

Abstract
The purpose of this study was to determine whether retinol palmitate could protect against myocardial ischemia/reperfusion (I/R) injury and explore the underlying mechanism. Retinol palmitate reduced the level of reactive oxygen species and prevented cellular apoptosis. In vivo, retinol palmitate increased superoxide dismutase (SOD) activity and reduced the level of malondialdehyde in I/R mice. Retinol palmitate also decreased myocardial infarct size and reduced cellular apoptosis by suppressing the expression of proapoptotic-related proteins and increasing that of SOD-related proteins. Our results suggest that retinol palmitate pretreatment has a protective effect against myocardial I/R injury by maintaining the balance between intracellular oxidants and antioxidants.
Keywords: Retinol palmitate, antioxidant, apoptosis, myocardial ischemia reperfusion injury
Introduction
Acute myocardial infarction (AMI) is characterized by irreversible myocardial necrosis due to an imbalanced blood supply [1]. Although a variety of treatments have been proposed, AMI remains the major cause of disability and mortality [2-4]. Rapid reperfusion is an important goal of the AMI treatment. However, reperfusion itself can lead to the damage and death of cardiomyocytes [5].
The mechanisms of ischemia/reperfusion (I/R) injury are complex, including generation of reactive oxygen species (ROS), calcium overload, leukocyte activation, apoptosis, and endothelial dysfunction [6].
In some experimental models, ischemia results in an increased level of myocardial ROS, followed by a reperfusion-induced myocardial ROS surge. In the past few decades, it has been documented that myocardial ROS generation is accelerated following myocardial I/R, which plays an important role in I/R-induced myocardial damage [7,8]. Several studies have reported that antioxidants reduce free radical production and may thereby inhibit thrombosis and decrease myocardial infarct size and arrhythmia in AMI [9,10]. Antioxidants act via the following mechanisms: (I) capturing ROS or their precursors, (II) suppressing ROS formation, (III) decreasing the catalysis of ROS generation by binding to metal ions, (IV) augmenting endogenous antioxidant capacity, and (V) upregulating Bcl-2 to decrease apoptosis [11].
Apoptosis is a unique form of cell damage involving enzymes that synthesize and degrade their own DNA. Researchers have revealed that myocardial I/R is accompanied by increased apoptotic cells [12-15]. Furthermore, oxidative stress triggers cardiomyocyte apoptosis during myocardial I/R [14,16-18].
Recent studies have found that carotenoids, vitamin A, and provitamin A carotenoids can be used to suppress the occurrence and progression of cardiac diseases because of their effective antioxidant functions. Retinol acts as an effective peroxyl radical scavenger by suppressing peroxidation in a homogeneous solution of methyl linoleate and in phosphatidylcholine liposomes [19]. A 28-year study of an Israeli population showed that an increase in dietary vitamin A is closely related to a reduction in mortality of patients with ischemic heart disease [20]. Vitamin A deficiency aggravates cardiac function and myocardial remodeling following AMI in rats [21].
Retinol palmitate, an analog of vitamin A, has been shown to promote axonal growth, neuronal differentiation, and neural patterning, and is a potential neuroprotective agent in cerebral I/R injury [22,23]. The purpose of our research was to determine whether retinol palmitate could prevent myocardial I/R injury and explore its underlying mechanism.
Materials and methods
Animals and H9C2 cells culture
All experimental procedures were approved by the Committee on the Ethics of Animal Experiments of Wenzhou Medical University. Male C57BL/6 mice (20-25 g, 6-7 weeks old) were provided by the SLAC Laboratory Animal Center of Shanghai. Mice were housed in a specific pathogen-free animal room at Wenzhou Medical University Animal Center and fed a free diet and water at a constant room temperature with a normal circadian rhythm.
H9C2 cells were purchased from the American Type Culture Collection (Manassas, VA, USA). H9C2 cells were cultured in high-glucose Dulbecco’s Modified Eagle Medium (DMEM; HyClone Laboratories, Inc., Erie, UK) supplemented with 10% (v/v) fetal bovine serum (FBS; Gibco Laboratories, Gaithersburg, MD, USA) and penicillin/streptomycin in humidified air (5% CO2, 21% O2) at 37°C. The medium was refreshed every 2 days. A stock of H9C2 cells was grown in a 100-mm Petri dish [24].
In vitro treatments
At the beginning of the experiment, retinol palmitate stock solution was dissolved in dimethyl sulfoxide (DMSO) and diluted with normal culture medium (final DMSO concentration: 0.1%). H9C2 cells were divided into five groups. In group C, cells were cultured in conventional culture solution. In the I/R group, cells were grown under simulated ischemic conditions for 2 h and subjected to reperfusion for 4 h in normal DMEM (hereafter referred to as I/R simulation culture). In the DMSO group, cells were pretreated with DMSO (0.1%) for 4 h, then followed by I/R simulation culture. In the I/R+L group, cells were pretreated with 0.1 μM of retinol palmitate for 4 h, then followed by I/R simulation culture. In the I/R+H group, cells were pretreated with 1 μM retinol palmitate for 4 h, then followed by I/R simulation culture.
I/R in vitro
H9C2 cells, a widely used cell line, were cultured in DMEM supplemented with 10% heat-inactivated FBS at 37°C in a humidified atmosphere containing 5% CO2. Simulated I/R treatment was carried out with physiological concentrations of potassium, hydrogen, and lactate as previously described [25]. Briefly, cardiomyocytes were exposed to ischemic buffer containing (in mmol/L) 137 NaCl, 12 KCl, 0.49 MgCl2, 0.9 CaCl2, 4 HEPES, 10 deoxyglucose, 0.75 sodium dithionate, and 20 lactate (pH 6.5) for 2 h in a humidified cell culture incubator (21% O2, 5% CO2, 37°C). Reperfusion was performed by returning the cells to normal culture medium for 4 h in a humidified cell culture incubator (21% O2, 5% CO2, 37°C).
Measurement of ROS
Cells were incubated with 10 μmol/L of the ROS sensitive dye 2’,7’-dichloruoresceindiacetate at 37°C for 20 min. A flow cytometry sorter (BD Biosciences, San Jose, CA, USA) was used to determine the level of ROS. Experiments were repeated three times.
Determination of the apoptotic ratio
In a dark environment, 5 μL annexin V-fluorescein isothiocyanate and 10 μL propidium iodide (PI; 20 μg/mL) were added to H9C2 cells (1.3 × 105) and incubated for 15 min. Flow cytometry was performed to analyze the fluorescence intensities of annexin V/PI-stained cells within 1 h. Apoptotic cells (annexin V+/PI-) were counted. The apoptotic ratio was calculated using BD FACS software. Experiments were repeated six times.
I/R in vivo
Mice were anesthetized with isoflurane, intubated, and ventilated with a respirator. A heating pad was used to maintain body temperature at 37°C. After left lateral thoracotomy, an 8-0 nylon suture was used to occlude the left anterior descending coronary artery (LAD) for 40 min, which was then released for reperfusion. Three days before ischemia, retinol palmitate (12 or 36 mg/kg) or vehicle (medium chain triglyceride 10 ml/kg) was administered via intraperitoneal injection. The development of ischemic ST-segment elevation was used to confirm successful coronary occlusion by electrocardiographic monitoring (AD Instruments, Sydney, Australia). After reperfusion for 40 min, mice were intraperitoneally injected with chloral sodium chloride (100 mg/kg) and euthanized by posterior cervical dislocation. Hearts were removed and the ischemic region of the left ventricle was separated before storing in liquid nitrogen. After reperfusion for 4 h, hearts were harvested and infarct size was measured by dual staining. The myocardium in the risk area was stained red with 1% triphenyl tetrazolium chloride and normal myocardium was stained blue with 2% Evans Blue. The necrotic tissue in the infarct zone was not stained. Hearts were fixed and sectioned into 1-mm slices, photographed using a Leica MZ95 microscope (Leica, Wetzlar, Germany), and analyzed with ImageJ software (NIH, Bethesda, MD, USA). The myocardial infarct ratio was calculated as follows: infarct area (INF)/area at ischemic risk (AAR).
In vivo treatments
Mice were randomly divided into four groups: (1) sham group: mice were anesthetized with isoflurane and a line was passed under the LAD without occlusion; (2) I/R group: mice were pretreated with 0.9% normal saline; (3) I/R+L group: mice were pretreated with retinol palmitate (12 mg/kg); and (4) I/R+H group: mice were pretreated with retinol palmitate (36 mg/kg). Retinol palmitate was given 30 min before ischemia induction. Coronary occlusion lasted for 40 min followed by reperfusion for 4 h. At the end of reperfusion, blood was collected from the common carotid artery and the heart was harvested.
Immunoblot analysis
Hearts were extracted approximately 4 h after heart reperfusion. The ischemic region of the left ventricle was homogenized in ice-cold lysis buffer (pH 7.4) containing 1% protease and phosphatase inhibitors, 20 mM Tris-HCl, 1% sodium deoxycholate, 10 mM NaF, 1% Triton X-100, 2.5 mM EDTA, and 0.1% SDS. The lysates were sonicated and centrifuged at 11,000 × g for 30 min. The supernatant was transferred to a 1.5-mL tube. The bovine serum albumin method was used to determine protein concentration. Western blot analysis was conducted as previously described [26]. The following primary antibodies were used: anti-cleaved caspase-3 (1:1000; Bioworld Technology, Inc., St. Louis Park, MN, USA), anti-Bcl-2 (1:500; Abcam, Cambridge, UK), anti-Bax (1:1000; Abcam), anti-superoxide dismutase (SOD)-1 (1:2000; Abcam), anti-SOD2 (1:5000; Abcam), and anti-glyceraldehyde 3-phosphate dehydrogenase (1:1000; Abcam). Membranes were washed with Tris-buffered saline containing Tween 20 and treated with the appropriate secondary antibodies for 2 h at room temperature.
Myocardial terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) staining
Approximately 4 h after reperfusion, mouse hearts were harvested for TUNEL staining. Formalin-fixed hearts of I/R mice were embedded in paraffin and sectioned (5-μm). Sections were deparaffinized and treated with 3% H2O2 for 10 min. TUNEL staining was performed according to the manufacturer’s instructions (Beyotime Biotechnology, Shanghai, China). Images were viewed and photographed with a fluorescence microscope (NIKON A1R/A1, Nikon, Tokyo, Japan).
SOD activity and malondialdehyde (MDA) levels
Myocardial SOD activity and MDA levels were measured using commercially available SOD and MDA kits according to the manufacturer’s instructions (Beyotime Biotechnology).
Analysis of serum creatine kinase CK-MB levels
A blood sample (0.5 mL) was collected from the arteria carotis into a tube containing EDTA. After centrifugation, plasma was collected and stored at -80°C until use. The plasma concentrations of CK-MB were measured using a commercial enzyme-linked immunosorbent assay CK-MB kit according to the manufacturer’s instructions (JianCheng Bioengineering Institute, Nanjing, China).
Statistical analysis
All data are expressed as the mean ± SD. Differences in all measured parameters were assessed by the one-way analysis of variance (ANOVA) and the Student-Newman-Keuls (SNK) test. Data were analyzed using SPSS22.0 software and GraphPad Prism-5 statistical software (GraphPad Software, Inc., La Jolla, CA, USA). P values < 0.05 were considered statistically significant.
Results
Pretreatment with retinol palmitate decreased ROS in H9C2 cells and increased SOD in H9C2 cells and myocardial tissue
The excessive generation of ROS is closely linked with myocardial I/R injury. The ROS level was significantly higher in H9C2 cardiomyocytes isolated from mice following I/R. However, the I/R-induced increased in ROS was significantly suppressed by retinol palmitate pretreatment in a dose-dependent manner (Figure 1). There was no significant difference between the I/R and DMSO groups. Tert-Butyl hydroperoxide lowered the expression of SOD, and pretreatment with high-dose retinol palmitate significantly enhanced the expression of SOD in both H9C2 cells and myocardial tissue. Thus, these data demonstrated that retinol palmitate could reduce the generation of ROS (Figure 2).
Figure 1.
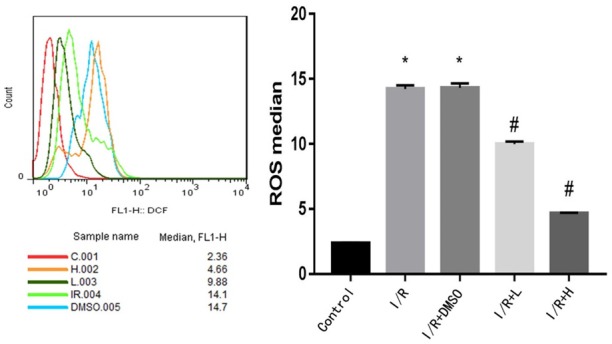
Retinol palmitate pretreatment reduced reactive oxygen species in IR-induced H9c2 cells. I/R: ischemia/reperfusion. DMSO: I/R+0.1% DMSO. I/R+L: IR+0.1 μM retinol palmitate. I/R+H: I/R+1 μM retinol palmitate. Data are expressed as mean ± SD (n = 3 per group). #P < 0.05, Retinol palmitate group versus IR group. *P < 0.05, I/R group and DMSO group versus control group.
Figure 2.
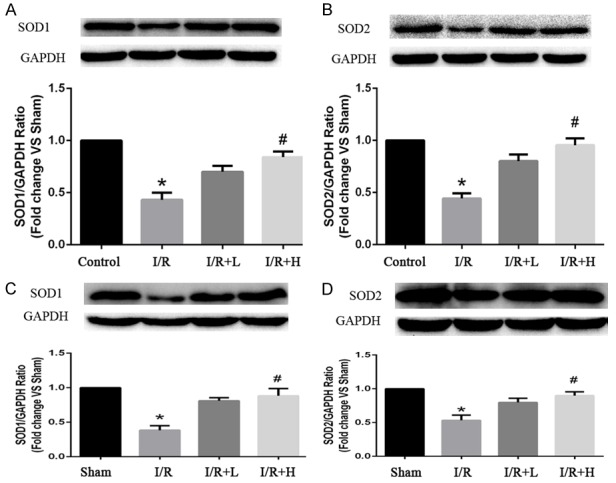
Effects of Retinol palmitate pretreatment on SOD1 and SOD2 expressions in H9c2 cells and myocardial tissue. (A) Quantitative analysis results of SOD1 in H9c2 cells. (B) Quantitative analysis results of SOD2 in H9c2 cells. (C) Quantitative analysis results of SOD1 in myocardial tissues. (D) Quantitative analysis results of SOD2 in myocardial tissues. I/R: ischemia/reperfusion. I/R+L (A, B): IR+0.1 μM retinol palmitate. I/R+H (A, B): I/R+1 μM retinol palmitate. I/R+L (C, D): IR+retinol palmitate (12 mg/kg). I/R+H (C, D): I/R+retinol palmitate (36 mg/kg). Data are expressed as mean ± SD (n = 6 per group). *P < 0.05, I/R group versus Control group or Sham group. #P < 0.05, I/R+L group. versus I/R group.
Retinol palmitate attenuated I/R-induced H9C2 cell apoptosis
Apoptosis plays an important role in myocardial I/R injury. Retinol palmitate (0.1 and 1 μM) ameliorated I/R-induced apoptosis in a concentration-dependent manner, indicating that retinol palmitate exerted a cardioprotective effect by inhibiting apoptosis (Figure 3A, 3B).
Figure 3.
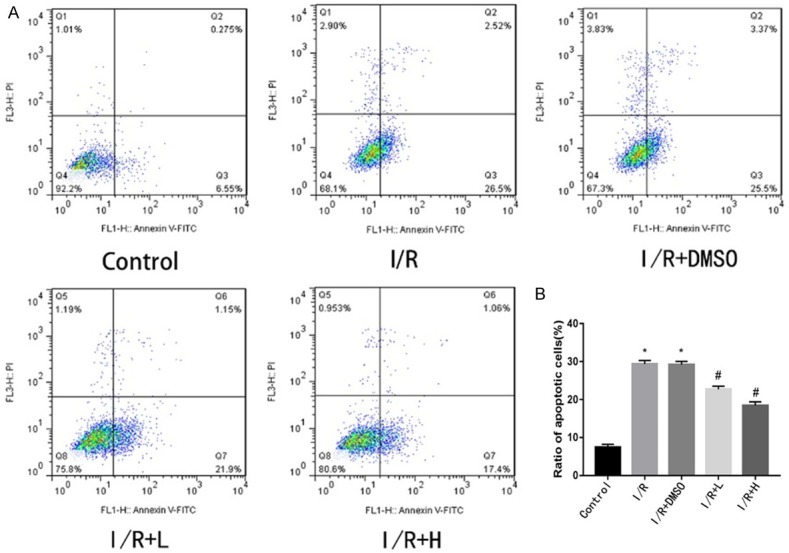
Effect of Retinol palmitate pretreatment on I/R-induced apoptosis in H9c2 cells. A. Typical images of flow cytometry. B. The apoptosis ratio was quantified by BD FACS software. Retinol palmitate pretreatment significantly reduced apoptosis compared with the IR group in a dose-dependent manner. I/R: ischemia/reperfusion. DMSO: I/R+0.1% DMSO. I/R+L: IR+0.1 μM retinol palmitate. I/R+H: I/R+1 μM retinol palmitate. Data are expressed as mean ± SD (n = 6 per group). *P < 0.05, IR group and DMSO group versus control group. #P < 0.05, Retinol palmitate group versus IR group.
Retinol palmitate diminished myocardial infract size and reduced serum CK-MB levels
Myocardial infarct area was shown in pale color in I/R (40 min/4 h) mice. Compared with the I/R group, the I/R+H group showed a significant improvement in the INF/AAR ratio, suggesting that pretreatment with retinol palmitate reduced the myocardial infarct area (Figure 4A, 4B). CK-MB is the primary marker of myocardial injury. I/R significantly increased serum CK-MB level, which was prevented by retinol palmitate (Figure 4C).
Figure 4.
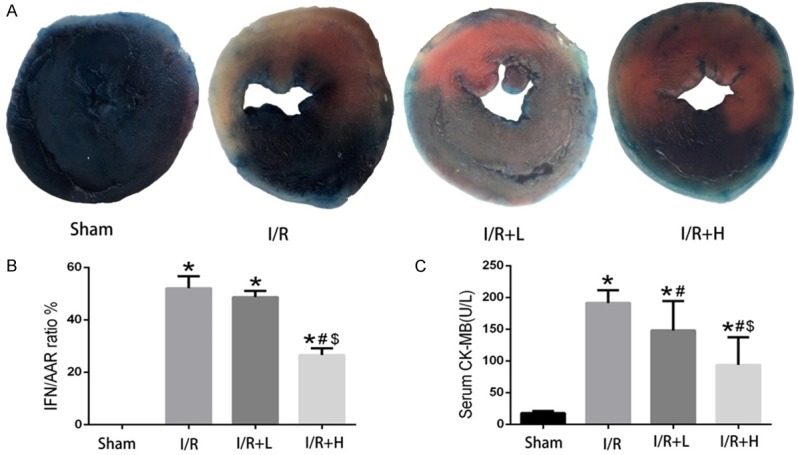
Retinol palmitate pretreatment reduced myocardial I/R injury. A. Representative images of the infract area (INF: white), area at risk (AAR: red and white), and normal area (blue). B. Quantitative analysis of myocardial infarct ratio (INF/AAR). C. Quantitative analysis of serum CK-MB activity. I/R: ischemia/reperfusion. I/R+L: IR+retinol palmitate (12 mg/kg). I/R+H: I/R+retinol palmitate (36 mg/kg). Data are expressed as mean ± SD (n = 6 per group). *P < 0.05, versus Sham group. #P < 0.05, versus I/R group. $P < 0.05, versus I/R+L group.
Effect of retinol palmitate on antioxidant- and apoptosis-related proteins in the myocardium
I/R enhanced the expression of cleaved caspase-3 and Bax compared with the sham group, whereas the expression of Bcl-2 was significantly decreased. Pretreatment with retinol palmitate (36 mg/kg) reduced the expression of cleaved caspase-3 and Bax and enhanced that of Bcl-2 (Figure 5).
Figure 5.
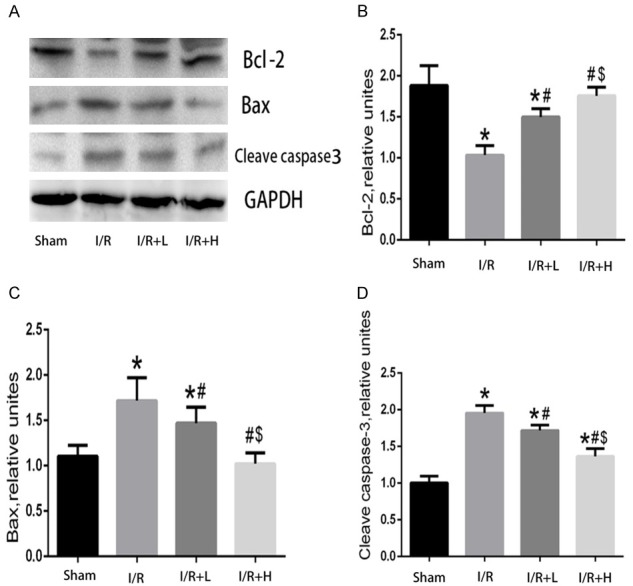
Effects of Retinol palmitate pretreatment on Bcl-2, Bax, cleaved caspase-3 expressions in myocardial tissues. A. Representative bands of Bcl-2, Bax, cleaved caspase-3. B. Quantitative analysis results of Bcl-2. C. Quantitative analysis results of Bax. D. Quantitative analysis results of cleaved caspase-3. I/R: ischemia/reperfusion. I/R+L: IR+retinol palmitate (12 mg/kg). I/R+H: I/R+retinol palmitate (36 mg/kg). Data are expressed as mean ± SD (n = 6 per group). *P < 0.05, versus Sham group. #P < 0.05, versus I/R group. $P < 0.05, versus I/R+L group.
Myocardial TUNEL staining
TUNEL staining is commonly used to evaluate apoptosis. As expected, I/R induced significant myocardial apoptosis. However, apoptosis was attenuated in I/R+H mice (Figure 6).
Figure 6.

The effect of Retinol palmitate pretreatment on myocardial TUNEL staining. A. Representative confocal microscopy images. DAPI staining (blue) indicates nucleus. Magnification: × 400. B. Percentages of TUNEL positive cells of total cells. I/R: ischemia/reperfusion. I/R+L: IR+retinol palmitate (12 mg/kg). I/R+H: I/R+retinol palmitate (36 mg/kg). Data are expressed as mean ± SD (n = 6 per group). *P < 0.05, versus Sham group. #P < 0.05, versus I/R group. $P < 0.05, versus I/R+L group.
Pretreatment with retinol palmitate enhanced SOD activity and decreased MDA levels in vivo
Pretreatment with retinol palmitate remarkably increased Myocardial SOD activity and reduced Myocardial MDA levels in a concentration-dependent manner in vivo (Figure 7).
Figure 7.
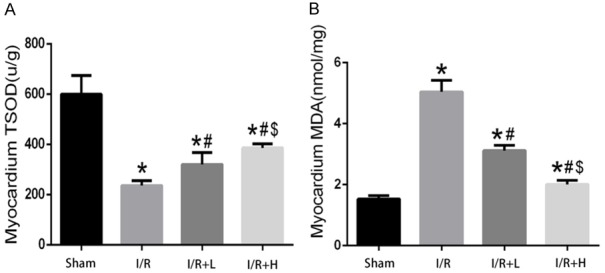
Retinol palmitate pretreatment increased SOD activities and alleviates MDA release. A. Myocardial SOD activities. B. Myocardial MDA leaves. I/R: ischemia/reperfusion. I/R+L: IR+retinol palmitate (12 mg/kg). I/R+H: I/R+retinol palmitate (36 mg/kg). Data are expressed as mean ± SD (n = 6 per group). *P < 0.05, versus Sham group. #P < 0.05, versus I/R group. $P < 0.05, versus I/R+L group.
Discussion
ROS is a major initiating factor for myocardial I/R injury [27]. Several studies have shown that ROS plays an essential role in the pathogenesis of myocardial I/R injury [28-32]. The generation of ROS following I/R injury and the increase of myocardial MDA promote lipid peroxidation, which accelerates the imbalance of oxidative stress and antioxidants [33]. The imbalance between oxidative stress and antioxidant defenses is also a major cause of myocardial damage.
The apoptotic signaling pathway refers to the activation of cell membrane death receptors (Fas), which leads to the activation of caspases (aspartate-specific proteases), DNA fragmentation, protein cleavage, and cell death. In the first few minutes after reflow, the overexpression of oxygen-free radicals, overload or redistribution of intracellular calcium, and the increase in MDA lead to a loss in mitochondrial membrane potential and the activation of caspase-3, which results in myocardial apoptosis and injury [34]. Myocardial I/R is associated with the upregulation of death receptors (Fas) and an increase in apoptotic cells [14,17,18]. Evidence has shown that the number of apoptotic cells in the myocardium may independently contribute to the prognosis of I/R injury [35]. Bcl-2 is an antioxidant oncogene with anti-apoptotic effects. A recent study showed that ischemic adaptation upregulated Bcl-2, thereby reducing DNA fragmentation and cellular apoptosis [14].
Several substances work as endogenous antioxidants, such as SOD [36], catalase, glutathione peroxidase, vitamin A, and vitamin E. In the cytoplasm and on the surface of endothelial cells, SOD is present in the form of copper SOD, zinc SOD, and manganese SOD [37]. Dismutation of the superoxide anion (O.-) is catalyzed into H2O2 by SOD. Subsequently, peroxidases (e.g., catalase or glutathione peroxidase) catalyze H2O2 into H2O and O2. In the ischemic preconditioning experiment, the activities and gene expression of Mn-SOD, catalase, and glutathione peroxidase in isolated hearts were significantly higher in mice subjected to four rounds of I/R than those subjected to one round of I/R [38]. Although the underlying mechanism through ischemic preconditioning protecting the myocardium is not completely understood, it is believed that increased endogenous antioxidant activity may be triggered by immediate and delayed preconditioning [39]. Therefore, antioxidants are likely to become new treatment for patients with AMI. Natural products such as tea polyphenols [40] and resveratrol [41,42] are good antioxidants, and have been used to protect myocardial I/R injury.
Vitamin A is a fat-soluble vitamin complex that includes vitamin A, retinal, retinoic acid, and retinyl esters, and is an important element in the maintenance of cellular function. Vitamin A exerts neuroprotective effects through a variety of mechanisms, such as inhibiting oxidative stress, activating the c-Jun N-terminal kinase pathway, and suppressing cyclooxygenase 2 [43]. Retinol palmitate, the ester of retinol and palmitic acid, plays multiple roles in the nervous system [22]. After a 17-year follow-up, Bellizzi et al. reported that men with high dietary beta carotene levels had a lower mortality of heart disease [44]. Similar results were reported in rabbits treated with 60,000 IU vitamin A and 150 mg vitamin E 24 h before myocardial I/R. With vitamins E and A pretreatment before I/R, rabbits displayed less mitochondrial damage and relative preservation of myocyte subcellular structures [45].
Therefore, we investigated whether retinol palmitate could protect against I/R-induced heart injury as an antioxidant. We studied the anti-oxidative properties of a vitamin A analog Retinol palmitate and its potential cardioprotective role against myocardial ischemia reperfusion injury. Oxidative stress is the major cause of myocardial injury during ischemia reperfusion, and antioxidant is considered to be an effective approach to limit cardiac damage. Given this, we studied the efficacy of Retinol palmitate as a strategy for reducing oxidative stress and preventing myocardial necrosis in response to ischemia reperfusion in vivo. Retinol palmitate increased SOD activity, reduced the serum creatine kinase and MDA levels in I/R mice. Also, Retinol palmitate decreased myocardial infarct size and reduced apoptosis by suppressing the expression of proapoptotic-related proteins and increasing that of SOD-related proteins. In vitro, retinol palmitate also reduced the level of ROS and prevented cellular apoptosis. Our study, for the first time, demonstrated the cardioprotective utility of the vitamin A analog Retinol palmitate against ischemia reperfusion injury.
The molecular mechanisms underlying the cardioprotective role of Retinol palmitate are anti-oxidative and anti-apoptosis. Increased ROS generation triggers Ca2+ overload and ER stress, which further leads to the loss of mitochondrial membrane potential and caspase-3 activation [46]. By reducing the ROS level, Retinol palmitate inhibits Bax and caspase-3 activation to prevent cell apoptosis against stress condition. Our results suggest that retinol palmitate pretreatment protects against myocardial I/R injury by maintaining the balance between intracellular oxidants and antioxidants.
Conclusions
Retinol palmitate prevents myocardial I/R injury by reducing the level of oxidative stress and inhibiting apoptosis.
Acknowledgements
We acknowledge the Second Affiliated and Yuying Children’s Hospital of Wenzhou Medical University for supporting the work of our study. We thank Prof LianMing Liao from Fujian Medical University Union Hospital for their technical support. This work was supported by the National Natural Science Foundation of China (No. 81573185), the Scientific Research Foundation of Wenzhou Municipal Science and Technology Bureau (No. Y20130211, H20140003, LY13H020004), and the Scientific Research Foundation of the Science and Technology Department of Taizhou City (No. 2017A33795).
Disclosure of conflict of interest
None.
References
- 1.Alla F, Zannad F, Filippatos G. Epidemiology of acute heart failure syndromes. Heart Fail Rev. 2007;12:91–95. doi: 10.1007/s10741-007-9009-2. [DOI] [PubMed] [Google Scholar]
- 2.Aronow WS. Epidemiology, pathophysiology, prognosis, and treatment of systolic and diastolic heart failure. Cardiol Rev. 2006;14:108–124. doi: 10.1097/01.crd.0000175289.87583.e5. [DOI] [PubMed] [Google Scholar]
- 3.Patel V, Upaganlawar A, Zalawadia R, Balaraman R. Cardioprotective effect of melatonin against isoproterenol induced myocardial infarction in rats: a biochemical, electrocardiographic and histoarchitectural evaluation. Eur J Pharmacol. 2010;644:160–168. doi: 10.1016/j.ejphar.2010.06.065. [DOI] [PubMed] [Google Scholar]
- 4.Thygesen K, Alpert JS, Jaffe AS, Simoons ML, Chaitman BR, White HD Joint ESC/ACCF/AHA/WHF Task Force for Universal Definition of Myocardial Infarction; Authors/Task Force Members Chairpersons; Thygesen K, Alpert JS, White HD Biomarker Subcommittee; Jaffe AS, Katus HA, Apple FS, Lindahl B, Morrow DA ECG Subcommittee; Chaitman BR, Clemmensen PM, Johanson P, Hod H Imaging Subcommittee; Underwood R, Bax JJ, Bonow JJ, Pinto F, Gibbons RJ Classification Subcommittee; Fox KA, Atar D, Newby LK, Galvani M, Hamm CW Intervention Subcommittee; Uretsky BF, Steg PG, Wijns W, Bassand JP, Menasche P, Ravkilde J Trials & Registries Subcommittee; Ohman EM, Antman EM, Wallentin LC, Armstrong PW, Simoons ML Trials & Registries Subcommittee; Januzzi JL, Nieminen MS, Gheorghiade M, Filippatos G Trials & Registries Subcommittee; Luepker RV, Fortmann SP, Rosamond WD, Levy D, Wood D Trials & Registries Subcommittee; Smith SC, Hu D, Lopez-Sendon JL, Robertson RM, Weaver D, Tendera M, Bove AA, Parkhomenko AN, Vasilieva EJ, Mendis S ESC Committee for Practice Guidelines (CPG); Bax JJ, Baumgartner H, Ceconi C, Dean V, Deaton C, Fagard R, Funck-Brentano C, Hasdai D, Hoes A, Kirchhof P, Knuuti J, Kolh P, McDonagh T, Moulin C, Popescu BA, Reiner Z, Sechtem U, Sirnes PA, Tendera M, Torbicki A, Vahanian A, Windecker S Document Reviewers. Morais J, Aguiar C, Almahmeed W, Arnar DO, Barili F, Bloch KD, Bolger AF, Botker HE, Bozkurt B, Bugiardini R, Cannon C, de Lemos J, Eberli FR, Escobar E, Hlatky M, James S, Kern KB, Moliterno DJ, Mueller C, Neskovic AN, Pieske BM, Schulman SP, Storey RF, Taubert KA, Vranckx P, Wagner DR. Third universal definition of myocardial infarction. J Am Coll Cardiol. 2012;60:1581–1598. [Google Scholar]
- 5.Li C, He J, Gao Y, Xing Y, Hou J, Tian J. Preventive effect of total flavones of choerospondias axillaries on ischemia/reperfusion-induced myocardial infarction-related MAPK signaling pathway. Cardiovasc Toxicol. 2014;14:145–152. doi: 10.1007/s12012-013-9238-7. [DOI] [PubMed] [Google Scholar]
- 6.Jennings RB. Historical perspective on the pathology of myocardial ischemia/reperfusion injury. Circ Res. 2013;113:428–438. doi: 10.1161/CIRCRESAHA.113.300987. [DOI] [PubMed] [Google Scholar]
- 7.Maxwell SR, Lip GY. Reperfusion injury: a review of the pathophysiology, clinical manifestations and therapeutic options. Int J Cardiol. 1997;58:95–117. doi: 10.1016/s0167-5273(96)02854-9. [DOI] [PubMed] [Google Scholar]
- 8.Petrosillo G, Ruggiero FM, Di Venosa N, Paradies G. Decreased complex III activity in mitochondria isolated from rat heart subjected to ischemia and reperfusion: role of reactive oxygen species and cardiolipin. FASEB J. 2003;17:714–716. doi: 10.1096/fj.02-0729fje. [DOI] [PubMed] [Google Scholar]
- 9.Grech ED, Jackson MJ, Ramsdale DR. Reperfusion injury after acute myocardial infarction. BMJ. 1995;310:477–478. doi: 10.1136/bmj.310.6978.477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bernier M, Hearse DJ, Manning AS. Reperfusion-induced arrhythmias and oxygen-derived free radicals. Studies with “anti-free radical” interventions and a free radical-generating system in the isolated perfused rat heart. Circ Res. 1986;58:331–340. doi: 10.1161/01.res.58.3.331. [DOI] [PubMed] [Google Scholar]
- 11.Maulik N, Engelman RM, Rousou JA, Flack JE 3rd, Deaton D, Das DK. Ischemic preconditioning reduces apoptosis by upregulating anti-death gene Bcl-2. Circulation. 1999;100(Suppl):II369–375. doi: 10.1161/01.cir.100.suppl_2.ii-369. [DOI] [PubMed] [Google Scholar]
- 12.Yeh ET. Life and death in the cardiovascular system. Circulation. 1997;95:782–786. doi: 10.1161/01.cir.95.4.782. [DOI] [PubMed] [Google Scholar]
- 13.Maulik N, Kagan VE, Tyurin VA, Das DK. Redistribution of phosphatidylethanolamine and phosphatidylserine precedes reperfusion-induced apoptosis. Am J Physiol. 1998;274:H242–248. doi: 10.1152/ajpheart.1998.274.1.H242. [DOI] [PubMed] [Google Scholar]
- 14.Maulik N, Yoshida T, Engelman RM, Deaton D, Flack JE 3rd, Rousou JA, Das DK. Ischemic preconditioning attenuates apoptotic cell death associated with ischemia/reperfusion. Mol Cell Biochem. 1998;186:139–145. [PubMed] [Google Scholar]
- 15.Kajstura J, Cheng W, Reiss K, Clark WA, Sonnenblick EH, Krajewski S, Reed JC, Olivetti G, Anversa P. Apoptotic and necrotic myocyte cell deaths are independent contributing variables of infarct size in rats. Lab Invest. 1996;74:86–107. [PubMed] [Google Scholar]
- 16.Dallak MM, Mikhailidis DP, Haidara MA, Bin-Jaliah IM, Tork OM, Rateb MA, Yassin HZ, Al-Refaie ZA, Ibrahim IM, Elawa SM, Rashed LA, Afifi NA. Oxidative stress as a common mediator for apoptosis induced-cardiac damage in diabetic rats. Open Cardiovasc Med J. 2008;2:70–78. doi: 10.2174/1874192400802010070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Aikawa R, Komuro I, Yamazaki T, Zou Y, Kudoh S, Tanaka M, Shiojima I, Hiroi Y, Yazaki Y. Oxidative stress activates extracellular signal-regulated kinases through Src and Ras in cultured cardiac myocytes of neonatal rats. J Clin Invest. 1997;100:1813–1821. doi: 10.1172/JCI119709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Long X, Boluyt MO, Hipolito ML, Lundberg MS, Zheng JS, O’Neill L, Cirielli C, Lakatta EG, Crow MT. p53 and the hypoxia-induced apoptosis of cultured neonatal rat cardiac myocytes. J Clin Invest. 1997;99:2635–2643. doi: 10.1172/JCI119452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tesoriere L, Ciaccio M, Bongiorno A, Riccio A, Pintaudi AM, Livrea MA. Antioxidant activity of all-trans-retinol in homogeneous solution and in phosphatidylcholine liposomes. Arch Biochem Biophys. 1993;307:217–223. doi: 10.1006/abbi.1993.1581. [DOI] [PubMed] [Google Scholar]
- 20.Palgi A. Association between dietary changes and mortality rates: Israel 1949 to 1977; a trend-free regression model. Am J Clin Nutr. 1981;34:1569–1583. doi: 10.1093/ajcn/34.8.1569. [DOI] [PubMed] [Google Scholar]
- 21.Minicucci MF, Azevedo PS, Oliveira SA Jr, Martinez PF, Chiuso-Minicucci F, Polegato BF, Justulin LA Jr, Matsubara LS, Matsubara BB, Paiva SA, Zornoff LA. Tissue vitamin A insufficiency results in adverse ventricular remodeling after experimental myocardial infarction. Cell Physiol Biochem. 2010;26:523–530. doi: 10.1159/000322320. [DOI] [PubMed] [Google Scholar]
- 22.Shimada J, Taniguchi J, Mori M, Sato Y, Takuwa H, Ito H, Kuwabara S. Retinol palmitate prevents ischemia-induced cell changes in hippocampal neurons through the Notch1 signaling pathway in mice. Exp Neurol. 2013;247:182–187. doi: 10.1016/j.expneurol.2013.04.014. [DOI] [PubMed] [Google Scholar]
- 23.Olson JA. Recommended dietary intakes (RDI) of vitamin A in humans. Am J Clin Nutr. 1987;45:704–716. doi: 10.1093/ajcn/45.4.704. [DOI] [PubMed] [Google Scholar]
- 24.Pang L, Cai Y, Tang EH, Yan D, Kosuru R, Li H, Irwin MG, Ma H, Xia Z. Cox-2 inhibition protects against hypoxia/reoxygenation-induced cardiomyocyte apoptosis via Akt-dependent enhancement of iNOS expression. Oxid Med Cell Longev. 2016;2016:3453059. doi: 10.1155/2016/3453059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang Y, Duan W, Jin Z, Yi W, Yan J, Zhang S, Wang N, Liang Z, Li Y, Chen W, Yi D, Yu S. JAK2/STAT3 activation by melatonin attenuates the mitochondrial oxidative damage induced by myocardial ischemia/reperfusion injury. J Pineal Res. 2013;55:275–286. doi: 10.1111/jpi.12070. [DOI] [PubMed] [Google Scholar]
- 26.Mo Y, Tang L, Ma Y, Wu S. Pramipexole pretreatment attenuates myocardial ischemia/reperfusion injury through upregulation of autophagy. Biochem Biophys Res Commun. 2016;473:1119–1124. doi: 10.1016/j.bbrc.2016.04.026. [DOI] [PubMed] [Google Scholar]
- 27.Duilio C, Ambrosio G, Kuppusamy P, DiPaula A, Becker LC, Zweier JL. Neutrophils are primary source of O2 radicals during reperfusion after prolonged myocardial ischemia. Am J Physiol Heart Circ Physiol. 2001;280:H2649–2657. doi: 10.1152/ajpheart.2001.280.6.H2649. [DOI] [PubMed] [Google Scholar]
- 28.Ferrari R, Ceconi C, Curello S, Guarnieri C, Caldarera CM, Albertini A, Visioli O. Oxygen-mediated myocardial damage during ischaemia and reperfusion: role of the cellular defences against oxygen toxicity. J Mol Cell Cardiol. 1985;17:937–945. doi: 10.1016/s0022-2828(85)80074-2. [DOI] [PubMed] [Google Scholar]
- 29.Garlick PB, Davies MJ, Hearse DJ, Slater TF. Direct detection of free radicals in the reperfused rat heart using electron spin resonance spectroscopy. Circ Res. 1987;61:757–760. doi: 10.1161/01.res.61.5.757. [DOI] [PubMed] [Google Scholar]
- 30.Grech ED, Dodd NJ, Jackson MJ, Morrison WL, Faragher EB, Ramsdale DR. Evidence for free radical generation after primary percutaneous transluminal coronary angioplasty recanalization in acute myocardial infarction. Am J Cardiol. 1996;77:122–127. doi: 10.1016/s0002-9149(96)90580-9. [DOI] [PubMed] [Google Scholar]
- 31.Kato K, Shao Q, Elimban V, Lukas A, Dhalla NS. Mechanism of depression in cardiac sarcolemmal Na+-K+-ATPase by hypochlorous acid. Am J Physiol. 1998;275:C826–831. doi: 10.1152/ajpcell.1998.275.3.C826. [DOI] [PubMed] [Google Scholar]
- 32.Musat S, Dhalla NS. Alteration in cardiac sarcolemmal ATP receptors by oxyradicals. Ann N Y Acad Sci. 1996;793:1–12. doi: 10.1111/j.1749-6632.1996.tb33500.x. [DOI] [PubMed] [Google Scholar]
- 33.Kumari SS, Menon VP. Changes in levels of lipid peroxides and activities of superoxide dismutase and catalase in isoproterenol induced myocardial infarction in rats. Indian J Exp Biol. 1987;25:419–423. [PubMed] [Google Scholar]
- 34.Lowenstein CJ. Myocardial reperfusion injury. N Engl J Med. 2007;357:2409–2410. [PubMed] [Google Scholar]
- 35.Zhao ZQ, Vinten-Johansen J. Myocardial apoptosis and ischemic preconditioning. Cardiovasc Res. 2002;55:438–455. doi: 10.1016/s0008-6363(02)00442-x. [DOI] [PubMed] [Google Scholar]
- 36.Fujimoto M, Masuzaki H, Tanaka T, Yasue S, Tomita T, Okazawa K, Fujikura J, Chusho H, Ebihara K, Hayashi T, Hosoda K, Nakao K. An angiotensin II AT1 receptor antagonist, telmisartan augments glucose uptake and GLUT4 protein expression in 3T3-L1 adipocytes. FEBS Lett. 2004;576:492–497. doi: 10.1016/j.febslet.2004.09.027. [DOI] [PubMed] [Google Scholar]
- 37.Ohta H, Adachi T, Hirano K. Internalization of human extracellular-superoxide dismutase by bovine aortic endothelial cells. Free Radic Biol Med. 1994;16:501–507. doi: 10.1016/0891-5849(94)90128-7. [DOI] [PubMed] [Google Scholar]
- 38.Das DK, Engelman RM, Kimura Y. Molecular adaptation of cellular defences following preconditioning of the heart by repeated ischaemia. Cardiovasc Res. 1993;27:578–584. doi: 10.1093/cvr/27.4.578. [DOI] [PubMed] [Google Scholar]
- 39.Hoshida S, Kuzuya T, Fuji H, Yamashita N, Oe H, Hori M, Suzuki K, Taniguchi N, Tada M. Sublethal ischemia alters myocardial antioxidant activity in canine heart. Am J Physiol. 1993;264:H33–39. doi: 10.1152/ajpheart.1993.264.1.H33. [DOI] [PubMed] [Google Scholar]
- 40.Gonzalez-Reyes S, Guzman-Beltran S, Medina-Campos ON, Pedraza-Chaverri J. Curcumin pretreatment induces Nrf2 and an antioxidant response and prevents hemin-induced toxicity in primary cultures of cerebellar granule neurons of rats. Oxid Med Cell Longev. 2013;2013:801418. doi: 10.1155/2013/801418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ray PS, Maulik G, Cordis GA, Bertelli AA, Bertelli A, Das DK. The red wine antioxidant resveratrol protects isolated rat hearts from ischemia reperfusion injury. Free Radic Biol Med. 1999;27:160–169. doi: 10.1016/s0891-5849(99)00063-5. [DOI] [PubMed] [Google Scholar]
- 42.Hung LM, Su MJ, Chu WK, Chiao CW, Chan WF, Chen JK. The protective effect of resveratrols on ischaemia-reperfusion injuries of rat hearts is correlated with antioxidant efficacy. Br J Pharmacol. 2002;135:1627–1633. doi: 10.1038/sj.bjp.0704637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Choi BK, Kim JH, Jung JS, Lee YS, Han ME, Baek SY, Kim BS, Kim JB, Oh SO. Reduction of ischemia-induced cerebral injury by all-trans-retinoic acid. Exp Brain Res. 2009;193:581–589. doi: 10.1007/s00221-008-1660-x. [DOI] [PubMed] [Google Scholar]
- 44.Palace VP, Khaper N, Qin Q, Singal PK. Antioxidant potentials of vitamin A and carotenoids and their relevance to heart disease. Free Radic Biol Med. 1999;26:746–761. doi: 10.1016/s0891-5849(98)00266-4. [DOI] [PubMed] [Google Scholar]
- 45.Llesuy S, Milei J, Picone V, Gonzalez Flecha B, Beigelman R, Boveris A. Effect of vitamins A and E on ischemia-reperfusion damage in rabbit heart. Mol Cell Biochem. 1995;145:45–51. doi: 10.1007/BF00925712. [DOI] [PubMed] [Google Scholar]
- 46.Yellon DM, Hausenloy DJ. Myocardial reperfusion injury. N Engl J Med. 2007;357:1121–1135. doi: 10.1056/NEJMra071667. [DOI] [PubMed] [Google Scholar]