

Abstract
Background: Endotoxin-induced acute inflammatory diseases such as sepsis, mediated by excessive production of various pro-inflammatory cytokines remain the leading cause of mortality in critically ill patients. Lipopolysaccharide (LPS), the characteristic endotoxin found in the outer membrane of Gram-negative bacteria, can induce the innate immunity system and through Mitogen activated protein kinase (MAPK) and Nuclear Factor-κB (NF-κB), increase the production of inflammatory mediators. Astaxanthin (ASX), a xanthophyll carotenoid, exerts beneficial effects against oxidation, inflammation, and cancer. But poor evidence has been reported that whether it has protective effects on LPS-induced injury. This study aims to investigate the effects of ASX on LPS-induced sepsis and acute lung injury and to demonstrate its mechanisms. Methods: Mouse prime macrophage (MPM) challenged with LPS were used for in vitro pharmacological activity and mechanistic studies. Inflammatory facors (tumor necrosis factor-alpha and interleukin-6 levels) in MPM were determined. The mouse models of LPS-induced sepsis and acute lung injury administrated with or without the compound were used for in vivo studies. Results: Pre-treatment of MPM with ASX inhibited MAPK/NF-κB signaling pathway, and attenuated LPS-increased inflammatory factors in vitro. In animal models of LPS-induced sepsis and acute lung injury, administration of ASX significantly improved survival and protected lung injury. Subsequently, ASX was shown to suppress LPS-induced inflammatory factors increase, MAPK phosphorylation, and NF-κB activation in vivo. Conclusions: ASX exerts impressively protective effects on LPS-induced injury in vitro and in vivo. Taken together, it might be used as a potential candidate for clinical sepsis.
Keywords: Astaxanthin, LPS, sepsis, acute lung injury, NF-κB
Introduction
Acute inflammatory diseases such as acute lung injury (ALI) and sepsis, which occur as a result of local or systemic inflammation, remain the most common cause of death inintensive care units worldwide [1]. Sepsis and acute lung injury (ALI) is a systemic inflammatory syndrome that is induced by infection and involves damage to multiple organs and tissues [2]. Despite extensive research, profound understanding of ALI pathogenesis and recent advances in therapeutic strategies being trialed, mortality rates from sepsis/ALI remain high at approximately 30% or 40% [3,4]. Therefore, highly effective pharmacotherapies are required urgently for the treatment of acute inflammatory diseases.
Research efforts in the field of sepsis and ALI have largely focused on the innate immune system and have conceptually viewed sepsis/ALI as a syndrome of hyperinflammation [5]. Increasing evidence indicates that dysregulation of cytokines in acute inflammation is an important step in mediating, amplifying, and perpetuating sepsis/ALI processes [6]. Lipopolysaccharide (LPS), known as a component in the outer membranes of Gram-negative bacteria, can trigger a biological inflammatory response and disrupt the immune function of various organs [7]. Microbial LPS binds to a surface receptor complex consisting of myeloid differentiation 2 (MD2) and toll-like receptor 4 (TLR4) in innate immune cells through ancillary proteins such as LPS-binding protein (LBP) and cluster of differentiation 14 (CD14) [8]. The LPS-TLR4/MD2 complex activates downstream pro-inflammatory signaling pathways, including the transcription factor nuclear factor kappa-light chain-enhancer of activated B cells (NF-κB) and mitogen activated protein kinases (MAPKs), triggering the production of a variety of inflammatory cytokines. The excessive release of various pro-inflammatory cytokines mainly including tumor necrosis factor-α (TNF-α), interleukin-6 (IL-6), IL-1β, IL-12 and IL-8 results in the acute cellular and/or organic injury to form sepsis or ALI [9,10]. Observations strongly suggest that the inflammatory processes occur in association with lung injury, and effective treatment should be given early in the development process of acute inflammatory diseases [11]. Therefore, the development of anti-inflammatory candidates to inhibit the excessive production of cytokines, including IL-6 and TNF-α, has been a promising strategy for the prevention and treatment of sepsis and ALI.
The discovery of novel anti-inflammatory agents derived from natural active products has attracted a lot of attention from medicinal chemists. Astaxanthin (ASX), 3,3’-two hydroxyl-4,4’-two ketone-beta, beta’-carotene, is a terpene unsaturated compound isolated from most of the crustacean and salmon family fish, plants, leaves, flowers, fruits, and Flamingo feathers. It is the most important of terpene unsaturated compound, and is regarded as a potential candidate drug for many diseases [12-15]. Recent studies have shown that ASX has a variety of pharmacological effects against inflammation, oxidation and cardiac damage [16-18]. It’s reported that ASX reduces the proinflammatory response and secondary brain injury after SAH, primarily by inhibiting the TLR4 signaling pathway [19]. ASX prevented the inflammatory cells infiltration, decreased free iron deposition, and fibrosis in liver of CCl-administered rats [20]. However, while there has been considerable investigation into these activities, the specific effects of ASX on acute inflammatory disease and the target remain inadequately unclear. In the present study, we investigated the protective effects of ASX and the underlying target on LPS-induced sepsis and ALI model. We found that ASX exerted significant protective effects on LPS-induced sepsis and ALI by inhibiting the production of inflammatory response. The anti-inflammatory property of ASX for the treatment of acute inflammatory diseases was derived from inhibiting MAPK/NF-κB signaling pathway. ASX was found to protect against LPS-induced lung tissue damage and to enhance the survival rate of LPS-challenged mice. Our results suggest that ASX might be a potential agent for the treatment of sepsis and ALI.
Methods
Cells and treatment
According to the methods described in the previous paper, Mouse primary peritoneal macrophage (MPM) were prepared from C57BL/6 mice and cultured in vitro [21]. At 37°C with 5% CO2, MPM were cultured in DMEM media (Gibco) supplemented with 10% FBS, 100 U/ml penicillin, and 100 mg/mL streptomycin. ASX was dissolved in DMSO solution and added to cells, with the final concentration of DMSO being 0.1%.
Animals
C57BL/6 (B6) mice purchased from the Animal Center of Wenzhou Medical University (Wenzhou, China) weighed 18-22 g. Animals were housed with a 12:12 h light-dark cycle at a constant room temperature, and a standard rodent diet and plenty of water were given. It was not until these animals had to be domesticated in the laboratory for at least 7 days that they were tested.
MTT assay for cell viability
Cells (1×105 cells/mL) were seeded into a 96-well plate and cultured overnight. The cells were pre-treated with various concentrations of ASX ranging from 0 to 200 μM for 1 h. ASX. MTT assay was used to determin the number of viable cells (Promega, Madison, WI). MTT dye solution was added into living cells and cultured with cells in CO2 incubator for 4 hours. Then, measure the absorbance at 490 nm to determine the number of living cells. For consistency, repeated the experiment three times.
siRNA-induced gene silencing
Specific siRNA sequences were used to achieve gene silencing in MPM. NF-κB siRNA was purchased from Gene Pharma Co. LTD. (Shanghai, China). Specific siRNA sequences were 5’-CUGGAUGACAUCUUAAACUTT-3’ for mouse NF-κB. According to the manufacturer’s instruction, Transfection siRNA into MPM was implement using LipofectAMINE™ 2000 (Invitrogen, Carlsbad, CA).
Determination of TNF-α and IL-6
After treatment, according to the manufacturer’s instructions, the enzyme-linked immunosorbent assay (ELISA) kit (eBioscience, San Diego, CA) was used to determine the levels of TNF-α and IL-6 in the medium.
Western immunoblot analysis
After treatment, collect and lyse the cell or lung tissue, then using 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis separate 30 μg of the whole cell lysates and electrotransfer them to a nitrocellulose membrane. Each membrane was preincubated for 1 h at room temperature in Tris-buffered saline, pH 7.6, containing 0.05% Tween-20 and 5% nonfat milk. Incubated each membrane with specific antibodies against p-p38, p38, p-JNK, JNK, p-ERK, ERK, IκB, and GAPDH from Cell Signaling (Danvers, MA, USA) and NF-κB p65, LaminB from Santa Cruz Technology (Santa Cruz, CA). Incubating with secondary antibody conjugated with horseradish peroxidase and using enhanced chemiluminescence reagents (Bio-Rad, Hercules, CA), then detect immunoreactive bands. Original images for all relevant western blots are presented in Figures S3, S4 and S5.
LPS-induced inflammatory mortality in B6 mice
Male C57BL/6 mice were randomly divided into 3 groups, committed Vehicle (n = 10), LPS (n = 10), and LPS + ASX (n = 10). First, ASX was dissolved in a water bath at 37°C with macrogol 15 hydroxystearate (a nonionic solubilizer for injection, from BASF [Ludwigshafen, Germany]), making the concentration of ASX 2 mg/mL. The solvent concentration in the final solution was 10%. Male C57BL/6 mice were intraperitoneally injected with ASX (1 mL, 100 mg/kg) solution 15 minutes before LPS (20 mg/kg) was injected into caudal vein. And the Vehicle group and LPS group were given the same volume of solvent. Mortality was recorded for 7 days.
LPS-induced ALI
Male C57BL/6 mice were randomly divided into 3 groups, committed Vehicle (n = 10), LPS (n = 10), and LPS + ASX (n = 10). According to the methods described in the previous paper [22], male C57BL/6 mice were given intraperitoneal injection of ASX solution (1 mL, 100 mg/kg) for 1 week, then intratracheal injection of LPS (10 mg/kg). LPS was induced for 6 hours and mice were euthanized with ketamine. Carefully open each animal’s chest and collect BALF. The BALF was centrifuged for 10 minutes at 1000 rpm at 4°C and then the supernatant was collected for subsequent cytokine determination and protein concentration detection.
Lung wet/dry ratio
The lung wet/dry weight ratio was determination for observing the pulmonary edema. Collect the middle lobe of right lung and record wet weight. Then the lung was heated for 72 hours at 65°C in a thermostat and weighed to ascertain the lung dry mass levels.
Histopathological study
The upper lobe of right lung was fixed in 4% paraformaldehyde and embedded in paraffin, then cut into 5 μm sections. According to standard protocol, hematoxylin and eosin was used to stain the sections which were observed by light microscopy.
Immunohistochemistry
Tissue sections (5 μm thickness) were used for immunohistochemistry, deparaffinized in xylene, and using an ethanol gradient hydrated. Repair of antigen with pressure cooker. To block endogenous peroxidase activity, the sections were incubated with 3% H2O2 for 10 min, then the sections were sealed in 5% bovine serum albumin (BSA) for 30 minutes and incubated overnight with anti-CD68/anti-TNF-α antibody at 4°C. The second antibody combined with horseradish peroxidase and DAB were used for detection.
Statistical analysis
Data are presented as means ± SD. The student’s t test or ANOVA multiple comparisons were used to obtain the statistical significance of differences between groups in GraphPadPro5.0 (GraphPad, San Diego, CA). The difference was considered to be significant when p < 0.05. All experiments were repeated at least three times.
Results
ASX inhibits LPS-induced secretion of pro-inflammatory cytokines in MPM
Initially, we investigated the toxic effects of ASX on RAW264.7 cell survival. Our MTT assay indicated that cell survival did not differ significantly when the cells were pretreated with ASX 0 to 200 μM (Figure 1A). As Figure S1 shown, LPS induced apoptosis of RAW264.7, but ASX had no obvious effect on viability of RAW264.7. Pretreatment of RAW264.7 with ASX (10 μM) significantly decreased apoptotic cells. TNF-α and IL-6 are pro-inflammatory cytokines that play core roles in the pathological process of sepsis. Our subsequent experiments showed that pre-treatment with ASX (10 μM) significantly decreased TNF-α and IL-6 secretion by LPS-stimulated MPM (Figure 1B and 1C, respectively).
Figure 1.

ASX inhibits LPS-induced secretion of TNF-α and IL-6 in mouse prime macrophage. (A) Cells were incubated with different concentrations of ASX for 24 h. Cell viability was determined by MTT assay. (B, C) Cells were incubated with different concentrations of ASX for 1 h and then stimulated with LPS for 24 h. Secreted TNF-α (B) and IL-6 (C) from the media were measured by ELISA (n = 4 independent experiments, ***P < 0.001, vs vehicle; ###P < 0.001, vs LPS).
ASX enhances the survival rate of LPS-challenged mice and inhibits the production of IL-6 and TNF-α in mouse serum
Overwhelming production of pro-inflammatory cytokines and mediators leads to tissue damage or death. In this experiment, mice were pre-treated with ASX (100 mg/kg, i.p.) for 1 h, and then injected with LPS (20 mg/kg, i.v.). The results showed that administration of ASX significantly decreased the production of IL-6 and TNF-α in serum in the 4 hours following LPS challenge (Figure 2A and 2B). Moreover, ASX also increased survival rate compared to LPS alone in the 7 days following LPS (Figure 2C).
Figure 2.
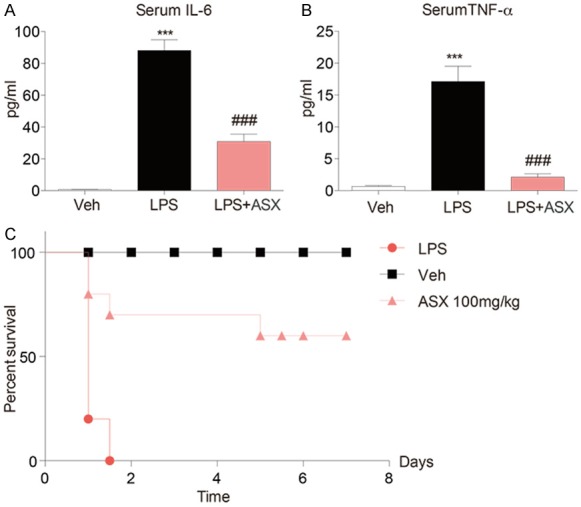
ASX enhances the survival rate of LPS-challenged mice and inhibits the production of IL-6 and TNF-α in mouse serum. Male C57BL/6 mice were injected with ASX (100 mg/kg intraperitoneally) or vehicle before LPS injection. Blood was sampled 4 h after LPS injection. (A, B) Serum IL-6 (A) and TNF-α (B) were measured by ELISA. (C) Survival was recorded at different intervals for 7 days. Each group contained 10 mice. Statistical significance was assessed by Log-Rank test and represented as follows (n = 10 independent experiments, ***P < 0.001, vs vehicle; ###P < 0.001, vs LPS).
ASX exhibits protective effects against LPS-induced lung injury in LPS-challenged mice
To further explore the protective effect of ASX on the sepsis, we also investigated the tissue injury in LPS-challenged mice. Mice were pre-treated with ASX (100 mg/kg, i.p.) for 1 h, and then injected with LPS (20 mg/kg, i.v.) to induce experimental sepsis. The lung samples were collected and examined by using Hematoxylin Eosin (H&E) staining and CD68/TNF-α. Administration of LPS significantly increased inflammatory cell infiltration in lung tissue samples (Figure 3). In the lung, alveolar wall thickness was increased and the number of pulmonary alveoli reduced in LPS-challenged mice compared with controls. Administration of ASX repressed alveolar wall swelling and attenuated the decline in the number of pulmonary alveoli in LPS-challenged mice (Figure 3).
Figure 3.
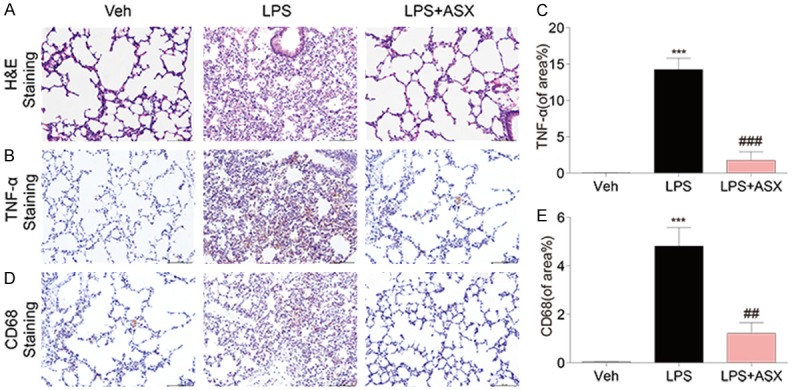
ASX exhibits protective effects against LPS-induced lung injury in LPS-challenged mice. Samples of lung tissue were harvested 20 h after LPS injection. (A) The results show H&E-staining of lung tissue sections from the indicated group. In the lung, alveolar wall thickness and the number of pulmonary alveoli were observed in LPS-challenged mice compared with controls. (B-E) Representative immunohistochemical staining and quantitative analysis for TNF-α (B and C) and CD68 (D and E). The expression of TNF-α and inflammatory cell infiltration were observed in lung tissue samples. The figure is a representative of three independent experiments (n = 10 independent experiments, ***P < 0.001, vs vehicle; ##P < 0.01, ###P < 0.001, vs LPS).
ASX inhibits the MAPK/NF-κB signaling pathway in LPS-induced sepsis and MPM
To elucidate the molecular mechanism underlying the anti-inflammatory action of ASX, we evaluated the MAPK/NF-κB signaling pathway related protein in lung of LPS-induced sepsis and LPS-induced MPM. Together with those results, the degradation of IκB-α and the phosphorylation of ERK1/2, P38 and JNK in lung of LPS-induced sepsis were markedly increased after LPS stimulation (Figures 4A and S2). However, pre-treatment with ASX significantly suppressed LPS-induced degradation of IκB-α and phosphorylation of ERK1/2, P38 and JNK in sepsis. Furthermore, the similar results were also observed in LPS-induced MPM (Figures 4B and S2). As Figure 4C shows, LPS increases the expression of NF-κB p65 in the nuclear. Pretreating with astaxanthin reduces LPS-induced the expression of NF-κB p65 in the nuclear (Figure 4C). To confirm the anti-inflammation action of ASX is NF-κB-dependent in LPS-challenged MPM, we knocked down the expression of NF-κB prior to LPS exposure. Compared with control group, transfection of cells with specific siRNA reduced protein abundance by more than 70% (Figure 4D). Silencing NF-κB down-regulate IL-6 (Figure 4E) and TNF-α (Figure 4F) secretion in LPS-stimulated MPM, while, ASX was not able to reduce LPS-induced secretion of IL-6 (Figure 4E) and TNF-α (Figure 4F) in NF-κB-knockdown MPM.
Figure 4.
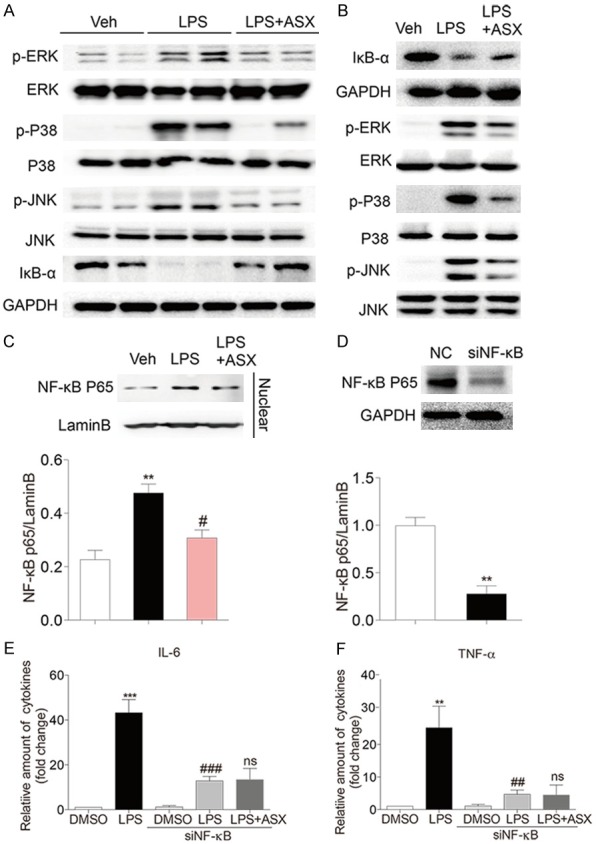
ASX inhibits the MAPK/NF-κB signaling pathway in LPS-induced sepsis and MPM. (A) Samples of lung tissue were harvested 20 h after LPS injection and total proteins were extracted to detect the phosphorylation of JNK, ERK1/2, and P38 and the degradation of IκB (n = 10 independent experiments). (B) MPM were incubated with ASX for 1 h, and then were treated with LPS for 30 min, and total proteins were extracted to detect the phosphorylation of JNK, ERK1/2, and P38 and the degradation of IκB. (C) Nuclear proteins were extracted to detect the expression of NF-κB p65. (D-F) Silencing NF-κB to verify that ASX attenuates LPS-induced inflammatory response through NF-κB. Western blot analysis for silencing NF-κB (D). Treating with or without ASX, NF-κB-knockdown MPM were induced by LPS and then the levels of IL-6 (E) and TNF-α (F) were determined by ELISA (n = 4 independent experiments, ***P < 0.001, **P < 0.05, vs vehicle or NC; ###P < 0.001, ##P < 0.01, #P < 0.05, vs LPS; ns, no significance, vs LPS + si NF-κB).
ASX attenuated LPS-induced acute lung injury in mice
ALI was induced in mice by intratracheal LPS instillation to further evaluate the protective ability of ASX against LPS-induced acute inflammation in vivo. As shown in Figure 5A, a marked increase in the protein concentration in mice bronchial alveolar lavage fluid (BALF) was observed after LPS (5 mg/kg) instillation, and the increase was significantly inhibited by ASX treatment. The lung wet/dry weight ratio, which is an index of lung edema, was significantly increased in the LPS alone group when compared to both the control saline group and the LPS + ASX group, indicating that ASX also suppressed LPS-induced lung edema (Figure 5B). Recruitment of inflammatory cells into the pulmonary compartment is an important feature of ALI. As shown in Figure 5C, LPS challenge caused a strong increase in the number of total cells in the BALF, while treatment with ASX significantly decreased the numbers of total cells in BALF from ALI mice. MPO activity in tissues is an indicator of neutrophil infiltration. Figure 5D shows that treatment with ASX significantly decreased LPS-induced MPO activity in mice lung tissues. To evaluate the histological changes, mice lung tissues were proceeded to hematoxylin and eosin (H&E) staining (Figure 5E). Mice in the control group exhibited a normal structure of lung tissues under a light microscope, while LPS instillation resulted insignificant pulmonary congestion, infiltration of inflammatory cells, thickening of the alveolar wall, and interstitial edema. These pathological changes induced by LPS were remarkably reduced by ASX treatment. LPS exposure also increased the secretion of inflammatory cytokines such as IL-6 and TNF-α in mice serum, which were all significantly reduced by ASX treatment (Figure 6A, 6B). The similar results were also observed in mice BALF (Figure 6C, 6D). Furthermore, Figure 6E validated the inhibitory effect of ASX against macrophage infiltration into the lung, as evidenced by the reduced CD68 expression in LPS + ASX group. We also observed LPS exposure also increased the production of TNF-α in mice lung tissues, which were significantly reduced by ASX treatment (Figure 6F).
Figure 5.
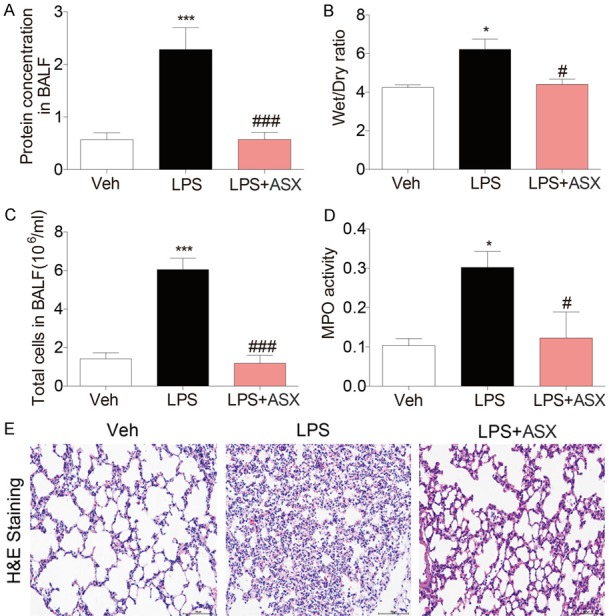
ASX attenuated LPS-induced acute lung injury in mice. Mice were then euthanized with ketamine 6 h after LPS induction. A. ASX inhibits the increase of the protein concentration in mouse BALF. B. ASX suppressed LPS-induced lung edema. C. Treatment with ASX significantly reduced the numbers of total cells in BALF. D. ASX significantly decreased LPS-induced MPO activity in mice lung tissues. E. H&E staining showed that ASX attenuated LPS-induced pathological changes (n = 10 independent experiments, ***P < 0.001, *P < 0.05, vs vehicle; ###P < 0.001, #P < 0.05, vs LPS).
Figure 6.
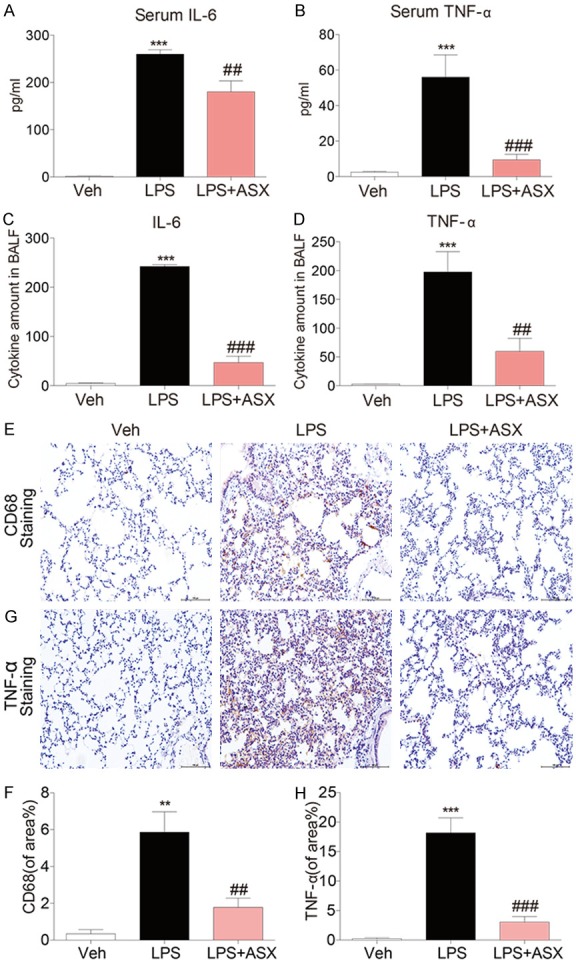
ASX attenuated the inflammatory response in LPS-induced acute lung injury. (A, B) ASX inhibits LPS-induced the secretion of IL-6 (A) and TNF-α (B) in mice serum. (C, D) ASX inhibits the production of IL-6 (C) and TNF-α (D) induced by LPS in mouse BALF. (E, F) Representative immunohistochemical staining for CD68 and quantitative analysis. ASX exhibited the inhibitory effect against macrophage infiltration. (G, H) Representative immunohistochemical staining for TNF-α and quantitative analysis. ASX inhibits the production of TNF-α in mice lung tissue (n = 10 independent experiments, **P < 0.01, ***P < 0.001, vs vehicle; ###P < 0.001, ##P < 0.01, vs LPS).
Finally, we investigated whether the pharmacological activity of ASX was associated with its inhibition of MAPK/NF-κB signaling pathway. As shown in Figures 7 and S2, compared with the control group, LPS treatment significantly induced the degradation of IκB-α and the phosphorylation of ERK1/2, P38 and JNK in mouse lung tissues, while ASX administration reduced the level of degradation of IκB-α and phosphorylation of ERK1/2, P38 and JNK. These data indicate that ASX exerts an anti-inflammatory and anti-ALI activity in LPS-induced mice lungs.
Figure 7.
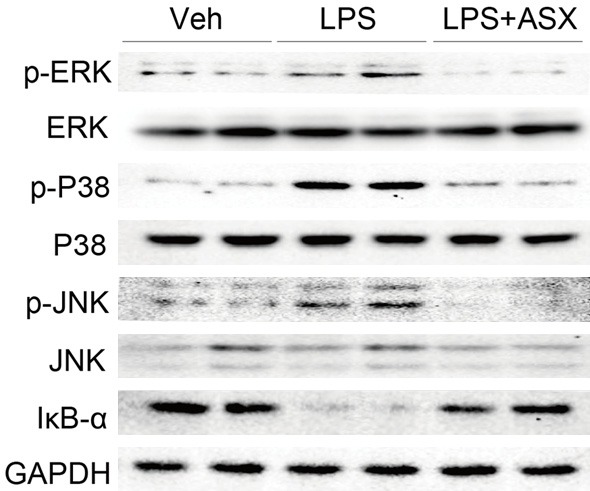
ASX inhibits the MAPK/NF-κB signaling pathway in LPS-induced acute lung injury. Samples of lung tissue were harvested 6h after LPS injection and total proteins were extracted to detect the phosphorylation of JNK, ERK1/2, and P38 and the degradation of IκB (n = 10 independent experiments).
Discussion and conclusion
Acute inflammatory diseases remain the main cause of death in intensive care units worldwide and still lack an effective therapy in clinical treatments. Steroidal and nonsteroidal anti-inflammatory drugs have been found to be therapeutically limited in the treatment of sepsis and ALI [23,24]. More than 30 pharmaceutical candidates for the treatment of sepsis have been developed or are currently in the developmental stage [25,26]; however, most treatments brought to clinical use have failed because of the complicated nature of sepsis, which remains the most common cause of death in intensive care units [27,28]. Therefore, there is an urgent need to find novel and effective therapeutic approaches for sepsis.
One potential approach to treating and preventing septic shock and its associated diseases is the intervention of the NF-κB-mediated inflammatory response [29,30]. LPS is presented to TLR4/MD2 complex via the LPS-binding protein and CD14. The formation of the TLR4/MD2/LPS complex leads to the phosphorylation of MAPK and activates the downstream NF-κB. Both TLR4/MD2 and MAPK/NF-κB are essential for the LPS-induced inflammatory response and sepsis. However, the clinical trials of TAK-242, a TLR4-specific inhibitor, have failed to treat severe sepsis and related respiratory disease in patients [27]. In addition, blocking TLRs can lead to severe side effects, “inappropriate” immune responses, such as allergic Th2 responses, or immunologic tolerance [31,32]. On the other hand, many compounds inhibit NF-κB in a variety of ways, showing treatment or relief of lung inflammation. It suggests that it may be a target for drug treatment of acute inflammatory response.
Recently, some natural active compounds that do not contain the structure of lipid A or fatty acids have been found to be able to anti-inflammatory effect. These small molecules, such as kaempferol, quercetin, and rutin, inhibit directly to the MAPK, and block the activation of NF-κB, resulting in the prevention of pro-inflammatory signaling and septic shock. Although their specificities for targeting other proteins remain to be defined, these natural compounds provide us the important information for the discovery of new anti-inflammatory agent.
Excessive expression of the pro-inflammatory cytokines, TNF-α and IL-6, characterize the initial phase of acute inflammatory disease [33]. They rapidly initiate a systemic inflammatory response leading to stimulation of an adaptive immune response and a cytokine storm [34,35]. As shown in Figure 1, we investigated the effect of ASX on LPS-induced TNF-α and IL-6 production using macrophages in non-cytotoxic doses. We found that ASX reduced production of both TNF-α and IL-6 in LPS-stimulated MPM (Figure 1). Similar findings were observed for the pro-inflammatory cytokines, TNF-α and IL-6 in the animal model (Figure 2A and 2B). Therefore, we proved that ASX inhibited the inflammatory process in LPS-induced MPM and animal model by reducing the expression of pro-inflammatory cytokines. Furthermore, we examined whether ASX affected the death rate in LPS-induced sepsis. Treatment with ASX markedly increased the survival of LPS-challenged mice (Figure 2). As lung tissues might be injured from the severe inflammatory response induced by LPS, we investigated the protective effects of ASX on lung tissues. ASX (100 mg/kg intraperitoneally) decreased alveolar wall swelling and lessened the decline in the number of pulmonary alveoli in lung tissue (Figure 3).
MAPKs and NF-κB have been demonstrated to be the main mediators in the LPS-TLR4/MD2 pro-inflammatory signaling pathway. Briefly, LPS stimulates and binds to TLR4, and induces the activation of MAPK-dependent intracellular signaling [9,36,37]. ASX prevented MAPKs and NF-κB activation in LPS-stimulated MPM and septic mouse, as evidenced by a decrease in the levels of IκB degradation, and ERK/p38/JNK phosphorylation (Figure 4). In our research, phosphorylation of ERK/p38/JNK, degradation of IκB and Nuclear transcription of NF-κB were markedly increased after LPS stimulation, but were significantly suppressed by ASX. Similar findings were observed in the ALI (Figure 7). Taken together, these results indicated that the potential mechanism for the protective effects of ASX was related to inhibition of the MAPK/NF-κB signaling pathway. Adult respiratory distress syndrome (ARDS) is characterized by acute lung injury with a high mortality rate [38]. Sepsis syndrome is the most frequent causes of ARDS, leading to increased lung permeability, enhanced polymorphonuclear neutrophil (PMN) sequestration and respiratory failure [38]. Figures 4 and 5 further shows the inhibitory effects of ASX on LPS-induced inflammatory cytokine overexpression in a murine model of acute lung injury induced by LPS. In vivo, lung histologic changes in the LPS-injected mice were also suppressed by ASX pretreatment (Figure 6).
In conclusion, we found that ASX exerted significant protective effects against LPS-induced acute inflammatory disease by the expression of pro-inflammatory cytokines through the MAPK/NF-κB signaling pathway. ASX may also protect against LPS-induced tissue damage and enhance the survival rate of LPS-challenged mice. Our results provide evidence that ASX might be a potential agent for the treatment of acute inflammatory disease.
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Opal SM, Laterre PF, Francois B, LaRosa SP, Angus DC, Mira JP, Wittebole X, Dugernier T, Perrotin D, Tidswell M, Jauregui L, Krell K, Pachl J, Takahashi T, Peckelsen C, Cordasco E, Chang CS, Oeyen S, Aikawa N, Maruyama T, Schein R, Kalil AC, Van Nuffelen M, Lynn M, Rossignol DP, Gogate J, Roberts MB, Wheeler JL, Vincent JL ACCESS Study Group. Effect of eritoran, an antagonist of MD2-TLR4, on mortality in patients with severe sepsis: the ACCESS randomized trial. JAMA. 2013;309:1154–1162. doi: 10.1001/jama.2013.2194. [DOI] [PubMed] [Google Scholar]
- 2.Walley KR. Biomarkers in sepsis. Curr Infect Dis Rep. 2013;15:413–420. doi: 10.1007/s11908-013-0357-x. [DOI] [PubMed] [Google Scholar]
- 3.Singer M, Deutschman CS, Seymour CW, Shankar-Hari M, Annane D, Bauer M, Bellomo R, Bernard GR, Chiche JD, Coopersmith CM, Hotchkiss RS, Levy MM, Marshall JC, Martin GS, Opal SM, Rubenfeld GD, van der Poll T, Vincent JL, Angus DC. The third international consensus definitions for sepsis and septic shock (Sepsis-3) JAMA. 2016;315:801–810. doi: 10.1001/jama.2016.0287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Levitt JE, Matthay MA. Clinical review: early treatment of acute lung injury--paradigm shift toward prevention and treatment prior to respiratory failure. Crit Care. 2012;16:223. doi: 10.1186/cc11144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ho MS, Mei SH, Stewart DJ. The immunomodulatory and therapeutic effects of mesenchymal stromal cells for acute lung injury and sepsis. J Cell Physiol. 2015;230:2606–2617. doi: 10.1002/jcp.25028. [DOI] [PubMed] [Google Scholar]
- 6.Deng JC, Standiford TJ. Growth factors and cytokines in acute lung injury. Compr Physiol. 2011;1:81–104. doi: 10.1002/cphy.c090011. [DOI] [PubMed] [Google Scholar]
- 7.Raetz CR, Whitfield C. Lipopolysaccharide endotoxins. Annu Rev Biochem. 2002;71:635–700. doi: 10.1146/annurev.biochem.71.110601.135414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Viladés C, Escoté X, López-Dupla M, Martinez E, Domingo P, Asensi V, Leal M, Peraire J, Inza MI, Arnedo M, Gutiérrez M, Valle-Garay E, Ferrando-Martinez S, Olona M, Alba V, Sirvent JJ, Gatell JM, Vidal F HALS Study Group. Involvement of the LPS-LPB-CD14-MD2-TLR4 inflammation pathway in HIV-1/HAART-associated lipodystrophy syndrome (HALS) J Antimicrob Chemother. 2014;69:1653–1659. doi: 10.1093/jac/dku032. [DOI] [PubMed] [Google Scholar]
- 9.Lu YC, Yeh WC, Ohashi PS. LPS/TLR4 signal transduction pathway. Cytokine. 2008;42:145–151. doi: 10.1016/j.cyto.2008.01.006. [DOI] [PubMed] [Google Scholar]
- 10.Yamamoto M, Akira S. Lipid A receptor TLR4-mediated signaling pathways. Adv Exp Med Biol. 2010;667:59–68. doi: 10.1007/978-1-4419-1603-7_6. [DOI] [PubMed] [Google Scholar]
- 11.Merrill JC, You J, Constable C, Leeman SE, Amar S. Whole-body deletion of LPS-induced TNF-alpha factor (LITAF) markedly improves experimental endotoxic shock and inflammatory arthritis. Proc Natl Acad Sci U S A. 2011;108:21247–21252. doi: 10.1073/pnas.1111492108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang Y, Bae M, Kim B, Park YK, Koo SI, Lee JY. Astaxanthin prevents and reverses the activation of mouse primary hepatic stellate cells. J Nutr Biochem. 2016;29:21–26. doi: 10.1016/j.jnutbio.2015.11.005. [DOI] [PubMed] [Google Scholar]
- 13.Otsuka T, Shimazawa M, Inoue Y, Nakano Y, Ojino K, Izawa H, Tsuruma K, Ishibashi T, Hara H. Astaxanthin protects against retinal damage: evidence from in vivo and in vitro retinal ischemia and reperfusion models. Current Eye Research. 2016;41:1465–1472. doi: 10.3109/02713683.2015.1127392. [DOI] [PubMed] [Google Scholar]
- 14.Kim B, Farruggia C, Ku CS, Pham TX, Yang Y, Bae M, Wegner CJ, Farrell NJ, Harness E, Park YK. Astaxanthin inhibits inflammation and fibrosis in the liver and adipose tissue of mouse models of diet-induced obesity and nonalcoholic steatohepatitis. J Nutr Biochem. 2017;43:27–35. doi: 10.1016/j.jnutbio.2016.01.006. [DOI] [PubMed] [Google Scholar]
- 15.Jia Y, Wu C, Kim J, Kim B, Lee SJ. Astaxanthin reduces hepatic lipid accumulations in high-fat-fed C57BL/6J mice via activation of peroxisome proliferator-activated receptor (PPAR) alpha and inhibition of PPAR gamma and Akt. J Nutr Biochem. 2016;28:9–18. doi: 10.1016/j.jnutbio.2015.09.015. [DOI] [PubMed] [Google Scholar]
- 16.Li W, Hellsten A, Jacobsson LS, Blomqvist HM, Olsson AG, Yuan XM. Alpha-tocopherol and astaxanthin decrease macrophage infiltration, apoptosis and vulnerability in atheroma of hyperlipidaemic rabbits. J Mol Cell Cardiol. 2004;37:969–978. doi: 10.1016/j.yjmcc.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 17.Shen H, Kuo CC, Chou J, Delvolve A, Jackson SN, Post J, Woods AS, Hoffer BJ, Wang Y, Harvey BK. Astaxanthin reduces ischemic brain injury in adult rats. FASEB J. 2009;23:1958–1968. doi: 10.1096/fj.08-123281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aoi W, Naito Y, Sakuma K, Kuchide M, Tokuda H, Maoka T, Toyokuni S, Oka S, Yasuhara M, Yoshikawa T. Astaxanthin limits exercise-induced skeletal and cardiac muscle damage in mice. Antioxid Redox Signal. 2003;5:139. doi: 10.1089/152308603321223630. [DOI] [PubMed] [Google Scholar]
- 19.Zhang X, Lu Y, Wu Q, Dai H, Li W, Lv S, Zhou X, Zhang X, Hang C, Wang J. Astaxanthin mitigates subarachnoid hemorrhage injury primarily by increasing sirtuin 1 and inhibiting the Toll-like receptor 4 signaling pathway. FASEB J. 2019;33:722–737. doi: 10.1096/fj.201800642RR. [DOI] [PubMed] [Google Scholar]
- 20.Islam MA, Al Mamun MA, Faruk M, Ul Islam MT, Rahman MM, Alam MN, Rahman A, Reza HM, Alam MA. Astaxanthin ameliorates hepatic damage and oxidative stress in carbon tetrachloride-administered rats. Pharmacognosy Res. 2017;9:S84–S91. doi: 10.4103/pr.pr_26_17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wu J, Li J, Cai Y, Pan Y, Ye F, Zhang Y, Zhao Y, Yang S, Li X, Liang G. Evaluation and discovery of novel synthetic chalcone derivatives as anti-inflammatory agents. J Med Chem. 2011;54:8110–8123. doi: 10.1021/jm200946h. [DOI] [PubMed] [Google Scholar]
- 22.Chen G, Zhang Y, Liu X, Fang Q, Wang Z, Fu L, Liu Z, Wang Y, Zhao Y, Li X, Liang G. Discovery of a new inhibitor of myeloid differentiation 2 from cinnamamide derivatives with anti-inflammatory activity in sepsis and acute lung injury. J Med Chem. 2016;59:2436–2451. doi: 10.1021/acs.jmedchem.5b01574. [DOI] [PubMed] [Google Scholar]
- 23.Bernard GR, Wheeler AP, Russell JA, Schein R, Summer WR, Steinberg KP, Fulkerson WJ, Wright PE, Christman BW, Dupont WD, Higgins SB, Swindell BB. The effects of ibuprofen on the physiology and survival of patients with sepsis. The Ibuprofen in Sepsis Study Group. N Engl J Med. 1997;336:912–918. doi: 10.1056/NEJM199703273361303. [DOI] [PubMed] [Google Scholar]
- 24.Watanabe T, Higuchi K, Kobata A, Nishio H, Tanigawa T, Shiba M, Tominaga K, Fujiwara Y, Oshitani N, Asahara T, Nomoto K, Takeuchi K, Arakawa T. Non-steroidal anti-inflammatory drug-induced small intestinal damage is toll-like receptor 4 dependent. Gut. 2008;57:181–187. doi: 10.1136/gut.2007.125963. [DOI] [PubMed] [Google Scholar]
- 25.Zhao X, Liu D, Gong W, Zhao G, Liu L, Yang L, Hou Y. The toll-like receptor 3 ligand, poly(I:C), improves immunosuppressive function and therapeutic effect of mesenchymal stem cells on sepsis via inhibiting MiR-143. Stem Cells. 2014;32:521–533. doi: 10.1002/stem.1543. [DOI] [PubMed] [Google Scholar]
- 26.Kwon OK, Lee W, Kim SJ, Lee YM, Lee JY, Kim JY, Bae JS, Lee S. In-depth proteomics approach of secretome to identify novel biomarker for sepsis in LPS-stimulated endothelial cells. Electrophoresis. 2015;36:2851–2858. doi: 10.1002/elps.201500198. [DOI] [PubMed] [Google Scholar]
- 27.Rice TW, Wheeler AP, Bernard GR, Vincent JL, Angus DC, Aikawa N, Demeyer I, Sainati S, Amlot N, Cao C, Ii M, Matsuda H, Mouri K, Cohen J. A randomized, double-blind, placebo-controlled trial of TAK-242 for the treatment of severe sepsis. Crit Care Med. 2010;38:1685–1694. doi: 10.1097/CCM.0b013e3181e7c5c9. [DOI] [PubMed] [Google Scholar]
- 28.Gaieski DF, Goyal M. What is sepsis? What is severe sepsis? What is septic shock? Searching for objective definitions among the winds of doctrines and wild theories. Expert Rev Anti Infect Ther. 2013;11:867–871. doi: 10.1586/14787210.2013.829633. [DOI] [PubMed] [Google Scholar]
- 29.Bohrer H, Qiu F, Zimmermann T, Zhang Y, Jllmer T, Mannel D, Bottiger BW, Stern DM, Waldherr R, Saeger HD, Ziegler R, Bierhaus A, Martin E, Nawroth PP. Role of NFkappaB in the mortality of sepsis. J Clin Invest. 1997;100:972–985. doi: 10.1172/JCI119648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen H, Wang X, Yan X, Cheng X, He X, Zheng W. LncRNA MALAT1 regulates sepsis-induced cardiac inflammation and dysfunction via interaction with miR-125b and p38 MAPK/NFkappaB. Int Immunopharmacol. 2018;55:69–76. doi: 10.1016/j.intimp.2017.11.038. [DOI] [PubMed] [Google Scholar]
- 31.Ishii KJ, Uematsu S, Akira S. ‘Toll’ gates for future immunotherapy. Curr Pharm Des. 2006;12:4135–4142. doi: 10.2174/138161206778743484. [DOI] [PubMed] [Google Scholar]
- 32.Nakamoto N, Kanai T. Role of toll-like receptors in immune activation and tolerance in the liver. Front Immunol. 2014;5:221. doi: 10.3389/fimmu.2014.00221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kurt AN, Aygun AD, Godekmerdan A, Kurt A, Dogan Y, Yilmaz E. Serum IL-1beta, IL-6, IL-8, and TNF-alpha levels in early diagnosis and management of neonatal sepsis. Mediators Inflamm. 2007;2007:31397. doi: 10.1155/2007/31397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wu LC, Fan NC, Lin MH, Chu IR, Huang SJ, Hu CY, Han SY. Anti-inflammatory effect of spilanthol from Spilanthes acmella on murine macrophage by down-regulating LPS-induced inflammatory mediators. J Agric Food Chem. 2008;56:2341–2349. doi: 10.1021/jf073057e. [DOI] [PubMed] [Google Scholar]
- 35.Zhang L, Wang CC. Inflammatory response of macrophages in infection. Hepatobiliary Pancreat Dis Int. 2014;13:138–152. doi: 10.1016/s1499-3872(14)60024-2. [DOI] [PubMed] [Google Scholar]
- 36.Kang JY, Lee JO. Structural biology of the toll-like receptor family. Annu Rev Biochem. 2011;80:917–941. doi: 10.1146/annurev-biochem-052909-141507. [DOI] [PubMed] [Google Scholar]
- 37.He H, Genovese KJ, Nisbet DJ, Kogut MH. Profile of toll-like receptor expressions and induction of nitric oxide synthesis by toll-like receptor agonists in chicken monocytes. Mol Immunol. 2006;43:783–789. doi: 10.1016/j.molimm.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 38.Nagase T, Uozumi N, Ishii S, Kume K, Izumi T, Ouchi Y, Shimizu T. Acute lung injury by sepsis and acid aspiration: a key role for cytosolic phospholipase A2. Nat Immunol. 2000;1:42. doi: 10.1038/76897. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.