

Abstract
Previous findings supported that fetuin B, a new hepatokine, may be involved in the development of hepatic steatosis, but the mechanism is still unknown. This study aims to investigate the role of fetuin B in hepatic steatosis in C57BL/6 mice and HepG2 cells. 1) We found that the administration of recombinant fetuin B aggravated hepatic lipid accumulation caused by free fatty acids (FFAs) or high fat diet, in vivo and in vitro. It lowered the phosphorylated AMPK levels and activated the LXR-SREBP1c pathway, accompanied by the downregulation of fatty acid oxidation and upregulation of lipogenesis. Furthermore, in HepG2 cells exposed to recombinant fetuin B and FFAs, the AMPK agonist depressed the LXR-SREBP1c pathway and alleviated lipid accumulation. The knockdown of LXR protected against steatosis but failed to change phosphorylated AMPK. 2) The knockdown of fetuin B by siRNA or shRNA alleviated lipid accumulation, in vivo and in vitro. It enhanced phosphorylated AMPK and depressed the LXR-SREBP1c pathway, accompanied by upregulation of fatty acid oxidation and downregulation of lipogenesis. Moreover, in HepG2 cells exposed to fetuin B siRNA and FFAs, LXR agonist aggravated lipid accumulation but failed to influence AMPK. This study indicated that fetuin B aggravated LXR-mediated hepatic steatosis through AMPK. It might offer new insights into clinical management and biomarker research on fatty liver.
Keywords: AMP-activated protein kinase (AMPK), fetuin B, hepatic steatosis, liver X receptor (LXR), Non-alcoholic fatty liver disease (NAFLD)
Introduction
Non-alcoholic fatty liver disease (NAFLD) is becoming a leading cause of chronic liver disease and a major public health concern [1-3]. The disease affects approximately 25% of the world’s population [4,5]. Despite recent progress in the field of NAFLD, the pathogenesis of steatosis and advancement to steatohepatitis is not yet understood [6].
A series of novel hormones, including adipokines, hepatokines and others, have been recognized to be involved in hepatic steatosis [7]. Fetuin is an endogenous inhibitor of the insulin receptor tyrosine kinase [8]. Fetuin B shares 22% homology with fetuin A, which has been found to cause insulin resistance by inducing inflammatory signaling [9-11]. Meex et al. [12] reported that fetuin B was a hepatokine, and its concentrations increased in obese individuals with T2D and NAFLD compared to NAFLD- and diabetes-free obese controls. They found that plasma fetuin B was associated positively with insulin resistance in rodents and that the effect of fetuin B on insulin function was dose- and time-dependent. The knockdown of fetuin B improved glucose tolerance.
AMP-activated protein kinase (AMPK) is a widely accepted sensor of energy status and is composed of heterotrimeric complexes that include catalytic α subunits and regulatory β and γ subunits. In terms of its downstream target, AMPK phosphorylates and inhibits acetyl-CoA carboxylase (ACC) and diminishes its synthesis at the level of transcription [13]. Recent advances have revealed that dysfunction of hepatic AMPK is associated with an essential mechanism for hepatic lipid accumulation and hyperlipidemia [14,15].
The imbalance between lipid deposition and elimination leads to lipid accumulation in liver. De novo lipogenesis is the essential procedure for FFA synthesis and is associated with obesity and type 2 diabetes [16]. It is also controlled by several nuclear receptors and transcription factors, e.g., liver X receptor (LXR) [17]. It has been shown that LXR together with sterol regulatory element binding transcription factor 1 (SREBP1c) belongs to a network of nutrient sensors that are associated with the regulation of fatty acid synthesis and TG accumulation [18,19]. Furthermore, LXR, especially LXRα, has been regarded as the upstream target of lipogenesis proteins, e.g., fatty acid synthase (FAS), ACC and stearoyl-CoA desaturase-1 (SCD1) [20-22]. Recent evidence shows that the LXR-SREBP1c axis plays an important role in promoting de novo lipogenesis in response to a high fat diet. Previous studies have shown that polyphenols and hormones were involved in LXR-mediated steatosis through AMPK in human and mouse hepatocytes [23,24].
In this study, we tried to confirm the role of fetuin B in hepatic steatosis and its association with hepatic de novo lipogenesis and fatty acid oxidation in vivo and in vitro. Moreover, as key metabolic regulators, AMPK and LXR signaling were studied for their roles in the effects of fetuin B in NAFLD.
Materials and methods
Reagents
Rabbit monoclonal phospho-Thr172, AMPKα (4188) and AMPKα (2603) antibodies were purchased from Cell Signaling Technology (Beverly, MA). Rabbit monoclonal GAPDH (ab128915), fetuin B (ab191569), Tubulin (ab7291) and LXRα (ab24362) antibodies were purchased from Abcam Inc. (Cambridge, MA). Rabbit polyclonal phospho-Ser79 ACC (3661), ACC (3662), and SREBP1c (ab28481) antibodies were purchased from Abcam Inc. (Cambridge, MA). Anti-rabbit IgG, HRP-linked Antibody (7074) and Anti-mouse IgG, HRP-linked Antibody (7076) were purchased from Cell Signaling Technology (Beverly, MA). Dulbecco’s modified Eagle’s medium (DMEM), fetal bovine serum, and antibiotics were obtained from Gibco (Shanghai, China). Oleate (O7501) and palmitate (P9767) were provided from Sigma-Aldrich Co. (St. Louis, MO). AICAR (S1802) and GW3965 (S2630) were purchased from Selleck Chemicals (Houston, TX).
ELISA kits
Blood samples from mice were taken for measurement of serum fetuin B levels using an enzyme-linked immunosorbent assay (Catalogue No. CSB-EL008598MO; CUSABIO Corp., Wuhan, China). The intra- and interassay variations were 9.3% and 10.6%, respectively.
HepG2 cell culture supernatants were taken for the measurement of fetuin B levels using an enzyme-linked immunosorbent assay (Catalogue No. SEB860Hu; Cloud-Clone Corp., Houston, TX, USA). The intra- and interassay variations were 5.6% and 7.7%, respectively.
Western blot analysis
Proteins were extracted using RIPA buffer (Applygen Technologies Inc., Beijing, China) with added protein and phosphatase inhibitor (Sigma), separated by SDS-PAGE, and electrophoretically transferred to PVDF membranes (Millipore). After blocking with 5% skim milk in TBST, the membranes were probed with primary and secondary antibodies. Chemiluminescence was visualized using an ECL kit (Lianke, Hangzhou, China).
Real-time quantitative PCR
Total RNA was extracted using Trizol reagent according to the manufacturer’s instructions (Takara). Then, RNA was reverse-transcribed to cDNA using a TaqMan Reverse Transcription Reagent Kit (Takara) and further analyzed with a real-time quantitative PCR system to evaluate relative mRNA levels. GAPDH and β-actin served as controls. Primer sequences are indicated in the Supplementary Table 1.
Cell culture and treatment
The human hepatocellular carcinoma cell line (HepG2 cells) was from the Chinese Academy of Sciences (Shanghai, China) and cultured in DMEM supplemented with 10% FBS, 100 U/mL penicillin, and 100 μg/mL streptomycin at 37°C in 5% CO2. To establish a cellular model of hepatic steatosis, HepG2 cells were exposed to a mixture of free fatty acids (FFA), including oleate and palmitate at a final ratio of 2:1 and a final concentration of 1 mM for 24 hours [25]. The medium was further diluted to contain 150 µM recombinant fetuin B protein for 24 h of culture (RPB860Hu01, Cloud-Clone Corp., Houston, TX) according to a previous study [12].
Small interfering RNA transfection (siRNA)
siRNAs were synthesized by Qiagen. HepG2 cells were transfected with fetuin B siRNA, LXR siRNA or scramble siRNA as the negative control (NC) using Lipofectamine 2000 (Invitrogen, Shanghai, China). The siRNA sense sequence of fetuin B was 5’-TGACTCAGATGTGCTGGCAGTTGCA-3’. The siRNA sense sequence of LXR was 5’-CCACGGGUACUUCUGUCAGCUGAUU-3’. The efficacy of knockdown was determined by real-time PCR and western blot.
Triglyceride assay
Intracellular (HepG2) and intrahepatic (mice) triglycerides (TGs) were measured using a TG assay kit (E1013; Applygen Technologies Inc., Beijing, China) according to the manufacturer’s instructions and a previous study [25]. For the measurement of intracellular TG, first, cells were scraped from 6-well culture plates and washed with PBS twice. Then, 100 µl of cell lysis buffer was added and incubated at 70°C for 10 min. Afterwards, the sample was centrifuged at 2000 rpm for 5 min, and the supernatant was collected and lastly evaluated with the BCA method.
Animal procedures
Eight- to ten-week-old female, specific pathogen-free C57BL/6 mice were provided by and maintained at the Experimental Animal Center of Zhejiang Province (Hangzhou, China) with ad libitum access to food and water. Mice were randomly divided into six groups of six, including (1) feeding a standard chow diet (SCD) vs. a high fat diet (HFD), (2) HFD vs. HFD + Recombinant fetuin B protein, and (3) HFD vs. HFD + short hairpin RNA (shRNA) fetuin B. The HFD (D12492; Research Diets, New Brunswick, NJ) contains 60% of kcal from fat, 20% from carbohydrates and 20% from protein. Food intake was measured every day for 8 weeks. All mice were housed in an air-conditioned (temperature 24 ± 2°C and humidity 50 ± 10%) and light-controlled (12 h light-12 h dark cycle, lights on from 08:00 to 20:00) animal room. Mice were anesthetized and killed after the experiment. Liver tissue was collected for further analyses. All animal studies were approved by the Animal Care and Use Committee of Zhejiang University in accordance with the Chinese guidelines for the care and use of laboratory animals. Mice were randomized into two groups (n = 6) with recombinant fetuin B protein delivered at a dose rate of 0 (vehicle) or 0.5 µg/g body mass per day according to a previous study [12]. Vehicle (20 mM Tris-HCL, Invitrogen, Carlsbad, CA) or recombinant mouse fetuin B (RPB860Mu0, Cloud-Clone Corp., Houston, TX) was infused using s.c. implanted osmotic pumps (Alzet #1002, Durect Corp., CA) for the 10-day duration.
Lentivirus target screening for RNAi
An RNAi target sequence was created within the mouse fetuin B gene along with a shRNA with the sequence shRNA-fetuin B, 5’-GGGCTGCAATGACTCAGATGT-3’. The fetuin B RNAi sequence was inserted into a pShuttle vector and co-transduced into 293T cells to produce the lentivirus. Mice were injected via the tail vein with 100 μL of lentivirus expression shRNA-fetuin B or the negative control on the 4th week before further experiment.
Histological analysis
The liver samples were fixed in 10% neutral formalin, embedded in paraffin, sectioned and then stained with hematoxylin and eosin (H&E) for histological examination. To determine hepatic fat accumulation, frozen liver sections (8 μm) were stained with freshly diluted Oil Red O solution for 10 min, washed, and counterstained with hematoxylin for 5 min. Cells in 6-well plates were washed twice with PBS, fixed with 10% neutral formalin for 15 min, washed and stained with freshly diluted Oil Red O solution for 10 min, followed by another two washes with PBS and stained with hematoxylin for 5 min. Representative photomicrographs were captured at 400 × magnification using a microscope (BX53F, Olympus, Japan).
Statistical analyses
Statistical analyses were performed using SPSS 21.0 for Windows (SPSS, Chicago, IL). The results are expressed as means ± standard deviations. The significance of the difference in means was determined by a two-tailed Student’s t test and one-way ANOVA. A p value < 0.05 (two-tailed) was considered statistically significant.
Results
Expression of fetuin B in HepG2 cells and mice
The mRNA level of fetuin B increased in human HepG2 cells exposed to FFAs compared with controls (Figure 1A). The protein levels of fetuin B in HepG2 cells and cell culture supernatants also increased with FFAs (Figure 1B and 1C). The mRNA level of hepatic fetuin B increased in mice exposed to a HFD compared with a SCD (Figure 1D). The protein levels in hepatocytes and serum also increased with the HFD (Figure 1E and 1F).
Figure 1.
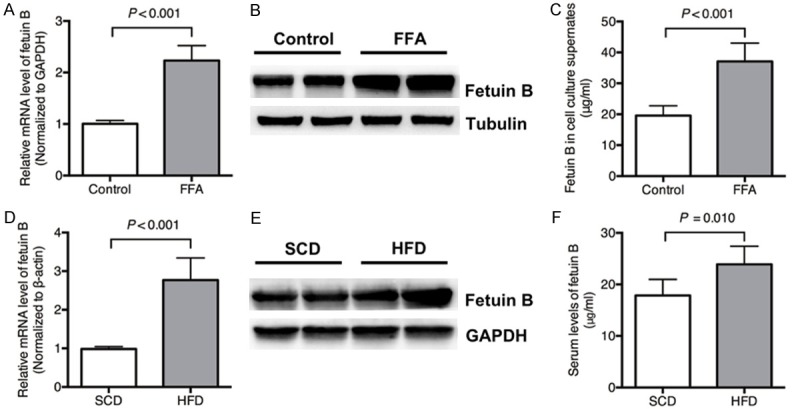
Expression of fetuin B in HepG2 cells and mice. The levels of fetuin B mRNA and protein in HepG2 cells and cell culture supernatants increased with FFAs compared with controls. The levels of fetuin B mRNA and protein in mice increased in the HFD group compared with the SCD group. A: The mRNA levels: Control vs. FFAs (P < 0.001); B: The protein expression of fetuin B in HepG2 cells; C: The protein levels in the culture supernatants of HepG2 cells (P < 0.001); (HepG2 experiment) Results are the mean ± SD of three independent experiments, each repeated in triplicate. D: The hepatic mRNA level: SCD vs. HFD (P < 0.001); E: The hepatic protein expression; F: The serum levels (SCD vs. HFD, P = 0.010); (Mouse experiment) Results are the mean ± SD of three independent experiments (n = 6, each group).
Recombinant fetuin B aggravated lipid accumulation in HepG2 cells
As shown in Figure 2A, Oil Red O staining showed that the lipid droplets caused by FFAs were aggravated in HepG2 cells treated with recombinant fetuin B. The intercellular TG content was markedly increased in the FFA + Rec FetB group compared with FFAs alone (Figure 2B, P = 0.001).
Figure 2.
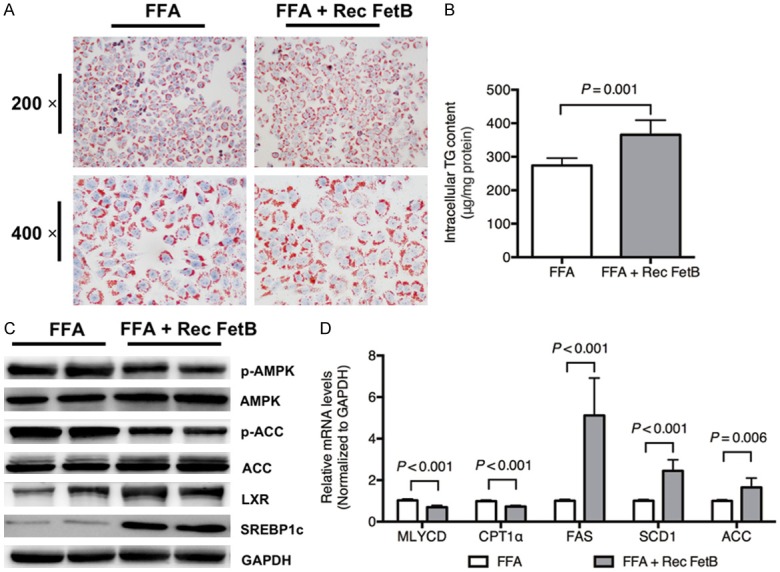
Recombinant fetuin B aggravated hepatic lipid accumulation in HepG2 cells. Hepatocellular lipid accumulation and AMPK-/LXR-pathways were evaluated after the administration of recombinant fetuin B in HepG2 cells exposed to FFAs. A: Oil Red O staining: FFA vs. FFA + Rec FetB (200 × and 400 ×); B: Intercellular TG content (FFA vs. FFA + Rec FetB, P = 0.001); C: The expression of key proteins in the AMPK and LXR pathways; D: The mRNA levels of lipid metabolic enzymes; Rec FetB, Recombinant fetuin B. Results are the mean ± SD of three independent experiments, each repeated in triplicate.
To identify the possible pathway through which fetuin B aggravates hepatocellular lipid accumulation, the protein expression of the AMPK and LXR pathways was evaluated. As revealed in Figure 2C, the administration of recombinant fetuin B decreased phosphorylated AMPK and ACC and upregulated LXR and SREBP1c. Furthermore, it led to an upregulation of mRNA levels of lipogenesis-related enzymes (FAS, SCD1 and ACC) and a downregulation of fatty acid oxidation enzymes (Malonyl-CoA decarboxylase, i.e., MLYCD and Carnitine palmitoyltransferase 1α, i.e., CPT1α), as shown in Figure 2D.
LXR is a downstream target of AMPK
To clarify the relation between AMPK and LXR, pretreatment of LXR siRNA was applied before the administration of recombinant fetuin B in HepG2 cells exposed to FFAs. First, the intercellular TG content decreased in the FFA + Rec FetB + siLXR group compared with the FFA + Rec FetB group (Figure 3A, P = 0.003). Furthermore, LXR knockdown downregulated SREBP1c in the FFA + Rec FetB group, but it failed to influence phosphorylated AMPK and ACC (Figure 3B). Lastly, LXR siRNA downregulated mRNA levels of lipogenesis-related enzymes rather than fatty acid oxidation enzymes (Figure 3C). Pretreatment with AMPK agonist, AICAR, was also applied to confirm if AMPK is the upstream target of LXR. AICAR attenuated the TG content in HepG2 cells exposed to FFAs and recombinant fetuin B (Figure 3D, P < 0.001). Moreover, it upregulated phosphorylated AMPK and ACC and downregulated LXR and SREBP1c (Figure 3E). It decreased the mRNA levels of lipogenesis-related enzymes and increased fatty acid oxidation enzymes (Figure 3F).
Figure 3.
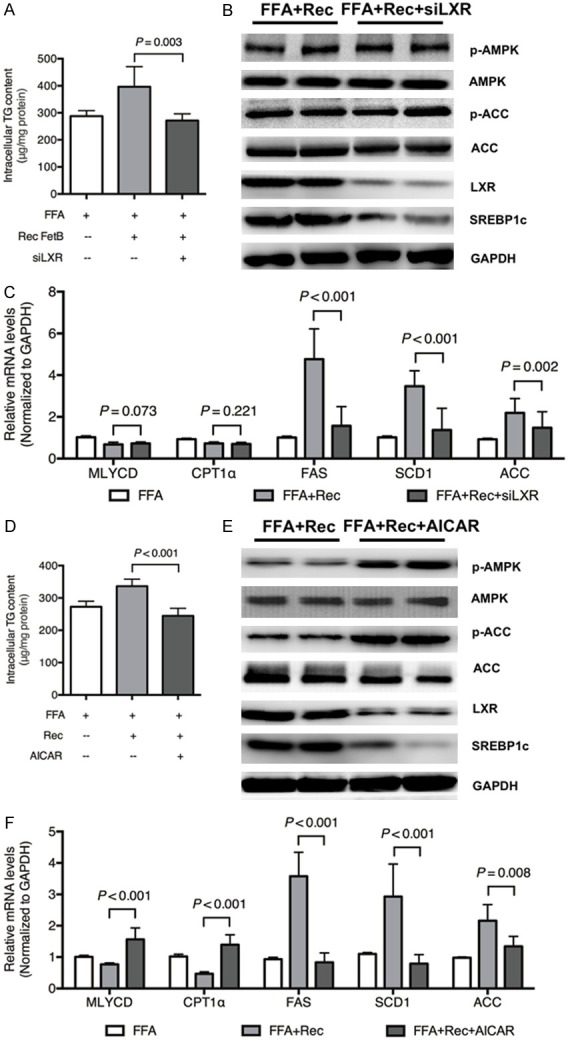
LXR is a downstream target of AMPK. To clarify the relation between AMPK and LXR, pretreatment with LXR siRNA was applied in HepG2 cells exposed to FFA + recombinant fetuin B. Pretreatment with AMPK agonist (AICAR, 1 mM) was applied to HepG2 cells exposed to FFA + recombinant fetuin B. A: Intercellular TG content: FFA + Rec vs. FFA + Rec + siLXR (P = 0.003); B: The expression of key proteins in the AMPK and LXR pathways; C: The mRNA levels of lipid metabolic enzymes; D: Intercellular TG content: FFA + Rec FetB vs. FFA + Rec FetB + AICAR (P < 0.001); E: The expression of key proteins in the AMPK AND LXR pathways; F: The mRNA levels of lipid metabolic enzymes; Rec, Recombinant fetuin B; siLXR, siRNA LXR. Results are the mean ± SD of three independent experiments, each repeated in triplicate.
Fetuin B knockdown attenuated hepatic lipid accumulation in HepG2 cells
As shown in Figure 4A, lipid accumulation was attenuated by fetuin B knockdown. The intercellular TG content was markedly decreased in the FFA + siFetB grouped compared with FFAs alone (Figure 4B, P = 0.004).
Figure 4.
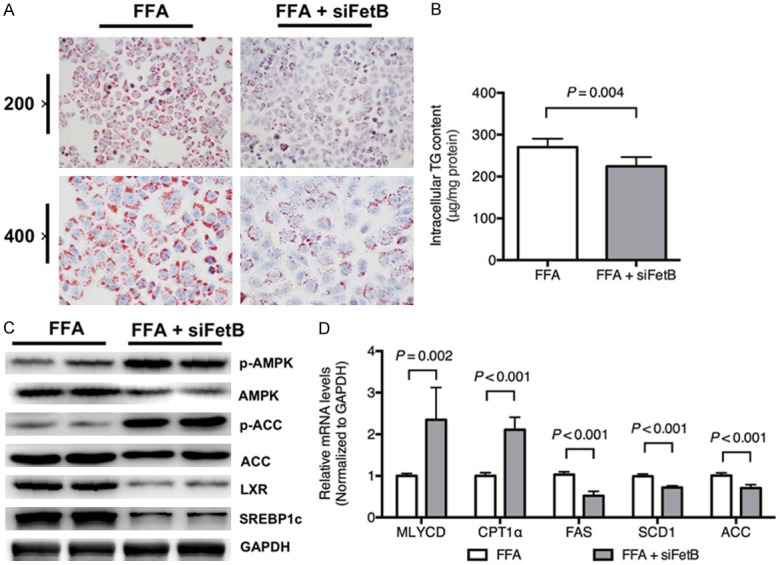
Fetuin B knockdown attenuated hepatic lipid accumulation in HepG2 cells. Hepatocellular lipid accumulation and the AMPK-/LXR-pathways were evaluated after fetuin B knockdown in HepG2 cells exposed to FFAs. A: Oil Red O staining: FFA vs. FFA + siFetB (200 × and 400 ×); B: Intercellular TG content (P = 0.004); C: The expression of key proteins in the AMPK and LXR pathways; D: The mRNA levels of lipid metabolic enzymes; siFetB, siRNA fetuin B. Results are the mean ± SD of three independent experiments, each repeated in triplicate.
As is revealed in Figure 4C, the administration of fetuin B siRNA increased phosphorylated AMPK and ACC and downregulated LXR and SREBP1c. Moreover, it upregulated the mRNA levels of MLYCD and CPT1α but downregulated FAS, SCD1 and ACC, as shown in Figure 4D.
LXR is a downstream target of AMPK
To confirm that LXR is a downstream target of AMPK, pretreatment with an LXR agonist, GW3965, was applied before the administration of Fetuin B siRNA to HepG2 cells exposed to FFAs. First, the intercellular TG content increased in the FFA + siFetB + GW3965 group compared with the FFA + siFetB group (Figure 5A, P = 0.005). Furthermore, the LXR agonist upregulated LXR and SREBP1c but not phosphorylated AMPK and ACC (Figure 5B). Lastly, it upregulated the mRNA levels of lipogenesis-related enzymes, but it had no influence on fatty acid oxidation enzymes (Figure 5C).
Figure 5.
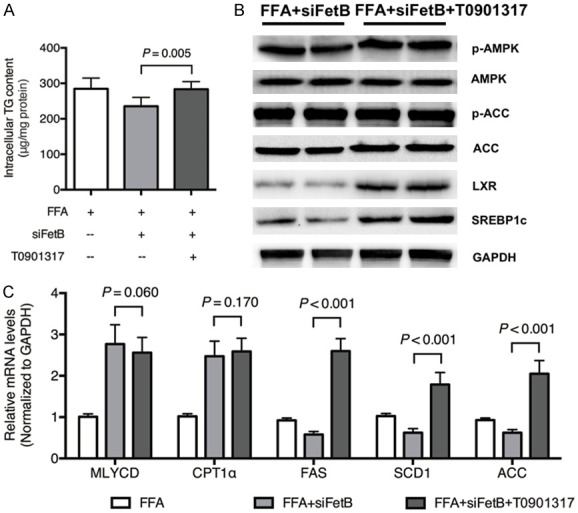
LXR is a downstream target of AMPK. To confirm that LXR is a downstream target of AMPK, pretreatment with LXR agonist (GW3965, 5 μM) was applied to HepG2 cells exposed to FFA + Fetuin B siRNA. A: Intercellular TG content: FFA + Rec FetB vs. FFA + siFetB + GW3965 (P = 0.005); B: The expression of key proteins in the AMPK and LXR pathways; C: The mRNA levels of lipid metabolic enzymes; siFetB, siRNA fetuin B. Results are the mean ± SD of three independent experiments, each repeated in triplicate.
Fetuin B knockdown attenuated hepatic lipogenesis in HFD-fed mice
Mice fed eight weeks of a HFD had significantly higher TG contents, whereas fetuin B knockdown by shRNA significantly attenuated the intrahepatic and serum TG contents. H&E and Oil Red O staining showed that the increase in lipid droplets caused by HFD was markedly attenuated in mice with fetuin B knockdown, as shown in Figure 6A-C. Fetuin B knockdown alleviated liver enzymes, serum alanine transaminase (ALT) and aspartate transaminase (AST) (Figure 6D).
Figure 6.
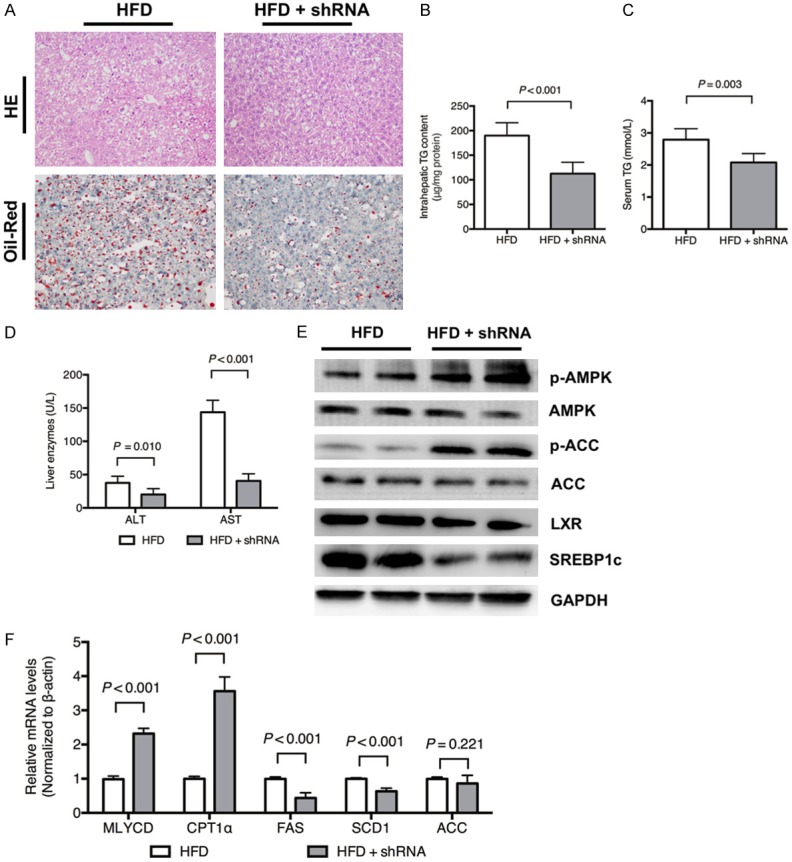
Fetuin B knockdown attenuated hepatic lipogenesis in mice fed a HFD. Hepatic lipid accumulation and the AMPK-/LXR-pathways were evaluated after fetuin B knockdown in mice exposed to a HFD. A: H&E staining and Oil Red O staining: HFD vs. HFD + shRNA (400 ×); B: Intrahepatic TG content (P < 0.001); C: Serum TG (P = 0.003); D: Liver enzymes; E: Key protein expression of the AMPK and LXR pathways in mouse liver; F: The mRNA levels of lipid metabolic enzymes; ALT, alanine transaminase; AST, aspartate transaminase. Results are the mean ± SD of three independent experiments (n = 6, each group).
As shown in Figure 6E, the hepatic expression of LXR and SREBP1c significantly decreased after fetuin B knockdown, while phosphorylated AMPK and ACC increased. Furthermore, fetuin knockdown downregulated the mRNA levels of lipogenesis enzymes (FAS and SCD1) and upregulated fatty acid oxidation enzymes (MLYCD and CPT1α) (Figure 6F).
Recombinant fetuin B aggravated hepatic lipogenesis in HFD-fed mice
Recombinant fetuin B aggravated the intrahepatic TG content, lipid droplets, serum TG and serum liver enzymes, as shown in Figure 7A-D. As shown in Figure 7E, recombinant fetuin B enhanced hepatic expression of LXR and SREBP1c and lowered the phosphorylation of AMPK and ACC. Moreover, recombinant fetuin B upregulated the mRNA levels of FAS, SCD1 and ACC, while it downregulated MLYCD and CPT1α (Figure 7F).
Figure 7.
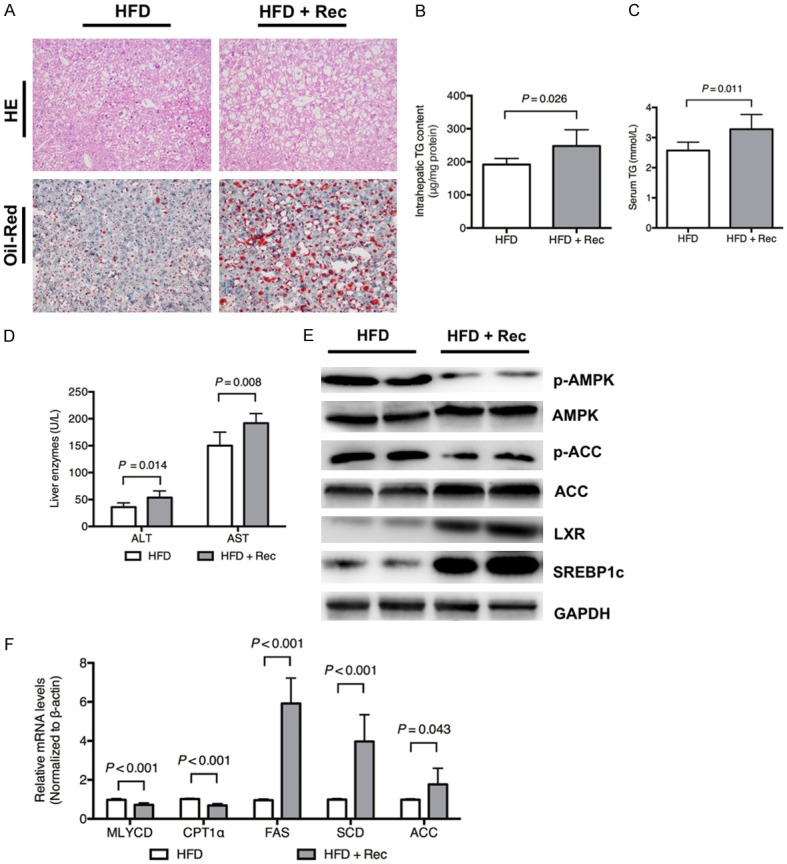
Recombinant fetuin B aggravated hepatic lipogenesis in HFD-fed mice. Hepatic lipid accumulation and the AMPK-/LXR-pathways were evaluated after the administration of recombinant fetuin B in mice exposed to a HFD. A: H&E staining and Oil Red O staining: HFD vs. HFD + Rec (400 ×); B: Intrahepatic TG content (P = 0.026); C: Serum TG (P = 0.011); D: Liver enzymes; E: Key protein expression of the AMPK and LXR pathways in mouse liver; F: The mRNA levels of lipid metabolic enzymes; ALT, alanine transaminase; AST, aspartate transaminase; Rec, Recombinant fetuin B. Results are the mean ± SD of three independent experiments (n = 6, each group).
Discussion
The present study shows that fetuin B, a new hepatokine, aggravates hepatic lipid accumulation. Mechanistically, it decreases the phosphorylated AMPK that depresses fatty acid oxidation and then activates LXR-SREBP1c, which upregulates lipogenesis. The results suggest that AMPK-dependent LXR activation represents a mechanism for the aggravated effects of fetuin B on hepatocyte lipid metabolism.
A hospital-based case-control study by Zhu et al. [26] indicated that serum levels of fetuin B increased in subjects with NAFLD. Wang et al. [27] performed a large cross-sectional study that found that the intrahepatic TG content was independently and positively correlated with serum fetuin B levels using magnetic resonance spectroscopy. Li et al. [28] investigated the association of genetic variants with FETUB and insulin resistance in 1,148 obese adults. They found that higher serum fetuin-B levels were correlated with increased homeostasis model assessment of insulin resistance, although the results did not support the association between fetuin B and insulin resistance. The previous findings supported that fetuin B might be involved in the development of hepatic steatosis, but the mechanism is still unknown.
In our study, the results reveal that the administration of recombinant fetuin B lowered the phosphorylated AMPK that is associated with lipid oxidation in HepG2 cells, while it activated the LXR-SREBP1c pathway that is closely related to lipogenesis. Furthermore, LXR knockdown decreased SREBP1c, rather than AMPK, and then protected against lipid accumulation. The administration of AMPK agonist upregulated phosphorylated AMPK and depressed LXR/SREBP1c-dependent lipogenesis, which resulted in the alleviation of hepatic lipid accumulation.
Fetuin B knockdown activated the phosphorylation of AMPK and then depressed the LXR-SREBP1c pathway. Moreover, LXR agonist increased the activities of the LXR-SREBP1c pathway, rather than AMPK, and then worsened the lipid accumulation protected by fetuin B knockdown. Thus, fetuin B-triggered depression of AMPK (downstream: lipid oxidation enzymes, i.e., MLYCD and CPT1α)-modulated hepatic LXR-SREBP1c activity (downstream: lipogenesis enzymes, i.e., FAS, SCD1 and ACC), which supports that AMPK functions as an upstream regulator of LXR signaling in the effect of fetuin B.
The mechanism based on cellular experiments was consistent with the observations in mice. In vivo, the administration of recombinant fetuin B lowered phosphorylated AMPK and its downstream lipid oxidation and then activated LXR-SREBP1c and lipogenesis, which led to the aggravation of hepatic lipid accumulation. On the contrary, hepatic knockdown of fetuin B activated the AMPK pathway and then depressed LXR-SREBP1c-dependent lipogenesis.
Our study has some limitations. It failed to investigate the way that fetuin B affects hepatocytes, either through autocrine or paracrine effects. Further studies should be performed to clarify the membrane receptors involved. The experiment involving AMPK knockdown and inhibition could be beneficial for identifying the role of AMPK in the pathway.
In conclusion, this study shows that fetuin B aggravated LXR-mediated hepatic steatosis through AMPK. Our findings may potentially have a translational impact on the diagnosis and treatment of NAFLD in humans.
Acknowledgements
This work was supported by the Foundation of Zhejiang Provincial Department of Health (2015KYB158) and the National Natural Science Foundation of China (No. 81771498).
Disclosure of conflict of interest
None.
Abbreviations
- ACC
Acetyl-CoA Carboxylase
- AMPK
AMP-activated protein kinase
- CPT1α
Carnitine palmitoyltransferase 1α
- FAS
Fatty acid synthase
- FFAs
Free fatty acids
- HFD
High-fat diet
- H&E
Hematoxylin and eosin
- LXR
Liver X receptor
- MLYCD
Malonyl-CoA decarboxylase
- NAFLD
Non-alcoholic fatty liver disease
- SCD
Standard chow diet
- SCD1
Stearoyl-CoA desaturase-1
- SREBP1c
Sterol regulatory element binding transcription factor 1
- TG
Triglyceride
Supporting Information
References
- 1.Zhu JZ, Dai YN, Wang YM, Zhou QY, Yu CH, Li YM. Prevalence of nonalcoholic fatty liver disease and economy. Dig Dis Sci. 2015;60:3194–3202. doi: 10.1007/s10620-015-3728-3. [DOI] [PubMed] [Google Scholar]
- 2.Musso G, Gambino R, Cassader M, Pagano G. Meta-analysis: natural history of non-alcoholic fatty liver disease (NAFLD) and diagnostic accuracy of non-invasive tests for liver disease severity. Ann Med. 2011;43:617–649. doi: 10.3109/07853890.2010.518623. [DOI] [PubMed] [Google Scholar]
- 3.Nascimbeni F, Pais R, Bellentani S, Day CP, Ratziu V, Loria P, Lonardo A. From NAFLD in clinical practice to answers from guidelines. J Hepatol. 2013;59:859–871. doi: 10.1016/j.jhep.2013.05.044. [DOI] [PubMed] [Google Scholar]
- 4.Williams CD, Stengel J, Asike MI, Torres DM, Shaw J, Contreras M, Landt CL, Harrison SA. Prevalence of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis among a largely middle-aged population utilizing ultrasound and liver biopsy: a prospective study. Gastroenterology. 2011;140:124–131. doi: 10.1053/j.gastro.2010.09.038. [DOI] [PubMed] [Google Scholar]
- 5.Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, Wymer M. Global epidemiology of nonalcoholic fatty liver disease-meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology. 2016;64:73–84. doi: 10.1002/hep.28431. [DOI] [PubMed] [Google Scholar]
- 6.Bril F, Barb D, Portillo-Sanchez P, Biernacki D, Lomonaco R, Suman A, Weber MH, Budd JT, Lupi ME, Cusi K. Metabolic and histological implications of intrahepatic triglyceride content in nonalcoholic fatty liver disease. Hepatology. 2017;65:1132–1144. doi: 10.1002/hep.28985. [DOI] [PubMed] [Google Scholar]
- 7.Meex RCR, Watt MJ. Hepatokines: linking nonalcoholic fatty liver disease and insulin resistance. Nat Rev Endocrinol. 2017;13:509–520. doi: 10.1038/nrendo.2017.56. [DOI] [PubMed] [Google Scholar]
- 8.Olivier E, Soury E, Ruminy P, Husson A, Parmentier F, Daveau M, Salier JP. Fetuin-B, a second member of the fetuin family in mammals. Biochem J. 2000;350:589–597. [PMC free article] [PubMed] [Google Scholar]
- 9.Denecke B, Graber S, Schafer C, Heiss A, Woltje M, Jahnen-Dechent W. Tissue distribution and activity testing suggest a similar but not identical function of fetuin-B and fetuin-A. Biochem J. 2003;376:135–145. doi: 10.1042/BJ20030676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pal D, Dasgupta S, Kundu R, Maitra S, Das G, Mukhopadhyay S, Ray S, Majumdar SS, Bhattacharya S. Fetuin-A acts as an endogenous ligand of TLR4 to promote lipid-induced insulin resistance. Nat Med. 2012;18:1279–1285. doi: 10.1038/nm.2851. [DOI] [PubMed] [Google Scholar]
- 11.Stefan N, Haring HU. Circulating fetuin-A and free fatty acids interact to predict insulin resistance in humans. Nat Med. 2013;19:394–395. doi: 10.1038/nm.3116. [DOI] [PubMed] [Google Scholar]
- 12.Meex RC, Hoy AJ, Morris A, Brown RD, Lo JC, Burke M, Goode RJ, Kingwell BA, Kraakman MJ, Febbraio MA, Greve JW, Rensen SS, Molloy MP, Lancaster GI, Bruce CR, Watt MJ. Fetuin B is a secreted hepatocyte factor linking steatosis to impaired glucose metabolism. Cell Metab. 2015;22:1078–1089. doi: 10.1016/j.cmet.2015.09.023. [DOI] [PubMed] [Google Scholar]
- 13.Ruderman N, Prentki M. AMP kinase and malonyl-CoA: targets for therapy of the metabolic syndrome. Nat Rev Drug Discov. 2004;3:340–351. doi: 10.1038/nrd1344. [DOI] [PubMed] [Google Scholar]
- 14.Hardie DG. AMPK: positive and negative regulation, and its role in whole-body energy homeostasis. Curr Opin Cell Biol. 2015;33:1–7. doi: 10.1016/j.ceb.2014.09.004. [DOI] [PubMed] [Google Scholar]
- 15.Hardie DG. AMPK--sensing energy while talking to other signaling pathways. Cell Metab. 2014;20:939–952. doi: 10.1016/j.cmet.2014.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Donnelly KL, Smith CI, Schwarzenberg SJ, Jessurun J, Boldt MD, Parks EJ. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Invest. 2005;115:1343–1351. doi: 10.1172/JCI23621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ducheix S, Montagner A, Theodorou V, Ferrier L, Guillou H. The liver X receptor: a master regulator of the gut-liver axis and a target for non alcoholic fatty liver disease. Biochem Pharmacol. 2013;86:96–105. doi: 10.1016/j.bcp.2013.03.016. [DOI] [PubMed] [Google Scholar]
- 18.Repa JJ, Liang G, Ou J, Bashmakov Y, Lobaccaro JM, Shimomura I, Shan B, Brown MS, Goldstein JL, Mangelsdorf DJ. Regulation of mouse sterol regulatory element-binding protein-1c gene (SREBP-1c) by oxysterol receptors, LXRalpha and LXRbeta. Genes Dev. 2000;14:2819–2830. doi: 10.1101/gad.844900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liang G, Yang J, Horton JD, Hammer RE, Goldstein JL, Brown MS. Diminished hepatic response to fasting/refeeding and liver X receptor agonists in mice with selective deficiency of sterol regulatory element-binding protein-1c. J Biol Chem. 2002;277:9520–9528. doi: 10.1074/jbc.M111421200. [DOI] [PubMed] [Google Scholar]
- 20.Joseph SB, Laffitte BA, Patel PH, Watson MA, Matsukuma KE, Walczak R, Collins JL, Osborne TF, Tontonoz P. Direct and indirect mechanisms for regulation of fatty acid synthase gene expression by liver X receptors. J Biol Chem. 2002;277:11019–11025. doi: 10.1074/jbc.M111041200. [DOI] [PubMed] [Google Scholar]
- 21.Talukdar S, Hillgartner FB. The mechanism mediating the activation of acetyl-coenzyme A carboxylase-alpha gene transcription by the liver X receptor agonist T0-901317. J Lipid Res. 2006;47:2451–2461. doi: 10.1194/jlr.M600276-JLR200. [DOI] [PubMed] [Google Scholar]
- 22.Chu K, Miyazaki M, Man WC, Ntambi JM. Stearoyl-coenzyme A desaturase 1 deficiency protects against hypertriglyceridemia and increases plasma high-density lipoprotein cholesterol induced by liver X receptor activation. Mol Cell Biol. 2006;26:6786–6798. doi: 10.1128/MCB.00077-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Park KG, Min AK, Koh EH, Kim HS, Kim MO, Park HS, Kim YD, Yoon TS, Jang BK, Hwang JS, Kim JB, Choi HS, Park JY, Lee IK, Lee KU. Alpha-lipoic acid decreases hepatic lipogenesis through adenosine monophosphate-activated protein kinase (AMPK)-dependent and AMPK-independent pathways. Hepatology. 2008;48:1477–1486. doi: 10.1002/hep.22496. [DOI] [PubMed] [Google Scholar]
- 24.Huang J, Kang S, Park SJ, Im DS. Apelin protects against liver X receptor-mediated steatosis through AMPK and PPARalpha in human and mouse hepatocytes. Cell Signal. 2017;39:84–94. doi: 10.1016/j.cellsig.2017.08.003. [DOI] [PubMed] [Google Scholar]
- 25.Xu C, Wan X, Xu L, Weng H, Yan M, Miao M, Sun Y, Xu G, Dooley S, Li Y, Yu C. Xanthine oxidase in non-alcoholic fatty liver disease and hyperuricemia: one stone hits two birds. J Hepatol. 2015;62:1412–1419. doi: 10.1016/j.jhep.2015.01.019. [DOI] [PubMed] [Google Scholar]
- 26.Zhu J, Wan X, Wang Y, Zhu K, Li C, Yu C, Li Y. Serum fetuin B level increased in subjects of nonalcoholic fatty liver disease: a case-control study. Endocrine. 2017;56:208–211. doi: 10.1007/s12020-016-1112-5. [DOI] [PubMed] [Google Scholar]
- 27.Wang D, Liu Y, Liu S, Lin L, Liu C, Shi X, Chen Z, Lin M, Yang S, Li Z, Li X. Serum fetuin-B is positively associated with intrahepatic triglyceride content and increases the risk of insulin resistance in obese Chinese adults: a cross-sectional study. J Diabetes. 2018;10:581–588. doi: 10.1111/1753-0407.12632. [DOI] [PubMed] [Google Scholar]
- 28.Li Z, Liu C, Shi X, Chen Z, Wang D, Li L, Tu Y, Lin M, Liu S, Yang S, Li X. Common genetic variants in the FETUB locus, genetically predicted fetuin-B levels, and risk of insulin resistance in obese Chinese adults. Medicine (Baltimore) 2017;96:e9234. doi: 10.1097/MD.0000000000009234. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.