udelson JE. Left ventricular remodeling in heart failure: current concepts in clinical significance and assessment. JACC Cardiovasc Imaging. 2011;4:98–108. doi: 10.1016/j.jcmg.2010.10.008. [DOI] [PubMed] [Google Scholar]
- 69.van Westering T, Betts C, Wood M. Current understanding of molecular pathology and treatment of cardiomyopathy in Duchenne muscular dystrophy. Molecules. 2015;20:8823–8855. doi: 10.3390/molecules20058823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Shirokova N, Niggli E. Cardiac phenotype of Duchenne muscular dystrophy: insights from cellular studies. J Mol Cell Cardiol. 2013;58:217–224. doi: 10.1016/j.yjmcc.2012.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Fanchaouy M, Polakova E, Jung C, Ogrodnik J, Shirokova N, Niggli E. Pathways of abnormal stress-induced Ca2+ influx into dystrophic mdx cardiomyocytes. Cell Calcium. 2009;46:114–121. doi: 10.1016/j.ceca.2009.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Nakayama H, Chen X, Baines CP, Klevitsky R, Zhang X, Zhang H, Jaleel N, Chua BHL, Hewett TE, Robbins J, Houser SR, Molkentin JD. Ca2+- and mitochondrial-dependent cardiomyocyte necrosis as a primary mediator of heart failure. J Clin Invest. 2007;117:2431–2444. doi: 10.1172/JCI31060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mosqueira M, Zeiger U, Förderer M, Brinkmeier H, Fink RH. Cardiac and respiratory dysfunction in Duchenne muscular dystrophy and the role of second messengers. Med Res Rev. 2013;33:1174–1213. doi: 10.1002/med.21279. [DOI] [PubMed] [Google Scholar]
- 74.Berridge MJ, Lipp P, Bootman MD. The versatility and universality of calcium signalling. Nat Rev Mol Cell Biol. 2000;1:11–21. doi: 10.1038/35036035. [DOI] [PubMed] [Google Scholar]
- 75.Vallejo-Illarramendi A, Toral-Ojeda I, Aldanondo G, López de Munain A. Dysregulation of calcium homeostasis in muscular dystrophies. Expert Rev Mol Med. 2014;16:e16. doi: 10.1017/erm.2014.17. [DOI] [PubMed] [Google Scholar]
- 76.Zorov DB, Juhaszova M, Sollott SJ. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol Rev. 2014;94:909–950. doi: 10.1152/physrev.00026.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gorski PA, Ceholski DK, Hajjar RJ. Altered myocardial calcium cycling and energetics in heart failure - a rational approach for disease treatment. Cell Metab. 2015;21:183–194. doi: 10.1016/j.cmet.2015.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Fong P, Turner PR, Denetclaw WF, Steinhardt RA. Increased activity of calcium leak channels in myotubes of Duchenne human and mdx mouse origin. Science. 1990;250:673–676. doi: 10.1126/science.2173137. [DOI] [PubMed] [Google Scholar]
- 79.Franco A, Lansman JB. Calcium entry through stretch-inactivated ion channels in mdx myotubes. Nature. 1990;344:670–673. doi: 10.1038/344670a0. [DOI] [PubMed] [Google Scholar]
- 80.Reid MB. Role of nitric oxide in skeletal muscle: synthesis, distribution and functional importance. Acta Physiol Scand. 1998;162:401–409. doi: 10.1046/j.1365-201X.1998.0303f.x. [DOI] [PubMed] [Google Scholar]
- 81.Stamler JS, Meissner G. Physiology of nitric oxide in skeletal muscle. Physiol Rev. 2001;81:209–237. doi: 10.1152/physrev.2001.81.1.209. [DOI] [PubMed] [Google Scholar]
- 82.Raman SV, Hor KN, Mazur W, Halnon NJ, Kissel JT, He X, Tran T, Smart S, McCarthy B, Taylor MD, Jefferies JL, Rafael-Fortney JA, Lowe J, Roble SL, Cripe LH. Eplerenone for early cardiomyopathy in Duchenne muscular dystrophy: a randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2015;14:153–161. doi: 10.1016/S1474-4422(14)70318-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Mendell JR, Moxley RT, Griggs RC, Brooke MH, Fenichel GM, Miller JP, King W, Signore L, Pandya S, Florence J, Schierbecker J, Robison J, Kaiser K, Mandel S, Arfken C, Gilder B. Randomized, double-blind six-month trial of prednisone in Duchenne’s muscular dystrophy. N Engl J Med. 1989;320:1592–1597. doi: 10.1056/NEJM198906153202405. [DOI] [PubMed] [Google Scholar]
- 84.Dec GW. Steroid therapy effectively delays Duchenne’s cardiomyopathy. J Am Coll Cardiol. 2013;61:955–956. doi: 10.1016/j.jacc.2012.12.011. [DOI] [PubMed] [Google Scholar]
- 85.Wu RS, Gupta S, Brown RN, Yancy CW, Wald JW, Kaiser P, Kirklin NM, Patel PC, Markham DW, Drazner MH, Garry DJ, Mammen PP. Clinical outcomes after cardiac transplantation in muscular dystrophy patients. J Heart Lung Transplant. 2010;29:432–438. doi: 10.1016/j.healun.2009.08.030. [DOI] [PubMed] [Google Scholar]
- 86.Ryan TD, Jefferies JL, Sawnani H, Wong BL, Gardner A, Del Corral M, Lorts A, Morales DL. Implantation of the HeartMate II and HeartWare left ventricular assist devices in patients with Duchenne muscular dystrophy: lessons learned from the first applications. ASAIO J. 2014;60:246–248. doi: 10.1097/MAT.0000000000000050. [DOI] [PubMed] [Google Scholar]
- 87.Stoller D, Araj F, Amin A, Fitzsimmons C, Morlend R, Thibodeau JT, Ramaciotti C, Drazner MH, Meyer DM, Mammen PPA. Implantation of a left ventricular assist device to provide long-term support for end-stage Duchenne muscular dystrophy-associated cardiomyopathy. ESC Hear Fail. 2017;4:379–383. doi: 10.1002/ehf2.12160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Duboc D, Meune C, Pierre B, Wahbi K, Eymard B, Toutain A, Berard C, Vaksmann G, Weber S, Bécane HM. Perindopril preventive treatment on mortality in Duchenne muscular dystrophy: 10 years’ follow-up. Am Heart J. 2007;154:596–602. doi: 10.1016/j.ahj.2007.05.014. [DOI] [PubMed] [Google Scholar]
- 89.Viollet L, Thrush PT, Flanigan KM, Mendell JR, Allen HD. Effects of angiotensin-converting enzyme inhibitors and/or beta blockers on the cardiomyopathy in Duchenne muscular dystrophy. Am J Cardiol. 2012;110:98–102. doi: 10.1016/j.amjcard.2012.02.064. [DOI] [PubMed] [Google Scholar]
- 90.Allen HD, Flanigan KM, Thrush PT, Dvorchik I, Yin H, Canter C, Connolly AM, Parrish M, McDonald CM, Braunlin E, Colan SD, Day J, Darras B, Mendell JR. A randomized, double-blind trial of lisinopril and losartan for the treatment of cardiomyopathy in Duchenne muscular dystrophy. PLoS Curr. 2013:5. doi: 10.1371/currents.md.2cc69a1dae4be7dfe2bcb420024ea865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kajimoto H, Ishigaki K, Okumura K, Tomimatsu H, Nakazawa M, Saito K, Osawa M, Nakanishi T. Beta-blocker therapy for cardiac dysfunction in patients with muscular dystrophy. Circ J. 2006;70:991–994. doi: 10.1253/circj.70.991. [DOI] [PubMed] [Google Scholar]
- 92.Matsumura T, Tamura T, Kuru S, Kikuchi Y, Kawai M. Carvedilol can prevent cardiac events in Duchenne muscular dystrophy. Intern Med. 2010;49:1357–1363. doi: 10.2169/internalmedicine.49.3259. [DOI] [PubMed] [Google Scholar]
- 93.Schram G, Fournier A, Leduc H, Dahdah N, Therien J, Vanasse M, Khairy P. All-cause mortality and cardiovascular outcomes with prophylactic steroid therapy in Duchenne muscular dystrophy. J Am Coll Cardiol. 2013;61:948–954. doi: 10.1016/j.jacc.2012.12.008. [DOI] [PubMed] [Google Scholar]
- 94.Buyse GM, Goemans N, Van Den Hauwe M, Meier T. Effects of glucocorticoids and idebenone on respiratory function in patients with Duchenne muscular dystrophy. Pediatr Pulmonol. 2013;48:912–920. doi: 10.1002/ppul.22688. [DOI] [PubMed] [Google Scholar]
- 95.Tandon A, Villa CR, Hor KN, Jefferies JL, Gao Z, Towbin JA, Wong BL, Mazur W, Fleck RJ, Sticka JJ, Benson DW, Taylor MD. Myocardial fibrosis burden predicts left ventricular ejection fraction and is associated with age and steroid treatment duration in Duchenne muscular dystrophy. J Am Heart Assoc. 2015;4:e001338. doi: 10.1161/JAHA.114.001338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Fenichel GM, Florence JM, Pestronk A, Mendell JR, Moxley RT, Griggs RC, Brooke MH, Miller JP, Robison J, King W, Signore L, Pandya S, Schierbecker J, Wilson B. Long-term benefit from prednisone therapy in Duchenne muscular dystrophy. Neurology. 1991;41:1874–1877. doi: 10.1212/wnl.41.12.1874. [DOI] [PubMed] [Google Scholar]
- 97.McMurray JJ, Östergren J, Swedberg K, Granger CB, Held P, Michelson EL, Olofsson B, Yusuf S, Pfeffer MA. Effects of candesartan in patients with chronic heart failure and reduced left-ventricular systolic function taking angiotensin-converting-enzyme inhibitors: the charm-added trial. Lancet. 2003;362:767–771. doi: 10.1016/S0140-6736(03)14283-3. [DOI] [PubMed] [Google Scholar]
- 98.Wu H, Lima WF, Zhang H, Fan A, Sun H, Crooke ST. Determination of the role of the human RNase H1 in the pharmacology of dna-like antisense drugs. J Biol Chem. 2004;279:17181–9. doi: 10.1074/jbc.M311683200. [DOI] [PubMed] [Google Scholar]
- 99.Lee J, Yokota T. Antisense therapy in neurology. J Pers Med. 2013;3:144–176. doi: 10.3390/jpm3030144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kole R, Krieg AM. Exon skipping therapy for Duchenne muscular dystrophy. Adv Drug Deliv Rev. 2015;87:104–107. doi: 10.1016/j.addr.2015.05.008. [DOI] [PubMed] [Google Scholar]
- 101.Guncay A, Yokota T. Antisense oligonucleotide drugs for Duchenne muscular dystrophy: how far have we come and what does the future hold? Future Med Chem. 2015;7:1631–1635. doi: 10.4155/fmc.15.116. [DOI] [PubMed] [Google Scholar]
- 102.Nguyen Q, Yokota T. Immortalized muscle cell model to test the exon skipping efficacy for Duchenne muscular dystrophy. J Pers Med. 2017;7:E13. doi: 10.3390/jpm7040013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Dias N, Stein CA. Antisense oligonucleotides: basic concepts and mechanisms. Mol Cancer Ther. 2002;1:347–355. [PubMed] [Google Scholar]
- 104.Eder PS, DeVine RJ, Dagle JM, Walder JA. Substrate specificity and kinetics of degradation of antisense oligonucleotides by a 3’ exonuclease in plasma. Antisense Res Dev. 1991;1:141–151. doi: 10.1089/ard.1991.1.141. [DOI] [PubMed] [Google Scholar]
- 105.Geary RS, Norris D, Yu R, Bennett CF. Pharmacokinetics, biodistribution and cell uptake of antisense oligonucleotides. Adv Drug Deliv Rev. 2015;87:46–51. doi: 10.1016/j.addr.2015.01.008. [DOI] [PubMed] [Google Scholar]
- 106.Juliano RL, Carver K. Cellular uptake and intracellular trafficking of oligonucleotides. Adv Drug Deliv Rev. 2015;87:35–45. doi: 10.1016/j.addr.2015.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.De Clercq E, Eckstein E, Merigan TC. Interferon induction increased through chemical modification of a synthetic polyribonucleotide. Science. 1969;165:1137–1139. doi: 10.1126/science.165.3898.1137. [DOI] [PubMed] [Google Scholar]
- 108.Rifai A, Brysch W, Fadden K, Clark J, Schlingensiepen KH. Clearance kinetics, biodistribution, and organ saturability of phosphorothioate oligodeoxynucleotides in mice. Am J Pathol. 1996;149:717–725. [PMC free article] [PubMed] [Google Scholar]
- 109.Watanabe TA, Geary RS, Levin AA. Plasma protein binding of an antisense oligonucleotide targeting human ICAM-1 (ISIS 2302) Oligonucleotides. 2006;16:169–180. doi: 10.1089/oli.2006.16.169. [DOI] [PubMed] [Google Scholar]
- 110.Freier S, Altmann KH. The ups and downs of nucleic acid duplex stability: structure-stability studies on chemically-modified DNA: RNA duplexes. Nucleic Acids Res. 1997;25:4429–4443. doi: 10.1093/nar/25.22.4429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Lubini P, Zürcher W, Egli M. Stabilizing effects of the RNA 2’-substituent: crystal structure of an oligodeoxynucleotide duplex containing 2’-O-methylated adenosines. Chem Biol. 1994;1:39–45. doi: 10.1016/1074-5521(94)90039-6. [DOI] [PubMed] [Google Scholar]
- 112.McKay RA, Miraglia LJ, Cummins LL, Owens SR, Sasmor H, Dean NM. Characterization of a potent and specific class of antisense oligonucleotide inhibitor of human protein kinase C-alpha expression. J Biol Chem. 1999;274:1715–1722. doi: 10.1074/jbc.274.3.1715. [DOI] [PubMed] [Google Scholar]
- 113.Hamm S, Latz E, Hangel D, Müller T, Yu P, Golenbock D, Sparwasser T, Wagner H, Bauer S. Alternating 2’-O-ribose methylation is a universal approach for generating non-stimulatory siRNA by acting as TLR7 antagonist. Immunobiology. 2010;215:559–569. doi: 10.1016/j.imbio.2009.09.003. [DOI] [PubMed] [Google Scholar]
- 114.Nielsen PE, Egholm M, et al. An introduction to peptide nucleic acid. Curr Issues Mol Biol. 1999;1:89–104. [PubMed] [Google Scholar]
- 115.Summerton J, Stein D, Huang B, Matthews P, Weller D, Partridge M. Morpholino and phosphorothioate antisense oligomers compared in cell-free and in-cell systems. Antisense Nucleic Acid Drug Dev. 1997;7:63–70. doi: 10.1089/oli.1.1997.7.63. [DOI] [PubMed] [Google Scholar]
- 116.Hudziak RM, Barofsky E, Barofski DF, Weller DL, Huang SB, Weller DD. Resistance of morpholino phosphorodiamidate oligomers to enzymatic degradation. Antisense Nucleic Acid Drug Dev. 1996;6:267–272. doi: 10.1089/oli.1.1996.6.267. [DOI] [PubMed] [Google Scholar]
- 117.Moulton JD. Guide for morpholino users: toward therapeutics. J Drug Discov Dev Deliv. 2016;3:1023. [Google Scholar]
- 118.U.S. Food and drug administration. FDA grants accelerated approval to first drug for Duchenne muscular dystrophy. FDA News Release. 2016 [Google Scholar]
- 119.Aartsma-Rus A, Krieg AM. FDA approves eteplirsen for Duchenne muscular dystrophy: the next chapter in the eteplirsen saga. Nucleic Acid Ther. 2017;27:1–3. doi: 10.1089/nat.2016.0657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Lim KR, Maruyama R, Yokota T. Eteplirsen in the treatment of Duchenne muscular dystrophy. Drug Des Devel Ther. 2017;11:533–545. doi: 10.2147/DDDT.S97635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Komaki H, Nagata T, Saito T, Masuda S, Takeshita E, Sasaki M, Tachimori H, Nakamura H, Aoki Y, Takeda S. Systemic administration of the antisense oligonucleotide NS-065/NCNP-01 for skipping of exon 53 in patients with Duchenne muscular dystrophy. Sci Transl Med. 2018;10:eaan0713. doi: 10.1126/scitranslmed.aan0713. [DOI] [PubMed] [Google Scholar]
- 122.Yokota T, Lu QL, Partridge T, Kobayashi M, Nakamura A, Takeda S, Hoffman E. Efficacy of systemic morpholino exon-skipping in Duchenne dystrophy dogs. Ann Neurol. 2009;65:667–676. doi: 10.1002/ana.21627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Maruyama R, Echigoya Y, Caluseriu O, Aoki Y, Takeda S, Yokota T. Systemic delivery of morpholinos to skip multiple exons in a dog model of Duchenne muscular dystrophy. Methods Mol Biol. 2017;1565:201–213. doi: 10.1007/978-1-4939-6817-6_17. [DOI] [PubMed] [Google Scholar]
- 124.Malerba A, Boldrin L, Dickson G. Long-term systemic administration of unconjugated morpholino oligomers for therapeutic expression of dystrophin by exon skipping in skeletal muscle: implications for cardiac muscle integrity. Nucleic Acid Ther. 2011;21:293–298. doi: 10.1089/nat.2011.0306. [DOI] [PubMed] [Google Scholar]
- 125.Townsend DW, Yasuda S, Li S, Chamberlain JS, Metzger JM. Emergent dilated cardiomyopathy caused by targeted repair of dystrophic skeletal muscle. Mol Ther. 2008;16:832–835. doi: 10.1038/mt.2008.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Margus H, Padari K, Pooga M. Insights into cell entry and intracellular trafficking of peptide and protein drugs provided by electron microscopy. Adv Drug Deliv Rev. 2013;65:1031–1038. doi: 10.1016/j.addr.2013.04.013. [DOI] [PubMed] [Google Scholar]
- 127.Cleal K, He L, Watson PD, Jones AT. Endocytosis, intracellular traffic and fate of cell penetrating peptide based conjugates and nanoparticles. Curr Pharm Des. 2013;19:2878–2894. doi: 10.2174/13816128113199990297. [DOI] [PubMed] [Google Scholar]
- 128.El-Sayed A, Harashima H. Endocytosis of gene delivery vectors: from clathrin-dependent to lipid raft-mediated endocytosis. Mol Ther. 2013;21:1118–1130. doi: 10.1038/mt.2013.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Juliano RL. The delivery of therapeutic oligonucleotides. Nucleic Acids Res. 2016;44:6518–6548. doi: 10.1093/nar/gkw236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.El-Sayed A, Futaki S, Harashima H. Delivery of macromolecules using arginine-rich cell-penetrating peptides: ways to overcome endosomal entrapment. AAPS J. 2009;11:13–22. doi: 10.1208/s12248-008-9071-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Lehto T, Alvarez AC, Gauck S, Gait MJ, Coursindel T, Wood MJ, Lebleu B, Boisguerin P. Cellular trafficking determines the exon skipping activity of Pip6a-PMO in mdx skeletal and cardiac muscle cells. Nucleic Acids Res. 2014;42:3207–3217. doi: 10.1093/nar/gkt1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Steffens R, Leumann CJ. Synthesis and thermodynamic and biophysical properties of tricyclo-DNA. J Am Chem Soc. 1999;121:3249–3255. [Google Scholar]
- 133.Steffens R, Leumann CJ. Tricycio-DNA: a phosphodiester-backbone based DNA analog exhibiting strong complementary base-pairing properties [7] . J Am Chem Soc. 1997;119:11548–11549. [Google Scholar]
- 134.Tarköy M, Leumann C. Synthesis and pairing properties of Decanucleotides from (3’S,5’R)-2’-Deoxy-3’,5’-ethanoβ-D-ribofuranosyladenine and -thymine. Angew Chemie Int Ed English. 1993;32:1432–1434. [Google Scholar]
- 135.Aupy P, Echevarría L, Relizani K, Goyenvalle A. The use of Tricyclo-DNA oligomers for the treatment of genetic disorders. Biomedicines. 2017;6:E2. doi: 10.3390/biomedicines6010002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Goyenvalle A, Leumann C, Garcia L. Therapeutic potential of tricyclo-DNA antisense oligonucleotides. J Neuromuscul Dis. 2016;3:157–167. doi: 10.3233/JND-160146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Renneberg D, Bouliong E, Reber U, Schuümperli D, Leumann CJ. Antisense properties of tricyclo-dna. Nucleic Acids Res. 2002;30:2751–2757. doi: 10.1093/nar/gkf412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Relizani K, Griffith G, Echevarría L, Zarrouki F, Facchinetti P, Vaillend C, Leumann C, Garcia L, Goyenvalle A. Efficacy and safety profile of tricyclo-DNA antisense oligonucleotides in Duchenne muscular dystrophy mouse model. Mol Ther Nucleic Acids. 2017;8:144–157. doi: 10.1016/j.omtn.2017.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Goyenvalle A, Griffith G, Babbs A, El Andaloussi S, Ezzat K, Avril A, Dugovic B, Chaussenot R, Ferry A, Voit T, Amthor H, Bühr C, Schürch S, Wood MJ, Davies KE, Vaillend C, Leumann C, Garcia L. Functional correction in mouse models of muscular dystrophy using exon-skipping tricyclo-DNA oligomers. Nat Med. 2015;21:270–275. doi: 10.1038/nm.3765. [DOI] [PubMed] [Google Scholar]
- 140.Ezzat K, Aoki Y, Koo T, McClorey G, Benner L, Coenen-Stass A, O’Donovan L, Lehto T, Garcia-Guerra A, Nordin J, Saleh AF, Behlke M, Morris J, Goyenvalle A, Dugovic B, Leumann C, Gordon S, Gait MJ, El-Andaloussi S, Wood MJ. Self-Assembly into nanoparticles is essential for receptor mediated uptake of therapeutic antisense oligonucleotides. Nano Lett. 2015;15:4364–4373. doi: 10.1021/acs.nanolett.5b00490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Nel AE, Mädler L, Velegol D, Xia T, Hoek EM, Somasundaran P, Klaessig F, Castranova V, Thompson M. Understanding biophysicochemical interactions at the nano-bio interface. Nat Mater. 2009;8:543–557. doi: 10.1038/nmat2442. [DOI] [PubMed] [Google Scholar]
- 142.Pooga M, Langel Ü. Classes of cell-penetrating peptides. Methods Mol Biol. 2015;1324:3–28. doi: 10.1007/978-1-4939-2806-4_1. [DOI] [PubMed] [Google Scholar]
- 143.Heitz F, Morris MC, Divita G. Twenty years of cell-penetrating peptides: from molecular mechanisms to therapeutics. Br J Pharmacol. 2009;157:195–206. doi: 10.1111/j.1476-5381.2009.00057.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Futaki S, Hirose H, Nakase I. Arginine-rich peptides: methods of translocation through biological membranes. Curr Pharm Des. 2013;19:2863–2868. doi: 10.2174/1381612811319160003. [DOI] [PubMed] [Google Scholar]
- 145.Lehto T, Ezzat K, Wood MJ, EL Andaloussi S. Peptides for nucleic acid delivery. Adv Drug Deliv Rev. 2016;106:172–182. doi: 10.1016/j.addr.2016.06.008. [DOI] [PubMed] [Google Scholar]
- 146.Yin H, Moulton HM, Seow Y, Boyd C, Boutilier J, Iverson P, Wood MJ. Cell-penetrating peptide-conjugated antisense oligonucleotides restore systemic muscle and cardiac dystrophin expression and function. Hum Mol Genet. 2008;17:3909–3918. doi: 10.1093/hmg/ddn293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Abes R, Arzumanov AA, Moulton HM, Abes S, Ivanova GD, Iversen PL, Gait MJ, Lebleu B. Cell-penetrating-peptide-based delivery of oligonucleotides: an overview. Biochem Soc Trans. 2007;35:775–779. doi: 10.1042/BST0350775. [DOI] [PubMed] [Google Scholar]
- 148.Amantana A, Moulton HM, Cate ML, Reddy MT, Whitehead T, Hassinger JN, Youngblood DS, Iversen PL. Pharmacokinetics, biodistribution, stability and toxicity of a cell-penetrating peptide - Morpholino oligomer conjugate. Bioconjug Chem. 2007;18:1325–1331. doi: 10.1021/bc070060v. [DOI] [PubMed] [Google Scholar]
- 149.Abes S, Moulton HM, Clair P, Prevot P, Youngblood DS, Wu RP, Iversen PL, Lebleu B. Vectorization of morpholino oligomers by the (R-Ahx-R)4peptide allows efficient splicing correction in the absence of endosomolytic agents. J Control Release. 2006;116:304–313. doi: 10.1016/j.jconrel.2006.09.011. [DOI] [PubMed] [Google Scholar]
- 150.Wu B, Moulton HM, Iversen PL, Jiang J, Li J, Li J, Spurney CF, Sali A, Guerron AD, Nagaraju K, Doran T, Lu P, Xiao X, Lu QL. Effective rescue of dystrophin improves cardiac function in dystrophin-deficient mice by a modified morpholino oligomer. Proc Natl Acad Sci U S A. 2008;105:14814–14819. doi: 10.1073/pnas.0805676105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Echigoya Y, Nakamura A, Nagata T, Urasawa N, Lim KR, Trieu N, Panesar D, Kuraoka M, Moulton HM, Saito T, Aoki Y, Iversen P, Sazani P, Kole R, Maruyama R, Partridge T, Takeda S, Yokota T. Effects of systemic multiexon skipping with peptide-conjugated morpholinos in the heart of a dog model of Duchenne muscular dystrophy. Proc Natl Acad Sci U S A. 2017;114:4213–4218. doi: 10.1073/pnas.1613203114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Ivanova GD, Arzumanov A, Abes R, Yin H, Wood MJ, Lebleu B, Gait MJ. Improved cell-penetrating peptide-PNA conjugates for splicing redirection in HeLa cells and exon skipping in mdx mouse muscle. Nucleic Acids Res. 2008;36:6418–6428. doi: 10.1093/nar/gkn671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Yin H, Saleh AF, Betts C, Camelliti P, Seow Y, Ashraf S, Arzumanov A, Hammond S, Merritt T, Gait MJ, Wood MJ. Pip5 transduction peptides direct high efficiency oligonucleotide-mediated dystrophin exon skipping in heart and phenotypic correction in mdx mice. Mol Ther. 2011;19:1295–1303. doi: 10.1038/mt.2011.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Betts C, Saleh AF, Arzumanov AA, Hammond SM, Godfrey C, Coursindel T, Gait MJ, Wood MJ. Pip6-PMO, a new generation of peptide-oligonucleotide conjugates with improved cardiac exon skipping activity for DMD treatment. Mol Ther Nucleic Acids. 2012;1:e38. doi: 10.1038/mtna.2012.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Jirka SM, Heemskerk H, Tanganyika-de Winter CL, Muilwijk D, Pang KH, de Visser PC, Janson A, Karnaoukh TG, Vermue R, ‘t Hoen PA, van Deutekom JC, Aguilera B, Aartsma-Rus A. Peptide conjugation of 2’-O-methyl phosphorothioate antisense oligonucleotides enhances cardiac uptake and exon skipping in mdx mice. Nucleic Acid Ther. 2014;24:25–36. doi: 10.1089/nat.2013.0448. [DOI] [PubMed] [Google Scholar]
- 156.Moulton HM, Wu B, Jearawiriyapaisarn N, Sazani P, Lu QL, Kole R. Peptide-morpholino conjugate: a promising therapeutic for Duchenne muscular dystrophy. Ann N Y Acad Sci. 2009;1175:55–60. doi: 10.1111/j.1749-6632.2009.04976.x. [DOI] [PubMed] [Google Scholar]
- 157.Moulton HM, Moulton JD. Morpholinos and their peptide conjugates: therapeutic promise and challenge for Duchenne muscular dystrophy. Biochim Biophys Acta Biomembr. 2010;1798:2296–2303. doi: 10.1016/j.bbamem.2010.02.012. [DOI] [PubMed] [Google Scholar]
- 158.Wu B, Lu P, Cloer C, Shaban M, Grewal S, Milazi S, Shah SN, Moulton HM, Lu QL. Long-term rescue of dystrophin expression and improvement in muscle pathology and function in dystrophic mdx mice by peptide-conjugated morpholino. Am J Pathol. 2012;181:392–400. doi: 10.1016/j.ajpath.2012.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Futaki S, Nakase I, Suzuki T, Youjun Z, Sugiura Y. Translocation of branched-chain arginine peptides through cell membranes: flexibility in the spatial disposition of positive charges in membrane-permeable peptides. Biochemistry. 2002;41:7925–7930. doi: 10.1021/bi0256173. [DOI] [PubMed] [Google Scholar]
- 160.Li YF, Morcos PA. Design and synthesis of dendritic molecular transporter that achieves efficient in vivo delivery of morpholino antisense oligo. Bioconjug Chem. 2008;19:1464–1470. doi: 10.1021/bc8001437. [DOI] [PubMed] [Google Scholar]
- 161.Wu B, Li Y, Morcos PA, Doran TJ, Lu P, Lu QL. Octa-guanidine morpholino restores dystrophin expression in cardiac and skeletal muscles and ameliorates pathology in dystrophic mdx mice. Mol Ther. 2009;17:864–871. doi: 10.1038/mt.2009.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Yokota T, Nakamura A, Nagata T, Saito T, Kobayashi M, Aoki Y, Echigoya Y, Partridge T, Hoffman EP, Takeda S. Extensive and prolonged restoration of dystrophin expression with vivo-morpholino-mediated multiple exon skipping in dystrophic dogs. Nucleic Acid Ther. 2012;22:306–315. doi: 10.1089/nat.2012.0368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Tsutsui JM, Xie F, Porter RT. The use of microbubbles to target drug delivery. Cardiovasc Ultrasound. 2004;2:23. doi: 10.1186/1476-7120-2-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Taniyama Y, Tachibana K, Hiraoka K, Namba T, Yamasaki K, Hashiya N, Aoki M, Ogihara T, Yasufumi K, Morishita R. Local delivery of plasmid DNA into rat carotid artery using ultrasound. Circulation. 2002;105:1233–1239. doi: 10.1161/hc1002.105228. [DOI] [PubMed] [Google Scholar]
- 165.Chen S, Shohet RV, Bekeredjian R, Frenkel P, Grayburn PA. Optimization of ultrasound parameters for cardiac gene delivery of adenoviral or plasmid deoxyribonucleic acid by ultrasound-targeted microbubble destruction. J Am Coll Cardiol. 2003;42:301–308. doi: 10.1016/s0735-1097(03)00627-2. [DOI] [PubMed] [Google Scholar]
- 166.Shohet RV, Chen S, Zhou YT, Wang Z, Meidell RS, Unger RH, Grayburn PA. Echocardiographic destruction of albumin microbubbles directs gene delivery to the myocardium. Circulation. 2000;101:2554–2556. doi: 10.1161/01.cir.101.22.2554. [DOI] [PubMed] [Google Scholar]
- 167.Mukherjee D, Wong J, Griffin B, Ellis SG, Porter T, Sen S, Thomas JD. Ten-fold augmentation of endothelial uptake of vascular endothelial growth factor with ultrasound after systemic administration. J Am Coll Cardiol. 2000;35:1678–1686. doi: 10.1016/s0735-1097(00)00575-1. [DOI] [PubMed] [Google Scholar]
- 168.Chômas JE, Dayton P, Alien J, Morgan K, Ferrara KW. Mechanisms of contrast agent destruction. IEEE Trans Ultrason Ferroelectr Freq Control. 2001;48:232–248. doi: 10.1109/58.896136. [DOI] [PubMed] [Google Scholar]
- 169.Chomas JE, Dayton P, May D, Ferrara K. Threshold of fragmentation for ultrasonic contrast agents. J Biomed Opt. 2001;6:141–150. doi: 10.1117/1.1352752. [DOI] [PubMed] [Google Scholar]
- 170.Deng CX, Sieling F, Pan H, Cui J. Ultrasound-induced cell membrane porosity. Ultrasound Med Biol. 2004;30:519–526. doi: 10.1016/j.ultrasmedbio.2004.01.005. [DOI] [PubMed] [Google Scholar]
- 171.Alter J, Sennoga CA, Lopes DM, Eckersley RJ, Wells DJ. Microbubble stability is a major determinant of the efficiency of ultrasound and microbubble mediated in vivo gene transfer. Ultrasound Med Biol. 2009;35:976–984. doi: 10.1016/j.ultrasmedbio.2008.12.015. [DOI] [PubMed] [Google Scholar]
- 172.Rimessi P, Sabatelli P, Fabris M, Braghetta P, Bassi E, Spitali P, Vattemi G, Tomelleri G, Mari L, Perrone D, Medici A, Neri M, Bovolenta M, Martoni E, Maraldi NM, Gualandi F, Merlini L, Ballestri M, Tondelli L, Sparnacci K, Bonaldo P, Caputo A, Laus M, Ferlini A. Cationic PMMA nanoparticles bind and deliver antisense oligoribonucleotides allowing restoration of dystrophin expression in the mdx mouse. Mol Ther. 2009;17:820–827. doi: 10.1038/mt.2009.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Ferlini A, Sabatelli P, Fabris M, Bassi E, Falzarano S, Vattemi G, Perrone D, Gualandi F, Maraldi NM, Merlini L, Sparnacci K, Laus M, Caputo A, Bonaldo P, Braghetta P, Rimessi P. Dystrophin restoration in skeletal, heart and skin arrector pili smooth muscle of mdx mice by ZM2 NP-AON complexes. Gene Ther. 2010;17:432–438. doi: 10.1038/gt.2009.145. [DOI] [PubMed] [Google Scholar]
- 174.Wang M, Wu B, Lu P, Cloer C, Tucker JD, Lu Q. Polyethylenimine-modified pluronics (PCMs) improve morpholino oligomer delivery in cell culture and dystrophic mdx mice. Mol Ther. 2013;21:210–216. doi: 10.1038/mt.2012.236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Wang M, Wu B, Tucker JD, Shah SN, Lu P, Bollinger LE, Lu Q. Tween 85-Modified Low molecular weight PEI enhances exon-skipping of antisense morpholino oligomer in vitro and in mdx mice. Mol Ther Nucleic Acids. 2017;9:120–131. doi: 10.1016/j.omtn.2017.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]