

Abstract
Alzheimer’s disease (AD) is a devastating neurodegenerative disease with limited treatments and no cure. Neurotropin (NTP) is an analgesic drug widely prescribed for neuropathic pain. Increasing evidence suggests that NTP may also protect against neurodegeneration, but NTP’s treatment potential against memory impairments of AD remains to be explored. APP/PS1 mice, which model AD, were given NTP for three months then cognitively tested with the Morris water maze. Their Aβ burden, microglial and astrocytic activation, and BDNF levels were compared to untreated controls using immunofluorescent staining. Expression of pro-inflammatory cytokines (IL-1β, IL-6 and TNF-α) and NF-κB pathway related proteins (p65 and IκB-α) were examined by ELISA or Western blots in vivo and in vitro in the microglia cell line. Lastly, BV-2 cells were pre-treated with the selective BDNF inhibitor ANA-12 and with NTP to examine mechanistic pathways. Taken together, NTP treatment reduced cognitive impairment, Aβ deposits, and glial activation in cortex and hippocampus APP/PS1 mice. IL-1β, IL-6 and TNF-α also decreased after NTP treatment in vivo and in vitro, and BDNF levels rose. Also, NTP reduced p65 and IκB-α activation and the effect of NTP on pro-inflammatory cytokines and NF-κB pathway related proteins was abolished by BDNF inhibitor. Our results indicate that NTP reduces neuroinflammation and improves the cognitive deficits in APP/PS1 mice possibly via BDNF/NF-κB pathway. NTP may be a new promising drug candidate for patients with AD.
Keywords: Alzheimer’s disease, neurotropin, neuroinflammation, brain derived neurotrophic factor, memory impairment, NF-κB
Introduction
Alzheimer’s disease (AD) is the most prevalent cause of dementia, which affects 1 in 10 people over 65 years of age [1,2]. AD characteristically causes extracellular accumulation of amyloid β (Aβ) [3], which forms plaques. There is convincing evidence that Aβ binds to inflammatory receptors (such as TNFR1 and IL-1R) and activates inflammation during AD. Immune-related receptors play an important role on learning and memory formation and excessive neuroinflammation can result in direct cognition impairment [4]. Importantly, synaptic pruning can be regulated by inflammatory signals [5] and chronic neuroinflammation can lead to synaptic-associated proteins loss. Also, it was reported that microglia caused synaptic pruning dysfunction and synaptic loss [6,7].
As projected lifespans increase worldwide, treating AD has become an urgent international health priority [8]. The two main AD drugs currently available are donepezil, a cholinesterase inhibitor that increases synaptic acetylcholine (Ach) to enhance cognition in mild AD, and memantine, an N-methyl-D-aspartate receptor (NMDAR) antagonist that reduces excitotoxic neuroinflammation in severe AD. Neither drug stops the progressive cognitive decline, and since 2003 no new drugs have been approved by the Food and Drug Administration (FDA) [9].
Neurotropin (NTP) is a well-known analgesic derived from inflamed rabbit skin inoculated with vaccinia virus [10]. For the past 50 years, NTP has been prescribed for neuropathic pain, and its safety is well-established [11-13]. More recent animal experiments suggest it may have significant neuroprotective effects as well. Three months of NTP treatment rescued the spatial cognitive impairment of Ts65Dn mice, a Downs Syndrome model with triplication of 65% of human trisomy-21 genes [14]. NTP treatment also reduced the volume of infarcted lesions, brain edema, and the resulting neurological deficits, and enhanced spatial learning in C57BL/6J mice [15]. Our recent work showed that NTP could alleviate oxidative stress in APP/PS1 mice, an AD model, and inhibits neuroinflammation in BV-2 cells [10,16]. However, NTP’s treatment potential in memory impairment and neuroinflammation during AD has not yet been evaluated.
BDNF plays a pivotal role in modulation of synaptic plasticity, neuronal maintenance, cell survival, neurotransmitter and neurogenesis, and thus in the maintenance of learning and memory [17]. Patients with Alzheimer’s disease often have reduced BDNF concentration in their blood and cerebrospinal fluid [18]. Evidence showed that the analgesic effect of NTP probably involved the descending pain inhibitory system via the induction of BDNF [11]. Also, growing evidence has shown that BDNF has modulatory functions on neuroinflammation [17]. NF-κB is a ubiquitous transcriptional factor and it can modulate the expression of inflammatory molecules by translocating into the nucleus and triggering transcription of target genes [19]. There is evidence that responsive sites for immune-related transcriptional factors including NF-κB are in the regulatory promoter region of the genes controlling the expression of APP [20]. In a mice experiment, genetic knockout of the TNF receptor reduces β-secretase 1 (BACE1) expression which is mediated by NF-κB. Interestingly, this process is also associated with reduced Aβ and enhanced cognitive function [21].
This study evaluates NTP’s effects on cognitive dysfunction and neuroinflammation in an AD transgenic mouse model, and examines molecular mechanisms involved.
Materials and methods
Mice and drug administration
APPswe/PS1dE9 (APP/PS1) double transgenic mice were purchased from the Model Animal Research Center of Nanjing University (Nanjing, China). These mice model AD through the chimeric insertion of human amyloid precursor protein (APP) and human presenilin 1 (PS1) genes, which are overexpressed in patients with early-onset AD. 24 6-month-old APP/PS1 males and 24 wild-type litter-mate controls were housed in specific pathogen free (SPF) conditions on a 12 h light/dark cycle with free access to food and water, and all were handled according to the protocols of the Institutional Animal Care and Use Committee of Sun Yat-sen University, Guangzhou, China. Half of the mice from each genotype were randomly chosen to receive 200 NU/kg NTP or 0.9% NaCl placebo, given by daily oral gavage for three months (n=12 in each group). After treatment, when they were 9 months old, the mice were behaviorally tested and then sacrificed to analyse biochemically.
Cell culture
Immortal BV-2 murine microglial cells, a gift from Dr. Ying Chen of Sun Yat-sen Memorial Hospital, Sun Yat-sen University were cultured as described [16]. BV-2 cultures were treated with 0.1 NU/mL NTP, then given lipopolysaccharides (1000 ng/mL, LotL2880, O55:B5, Sigma-Aldrich, St. Louis, MO, USA) 12 h later. Some cultures were pre-treated with 10 uM of selective non-competitive BDNF receptor agonist ANA-12 (Sigma-Aldrich) 1 h before NTP, to demonstrate NTP’s action through BDNF pathways [22,23].
Morris water maze (MWM)
After three months of NTP or vehicle treatment, the mice were tested for spatial learning and memory in the Morris water maze as previously described [24]. Briefly, they were given four consecutive trials per day, starting in a different quadrant for each trial. Trials lasted 90 seconds and ended when the mice successfully reached the platform and stayed there for 5 s. If mice could not find the platform in 90 s, the experimenter manually set them there and let them stay for 20 s.
Each mouse’s time to find the platform on the first day was normalized at 1, then used to normalize the and platform times on subsequent days were normalized to the previous day (latency day n/latency day n-1), to calculate a learning trend. The relative escape latencies in the following training day to that of the first day were analyzed (escape latency in the following day/escape latency in the first day) and labeled as learning trend. The probe trial was conducted 24 h after the end of the acquisition trial when the platform was removed. In our experiment, the latency to the primary target site, the time spent in the target quadrant, and the numbers of platform-site crossovers within 60 s were recorded.
Bielschowsky silver staining and immunofluorescent staining
Bielschowsky silver staining and immunofluorescent staining were performed on fixed sections as described previously [25,26]. Bielschowsky silver staining was used to assess Aβ and immunofluorescence was used to evaluate levels of Aβ deposits, BDNF expression, and the area of GFAP+ and Iba1+ cells in the hippocampus and cortex of each group. The primary antibodies used in immunofluorescent staining were as following: rabbit anti-Aβ (1:100, Abcam, MA, USA), rabbit anti-BDNF (1:500; Millipore, MA, USA), goat anti-GFAP (1:1000; Abcam, MA, USA), goat anti-Iba1 (1:500; Abcam, MA, USA). DAPI (Invitrogen, CA, USA) was used to detect nuclei. Images were acquired from a fluorescent microscope. The area of Aβ plaques, GFAP+ cells, and Iba1+ cells in the cortex and hippocampus in each image were quantified by Image J (National Institutes of Health, MD, USA).
Enzyme-linked immunosorbent assay (ELISA)
The brain samples (separated into the cortex and the hippocampus) were stored at -80°C till analysis. We measured the concentration of Aβ1-40, Aβ1-42, BDNF, NGF, NT-3, IL-1β, IL-6 and TNF-α with the ELISA method at 9 months of age, which have been administrated with NTP for 3 months [16]. The assays were performed using commercially available ELISA kits (Invitrogen for Aβ1-40, Aβ1-42, IL-6, IL-1β and TNF-α, Promega for BDNF, and CUSABIO for NGF and NT-3) according to the manufacturer’s instructions. The total protein concentration was determined using the BCA Protein Assay kit (Thermo Scientific, USA). Absorbance of the samples was detected with a multifunctional microplate reader (SpectraMax M5, Sunnyvale, CA, USA).
Western blot analysis
Western blotting and semi-quantitative analyses were performed following previously described procedures [27]. In brief, proteins in cerebral cortex and hippocampus were extracted with lysis buffer for 30 min, followed by centrifugation at 14,000 rpm for 15 min at 4°C to obtain the supernatant for western blot analysis. Primary antibodies and dilution rates used were listed as follow: NF-κB (p65), 1:1000; p-IκBα, 1:500 and β-actin, 1:1000. Primary antibodies against NF-κB (p65), p-IκBα and β-actin were purchased from Cell Signaling Technology Inc (MA, USA). Horseradish peroxidase-conjugated secondary antibodies were used, and the bands were fixed and visualized by an ECL advanced kit. β-actin was utilized as an internal control for protein loading and transfer efficiency. Western blot assay results reported here are representative of at least 3 experiments. The quantification of protein expression was analyzed by Image J (National Institutes of Health, MD, USA).
CCK-8 assay for cell viability
The effects of ANA-12 on BV-2 cells viability were detected by CCK-8 assay [22]. In brief, cells were cultured on a 96-well plate at a density of 1 × 104 per well for 24 h and then administrated with ANA12 (5 uM, 10 uM, 15 uM) for another 24 h. Then the cells were incubated at 37 C for 2 h and the absorbance values of the samples were measured at 450 nm by a multifunctional microplate reader (SpectraMax M5, Sunnyvale, CA, USA).
Statistical analysis
SPSS 16.0 for Windows (SPSS Inc., Chicago, IL, USA) was used to carry out the statistical analyses. Two-way analysis of variance (ANOVA) with repeated measures was used to analyze the MWM data. Other statistical tests were conducted using one-way ANOVA and Student’s t-test for comparisons between groups. The data were expressed as the mean ± SE, and differences were considered statistical significance at P < 0.05.
Results
Chronic NTP treatment attenuates cognitive deficits of APP/PS1 mice in the Morris water maze
Morris water maze test was performed to evaluate whether NTP could attenuate the cognitive deficits in the APP/PS1 transgenic mice at 9 months of age (Figure 1). The NTP-treated APP/PS1 mice were administrated with NTP at 6 months of age for 3 months by oral gavage delivery. The control APP/PS1 mice were administrated with saline (0.9% NaCl). During the hidden platform tests, control WT mice showed progressively decreased in the escape latencies over the consecutive 5 days of training. Control APP/PS1 mice had a slight decline in the escape latencies during the entire training periods, but there was a significant extent in escape latency time compared with WT mice (P < 0.01, Figure 1A). To control individual differences in swimming speed, we also normalized the escape latencies of each group in the first trial day to 1.0 (Figure 1B). Compared with WT mice, control APP/PS1 mice still show a failure in learning trend, indicating impaired learning ability (P < 0.01, Figure 1B). In contrast, NTP-treated APP/PS1 mice exhibited a comparable learning trend with WT mice. Similar to the escape latencies, NTP-treated APP/PS1 mice showed progressively decreased in the swimming length compared with control APP/PS1 mice (P < 0.01, Figure 1C and 1D). In the probe test, NTP-treated APP/PS1 mice tended to concentrate in the target area of the pool and cross over the target quadrant more times than control APP/PS1 mice (P < 0.01, Figure 1E). NTP-treated mice were similar to control WT mice and no significant differences were observed in escape latencies, path length, and numbers of platform area crossings. These results demonstrate that chronic NTP treatment can improve cognitive deficits in APP/PS1 mice.
Figure 1.
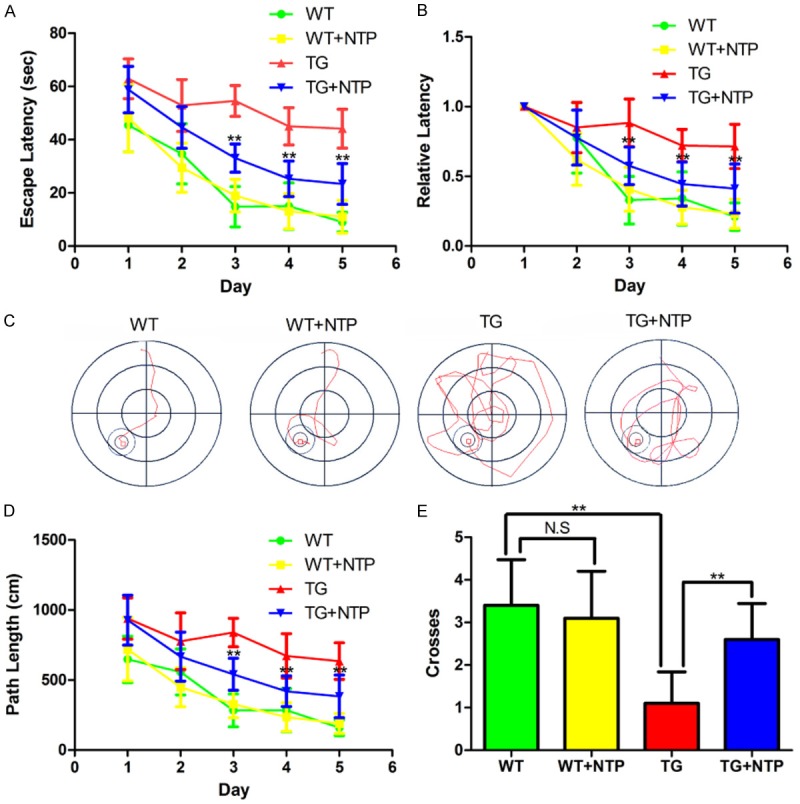
Neurotropin attenuates cognitive impairment of APP/PS1 mice. A. The escape latencies of the mice in each group of mice. B. The normalized escape latencies of each group of mice. C. Representative path images of the mice finding the platform. D. The average distances of the mice swimming to find the platform. E. The times of the mice swimming across the target quadrants. The results are presented as mean ± SE from at least eight mice in each group. **P < 0.01, and NS, nonsignificant.
Chronic NTP treatment reduces Aβ burden in APP/PS1 mice
To examine the potential function of NTP treatment on Aβ aggregation and to observe the morphologic changes after NTP treatment, the slices of the cortex and hippocampus of four groups of mice were stained using both Bielschowsky silver staining and immunofluorescent staining (Figure 2A and 2B). Quantification analysis revealed that the APP/PS1 mice treated with NTP showed significantly lower amyloid plaques in both the cortical and hippocampal areas than the control APP/PS1 mice (P < 0.01, Figure 2C and 2D). Moreover, previous studies have shown that APP/PS1 mice have age-related increased levels in both soluble and insoluble Aβ1-40 and Aβ1-42 [19,20]. Consistent with decreased Aβ burden, ELISA analysis demonstrated that NTP-treated APP/PS1 mice showed a significant decline in both soluble Aβ1-40 and Aβ1-42 levels compared with that in both the hippocampus and cortex of APP/PS1 mice (P < 0.05, Figure 2E, 2F). For insoluble Aβ1-40 and Aβ1-42, we also found a significant decrease in NTP-treated APP/PS1 mice (P < 0.05, Figure 2G, 2H). These results suggest that chronic treatment with NTP may be able to have an inhibitory effect on the generation and accumulation of Aβ plaques in the brain of APP/PS1 mice.
Figure 2.
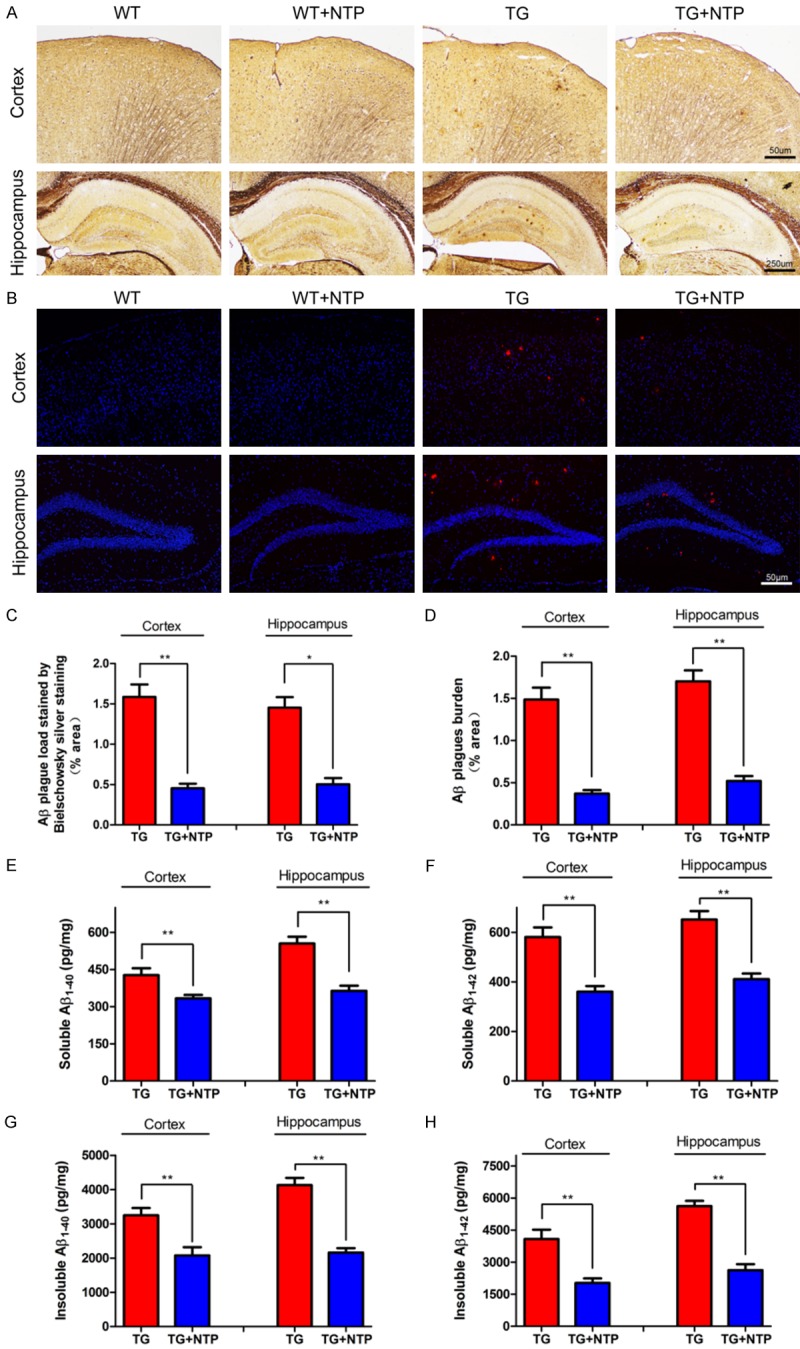
Neurotropin decreases Aβ accumulation of APP/PS1 mice. A. Aβ plaques were detected by Bielschowsky silver staining in the cortex and hippocampus. B. Aβ plaques were detected with immunofluorescent staining in the cortex and hippocampus. C. Quantification of Aβ plaque load using Bielschowsky silver staining. D. Statistical analysis of Aβ plaque burden with immunofluorescent staining. E and G. Soluble and insoluble Aβ1-40 in the brain of TG and TG+NTP mice. F and H. Soluble and insoluble Aβ1-42 in the brain of TG mice and TG+NTP mice. The results presented as means ± SE from six independent experiments. *P < 0.05 and **P < 0.01 versus TG mice.
Chronic NTP treatment inhibits glial activation in APP/PS1 mice
Activated microglia and astrocytes have been shown to be associated with Aβ accumulation, and they can promote the production of pro-inflammatory cytokines, resulting in synaptic dysfunction, neuronal death, and neurodegeneration. Therefore, we examined whether NTP treatment might alter glial activation in the cerebral cortex and hippocampus of APP/PS1 mice at 9 months of age, using immunofluorescent staining with antibodies against ionized calcium-binding adaptor molecule 1 (Iba-1) and glial fibrillary acidic protein (GFAP) to reveal changes in microgliosis and astrogliosis. We found that Αβ plaques were surrounded by Iba-1 immunoreactivity (IR) microglia (Figure 3A) and GFAP-IR astrocytes (Figure 3B), indicating both microglial and astrocytic activation in the cortex and the hippocampus of control APP/PS1 mice. In contrast, significant decreases in the area percentage of Iba-1-IR microglia astrocytes was observed accompanied with reduced Αβ burden in NTP-treated APP/PS1 mice (P < 0.01, Figure 3C). Consistently, the area percentage of GFAP-IR astrocytes also reduced after NTP treatment (P < 0.01, Figure 3D). These results show that chronic NTP treatment may suppress glial activation in APP/PS1 mice.
Figure 3.

Neurotropin alleviates astrocyte activation with attenuation of reactive gliosis and neuroinflammation. The coronal sections of the cortex and hippocampus in TG group and TG+NTP group of the mice were stained for (A). Aβ, Iba1 and DAPI, (B) Aβ, GFAP and DAPI. The percentage of the areas of microglial (C) and astrocytes (D) in the cortex and hippocampus. Analysis of the levels of IL-1β (E), IL-6 (F), and TNF (G) in the cortex and hippocampus of each group by ELISA. Data are presented as mean ± SE from six mice in each group. *P < 0.05, and **P < 0.01.
NTP treatment decreases pro-inflammatory cytokines in APP/PS1 mice
Persistent activated microglia and astrocytes can mediate neuroinflammation via releasing pro-inflammatory cytokines and facilitate Aβ deposition, leading to inflammatory neuronal damage. Furthermore, previous evidence has suggested that NTP was able to suppress inflammatory cytokine expression in hepatocytes. Thus, to explore whether chronic treatment with NTP could affect the production of inflammatory factors in 9-month APP/PS1 mice, we examined the levels of pro-inflammation cytokines including interleukin-1 beta (IL-1β), interleukin-6 (IL-6) and tumor necrosis factor-alpha (TNF-α) using ELISA tests. We observed that APP/PS1 mice had markedly higher levels of IL-1β than NTP-treated APP/PS1 mice (P < 0.05, Figure 3E). After NTP treatment, APP/PS1 mice showed decreased IL-6 level (P < 0.05, Figure 3F). Additionally, the level of TNF-α was lower in NTP-treated group when compared with APP/PS1 mice without NTP treatment (P < 0.05, Figure 3G). There was no difference in levels of IL-1β, IL-6 and TNF-α between WT and NTP-treated WT mice. These results demonstrate that NTP may effectively reduce inflammatory reaction, ameliorating neuroinflammation in APP/PS1 mice.
NTP treatment promotes BDNF expression in APP/PS1 mice
Brain-derived neurotrophic factor (BDNF), a member of the neurotrophin family, is vital for synaptic plasticity and neuronal survival, and it is critical for learning and memory. It has come to light that BDNF was able to attenuate proinflammatory cytokines production, demonstrating that BDNF may be correlated with homeostatic maintenance during neuroinflammation [17]. Thus, we further explored the changes of BDNF expression in each group of mice by immunostaining (Figure 4A and 4B) and ELISA (P < 0.01, Figure 4C).
Figure 4.
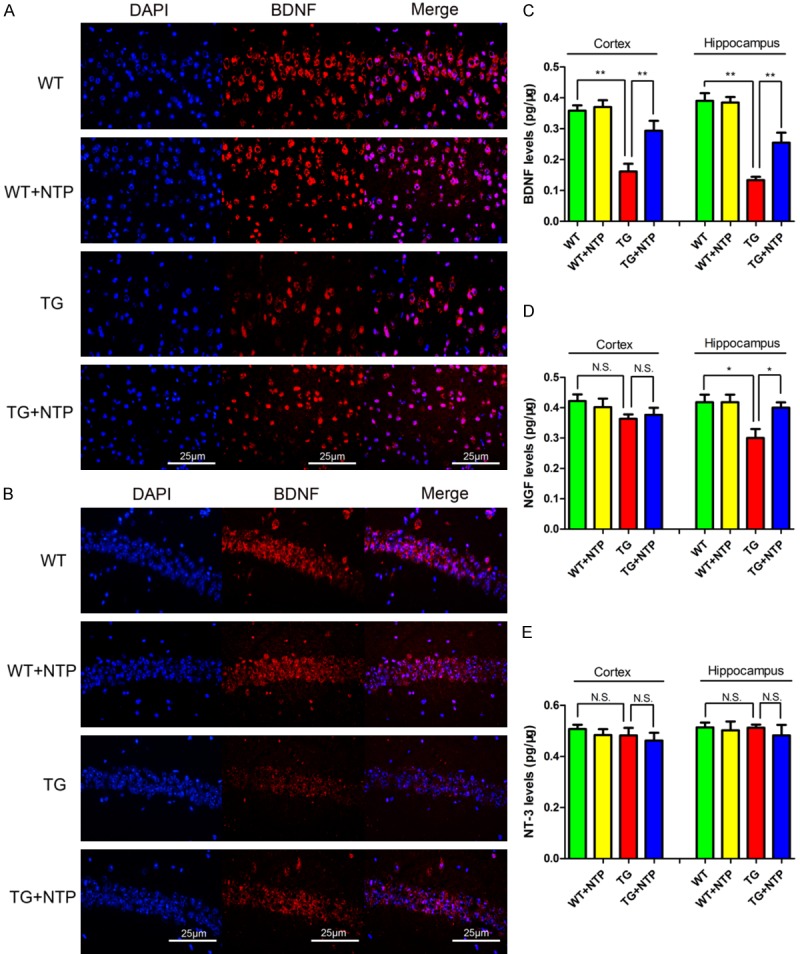
Neurotropin enhances levels of BDNF in APP/PS1 mice. BDNF was detected with immunofluorescent staining in the cortex (A) and the hippocampus (B) of each group. Analysis of the levels of BDNF (C), NGF (D), and NT-3 (E) in the cortex and the hippocampus with ELISA. Data are presented as mean ± SE from six mice in each group. *P < 0.05, **P < 0.01, and N.S., nonsignificant.
The results showed that 9-month old APP/PS1 mice had significantly lower levels of BDNF in both the cerebral cortex and hippocampus when compared to WT mice. In contrast, BDNF levels were significantly enhanced in the cortex and hippocampus of NTP-treated APP/PS1 mice compared with control APP/PS1 mice (P < 0.01, Figure 4A-C). In addition, upregulated levels of NGF were observed in the hippocampus but not the cortex of NTP-treated APP/PS1 mice compared with control APP/PS1 mice (P < 0.05, Figure 4D). However, we found that levels of neurotrophin-3 (NT-3) remained unchanged among all the groups of mice (P > 0.05, Figure 4E). Together, these findings reveal that chronic NTP treatment could markedly promote the expression of BDNF in the brain of APP/PS1 mice.
NTP regulates NF-κB pathway in vivo and in vitro
To further explore the underlying mechanism, we assessed the expression of NF-κB pathways related proteins by Western blot analysis. We found that protein expression p-p65 and p-IκB-α were significantly up-regulated in APP/PS1 mice when compared with the WT group. After NTP treatment, p-p65 and p-IκB-α in the APP/PS1 mice were significantly down-regulated (Figure 5). It suggests that NTP may inhibit neuroinflammation and improve cognitive impairment via BDNF/NF-κB pathway.
Figure 5.
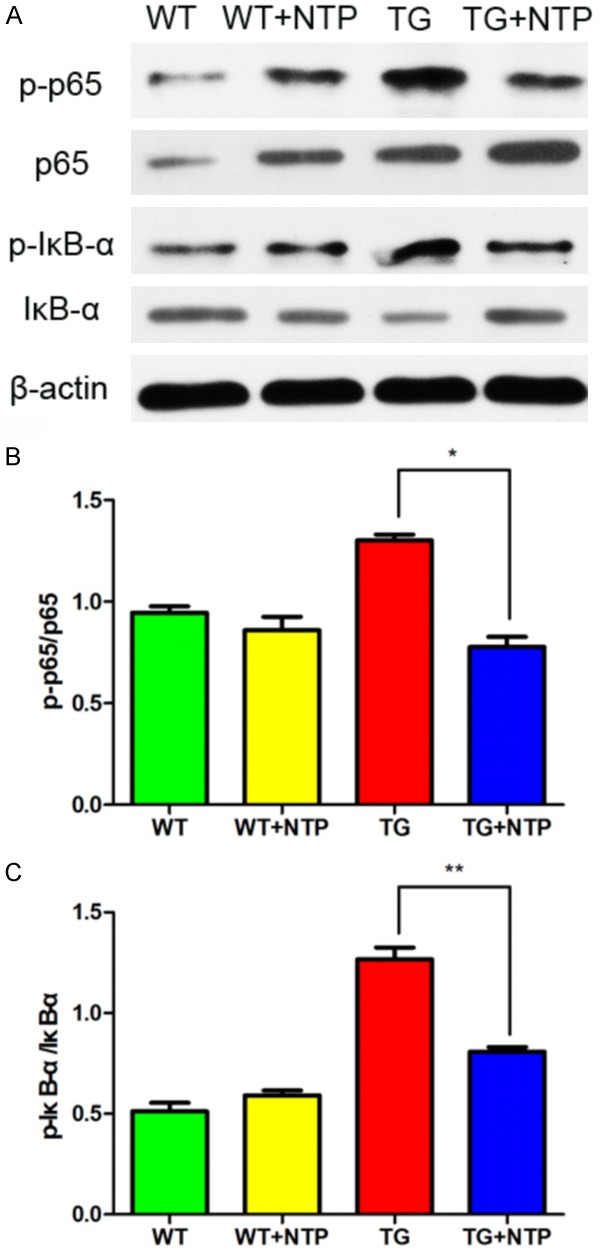
Chronic Neurotropin treatment regulates NF-κB pathways in APP/PS1 mice. A. Western blot analyses of the levels of p-65 and p-IκB. B and C. The relative levels of p-P65, p-IκB-α, and β-actin as a loading control in each group of mice, were quantified. Data are presented as mean ± SE from at least three mice in each group. *P < 0.05, and **P < 0.01.
To verify this mechanism, we used LPS to induce inflammation in BV-2 cell. It is shown that IL-1β, IL-6 and TNF-α were found highly expressed after LPS treatment (1000 ng/mL) by comparing with control group (Figure 6A). To further explore the link between BDNF and NF-κB, we used a selective, non-competitive BDNF receptor antagonist, ANA12, to inhibit BDNF pathway. As is shown in the Figure 6A, the expression of IL-1β, IL-6 and TNF-α decreased after NTP treatment but increased after ANA12 administration. Cell viability was assayed by CCK8 after treatment with ANA12 and there was no difference after ANA12 treatment at the concentration of 5 uM, 10 uM and 15 uM (Figure 6B). Additionally, BDNF level was detected after NTP and ANA12 treatment. LPS reduced BDNF level while NTP increased BDNF level. The effect of NTP on BDNF was abolished by ANA12 (Figure 6C). We also examined the expression of p-p65 and p-IκB-α on LPS-stimulated cells. Consistently, we found that both p-p65 and p-IκB-α were activated by LPS and reduced by NTP. Interestingly, the activation of p-P65 and p-IκB-α were shown to be abolished by ANA-12 (Figure 6D). Taken together, our results demonstrated that NTP regulated BDNF/NF-κB pathways in vivo and in vitro.
Figure 6.
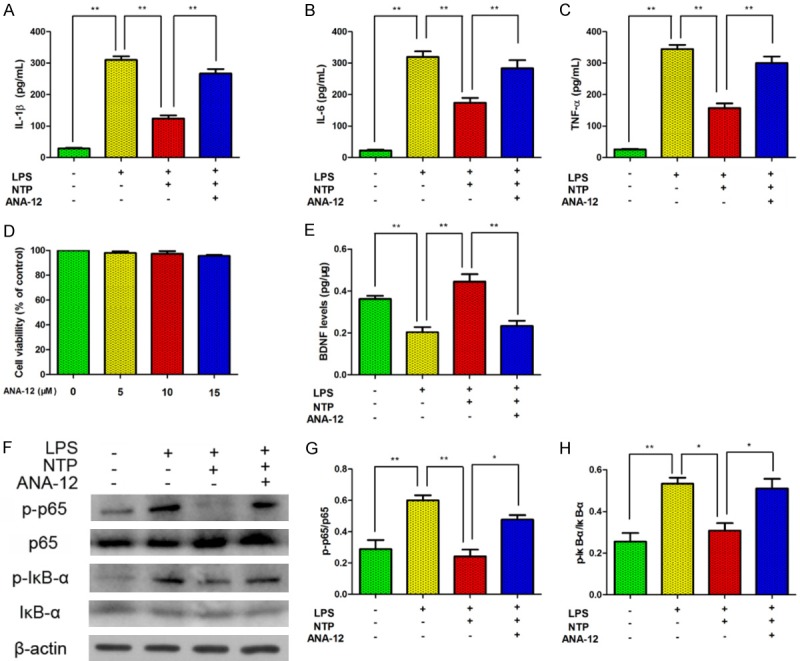
The effect of NTP on BV-2 cells was abolished by BDNF inhibitor. A-C. IL-1β, IL-6 and TNF-α were found highly expressed after LPS treatment by comparing with control group. IL-1β, IL-6 and TNF-α increased after a selective, non-competitive BDNF receptor antagonist, ANA12, administration. D. Cell viability was assayed by CCK8 after treatment with ANA12. E. BDNF level was detected after NTP and ANA12 treatment. F-H. Both p-p65 and p-IκB-α were activated by LPS and inactivated by NTP. The activation of p-p65 and p-IκB-α was abolished by ANA12.
Discussion
Neurotropin, a non-protein extract isolated from the inflamed skin of rabbits inoculated with vaccinia virus, is a widely used analgesic drug for the treatment of intractable neuropathic pain [25]. Recently, the potential therapeutic effects of NTP are rapidly expanding [28]. NTP showed capability of protecting the brain against ischemic stroke, accelerates the remyelination in demyelination disease and reduced muscular mechanical hyperalgesia [29,30]. However, there is still no evidence for the role of NTP play on cognitive function and inflammation in mouse model of AD, which is a multifactorial neurodegenerative disease without effective treatment.
NTP was demonstrated to have function of enhancing spatial learning of C57BL/6J mice [15]. In addition, NTP was found to facilitate cognitive improvement of Ts65Dn mice, a Down Syndrome mouse model [14]. However, there is still no evidence that NTP can have any influence on Alzheimer’s disease. Our study shown that chronic NTP treatment was sufficiently to improve cognitive deficits in APP/PS1 mice, which was assessed by Morris water maze test.
Neuroinflammation is a critical feature of AD [31] and activation of microglia and astrocytes by Aβ may promote the production of proinflammatory cytokines, enhancing neuroinflammation reactions [32]. In this study, we chose APP/PS1 mice as our AD transgenic model since this model steadily mimic the behavioral and pathological changes of AD and has been widely used in AD researches [33,34]. The present study highlighted the inhibition of NTP on neuroinflammation including microgliosis, astrogliosis, and pro-inflammation cytokines (IL-1β, IL-6, and TNF-α) in APP/PS1 mice.
In the present study, we observed that NTP treatment significantly increased the expression of BDNF and inhibitor of BDNF receptor could abolish this effect. It suggests that NTP may play the neuroprotective role in a BDNF dependent manner. Bdnf gene expression has been demonstrated to be regulated by physical activity or pathological stimuli like stress, trauma, infection and aging [35]. BDNF levels are reduced in plasma of patients with AD [17]. Normally, BDNF is translated as pro-neurotrophin (pro-BDNF) that can be cleaved into mature BDNF by endoproteases or metalloproteinases [36]. BDNF can be secreted and bind to the two different kinds of receptors, low affinity p75 neurotrophin receptor (p75NTR) and high-affinity receptor tyrosine kinase B (TrkB). Binding to these two different receptors potentially activates different pathways and leads to either cell death or survival [37]. However, the concentration of pro-BDNF was reported to be ten times lower than mature BDNF in animal model [37]. Therefore, we detected mature BDNF and used the TrkB inhibitor to block the BDNF pathway in the present study.
Microglia participate actively in the development of pathological neuroinflammatory process, which plays an important role in AD pathogenesis [38]. In this study, our results showed that chronic NTP treatment inhibited glial activation and decreased pro-inflammatory cytokines in APP/PS1 mice. In the nervous system, the main factor of neuronal inflammatory activation is NF-κB, a regulator of apoptosis, proliferation, and maturation of immune cells. NF-κB (p65) is bound to IκB as an inactive p65/IκB complex existing in the cytoplasm before its activation [39]. It is reported that activated NF-κB is found surrounding amyloid plaques in AD brain [40]. Frede and colleagues observed that bacterial LPS was able to induce NF-κB up-regulation [41]. In agreement, our recent studies have demonstrated that NTP can suppress the expression of NF-κB in lipopolysaccharide-stimulated BV2 cells [16]. Consistently, present results exhibited that supplementation of NTP markedly decreased the activation of p-p65 and p-IκB-α in APP/PS1 mouse model. However, it is reported that binding of BDNF to the TrkB could also induce the expression of NF-κB. NF-κB stimulated by BDNF might activate PLC-γ/PKC signaling via the kinases IKKα and IKKβ, which subsequently phosphorylates the NF-κB inhibitory unit IκBα. Consequently, binding of ubiquitin and degradation of IκBα by proteasomes induces the release of the NF-κB [42].
Qirui Bi et al. showed that venenum bufonis triggers neuroinflammation through NF-κB pathways, leading to an ultimate decrease in BDNF, but they did not directly link NF-κB cytokines with BDNF [43]. Cai et al. report that BDNF protects against IL-1β stimulation by modulating NF-κB signaling [44]. We used a specific BDNF receptor inhibitor, ANA12, to block the BDNF pathway in vitro [22]. This pre-treatment abolished NTP’s neuroprotective effects against LPS-stimulated inflammation, supporting the notion that NTP may protect the neuroinflammation via BDNF/NF-κB pathway.
However, the exact mechanism of BDNF functions on NF-κB still remains to be explored. Casein kinase II (CK2) is a highly conserved ubiquitous serine/threonine protein kinase which have been proved to activate NF-κB [45-47]. It is reported that BDNF upregulated NF-κB by CK2. Also, BDNF was demonstrated to produce neuroprotective effect via ERK1/2 signaling which is consistent with our previous researches [48]. BDNF activate NF-κB via CK2, which seems to be independent of ERK1/2 and PI3K [49]. As is shown by our previous research, NTP cloud decrease the translocation of p65 from cytoplasm to nuclear, which might be a novel mechanism for BDNF to regulate NF-κB activation [16]. Further research needs to be conducted to fully understand the mechanism of BDNF on NF-κB pathway.
Conclusions
These intriguing findings suggest that NTP can counteract neuroinflammation and rescue cognitive deficits of APP/PS1 mice by enhancing through the BDNF/NF-κB pathway. The results provide further insight into the interactions of NTP and neuroinflammation. NTP may be a new promising drug candidate for patients with AD. In addition, although NTP has established safe profiles in humans, it still requires large-scale clinical trials for further confirmation of its neuroprotective capability in both sporadic and familial AD.
Acknowledgements
This study was funded by Guangzhou Science and Technology Program key projects (Jun Liu, 201604020100, Songhua Xiao, 201803010013), Guangdong Science and Technology Program (Jun Liu, 2017A020211017) and the National Nature Science Foundation of China (Jun Liu, 81870836). Special thanks go to Prof. Qiulan Ma at University of California, Los Angeles for critical comments and helpful discussions on this manuscript.
Disclosure of conflict of interest
None.
Abbreviations
- AD
Alzheimer’s disease
- APP/PS1
APPswe/PSEN1dE9
- Aβ
Neurotropin
- NTP
linear dichroism
- BDNF
Brain derived neurotrophic factor
- ELISA
Enzyme-linked immunosorbent assay
- GFAP
Glial fibrillary acidic protein
- Iba-1
Ionized calcium-binding adaptor molecule-1
- IF
Immunofluorescent staining
- IL-1β
Interleukin-1 beta
- IL-6
Interleukin-6
- MWM
Morris water maze
- NGF
Nerve growth factor
- NF-κB
Nuclear factor-κB
- NT-3
neurotrophin-3
- SPF
Specific pathogen free
- TG mice
Transgenic mice
- TNF-α
Tumor necrosis factor-α
- TrkB
Tyrosine kinase receptor B
- WT mice
Wild-type mice
References
- 1.Cavedo E, Chiesa PA, Houot M, Ferretti MT, Grothe MJ, Teipel SJ, Lista S, Habert MO, Potier MC, Dubois B, Hampel H INSIGHT-preAD Study Group; Alzheimer Precision Medicine Initiative (APMI) Sex differences in functional and molecular neuroimaging biomarkers of Alzheimer’s disease in cognitively normal older adults with subjective memory complaints. Alzheimers Dement. 2018;14:1204–1215. doi: 10.1016/j.jalz.2018.05.014. [DOI] [PubMed] [Google Scholar]
- 2.Newcombe EA, Camats-Perna J, Silva ML, Valmas N, Huat TJ, Medeiros R. Inflammation: the link between comorbidities, genetics, and Alzheimer’s disease. J Neuroinflammation. 2018;15:276. doi: 10.1186/s12974-018-1313-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tournier BB, Tsartsalis S, Rigaud D, Fossey C, Cailly T, Fabis F, Pham T, Gregoire MC, Kovari E, Moulin-Sallanon M, Savioz A, Millet P. TSPO and amyloid deposits in sub-regions of the hippocampus in the 3xTgAD mouse model of Alzheimer’s disease. Neurobiol Dis. 2019;121:95–105. doi: 10.1016/j.nbd.2018.09.022. [DOI] [PubMed] [Google Scholar]
- 4.Marin I, Kipnis J. Learning and memory ... and the immune system. Learn Mem. 2013;20:601–606. doi: 10.1101/lm.028357.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schafer DP, Lehrman EK, Kautzman AG, Koyama R, Mardinly AR, Yamasaki R, Ransohoff RM, Greenberg ME, Barres BA, Stevens B. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron. 2012;74:691–705. doi: 10.1016/j.neuron.2012.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Spangenberg EE, Lee RJ, Najafi AR, Rice RA, Elmore MR, Blurton-Jones M, West BL, Green KN. Eliminating microglia in Alzheimer’s mice prevents neuronal loss without modulating amyloid-beta pathology. Brain. 2016;139:1265–1281. doi: 10.1093/brain/aww016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hong S, Beja-Glasser VF, Nfonoyim BM, Frouin A, Li S, Ramakrishnan S, Merry KM, Shi Q, Rosenthal A, Barres BA, Lemere CA, Selkoe DJ, Stevens B. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science. 2016;352:712–716. doi: 10.1126/science.aad8373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lai RY, Harrington CR, Wischik CM. Absence of a role for phosphorylation in the tau pathology of Alzheimer’s disease. Biomolecules. 2016;6 doi: 10.3390/biom6020019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cummings J, Lee G, Mortsdorf T, Ritter A, Zhong K. Alzheimer’s disease drug development pipeline: 2017. Alzheimers Dement (N Y) 2017;3:367–384. doi: 10.1016/j.trci.2017.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fang WL, Zhao DQ, Wang F, Li M, Fan SN, Liao W, Zheng YQ, Liao SW, Xiao SH, Luan P, Liu J. Neurotropin (R) alleviates hippocampal neuron damage through a HIF-1/MAPK pathway. CNS Neurosci Ther. 2017;23:428–437. doi: 10.1111/cns.12689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ishikawa T, Yasuda S, Minoda S, Ibuki T, Fukuhara K, Iwanaga Y, Ariyoshi T, Sasaki H. Neurotropin((R)) ameliorates chronic pain via induction of brain-derived neurotrophic factor. Cell Mol Neurobiol. 2015;35:231–241. doi: 10.1007/s10571-014-0118-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.De Reuck J, Decoo D, Vanderdonckt P, Dallenga A, Ceusters W, Kalala JP, De Meulemeester K, Abdullah J, Santens P, Huybrechts J, et al. A double-blind study of neurotropin in patients with acute ischemic stroke. Acta Neurol Scand. 1994;89:329–335. doi: 10.1111/j.1600-0404.1994.tb02643.x. [DOI] [PubMed] [Google Scholar]
- 13.Masuguchi K, Watanabe H, Kawashiri T, Ushio S, Ozawa N, Morita H, Oishi R, Egashira N. Neurotropin(R) relieves oxaliplatin-induced neuropathy via Gi protein-coupled receptors in the monoaminergic descending pain inhibitory system. Life Sci. 2014;98:49–54. doi: 10.1016/j.lfs.2013.12.229. [DOI] [PubMed] [Google Scholar]
- 14.Fukuda Y, Berry TL, Nelson M, Hunter CL, Fukuhara K, Imai H, Ito S, Granholm-Bentley AC, Kaplan AP, Mutoh T. Stimulated neuronal expression of brain-derived neurotrophic factor by neurotropin. Mol Cell Neurosci. 2010;45:226–233. doi: 10.1016/j.mcn.2010.06.013. [DOI] [PubMed] [Google Scholar]
- 15.Nakajo Y, Yang D, Takahashi JC, Zhao Q, Kataoka H, Yanamoto H. ERV enhances spatial learning and prevents the development of infarcts, accompanied by upregulated BDNF in the cortex. Brain Res. 2015;1610:110–123. doi: 10.1016/j.brainres.2015.03.042. [DOI] [PubMed] [Google Scholar]
- 16.Zheng Y, Fang W, Fan S, Liao W, Xiong Y, Liao S, Li Y, Xiao S, Liu J. Neurotropin inhibits neuroinflammation via suppressing NF-kappaB and MAPKs signaling pathways in lipopolysaccharide-stimulated BV2 cells. J Pharmacol Sci. 2018;136:242–248. doi: 10.1016/j.jphs.2018.02.004. [DOI] [PubMed] [Google Scholar]
- 17.Lima Giacobbo B, Doorduin J, Klein HC, Dierckx RAJO, Bromberg E, de Vries EFJ. Brain-derived neurotrophic factor in brain disorders: focus on neuroinflammation. Mol Neurobiol. 2018 doi: 10.1007/s12035-018-1283-6. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Leyhe T, Eschweiler GW, Stransky E, Gasser T, Annas P, Basun H, Laske C. Increase of BDNF serum concentration in lithium treated patients with early Alzheimer’s disease. J Alzheimers Dis. 2009;16:649–656. doi: 10.3233/JAD-2009-1004. [DOI] [PubMed] [Google Scholar]
- 19.Zhou J, Ping FF, Lv WT, Feng JY, Shang J. Interleukin-18 directly protects cortical neurons by activating PI3K/AKT/NF-kappaB/CREB pathways. Cytokine. 2014;69:29–38. doi: 10.1016/j.cyto.2014.05.003. [DOI] [PubMed] [Google Scholar]
- 20.Grilli M, Ribola M, Alberici A, Valerio A, Memo M, Spano P. Identification and characterization of a kappa B/Rel binding site in the regulatory region of the amyloid precursor protein gene. J Biol Chem. 1995;270:26774–26777. doi: 10.1074/jbc.270.45.26774. [DOI] [PubMed] [Google Scholar]
- 21.He P, Zhong Z, Lindholm K, Berning L, Lee W, Lemere C, Staufenbiel M, Li R, Shen Y. Deletion of tumor necrosis factor death receptor inhibits amyloid beta generation and prevents learning and memory deficits in Alzheimer’s mice. J Cell Biol. 2007;178:829–841. doi: 10.1083/jcb.200705042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fan D, Li J, Zheng B, Hua L, Zuo Z. Enriched environment attenuates surgery-induced impairment of learning, memory, and neurogenesis possibly by preserving BDNF expression. Mol Neurobiol. 2016;53:344–354. doi: 10.1007/s12035-014-9013-1. [DOI] [PubMed] [Google Scholar]
- 23.Liu S, Li X, Gao J, Liu Y, Shi J, Gong Q. Icariside II, a phosphodiesterase-5 inhibitor, attenuates beta-amyloid-induced cognitive deficits via BDNF/TrkB/CREB signaling. Cell Physiol Biochem. 2018;49:985. doi: 10.1159/000493232. [DOI] [PubMed] [Google Scholar]
- 24.Xiao SH, Zhou DY, Luan P, Gu BB, Feng LB, Fan SN, Liao W, Fang WL, Yang LH, Tao EX, Guo R, Liu J. Graphene quantum dots conjugated neuroprotective peptide improve learning and memory capability. Biomaterials. 2016;106:98–110. doi: 10.1016/j.biomaterials.2016.08.021. [DOI] [PubMed] [Google Scholar]
- 25.Knezovic A, Osmanovic-Barilar J, Curlin M, Hof PR, Simic G, Riederer P, Salkovic-Petrisic M. Staging of cognitive deficits and neuropathological and ultrastructural changes in streptozotocin-induced rat model of Alzheimer’s disease. J Neural Transm (Vienna) 2015;122:577–592. doi: 10.1007/s00702-015-1394-4. [DOI] [PubMed] [Google Scholar]
- 26.Liu J, Rasul I, Sun Y, Wu G, Li L, Premont RT, Suo WZ. GRK5 deficiency leads to reduced hippocampal acetylcholine level via impaired presynaptic M2/M4 autoreceptor desensitization. J Biol Chem. 2009;284:19564–19571. doi: 10.1074/jbc.M109.005959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liao W, Jiang MJ, Li M, Jin CL, Xiao SH, Fan SN, Fang WL, Zheng YQ, Liu J. Magnesium elevation promotes neuronal differentiation while suppressing glial differentiation of primary cultured adult mouse neural progenitor cells through ERK/CREB activation. Front Neurosci. 2017;11:87. doi: 10.3389/fnins.2017.00087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sakai D, Nakai T, Hiraishi S, Nakamura Y, Ando K, Naiki M, Watanabe M. Upregulation of glycosaminoglycan synthesis by neurotropin in nucleus pulposus cells via stimulation of chondroitin sulfate N-acetylgalactosaminyltransferase 1: a new approach to attenuation of intervertebral disc degeneration. PLoS One. 2018;13:e0202640. doi: 10.1371/journal.pone.0202640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Matsuoka H, Tanaka H, Sayanagi J, Iwahashi T, Suzuki K, Nishimoto S, Okada K, Murase T, Yoshikawa H. Neurotropin((R)) accelerates the differentiation of schwann cells and remyelination in a rat lysophosphatidylcholine-induced demyelination model. Int J Mol Sci. 2018;19 doi: 10.3390/ijms19020516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nasu T, Murase S, Takeda-Uchimura Y, Mizumura K. Intramuscularly injected neurotropin reduced muscular mechanical hyperalgesia induced by repeated cold stress in rats. Behav Pharmacol. 2018;29:261–269. doi: 10.1097/FBP.0000000000000313. [DOI] [PubMed] [Google Scholar]
- 31.Fu WY, Wang X, Ip NY. Targeting neuroinflammation as a therapeutic strategy for Alzheimer’s disease: mechanisms, drug candidates, and new opportunities. ACS Chem Neurosci. 2019;10:872–879. doi: 10.1021/acschemneuro.8b00402. [DOI] [PubMed] [Google Scholar]
- 32.Chen JJ, Wang T, An CD, Jiang CY, Zhao J, Li S. Brain-derived neurotrophic factor: a mediator of inflammation-associated neurogenesis in Alzheimer’s disease. Rev Neurosci. 2016;27:793–811. doi: 10.1515/revneuro-2016-0017. [DOI] [PubMed] [Google Scholar]
- 33.Oakley H, Cole SL, Logan S, Maus E, Shao P, Craft J, Guillozet-Bongaarts A, Ohno M, Disterhoft J, Van Eldik L, Berry R, Vassar R. Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: potential factors in amyloid plaque formation. J Neurosci. 2006;26:10129–10140. doi: 10.1523/JNEUROSCI.1202-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Joshi G, Gan KA, Johnson DA, Johnson JA. Increased Alzheimer’s disease-like pathology in the APP/PS1DeltaE9 mouse model lacking Nrf2 through modulation of autophagy. Neurobiol Aging. 2015;36:664–679. doi: 10.1016/j.neurobiolaging.2014.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McMurphy T, Huang W, Liu X, Siu JJ, Queen NJ, Xiao R, Cao L. Hypothalamic gene transfer of BDNF promotes healthy aging in mice. Aging Cell. 2018:e12846. doi: 10.1111/acel.12846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dieni S, Matsumoto T, Dekkers M, Rauskolb S, Ionescu MS, Deogracias R, Gundelfinger ED, Kojima M, Nestel S, Frotscher M, Barde YA. BDNF and its pro-peptide are stored in presynaptic dense core vesicles in brain neurons. J Cell Biol. 2012;196:775–788. doi: 10.1083/jcb.201201038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Matsumoto T, Rauskolb S, Polack M, Klose J, Kolbeck R, Korte M, Barde YA. Biosynthesis and processing of endogenous BDNF: CNS neurons store and secrete BDNF, not pro-BDNF. Nat Neurosci. 2008;11:131–133. doi: 10.1038/nn2038. [DOI] [PubMed] [Google Scholar]
- 38.Niraula A, Sheridan JF, Godbout JP. Microglia priming with aging and stress. Neuropsychopharmacology. 2017;42:318–333. doi: 10.1038/npp.2016.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shih RH, Wang CY, Yang CM. NF-kappaB signaling pathways in neurological inflammation: a mini review. Front Mol Neurosci. 2015;8:77. doi: 10.3389/fnmol.2015.00077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shi C, Zhu X, Wang J, Long D. Intromitochondrial IkappaB/NF-kappaB signaling pathway is involved in amyloid beta peptide-induced mitochondrial dysfunction. J Bioenerg Biomembr. 2014;46:371–376. doi: 10.1007/s10863-014-9567-7. [DOI] [PubMed] [Google Scholar]
- 41.Frede S, Stockmann C, Freitag P, Fandrey J. Bacterial lipopolysaccharide induces HIF-1 activation in human monocytes via p44/42 MAPK and NF-kappaB. Biochem J. 2006;396:517–527. doi: 10.1042/BJ20051839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mattson MP, Meffert MK. Roles for NF-kappaB in nerve cell survival, plasticity, and disease. Cell Death Differ. 2006;13:852–860. doi: 10.1038/sj.cdd.4401837. [DOI] [PubMed] [Google Scholar]
- 43.Bi QR, Hou JJ, Qi P, Ma CH, Shen Y, Feng RH, Yan BP, Wang JW, Shi XJ, Zheng YY, Wu WY, Guo D. Venenum Bufonis induces rat neuroinflammation by activiating NF-kappaB pathway and attenuation of BDNF. J Ethnopharmacol. 2016;186:103–110. doi: 10.1016/j.jep.2016.03.049. [DOI] [PubMed] [Google Scholar]
- 44.Cai Z, Zhang X, Wang G, Wang H, Liu Z, Guo X, Yang C, Wang X, Wang H, Shu C, Xiao L. BDNF attenuates IL-1beta-induced F-actin remodeling by inhibiting NF-kappaB signaling in hippocampal neurons. Neuro Endocrinol Lett. 2014;35:13–19. [PubMed] [Google Scholar]
- 45.Schaefer S, Guerra B. Protein kinase CK2 regulates redox homeostasis through NF-kappaB and Bcl-xL in cardiomyoblasts. Mol Cell Biochem. 2017;436:137–150. doi: 10.1007/s11010-017-3085-y. [DOI] [PubMed] [Google Scholar]
- 46.Huang J, Chen Z, Li J, Chen Q, Li J, Gong W, Huang J, Liu P, Huang H. Protein kinase CK2alpha catalytic subunit ameliorates diabetic renal inflammatory fibrosis via NF-kappaB signaling pathway. Biochem Pharmacol. 2017;132:102–117. doi: 10.1016/j.bcp.2017.02.016. [DOI] [PubMed] [Google Scholar]
- 47.Qaiser F, Trembley JH, Sadiq S, Muhammad I, Younis R, Hashmi SN, Murtaza B, Rector TS, Naveed AK, Ahmed K. Examination of CK2alpha and NF-kappaB p65 expression in human benign prostatic hyperplasia and prostate cancer tissues. Mol Cell Biochem. 2016;420:43–51. doi: 10.1007/s11010-016-2765-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Caviedes A, Lafourcade C, Soto C, Wyneken U. BDNF/NF-kappaB signaling in the neurobiology of depression. Curr Pharm Des. 2017;23:3154–3163. doi: 10.2174/1381612823666170111141915. [DOI] [PubMed] [Google Scholar]
- 49.Chao CC, Ma YL, Lee EH. Brain-derived neurotrophic factor enhances Bcl-xL expression through protein kinase casein kinase 2-activated and nuclear factor kappa B-mediated pathway in rat hippocampus. Brain Pathol. 2011;21:150–162. doi: 10.1111/j.1750-3639.2010.00431.x. [DOI] [PMC free article] [PubMed] [Google Scholar]