

Abstract
Circulating tumor DNA (ctDNA) carries genetic information consistent with tumor cells and has potential value for molecular diagnosis of tumors. The present study analysed the gene mutations of plasma circulating cell-free DNA (cfDNA) and tumor tissue DNA in hepatocellular carcinoma (HCC) patients and explored the clinical application value of plasma cfDNA as a tumor marker in HCC molecular diagnosis. Samples from 29 patients with primary HCC were collected. Hotspot mutations in 50 tumor-associated genes were analysed using amplicon sequencing technology and gene loci with a mutant allele frequency (MAF) >1% were analysed. 35 mutant genes in total were detected by deep sequencing method of which the genes with maximum mutation frequencies were TP53, ATM, and ALK. In addition, a total of 21 patients were found to have a consistent gene mutation in plasma cfDNA and tumor tissue DNA and 17 cases had consistent gene mutations in the paracancerous tissue and tumor tissue DNA. Further analysis showed that the MAFs in the TP53, CTNNB1, PIK3CA, and CDKN2A genes were higher in patients with tumor diameters >5 cm than those with tumor diameters <5 cm. And the MAFs in the TP53, RET, FGFR3 and APC genes were significantly higher in patients with multiple tumors or with metastasis than in single tumor patients. In conclusion, amplicon sequencing technology is highly sensitive for the detection of mutant genes in the plasma cfDNA of HCC patients. Plasma cfDNA might be an effective molecular marker for HCC molecular diagnosis.
Keywords: Hepatocellular carcinoma, circulating cell-free DNA, circulating tumor DNA, molecular diagnosis
Introduction
Primary hepatocellular carcinoma (HCC) is one of the most common malignancies worldwide and affects more than 700,000 individuals annually, thereby representing the second leading cause of tumor-related deaths after lung cancer [1-3]. HCC has complex pathogenic factors, a hidden onset, and atypical clinical symptoms. Therefore, some patients are diagnosed at an advanced stage of the disease and lose the chance for surgical treatment [4,5]. Surgical treatment combined with immunotherapy and molecular-targeted therapy has been highly recognized as a comprehensive treatment model for HCC, which is the future development direction of HCC treatment [6,7]. Tumor-targeted therapy is based on the precise molecular pathology of individual tumors. However, currently, there is no effective marker for the molecular diagnosis of HCC, while new and effective markers are essential to improve the early diagnosis of HCC [8]. In addition, the clinical study of targeted drugs for HCC has shown that the molecular pathological diagnosis of tumors is valuable in optimizing patient selection and improving the success rate of targeted drugs in clinical studies.
Similar to most malignancies, the development of HCC is accompanied by genetic and epigenetic variations. The application of high-throughput sequencing technology has revealed a plethora of information related to HCC pathogenesis such as gene mutations, copy number variations and methylation modification abnormalities and these pathogenesis suggest some potential molecular markers for the diagnosis and prognosis of HCC [9-13]. Moreover, some clinical studies on the targeted treatment of HCC have showed that tumor molecular pathology could guide the selection of appropriate targeted drugs for patients to improve the success rate of treatment, reduce the cost and decrease side effects. But the molecular diagnosis of HCC is a challenge because conventional puncture biopsy is easily to increase the risk of tumor metastasis while tissue puncture biopsy can not sufficiently reflect the tumor heterogeneity. The detection of circulating cell-free DNA (cfDNA) can overcome the limitations of tissue puncture and has potential application value in the diagnosis and monitoring of multiple solid tumors. cfDNA is primarily released from apoptotic and necrotic cells and can be detected at an early stage of tumor development. The cfDNA levels in tumor patients are significantly higher than those in healthy individuals. Thus, cfDNA might replace the tumor biopsy and become an important marker in the molecular diagnosis of tumors [14-17].
Molecular diagnosis of liver cancer gene mutations is the basis of targeted therapy, however, there is currently no useful markers for the molecular diagnosis of liver cancer. The detection of mutant genes in a tumor can be efficiently utilized to explore the molecular markers. Whole-genome sequencing and all-exon sequencing can provide additional comprehensive information regarding tumor gene mutations. But these strategies need higher sequencing costs, require large samples and have long detection periods. By contrast, the detection of plasma cfDNA has features including small initial sample size and short study cycle for targeted gene detection.
In this study, we detected mutant genes in common tumor hotspots by the amplicon sequencing technique and targeted amplicon sequencing of gene mutations in the plasma cfDNA and tumor tissue DNA of HCC patients to investigate the clinical value of plasma cfDNA as a tumor marker in HCC molecular diagnosis.
Materials and methods
Research subjects
Samples were collected from 29 patients enrolled in the Hepatobiliary Surgery Departments of Peking Union Medical College Hospital and Henan Provincial Cancer Hospital. All the patients, 24 males and 5 females with an average age of 57 (45-79) years, were diagnosed with primary HCC by imaging and histopathological examination. They were of Han nationality and without diabetes, hypertension, heart disease or autoimmune diseases. The plasma samples and peripheral white blood cells were collected 1 day before surgery. Tumor tissue specimens and paired specimens that were approximately 2 cm outside of the edge of the tumor were freshly resected during surgery and immediately preserved in liquid nitrogen. All the participants provided written informed consent.
Sample processing
Tumor tissue and paracancerous tissue blocks were obtained from the surgically resected specimens. The tissue specimens were collected and cryopreserved in liquid nitrogen and transported on dry ice at an ultra-low temperature. Peripheral venous blood samples of 2-3 mL were drawn from the patients on the day before surgery and were centrifuged at 1600 g for 10 min at 4°C to separate the plasma from. Then upper plasma layer was transferred to a new tube at 1 mL/tube (the blood cells were removed) and was centrifuged again at 16,000 g for 10 min at 4°C to remove the protein, after which it was transferred to a new tube and subsequently cryopreserved with fewer blood cells.
DNA extraction and library construction
The tissue DNA was extracted using the BioTeke Tissue DNA Extraction Kit (Cat# AU19011, BioTeke Corporation, Beijing, China) and a nucleic acid automatic extractor. The free DNA in plasma was isolated by a magnetic bead method using the Beaver Beads™ Circulating DNA Kit (Cat# 70404-100, Suzhou, China). The DNA extracted from tumor tissue, paracancerous tissue, and peripheral blood mononuclear cells (PBMCs) was quantified using the Qubit® dsDNA BR Assay Kit (Q32850, Thermo Fisher Scientific Co., MA, USA). The cfDNA extracted from the plasma was quantified using the Qubit® dsDNA HS Analysis Kit (Q32851, Thermo Fisher Scientific, MA, USA). A library was constructed using the GeneCast Kit (GeneCast Biotechnology Co., Beijing, China). The prepared amplicon library was quantified using the Qubit method as described above, and the size distribution and concentration of the amplicon fragments in the library were assessed using an Agilent Bioanalyzer 2100 instrument and the Agilent High Sensitivity DNA Kit. Based on the quantitative results, the libraries were mixed at the same concentration ratio (10 ng of each sample) to ensure the identical rates of coverage of the different samples in parallel sequencing. After mixing the library, we used the Agilent Bioanalyzer instrument to identify the fragment size of the hybrid library (approximately 280 bp in every library) to ensure library consistency. The Illumina HiSeq X Ten sequencing platform was used for the high-throughput sequencing.
Bioinformatics analysis and data filtering
The original data of the FASTQ file were sequenced for bioinformatics analysis and data filtering sequencing to verify the sample sequence, mutation sites, and calculate the mutant allele frequency (MAF). First, for each sample, the FASTQ sequencing files were compared to the human genome sequence (hg19) using BWA (Burrows-Wheeler Aligner, version 0.7.12) sequence alignment software. Subsequently, the data were filtered using Picard tools to generate SAM files and reordered using Samtools (version 1.3). The SAM files for all the samples were then subjected to single nucleotide polymorphism (SNP) processing to obtain information regarding single nucleotide mutations. The following functional annotation and further analyses were conducted for these sites and their genes:
Step 1: Comparison of the mutation trend of different genes, i.e., analysis of the mutant genes and mutation load rates in tumor tissue DNA, paracancerous tissue DNA and plasma cfDNA.
Step 2: Identification of valuable mutation sites/genes and comparisons of DNA mutations in tumor tissue, paracancerous tissue, blood cell samples and plasma samples. These mutations were divided into the following four categories: (1) The frequency of mutations was higher in the blood cell samples but lower or negative in tumor tissue, paracancerous tissue and plasma DNA indicating this mutation was not related to the tumor formation. (2) The mutation exhibited a higher frequency in tumor tissue and/or paracancerous tissues and a lower frequency in blood cell DNA and free DNA; such mutations may be tumor-related and were further analysed in the present study. (3) That the mutation frequency was higher in tumor tissue, paracancerous tissue and plasma DNA and lower or 0 frequency in blood cell DNA as well as which showed a small number of free tumor DNA fragments in the plasma tumor-related mutation analysis while almost all the blood cells are normal cells were the types that were further analysed in the present study. (4) The mutations exhibiting a low or 0 frequency in the tumor tissue DNA and blood cells but a high frequency in paracancerous tissues and plasma cfDNA might reflect a mutation in the plasma DNA that originated from tumor tissue but was not expressed in it because of limitations in the material. This type of mutation might be involved in tumorigenesis and was further analysed in the present study.
The mutations of types (2), (3) and (4) were further followed up and analysed.
Statistical analysis
Plots were constructed using GraphPad Prism 6 (La Jolla, CA, USA), and the statistical analysis was performed by SPSS 22.0 (Chicago, IL, USA) software. The normality test was performed to the counting data and the normally distributed data were expressed as the means ± standard error (SE). Student’s t-test was used to compare the differences between the groups. While the non-normally distributed data were presented as the means ± quartile spacing or the median. Comparisons between the groups were performed using the Mann-Whitney U test and the counting data were tested using Fisher’s exact test. P<0.05 was considered statistically significant.
Results
Clinicopathological features of HCC patients
Twenty-nine eligible patients with HCC were enrolled in this study (Tables 1 and S1). The patients were aged 45-79 years (median age 57 years) and primarily were males (82.8%). A majority of the patients presented hepatitis B-related HCC (75.9%) and more than a half of the patients had a history of cirrhosis (51.7%). Table 1 illustrates that only 48.3% (14/29) of the HCC patients were alpha-foetoprotein (AFP)-positive (>20 ng/mL) and that nearly a half of the patients (48.3%) had a tumor diameter >5 cm. According to Barcelona Clinic Liver Cancer (BCLC) liver staging criteria, 15 patients (51.7%) were categorized to stage A while 14 (48.3%) patients to stages B and C in this study.
Table 1.
Clinicopathological features of HCC patients
| Clinical features | Grouping | Number of cases | |
|---|---|---|---|
|
| |||
| n=29 | % | ||
| Age (median, range) | 57 (45-79) years | 29 | 100 |
| Gender | Male | 24 | 82.8 |
| Female | 5 | 17.2 | |
| HBsAg* (+/-) | + | 22 | 75.9 |
| - | 7 | 24.1 | |
| ALT (IU/L) | >80 | 3 | 10.3 |
| ≤80 | 26 | 89.7 | |
| AFP, ng/mL | >20 | 14 | 48.3 |
| ≤20 | 15 | 51.7 | |
| Cirrhosis (with/without) | with | 16 | 55.2 |
| without | 13 | 44.8 | |
| Maximum diameter of tumour (cm) | ≥5 | 14 | 48.3 |
| <5 | 15 | 51.7 | |
| Number of tumour nodules (s) | 1 | 24 | 82.8 |
| >1 | 5 | 17.2 | |
| BCLC staging | A | 15 | 51.7 |
| B | 9 | 31 | |
| C | 5 | 17.3 | |
HBsAg: hepatitis B surface antigen;
ALT: alanine aminotransferase, AFP: alpha-fetoprotein, BCLC: Barcelona clinic liver cancer.
Quality control of the amplicon sequencing
In the present study, 50 tumor-associated high-frequency mutations were detected in 29 HCC patients using amplicon sequencing technology. First, the sequencing data were subjected to a quality control analysis which scored the sequencing of each base position in the fragment, the A/T/C/G four-base distribution at each nucleotide position, the read length of the fragment and the depth of sequencing (Figure 1). The results of the nucleotide position sequencing showed satisfactory consistency with the sequencing data (Figure 1A), suggesting that the specimens were homogeneous and did not produce a significant bias in the construction of the database. In addition, the G/C and A/T distributions were not uniform in the 1-4 bp region of the sequencing reads, and the A/T/C/G nucleotides of the remaining base positions were evenly distributed (Figure 1B). The length of the sequencing fragment was normally distributed with an average of 150 bp (Figure 1C). The depth analysis of the sequencing data showed that the average sequencing depth of the plasma cfDNA samples was >4000×, and the average sequencing depths of the tissue DNA and PBMC DNA samples were >6000× each (Figure 1D).
Figure 1.

Representative results for quality control analysis of the data from amplicon sequencing. A. Sequencing quality control score of each base position in the sequencing fragment (yellow represents the quartile spacing, the red line represents the median, and the blue line represents the average); B. A/C/T/G four-base distributions in each base position of the sequencing fragments; C. Size distribution of the sequencing fragments; D. The average sequencing depth analysis of all the samples.
Detection of HCC-related mutant genes using amplicon sequencing
To clarify the mutation spectrum of the common tumor-related genes in tumor tissue, paracancerous tissue, and plasma cfDNA in HCC patients, we performed amplicon-depth sequencing using an amplicon sequencing kit containing 50 common tumor mutations. The results showed that single nucleotide mutations (SNVs) as well as synonymous and non-synonymous mutations were the most common mutation types, followed by partial insertions/deletions (Indels) and termination mutations (Figure 2 and Table S2). In the 29 cases, the number of mutant genes detected in the tumor tissue DNA, paracancerous tissue DNA, and plasma cfDNA respectively were 35, 33 and 50 of which TP53 had the highest mutation frequency. The comparative analysis of the mutant genes revealed that the mutation spectrum of the tumor tissue DNA in HCC patients was similar to that of the paracancerous tissue DNA, in contrast, the number of gene mutations in the plasma cfDNA was significantly higher. Moreover, there were some mutations detected in the plasma cfDNA but without in the tumor and paracancerous tissues (Figure 2).
Figure 2.
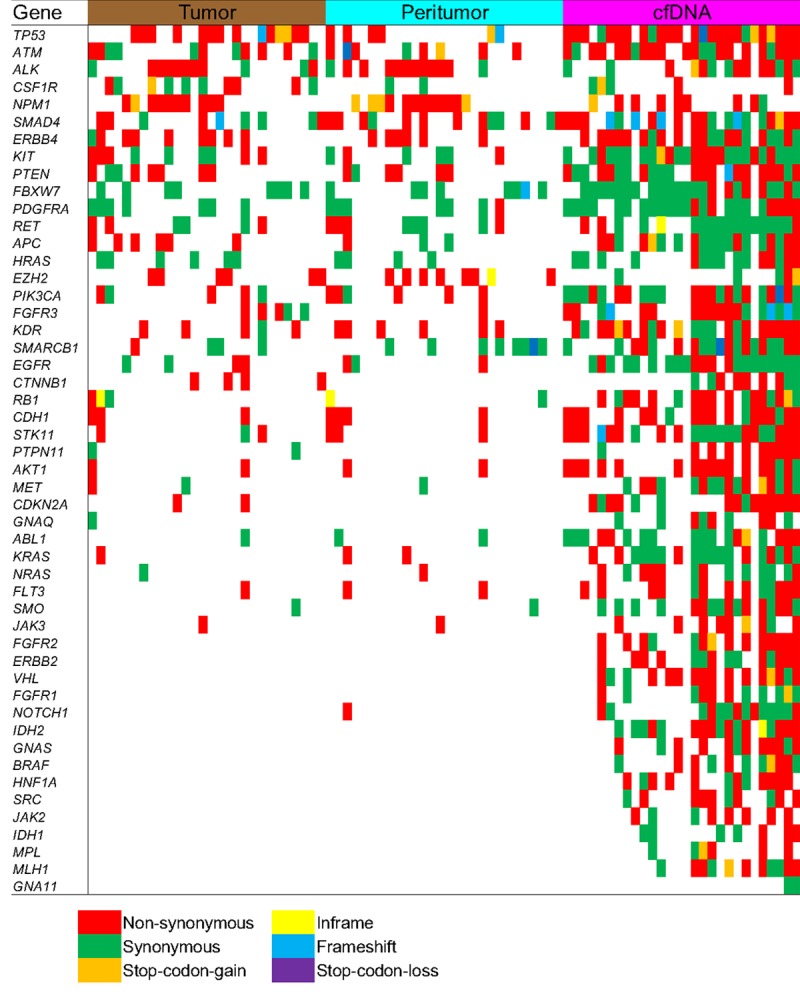
Summary results of amplicon sequencing detection of driver gene mutations for all paired samples (n=29). Different color indicates different mutation type in each sample, including tumor tissue (Tumor), paracancerous tissue (Peritumor), and plasma cfDNA for HCC patients.
Somatic mutations in tumor tissue detected by amplicon sequencing
In this study, 2800 COSMIC hotspots in 50 high-frequency mutations in tumor genes including TP53, PIK3CA, CTNNB1, PTEN, and other HCC-related genes were examined using amplicon deep sequencing technology. The DNA from 29 HCC tumor tissue samples was extracted and sequenced and 28 of them were obtained excluded one failure of library construction. The results showed the differences in the mutation spectra of the tumor tissues in different HCC patients. The number of mutant genes was ranged from 0 to 18 and there was 1 case detected no mutation (1/28, 3.6%) (Figure 3A). In the remaining 27 patients (27/28, 96.4%), 35 mutant genes in total were detected, of which 28 genes were detected in at least 2 patients (Figure 3B) and 7 genes were detected in only 1 patient. High-frequency mutant genes detected in the HCC tumor tissue samples were TP53, ATM, ALK, NPM1, CSF1R, KIT, ERBB4, SMAD4, FBXW7, and PTEN genes (Figure 3B), with mutation frequencies of 50% (14/28), 39% (9/28), 36% (11/28), 36% (10/28), 36% (10/28), 32% (9/28), 32% (9/28) 28), 29% (8/28), and 29% (8/28), respectively. In addition, the MAF of the same gene in different patients was variable. For example, the TP53 MAF was ranged 1.57-97.81% in different patients (Figure 3C). CTNNB1, another highly common driving gene in HCC, had an MAF ranging 23-45%. Notably, TP53 gene mutation was not detected in the 4 patients with CTNNB1 mutations.
Figure 3.

Detailed information of tumor-associated somatic mutations in tumor tissue from HCC patient. A. The number of mutant genes in the tumor tissue of the HCC patients (No mutations were detected in patient No. 10; Patient No. 27 was not assessed, possibly due to the necrosis of tumor tissue); B. The mutation frequency of the genes in tumor tissue from each specific patient; C. The gene mutation spectrum and mutation allele frequency (MAF) in tumor tissues from each specific patient (different colour indicates different mutation allele frequency).
Consistent gene mutations detected in the plasma cfDNA and tumor tissues of the HCC patients
After confirmation of the sensitivity of the amplicon-depth sequencing, we analysed the consistency of mutant genes in the plasma cfDNA and the tumor tissue of the HCC patients and results showed there were some gene mutations in common. Among 28 HCC patients, a total of 19 consistent mutations were detected both in tumor tissue DNA and plasma cfDNA samples in 21 patients. The mutation frequencies TP53, PDGFRA, and KIT genes was the highest accounting for 33% (7/21), 24% (5/21), and 19% (4/21) respectively in plasma cfDNA and tumor tissue DNA (Figure 4C). The sensitivity of the mutation detection in plasma cfDNA was 75% (Figure 4A, 4B). Then, further analysis of MAFs of mutant genes in the plasma cfDNA and tumor tissue revealed that the MAF of mutant genes in the plasma cfDNA was 80.9% which was close to the MAF of the paired tumor tissue. Additionally, TP53, PDGFRA, and KIT which had the highest mutation frequencies among the mutant genes, exhibited high MAFs in HCC tissues. Moreover, 19.1% of the genes had inconsistent MAFs in plasma cfDNA and the paired tumor tissue DNA (Figure 4C).
Figure 4.
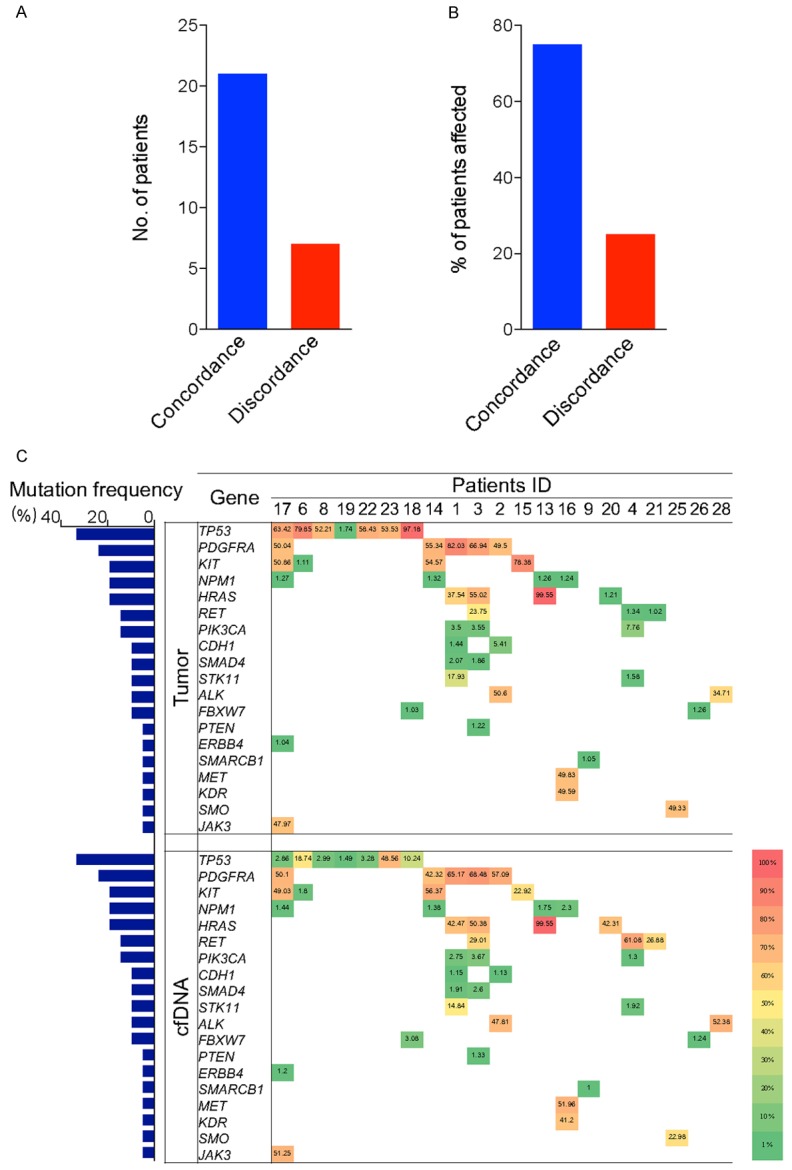
Consistent mutant genes in the plasma cfDNA and tumor tissues in the HCC patients. A, B. The number of cases with consistent and inconsistent gene mutations in paired tumor tissue and cfDNA along with the proportion of patients; C. Detailed information of consistent mutant genes and MAF detected in the tumor tissue and cfDNA from each specific patient.
Besides, 7/28 (25%) patients had inconsistent gene mutations in the preoperative plasma cfDNA and tumor tissue (Figure S1). In the plasma cfDNA, commonly mutated genes were RB1, KRAS, CDKN2A, FGFR2, ERBB2, NOTCH1, NRAS, and JAK2, a majority of which had lower MAFs (<5%).
Common gene mutations in tumor/paracancerous tissue and plasma cfDNA
Besides tumor cells, there were a large number of activated tumor-related stromal cells and the infiltration of immune cells in HCC paracancerous tissues, which provide the ‘soil’ for tumor cell proliferation. Therefore, in this study, we conducted a preliminary analysis of the gene mutation in paracancerous tissues. The comparative analysis of gene mutations in paired tumor and paracancerous tissues of 28 patients revealed that 17/28 (61%) had consistent gene mutations (Figure 5A). Among these genes, a total of 16 mutant genes were in common in the tumor tissue DNA and paracancerous tissue DNA, in which the rate of mutant genes respectively were 70% and 80% (Figure 5B). Next, the mutant genes in the tumor/paracancerous tissues were compared with those in the plasma cfDNA. The results demonstrated that 14/28 (50%) patients had completely consistent gene mutations in their plasma cfDNA with those in the tumor/paracancerous tissues (Figure 5C), including the mutations of PDGFRA, KIT, NPM1, HRAS, TP53, and PIK3CA.
Figure 5.

Consistent mutant genes in the plasma cfDNA, tumor tissue DNA, and paracancerous tissue DNA of the HCC patients. A, B. The number of consistent and inconsistent mutant genes in the tumor tissue and paracancerous tissues from the HCC patients and the ratio of total mutant genes; C. The consistent gene mutation spectrum and MAFs detected in plasma cfDNA, tumor tissue and paracancerous tissues of HCC patients.
Relationship between preoperative plasma cfDNA concentration and the clinical characteristics of the HCC patients
It is known that the plasma cfDNA concentration varies from the different extraction methods and nucleic acid quantitation methods and the cfDNA of the tumor cells is primarily obtained from the small fragments of 80-200 bp. In this study, the plasma cfDNA was extracted by the two-step magnetic method and small fragments of 200 bp were enriched in the plasma. The quantitative analysis of the plasma cfDNA showed a significant difference in concentrations ranging 1.5-62.02 ng/mL in different patients and the median was 5.39 ng/mL (Table 2). In addition, there was no significant correlation was not observed between the plasma cfDNA concentration and the general characteristics of the patients or the size, number, or staging of the tumor.
Table 2.
Correlation analysis of the preoperative plasma cfDNA concentration and the clinicopathological features of the HCC patients
| Clinical characteristics of patients | Grouping | Plasma cfDNA concentration (median ± IQR*, ng/mL) | P-valuea |
|---|---|---|---|
| Age | - | 5.39 | - |
| Gender | Male | 5.15±17.60 | 0.271 |
| Female | 27.33±50.61 | ||
| HBsAg | + | 9.86±17.09 | 0.282 |
| - | 2.90±8.17 | ||
| ALT (IU/L) | >80 | 11.47±19.46 | 0.909 |
| ≤80 | 5.40±22.58 | ||
| AFP (ng/mL) | >20 | 9.08±19.00 | 0.921 |
| ≤20 | 4.85±26.63 | ||
| Cirrhosis (with/without) | with | 6.7±19.80 | 0.914 |
| without | 5.15±22.43 | ||
| Maximum diameter of tumour | >5 | 5.64±17.36 | 0.490 |
| ≤5 | 3.79±24.53 | ||
| Number of tumour nodules (s) | 1 | 5.40±22.58 | 0.732 |
| >1 | 12.50±17.41 | ||
| BCLC staging | A+B | 4.47±20.87 | 0.521 |
| C | 9.86±45.06 |
IQR: interquartile range;
comparison between groups according to the Mann-Whitney U test.
HBsAg: hepatitis B surface antigen, ALT: alanine aminotransferase, AFP: alpha-foetoprotein.
Correlation analysis between the MAF of plasma circulating tumor DNA (ctDNA) and tumor diameter in the HCC patients
The above studies showed that the plasma cfDNA concentration was not significantly correlated with the clinicopathological features of the patients. Moreover, it is known that cfDNA can be released from all cells, therefore, we assumed the circulating tumor DNA (ctDNA) levels within the free DNA were the true reflection of the tumor cells. We determined the MAF of ctDNA within cfDNA and further analysed the relationship between MAF of ctDNA and the tumor load in the HCC patients. The MAF of several HCC-related high-frequency genes and the maximal diameter of the tumor were recorded. 23 patients with single-episode tumors were divided into two groups according to the tumor size: 11 patients with tumor diameters greater than 5 cm (D>5 cm) and 12 patients with tumor diameters of less than 5 cm (D<5 cm). the correlation analysis showed an increasing trend of the plasma ctDNA levels in the D>5 cm group compared with the D<5 cm group (Figure 6A), however, this difference was not statistically significant (Figure 6B, P>0.05, Student’s t-test).
Figure 6.

Correlation between tumor-associated mutations detected in cfDNA and tumor size in HCC patients. A. The general MAF levels of plasma ctDNA in each specific patient with different tumor diameters; B. The MAF levels of TP53, PIK3CA, CTNNB1, and CDKN2A genes were found moderately higher in patients with large tumor nodule (diameters >5 cm), but not statistically (P>0.05 for all genes, Student’s t-test).
Correlation analysis between plasma cfDNA levels and tumor number in the HCC patients
Clinically, it is challenging to assess the tumor volume and minimal residual tumors in patients with multifocal tumors and metastasis. In this study, the HCC patients were divided into single tumor group and multiple tumor or concomitant metastasis group, and the MAF values of mutant genes in the plasma cfDNA of both groups were analysed. The results showed that the MAFs of TP53, RET, APC and FGFR3 were significantly higher in patients with multiple tumors or HCC with tumor metastasis than in patients with single-tumor HCC (Figure 7).
Figure 7.

Correlation between MAF levels of specific mutant genes and tumor number in the HCC patients with single and multiple/metastatic tumor. A. The general MAF levels of the dedicated genes in plasma ctDNA from patients with different number of tumor nodules; B. The MAF levels of the TP53, FGFR3, RET and APC genes were found significantly higher in patients with multiple/metastasis tumor nodules than that in patients with single tumor nodules (P<0.05 for all genes, Student’s t-test).
Discussion
The present study showed that the amplicon sequencing technique could be used to detect tumor-related gene mutations in plasma cfDNA. This high-throughput and low-cost detection method is suitable for the routine clinical screening of molecular markers. In our study, the sensitivity of detecting mutant genes in plasma cfDNA was relatively high, even an MAF <1% was also detected. The consistency of gene mutation detection in the paired tumor tissues reached 75%, whereas in a previous whole-genome sequencing study, the sensitivity of the plasma cfDNA mutant gene detection was only 63% [18]. These data further confirmed the applicability of the amplicon sequencing technique and therefore, the detection of plasma cfDNA could be used for the diagnosis of molecular pathology and select patients the appropriate targeted drugs. In addition, plasma cfDNA can enable to assess the gene mutations in undetected tumor tissues, which might be the atypical tumors. In this study, the increase of tumor load in HCC patients was associated with an increased MAF of certain genes in plasma cfDNA, suggesting that the mutations in plasma cfDNA could serve as molecular markers for the diagnosis of HCC disease.
In the present study, plasma cfDNA mutations in 75% of the HCC patients were consistent with the tumor tissue DNA which suggested that plasma cfDNA-based detection was rather consistent with a tumor biopsy. In previous reports [19], patients with plasma ctDNA-positive HCC presented a higher risk of tumor recurrence and intrahepatic metastasis than those with plasma ctDNA-negative HCC. In our study, the AFP levels in ctDNA-positive patients were elevated, and patients with multiple or metastatic tumors exhibited an increased MAF in plasma cfDNA. Therefore, combining the results of previous reports and our study, the conclusion was drew that mutation detection based on plasma cfDNA had potential applicability to tumor diagnosis and prognosis.
Plasma cfDNA including all the tumor cell-derived plasma ctDNA was the DNA fragments released by all cells including primary tumor, metastases and circulating tumor cells [16]. Therefore, cfDNA-based detection can be a more comprehensive reflection of gene mutations in all tumor cells than conventional tissue biopsy. In this study, common gene mutations in tumor and paracancerous tissues were also detected in the plasma cfDNA of HCC patients, and simultaneously, the evaluation of the plasma cfDNA provided information of concerning gene mutations in a cancerous thrombus (1 case, not analysed in this study). Although the number of patients with portal vein thrombosis was small in our study, the results of metastatic breast cancer [20] were consistent with the findings that plasma cfDNA can replace tumor biopsy and may be considered a crucial approach for molecular detection of metastatic tumors [21]. Clinically, tissue biopsy in HCC patients can result in tumor bleeding, metastasis as well as other risks, and some patients with multiple and concomitant metastases face difficulties in undergoing tissue biopsy. Moreover, tumor heterogeneity results in the inability of conventional biopsy to provide all the information about the tumor [22-24]. In contrast, plasma cfDNA detection only requires the extraction of easily obtainable peripheral venous blood sample. The method is easily to operate, causes little trauma, can be used in cases where patients repeatedly undergo the process and can overcome the multiple limitations of tissue biopsy to achieve real-time tumor data as well as dynamic monitoring [17].
Furthermore, various factors can cause the inconsistency of gene mutations in the plasma cfDNA and tumor tissue DNA. Besides the tumor heterogeneity discussed above, plasma ctDNA fragments are primarily derived from apoptotic and dead tumor cells might be another reason might be another reason [25]. Detection in tumor tissue biopsy provides the mutation information for local tumor cells at the puncture site which might be similar to the gene mutation information in plasma cfDNA. The levels of circulating cfDNA are related to the degree of tumor differentiation and the size of tumors in patients [26]. Therefore, individuals with different tumors and at various stages of the disease exhibit inconsistent levels of circulating cfDNA. In addition, the concentration of plasma cfDNA and the sensitivity of mutation detection affect the positive rate of ctDNA detection in plasma cfDNA [3,22]. When the plasma cfDNA concentration is low, the content of ctDNA ranges 0.01-93% [16]. Thus, the results plasma of cfDNA mutation detection from different studies are different. There are various detection methods obtain the inconsistent results with the current use of digital polymerase chain reaction (PCR) in which targeted deep sequencing can detect mutations of ≤0.01% [27,28]. However, regardless of the mutation detection methods, sufficient plasma concentrations of cfDNA and ctDNA ratios are required, for example, in our study, the plasma cfDNA concentrations in patients ranged from <0.1 ng/mL to 145 mg/mL. In clinical research, increasing the initial plasma volume or optimizing the plasma cfDNA extraction efficiency can increase plasma cfDNA levels. However, the plasma cfDNA concentrations are also affected by the tumor type, tumor volume, and disease stage, such as the difference among patients with lung cancer and gastrointestinal cancer. In the case of patients with tumor metastasis, the detection rate of ctDNA is significantly higher than that in patients with head and neck tumors or early-stage tumors [3,29].
It is reported that plasma cfDNA-based detection is critical in HCC diagnosis. The serum levels of cfDNA predict the risk of tumor metastasis in patients [30,31]. Plasma cfDNA-based ZNF300, SLC22A20, and SHISA7 methylation tests can be used for the differential diagnosis of chronic hepatitis B (CHB), cirrhosis and HCC. The results of a recent meta-analysis showed that circulating cfDNA alone was not recommended for the diagnosis of HCC, whereas combination detection of AFP with circulating cfDNA significantly increased the diagnostic sensitivity of HCC [32]. Previous studies have demonstrated that HCC patients with plasma ctDNA-positive had high AFP levels and large tumor diameters and are prone to tumor metastasis [19]. A recent high-throughput methylation analysis of plasma ctDNA in patients with liver cancer using methylation CpG short-sequence amplification sequencing method improved the diagnostic sensitivity of AFP-negative HCC, suggesting this approach could be used for the early diagnosis of HCC in patients [15]. In another study, the whole-genome sequencing of preoperative plasma cfDNA, primary tumor tissue DNA, and postoperative plasma cfDNA in 4 HCC patients were performed to explore HCC-related gene mutations. The results presented a mutation in preoperative plasma cfDNA was consistent with tumor tissues but not detected in the postoperative plasma cfDNA [33]. It suggested that mutation detection based on plasma cfDNA could be used for the dynamic monitoring of tumor load. In addition, we further analysed the correlation between the MAFs of specific genes in plasma cfDNA and the tumor load and found that the MAF values for TP53, RET, APC, and FGFR3 were significantly higher in patients with multiple tumors or vascular metastasis than those with a single tumor. Therefore, we hypothesized that plasma ctDNA levels are more accurate than cfDNA levels in reflecting the tumor load, however, a multicentre study with a larger sample is imperative for validation.
The detection of plasma cfDNA mutations may be used to guide tumor-targeted therapy and individualized treatment. At Present, the treatment of HCC patients lacks the specific tumor molecular classification. For example, although sorafenib is an approved targeted drug for systemic HCC treatment, not all the patients receive good response to this drug, even there are some patients displaying side effects. Plasma cfDNA detection can clarify the presence of specific molecular therapeutic targets in patients and thereby guide the selection of specific targeted drug. MET gene activation is a major pathway for cell proliferation and tumor metastasis in HCC, thus, MET inhibitors have presented a new strategy for HCC-targeted therapy in recent years [34]. In a phase II clinical study of MET inhibitors for HCC patients, all patients were underwent the detection of MET gene mutations in the tumor tissue prior to treatment. The study showed that patients with high protein expression had significantly better response to drug treatment than those with low protein expression [35], thereby justifying the customized treatment model for HCC. Nevertheless, in this study, tumor molecular classification was based on tissue puncture biopsy, which did not reflect all tumor mutation information. Therefore, we used amplicon sequencing technology to detect drug-related gene mutations such as EGFR, PIK3CA, PDGFR, MET and the resistance-related gene BRAF in the plasma of HCC patients and results suggested that plasma cfDNA could be used to guide targeted drug therapy in patients.
In summary, the results of this study suggest that plasma cfDNA can be used to detect mutant genes in tumors. Amplicon sequencing technology is highly sensitive and can comprehensively reflect the data on tumor cell gene mutations. Thus, this technique might replace tumor tissue biopsy and become the main method in HCC mutant gene screening. In this study, the MAFs gene mutations in plasma cfDNA were significantly correlated with the tumor load suggesting that plasma cfDNA might serve as a molecular marker for HCC diagnoses.
Acknowledgements
We acknowledge Bo Du, Xinyuan Zhang and Xiangru Li from Beijing Genecast Biotechnology Co., Ltd. for their assistance of molecular technical support. We also thank Gaihua Zhang and Linlin Yan for their precious assistance and help with bioinformatics analysis. This work was supported by National Natural Science Foundation of China (91029741 and 81001072), National Key Sci-Tech Special Project of China (2012ZX10002011-006 and 2018ZX10302207), and Beijing Natural Science Foundation (7152151).
Disclosure of conflict of interest
None.
Figure S1
Tables S1, S2
References
- 1.Lozano R, Naghavi M, Foreman K, Lim S, Shibuya K, Aboyans V, Abraham J, Adair T, Aggarwal R, Ahn SY, Alvarado M, Anderson HR, Anderson LM, Andrews KG, Atkinson C, Baddour LM, Barker-Collo S, Bartels DH, Bell ML, Benjamin EJ, Bennett D, Bhalla K, Bikbov B, Bin Abdulhak A, Birbeck G, Blyth F, Bolliger I, Boufous S, Bucello C, Burch M, Burney P, Carapetis J, Chen H, Chou D, Chugh SS, Coffeng LE, Colan SD, Colquhoun S, Colson KE, Condon J, Connor MD, Cooper LT, Corriere M, Cortinovis M, de Vaccaro KC, Couser W, Cowie BC, Criqui MH, Cross M, Dabhadkar KC, Dahodwala N, De Leo D, Degenhardt L, Delossantos A, Denenberg J, Des Jarlais DC, Dharmaratne SD, Dorsey ER, Driscoll T, Duber H, Ebel B, Erwin PJ, Espindola P, Ezzati M, Feigin V, Flaxman AD, Forouzanfar MH, Fowkes FG, Franklin R, Fransen M, Freeman MK, Gabriel SE, Gakidou E, Gaspari F, Gillum RF, Gonzalez-Medina D, Halasa YA, Haring D, Harrison JE, Havmoeller R, Hay RJ, Hoen B, Hotez PJ, Hoy D, Jacobsen KH, James SL, Jasrasaria R, Jayaraman S, Johns N, Karthikeyan G, Kassebaum N, Keren A, Khoo JP, Knowlton LM, Kobusingye O, Koranteng A, Krishnamurthi R, Lipnick M, Lipshultz SE, Ohno SL, Mabweijano J, MacIntyre MF, Mallinger L, March L, Marks GB, Marks R, Matsumori A, Matzopoulos R, Mayosi BM, McAnulty JH, McDermott MM, McGrath J, Mensah GA, Merriman TR, Michaud C, Miller M, Miller TR, Mock C, Mocumbi AO, Mokdad AA, Moran A, Mulholland K, Nair MN, Naldi L, Narayan KM, Nasseri K, Norman P, O’Donnell M, Omer SB, Ortblad K, Osborne R, Ozgediz D, Pahari B, Pandian JD, Rivero AP, Padilla RP, Perez-Ruiz F, Perico N, Phillips D, Pierce K, Pope CA 3rd, Porrini E, Pourmalek F, Raju M, Ranganathan D, Rehm JT, Rein DB, Remuzzi G, Rivara FP, Roberts T, De Leon FR, Rosenfeld LC, Rushton L, Sacco RL, Salomon JA, Sampson U, Sanman E, Schwebel DC, Segui-Gomez M, Shepard DS, Singh D, Singleton J, Sliwa K, Smith E, Steer A, Taylor JA, Thomas B, Tleyjeh IM, Towbin JA, Truelsen T, Undurraga EA, Venketasubramanian N, Vijayakumar L, Vos T, Wagner GR, Wang M, Wang W, Watt K, Weinstock MA, Weintraub R, Wilkinson JD, Woolf AD, Wulf S, Yeh PH, Yip P, Zabetian A, Zheng ZJ, Lopez AD, Murray CJ, AlMazroa MA, Memish ZA. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet. 2012;380:2095–2128. doi: 10.1016/S0140-6736(12)61728-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen W, Zheng R, Baade PD, Zhang S, Zeng H, Bray F, Jemal A, Yu XQ, He J. Cancer statistics in China, 2015. Ca Cancer J Clin. 2016;66:115–132. doi: 10.3322/caac.21338. [DOI] [PubMed] [Google Scholar]
- 3.Bettegowda C, Sausen M, Leary RJ, Kinde I, Wang Y, Agrawal N, Bartlett BR, Wang H, Luber B, Alani RM, Antonarakis ES, Azad NS, Bardelli A, Brem H, Cameron JL, Lee CC, Fecher LA, Gallia GL, Gibbs P, Le D, Giuntoli RL, Goggins M, Hogarty MD, Holdhoff M, Hong SM, Jiao Y, Juhl HH, Kim JJ, Siravegna G, Laheru DA, Lauricella C, Lim M, Lipson EJ, Marie SK, Netto GJ, Oliner KS, Olivi A, Olsson L, Riggins GJ, Sartore-Bianchi A, Schmidt K, Shih lM, Oba-Shinjo SM, Siena S, Theodorescu D, Tie J, Harkins TT, Veronese S, Wang TL, Weingart JD, Wolfgang CL, Wood LD, Xing D, Hruban RH, Wu J, Allen PJ, Schmidt CM, Choti MA, Velculescu VE, Kinzler KW, Vogelstein B, Papadopoulos N, Diaz LA Jr. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci Transl Med. 2014;6:224ra24. doi: 10.1126/scitranslmed.3007094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.de Lope CR, Tremosini S, Forner A, Reig M, Bruix J. Management of HCC. J Hepatol. 2012;56:S75–S87. doi: 10.1016/S0168-8278(12)60009-9. [DOI] [PubMed] [Google Scholar]
- 5.Margini C, Dufour JF. The story of HCC in NAFLD: from epidemiology, across pathogenesis, to prevention and treatment. Liver Int. 2015;36:317–324. doi: 10.1111/liv.13031. [DOI] [PubMed] [Google Scholar]
- 6.Prieto J, Melero I, Sangro B. Immunological landscape and immunotherapy of hepatocellular carcinoma. Nat Rev Gastroenterol Hepatol. 2015;12:681–700. doi: 10.1038/nrgastro.2015.173. [DOI] [PubMed] [Google Scholar]
- 7.Bertino G, Demma S, Ardiri A, Proiti M, Gruttadauria S, Toro A, Malaguarnera G, Bertino N, Malaguarnera M, Malaguarnera M. Hepatocellular carcinoma: novel molecular targets in carcinogenesis for future therapies. Biomed Res Int. 2014;2014:203693. doi: 10.1155/2014/203693. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 8.Su YH, Lin SY, Song W, Jain S. DNA markers in molecular diagnostics for hepatocellular carcinoma. Expert Rev Mol Diagn. 2014;14:803–817. doi: 10.1586/14737159.2014.946908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cleary SP, Jeck WR, Zhao X, Chen K, Selitsky SR, Savich GL, Tan TX, Wu MC, Getz G, Lawrence MS. Identification of driver genes in hepatocellular carcinoma by exome sequencing. Hepatology. 2013;58:1693–1702. doi: 10.1002/hep.26540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guichard C, Amaddeo G, Imbeaud S, Ladeiro Y, Pelletier L, Maad IB, Calderaro J, Bioulacsage P, Letexier M, Degos F. Integrated analysis of somatic mutations and focal copy-number changes identifies key genes and pathways in hepatocellular carcinoma. Nat Genet. 2012;44:694–698. doi: 10.1038/ng.2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schulze K, Imbeaud S, Letouzé E, Alexandrov LB, Calderaro J, Rebouissou S, Couchy G, Meiller C, Shinde J, Soysouvanh F, Calatayud AL, Pinyol R, Pelletier L, Balabaud C, Laurent A, Blanc JF, Mazzaferro V, Calvo F, Villanueva A, Nault JC, Bioulac-Sage P, Stratton MR, Llovet JM, Zucman-Rossi J. Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat Genet. 2015;47:505–511. doi: 10.1038/ng.3252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Totoki Y, Tatsuno K, Covington KR, Ueda H, Creighton CJ, Kato M, Tsuji S, Donehower LA, Slagle BL, Nakamura H. Trans-ancestry mutational landscape of hepatocellular carcinoma genomes. Nat Genet. 2014;46:1267–1273. doi: 10.1038/ng.3126. [DOI] [PubMed] [Google Scholar]
- 13.Nakagawa H, Shibata T. Comprehensive genome sequencing of the liver cancer genome. Cancer Letters. 2013;340:234–240. doi: 10.1016/j.canlet.2012.10.035. [DOI] [PubMed] [Google Scholar]
- 14.Zhou J, Huang A, Yang XR. Liquid biopsy and its potential for management of hepatocellular carcinoma. J Gastrointest Cancer. 2016;47:157–167. doi: 10.1007/s12029-016-9801-0. [DOI] [PubMed] [Google Scholar]
- 15.Wen L, Li J, Guo H, Liu X, Zheng S, Zhang D, Zhu W, Qu J, Guo L, Du D. Genome-scale detection of hypermethylated CpG islands in circulating cell-free DNA of hepatocellular carcinoma patients. Cell Res. 2015;25:1376. doi: 10.1038/cr.2015.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Crowley E, Di NF, Loupakis F, Bardelli A. Liquid biopsy: monitoring cancer-genetics in the blood. Nat Rev Clin Oncol. 2013;10:472. doi: 10.1038/nrclinonc.2013.110. [DOI] [PubMed] [Google Scholar]
- 17.Esposito A, Criscitiello C, Locatelli M, Milano M, Curigliano G. Liquid biopsies for solid tumors: understanding tumor heterogeneity and real time monitoring of early resistance to targeted therapies. Pharmacol Ther. 2016;157:120–124. doi: 10.1016/j.pharmthera.2015.11.007. [DOI] [PubMed] [Google Scholar]
- 18.Jiang P, Chan CW, Chan KC, Cheng SH, Wong J, Wong VW, Wong GL, Chan SL, Mok TS, Chan HL. Lengthening and shortening of plasma DNA in hepatocellular carcinoma patients. Proc Natl Acad Sci U S A. 2015;112:E1317. doi: 10.1073/pnas.1500076112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ono A, Fujimoto A, Yamamoto Y, Akamatsu S, Hiraga N, Imamura M, Kawaoka T, Tsuge M, Abe H, Hayes CN. Circulating tumor DNA analysis for liver cancers and its usefulness as a liquid biopsy. Cell Mol Gastroenterol Hepatol. 2015;1:516. doi: 10.1016/j.jcmgh.2015.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rothe F, Laes JF, Lambrechts D, Smeets D, Vincent D, Maetens M, Fumagalli D, Michiels S, Drisis S, Moerman C, Detiffe JP, Larsimont D, Awada A, Piccart M, Sotiriou C, Ignatiadis M. Plasma circulating tumor DNA as an alternative to metastatic biopsies for mutational analysis in breast cancer. Ann Oncol. 2014;25:1959–1965. doi: 10.1093/annonc/mdu288. [DOI] [PubMed] [Google Scholar]
- 21.Ady JW, Heffner J, Mojica K, Johnsen C, Belin LJ, Love D, Chen CT, Pugalenthi A, Klein E, Chen NG, Yu YA, Szalay AA, Fong Y. Oncolytic immunotherapy using recombinant vaccinia virus GLV-1h68 kills sorafenib-resistant hepatocellular carcinoma efficiently. Surgery. 2014;156:263–269. doi: 10.1016/j.surg.2014.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Beaver JA, Jelovac D, Balukrishna S, Cochran RL, Croessmann S, Zabransky DJ, Wong HY, Valda TP, Cidado J, Blair BG. Detection of cancer DNA in plasma of patients with early-stage breast cancer. Clin Cancer Res. 2014;20:2643–2650. doi: 10.1158/1078-0432.CCR-13-2933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xue R, Li R, Guo H, Guo L, Su Z, Ni X, Qi L, Zhang T, Li Q, Zhang Z, Xie XS, Bai F, Zhang N. Variable intra-tumor genomic heterogeneity of multiple lesions in patients with hepatocellular carcinoma. Gastroenterology. 2016;150:998–1008. doi: 10.1053/j.gastro.2015.12.033. [DOI] [PubMed] [Google Scholar]
- 24.Friemel J, Rechsteiner M, Frick L, Böhm F, Struckmann K, Egger M, Moch H, Heikenwalder M, Weber A. Intratumor heterogeneity in hepatocellular carcinoma. Clin Cancer Res. 2015;21:1951. doi: 10.1158/1078-0432.CCR-14-0122. [DOI] [PubMed] [Google Scholar]
- 25.Jahr S, Hentze H, Englisch S, Hardt D, Fackelmayer FO, Hesch RD, Knippers R. DNA fragments in the blood plasma of cancer patients: quantitations and evidence for their origin from apoptotic and necrotic cells. Cancer Res. 2001;61:1659–1665. [PubMed] [Google Scholar]
- 26.Iizuka N, Sakaida I, Moribe T, Fujita N, Miura T, Stark M, Tamatsukuri S, Ishitsuka H, Uchida K, Terai S. Elevated levels of circulating cell-free DNA in the blood of patients with hepatitis C virus-associated hepatocellular carcinoma. Anticancer Res. 2006;26:4713. [PubMed] [Google Scholar]
- 27.Newman AM, Bratman SV, To J, Wynne JF, Eclov NC, Modlin LA, Liu CL, Neal JW, Wakelee HA, Merritt RE. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat Med. 2014;20:548. doi: 10.1038/nm.3519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Narayan A, Carriero NJ, Gettinger SN, Kluytenaar J, Kozak KR, Yock TI, Muscato NE, Ugarelli P, Decker RH, Patel AA. Ultrasensitive measurement of hotspot mutations in tumor DNA in blood using error-suppressed multiplexed deep sequencing. Cancer Res. 2012;72:3492–3498. doi: 10.1158/0008-5472.CAN-11-4037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jovelet C, Ileana E, Le DM, Motte N, Rosellini S, Romero A, Lefebvre C, Pedrero M, Pata-Merci N, Droin N. Circulating cell-free tumor DNA (cfDNA) analysis of 50-genes by next-generation sequencing (NGS) in the prospective MOSCATO trial. Clin Cancer Res. 2016;22:2960. doi: 10.1158/1078-0432.CCR-15-2470. [DOI] [PubMed] [Google Scholar]
- 30.Tokuhisa Y, Iizuka N, Sakaida I, Moribe T, Fujita N, Miura T, Tamatsukuri S, Ishitsuka H, Uchida K, Terai S. Circulating cell-free DNA as a predictive marker for distant metastasis of hepatitis C virus-related hepatocellular carcinoma. Br J Cancer. 2011;97:1399–1403. doi: 10.1038/sj.bjc.6604034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhao Y, Xue F, Sun J, Guo S, Zhang H, Qiu B, Geng J, Gu J, Zhou X, Wang W. Genome-wide methylation profiling of the different stages of hepatitis B virus-related hepatocellular carcinoma development in plasma cell-free DNA reveals potential biomarkers for early detection and high-risk monitoring of hepatocellular carcinoma. Clin Epigenetics. 2014;6:30. doi: 10.1186/1868-7083-6-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liao W, Mao Y, Ge P, Yang H, Xu H, Lu X, Sang X, Zhong S. Value of quantitative and qualitative analyses of circulating cell-free DNA as diagnostic tools for hepatocellular carcinoma: a meta-analysis. Medicine. 2015;94:e722. doi: 10.1097/MD.0000000000000722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chan KC, Jiang P, Zheng YW, Liao GJ, Sun H, Wong J, Siu SS, Chan WC, Chan SL, Chan AT. Cancer genome scanning in plasma: detection of tumor-associated copy number aberrations, single-nucleotide variants, and tumoral heterogeneity by massively parallel sequencing. Clin Chem. 2013;59:211–224. doi: 10.1373/clinchem.2012.196014. [DOI] [PubMed] [Google Scholar]
- 34.Giordano S, Columbano A. Met as a therapeutic target in HCC: facts and hopes. J Hepatol. 2014;60:442–452. doi: 10.1016/j.jhep.2013.09.009. [DOI] [PubMed] [Google Scholar]
- 35.Santoro A, Rimassa L, Borbath I, Daniele B, Salvagni S, Van Laethem JL, Van VH, Trojan J, Kolligs FT, Weiss A. Tivantinib for second-line treatment of advanced hepatocellular carcinoma: a randomised, placebo-controlled phase 2 study. Lancet Oncol. 2013;14:55. doi: 10.1016/S1470-2045(12)70490-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.