

Abstract
KCNQ1 (Kv7.1 or KvLQT1) is a potassium ion channel protein found in the heart, ear, and other tissues. In complex with the KCNE1 accessory protein, it plays a role during the repolarization phase of the cardiac action potential. Mutations in the channel have been associated with several diseases, including congenital deafness and long QT syndrome. Nuclear magnetic resonance (NMR) structural studies in detergent micelles and a cryo-electron microscopy structure of KCNQ1 from Xenopus laevis have shown that the voltage sensor domain (Q1-VSD) of the channel has four transmembrane helices, S1–S4, being overall structurally similar with other VSDs. In this study, we describe a reliable method for the reconstitution of Q1-VSD into (POPC/POPG) lipid bilayer vesicles. Site-directed spin labeling electron paramagnetic resonance spectroscopy was used to probe the structural dynamics and topology of several residues of Q1-VSD in POPC/POPG lipid bilayer vesicles. Several mutants were probed to determine their location and corresponding immersion depth (in angstroms) with respect to the membrane. The dynamics of the bilayer vesicles upon incorporation of Q1-VSD were studied using 31P solid-state NMR spectroscopy by varying the protein:lipid molar ratios confirming the interaction of the protein with the bilayer vesicles. Circular dichroism spectroscopic data showed that the α-helical content of Q1-VSD is higher for the protein reconstituted in vesicles than in previous studies using DPC detergent micelles. This study provides insight into the structural topology and dynamics of Q1-VSD reconstituted in a lipid bilayer environment, forming the basis for more advanced structural and functional studies.
Graphical Abstract
Human KCNQ1 (Q1, Kv7.1, or KvLQT1) is a voltage-gated potassium channel expressed in several tissues of the body and is known to regulate various physiological functions. It is a six-pass transmembrane protein, prominently involved in cardiac function in addition to regulating water and salt transport in the epithelial tissues.1–5 Q1 is involved in the repolarization phase of the cardiac action potential and was identified as the gene causing chromosome 11-linked long QT syndrome.3–6 Dysfunction of the channel has also been linked to other disease conditions like Romano-Ward syndrome, sudden infant death syndrome, congenital deafness, and familial atrial fibrillation.7 Like other voltage-gated ion channels, Q1 has a voltage-sensing domain, S1–S4 (VSD), and a pore domain, S5 and S6 (PD), connected by a linker region containing the selectivity filter.8 The unique property of Q1 is that it forms a channel that is responsive to a change in voltage and can be activated upon membrane depolarization or remain constitutively active regardless of the membrane potential.9,10 Q1 forms a complex with KCNE1 (E1 or minK), slowing the channel activation kinetics and giving rise to slowly activating Iks currents that are crucial during the repolarization of action potential in the heart.9–11 In the absence of E1, Q1 is rapidly activated to the open state of the channel, which has an only modest conductance.10
Some groups have reported the structural studies of the water-soluble segment of the cytosolic C-terminus of KCNQ1 using biophysical, electrophysiology, and in silico approaches.12–14 The Sanders group has published the purification and preliminary structural characterization of the voltage-sensing domain of human KCNQ1 (Q1-VSD) in lysophosphatidylglycerol detergent micelles using solution nuclear magnetic resonance (NMR) spectroscopic studies.15 Previous studies conducted on voltage-gated channels (e.g., KvAP) have shown that isolated VSDs can fold even in the absence of the pore domain (PD), suggesting that VSDs can adopt nativelike structure in a manner that is independent of the PD.16–18 The cryo-electron microscopy structure of the KCNQ1/CaM complex from Xenopus laevis was recently published, suggesting a transmembrane topology resembling that of Shaker-like voltage-gated K+ channels.3 Though the sequence of KCNQ1 of X. laevis is 78% identical with that of human KCNQ1, the study used KCNQ1 solubilized in n-Dodecyl-b-D-Maltopyranoside (DDM)/Cholesteryl hemisuccinate (CHS) detergent micelles.3 Several recent findings illustrate the crucial role of a lipid bilayer in maintaining the relative orientation of membrane proteins, thereby affecting their function16,19–25 leading to the hypothesis that the structural and functional properties of KCNQ1 can vary considerably from detergent micelles to lipid bilayer vesicles. Therefore, understanding the structural properties of KCNQ1 in a lipid bilayer environment is crucial.
In this study, we describe a reliable method for reconstituting human Q1-VSD into lipid vesicles to enable study of the dynamics and topology of spin-labeled Q1-VSD mutants using continuous wave electron paramagnetic resonance (CW-EPR) line shape analysis, circular dichroism (CD) spectroscopy, 31P solid-state NMR spectroscopy, and CW-EPR power saturation experiments. Q1-VSD has four transmembrane helices like other VSDs. However, the exact location of the amino acid residues with respect to the membrane bilayer needs to be determined. Structural and functional studies of Q1-VSD in a lipid bilayer environment are very important because ~40% of the >200 reported disease-related mutations arise from the amino acid substitutions in the VSD.26
MATERIALS AND METHODS
Site-Directed Mutagenesis, Protein Expression, and Purification.
The His tag expression vector (pET-16b) containing wild type Q1-VSD (residues 100–249) was expressed and purified using the Rosetta/C43(DE3) strain of Escherichia coli as previously described.15 Site-directed mutagenesis was performed to remove all of the native cysteines and generate single-cysteine mutants from the Cys-less vector using the QuickChange Lightning Site-Directed Mutagenesis Kit (Stratagene). The mutations were confirmed by DNA sequencing. Successful mutants were transformed into Rosetta/C43(DE3) E. coli cells for protein overexpression.15 Twenty-nine mutants were engineered across the Q1-VSD to probe the location of each residue with respect to the membrane.
A 5 mL preculture of LB containing 50 μg/mL ampicillin and 50 μg/mL chloramphenicol was inoculated with the Rosetta/C43(DE3) Q1-VSD mutant at 37 °C. A culture grown overnight was transferred to 1 L of sterile M9 minimal medium supplemented with MEM vitamin (Mediatech) and ZnCl2. The cells were grown in a shaker-incubator at 25 °C and 250 rpm. Protein expression was induced for 24 h by addition of isopropyl β-d-1-thiogalactopyranoside to a final concentration of 1 mM, once the OD600 reached 0.70–0.80.
Cells were harvested by centrifugation of the cultures at 6500 rpm for 20 min at 4 °C. The cell pellets were resuspended in lysis buffer [75 mM Tris-HCl (pH 7.8), 300 mM NaCl, and 0.2 mM EDTA] and tumbled at room temperature for 45 min after the addition of the LDR stock (100 mg/mL lysozyme, 10 mg/mL DNase, 10 mg/mL RNase, and 10% glycerol), phenylmethanesulfonyl fluoride (20 mg/mL), and magnesium acetate to a final concentration of 5 mM. The cells were then sonicated for 10 min (5 s on/off cycles, Fisher Scientific Sonic Dismembrator Model 500, amplitude of 40%) on ice. The inclusion bodies were pelleted at 20000 rpm for 30 min at 4 °C, and the pellet was washed with lysis buffer and homogenized with buffer A [40 mM HEPES (pH 7.5) and 300 mM NaCl] containing 2 mM tris(2-carboxyethyl)-phosphine (TCEP). n-dodecylphosphocholine (DPC) was added to a final concentration of 0.5% and rotated overnight at 4 °C to solubilize inclusion bodies. After detergent solubilization, the insoluble debris was removed by centrifugation at 20000 rpm for 30 min at 4 °C. The supernatant was incubated with Ni(II)-NTA superflow resin (Qiagen) for 45 min and tumbled at 4 °C. The protein was then purified using the gravity flow column by washing with 4–5 bed volumes of rinse buffer (buffer A with TCEP) containing DPC. Non-specific proteins were removed with wash buffer (buffer A with DPC, 50 mM imidazole, and TCEP). Q1-VSD was eluted with elution buffer (250 mM imidazole with 2 mM TCEP) containing DPC. The protein concentration was measured with a Nanodrop, and the purity was confirmed by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE).
Methanethiosulfonate Spin Label (MTSL) Labeling.
MTSL labeling was carried out as previously described.27–29 Briefly, a MTSL (Toronto Research Chemicals) stock (250 mM in methanol) was added to the protein (concentrated to a volume of 500 μL) at a 10:1 MTSL:protein molar ratio. The reaction mixture was incubated at room temperature for 30 min and then shaken at 37 °C for 3 h, and the final reaction was carried out at room temperature with vigorous shaking overnight in the dark. Buffer exchange was performed three times with phosphate buffer (pH 7.50) using centrifugal ultrafiltration to remove imidazole (from elution buffer), and the sample concentrated to 500 μL. Excess MTSL was removed by rebinding the labeled protein with Ni-NTA resin (which had been pre-equilibrated with phosphate buffer containing DPC) and washing by resuspending the resin in the same buffer. The spin-labeled protein was then eluted with 250 mM imidazole containing 0.5% DPC and concentrated using Amicon Ultra centrifugal filters (3K).
Reconstitution into Lipid Bilayers.
Lipids (1-palmitoyl-2-oleoyl-glycero-3-phosphocholine (POPC) and 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-(1′-rac-glycerol) (POPG) powder from Avanti Polar Lipids, Alabaster, AL) were dissolved with 100 mM imidazole, 0.1 mM EDTA, and 400 mM SDS at a lipid concentration of 70 mg/mL by repeating freeze–thaw sonication cycles until the solution became clear. Each spin-labeled protein was mixed with 35 mg (500 μL of the above stock) of dissolved lipid at a 1:250 (protein:lipid) molar ratio and frozen and thawed three times to equilibrate. SM2 Bio Beads (Bio-Rad) were washed with methanol followed by water using a gravity flow column prior to use. The equilibrated mixture was then added to 400 mg of wet Bio Beads and rotated at room temperature for 4 h. The resulting sample was centrifuged at 2000 rpm for 2 min to remove Bio Beads. The supernatant was centrifuged at 100000 rpm for 30 min to pellet the vesicles. The proteoliposome pellet was resuspended in phosphate buffer (pH 7.5) and used for spectroscopic studies within a week (for NMR studies, the proteoliposomes were resuspended in HEPES buffer).
EPR Spectroscopic Experiments.
All EPR measurements were conducted at the Ohio Advanced EPR Laboratory at Miami University. CW-EPR spectra were collected on a Bruker EMX CW-EPR instrument at X-band using an ER041xG microwave bridge and ER4119-HS cavity. Spectra were recorded by signal averaging 20–30 42 s scans with a central field of 3315 G, a sweep width of 100 G, and a field modulation of 1 G at a frequency of 100 kHz. All of the samples were collected at a microwave power of 10 mW at 296 K. The side chain mobility was determined by calculating the inverse central line width from each CW-EPR spectrum. The rotational correlation time (τc) was calculated using the empirical estimation method.30,31
Insertion of Q1-VSD into POPC/POPG bilayers was verified using CW-EPR power saturation at X-band. Power saturation experiments were performed on a Bruker EMX X-band CW-EPR spectrometer consisting of an ER 041XG microwave bridge coupled with an ER 4123D CW resonator (Bruker BioSpin) as previously described.23,27,32,33 Samples were loaded into gas permeable TPX capillary tubes with a total volume of 3–4 μL at a concentration of 50–100 μM. EPR data were collected using a modulation amplitude of 1 G and a microwave power varying from 0.4 to 100 mW. The scan range of all spectra was 100 G, and the final spectra were obtained by signal averaging three to five scans. The power saturation curves were obtained for the spin probes outside of the model membrane and inside the membrane for 20 residues on Q1-VSD under three conditions: (1) equilibrated with nitrogen as a control, (2) equilibrated with the lipid-soluble paramagnetic reagent 20% oxygen (air), and (3) equilibrated with nitrogen in the presence of a water-soluble paramagnetic reagent NiEDDA (2 mM) as previously synthesized.28 The samples were purged with gas for at least 60 min at a rate of 10 mL/min before each measurement was performed. High-purity nitrogen and house-supplied compressed air lines were used during the experiment. The resonator remained connected to the gas during all measurements, and the sample temperature was held at 296 K. The peak-to-peak amplitude of the first derivative mI = 0 resonance line (A) was measured and plotted against the square root of the incident microwave power. The data points were then fit using a Matlab software script according to eq 1:
| (1) |
where I is a scaling factor, P1/2 is the power where the first-derivative amplitude is reduced to half of its unsaturated value, and ε is a measure of the homogeneity of saturation of the resonance line. In eq 1, I, ε, and P1/2 are adjustable parameters and yield a characteristic P1/2 value. The corresponding Φ depth parameters were calculated using eq 2:32
| (2) |
where ΔP1/2(O2) is the difference in the P1/2 values of air- and nitrogen-exposed samples and ΔP/2(NiEDDA) is the difference in the P1/2 values of NiEDDA- and nitrogen-exposed samples.
The membrane depth (in angstroms) of the spin-labeled Q1-VSD mutants was calculated using eq 3:
| (3) |
The bilayer calibration equation given above was derived using standard n-doxyl spin-labeled POPCs (Avanti Polar Lipids) incorporated into POPC/POPG vesicles containing WT Q1-VSD. Φ values for 5-, 7-, 10-, and 12-POPC spin-labels were measured and plotted against the previously determined depths of the spin-labels.33,34 Linear regression analysis exhibited a linear dependence of Φ for various POPC spin-labels.
CD Spectroscopy.
CD measurements were performed on a model 435 Aviv circular dichroism spectrometer in a quartz cuvette. Data were collected from 190 to 260 nm by signal averaging 10 scans with a bandwidth of 1 nm. The cuvette path length was 1.00 mm, and spectra were recorded at 296 K. When necessary, samples were diluted in 50 mM NaH2PO4 (pH 7.5). Scattering effects produced by the sample matrix were found to be reduced when the vesicle samples were passed through a 100 nm pore size extruder. Spectra of empty vesicles without the protein were used to account for the background lipid absorbance and light scattering phenomenon and subtracted from lipid–protein spectra. For micelle samples, buffer exchange was performed [50 mM NaH2P04 (pH 7.5) and 0.5% DPC] using centrifugal ultrafiltration to remove imidazole from the elution step. Spectra were processed and analyzed on Dichroweb using CONTIN with the SMP180 reference set. A spectral width of 190–240 nm was used for data analysis 36–38
31P Solid-State NMR Measurements.
A Bruker Advance 500 MHz WB NMR spectrometer equipped with a CPMAS probe (Bruker, Billerica, MA) was used to record all of the NMR data. The 31P NMR spectra were collected at 200 MHz with proton decoupling with a 4 μs 90° pulse and a 4 s recycle delay. The spectra were collected overnight with the spectral line width set to 500 ppm. All 31P NMR spectra were referenced by assigning the 85% H3PO4 31P peak to 0 ppm. The data were analyzed as previously described 39–41
RESULTS AND DISCUSSION
Expression and Mutagenesis of Recombinant Q1-VSD (residues 100–249).
To better understand the structural topology and dynamics of the Q1-VSD using EPR spectroscopy, all native cysteines in the VSD were mutated using site-directed mutagenesis and single-cysteine mutants were generated from the Cys-less vector. Twenty-nine mutants were designed across the four TMDs of Q1-VSD. Twenty Cys mutants were successfully expressed and purified, while nine mutants suffered from lower expression levels or failed to yield structurally stable protein as confirmed by SDS–PAGE and CD experiments. For example, residue L236C migrated as two separate bands on the gel. This finding corroborates recently published data that showed that the L236P mutation led to a defective channel with respect to both surface expression level and channel properties.4 Also, mutants Y111C, L114C, P117C, and W120C in the S0 helical region and G179C toward the cytosolic end of helix S2 suffered from lower expression levels. The lower expression levels and/or protein yield is not surprising as point mutations in functionally active proteins have been reported to cause overall protein distortions and instability 4,42
Homogeneous Reconstitution of Q1VSD into POPC/POPG Vesicles.
To generate homogeneously reconstituted Q1-VSD in a lipid bilayer, POPC/POPG lipids were selected. These two lipids best mimic a phospholipid bilayer found in most mammalian cell membrane systems. 16,23,25 Furthermore, the POPC/POPG phase transition temperatures to the Lα liquid crystalline phase are <0 °C.23 The POPC:POPG molar ratio was 3:1, which mimics those of the anionic phospholipids commonly found in a mammalian cell membrane.16,23,25 Several reconstitution methods, including dialysis, SM2 Bio Beads, and gel filtration, were tested. SM2 Bio Beads resulted in homogeneous vesicles as confirmed by DLS.
To confirm the interaction of Q1-VSD with the phospholipid head groups of the membrane, we investigated the dynamic properties of the lipid bilayers after Q1-VSD incorporation using 31P solid-state NMR spectroscopy. Varying the protein:lipid molar ratio was used to titrate Q1-VSD in POPC/POPG bilayer vesicles. The static 31P NMR spectra collected at 25 °C displaying the perturbations of Q1-VSD on the dynamics of lipid bilayer vesicles with varying protein/lipid ratios between 0 and 4 mol % are shown in Figure 1. In the absence of protein, the 31P NMR spectrum at 25 °C showed a motionally averaged axially symmetric powder pattern with a chemical shift anisotropy (CSA) width of 37 ppm in the liquid crystalline phase (Lα).41 However, upon addition of Q1-VSD, the CSA spectral width decreased, when compared to that of empty POPC/POPG bilayers. The CSA line width was measured for empty POPC/POPG (Figure 1, 0 mol %, CSA = 37 ppm), 1 mol % (CSA = 35 ppm), 2 mol % (CSA = 32 ppm), and 4 mol % (CSA = 31 ppm) bilayers. The 31P line shape and CSA width data clearly indicate an increase in the membrane surface fluidity upon addition of Q1-VSD and its interaction with the membrane. A similar observed reduction in CSA line width with an increase in the amount of protein has been previously reported.39–41 Overall, the 31P line shape and CSA line width data show that the phospholipid head groups of POPC/POPG vesicles are perturbed in a linear fashion with an increase in protein concentration from 1 to 4 mol %, indicating that Q1-VSD interacts with the phospholipid head groups of POPC/POPG lipid bilayer vesicles.
Figure 1.
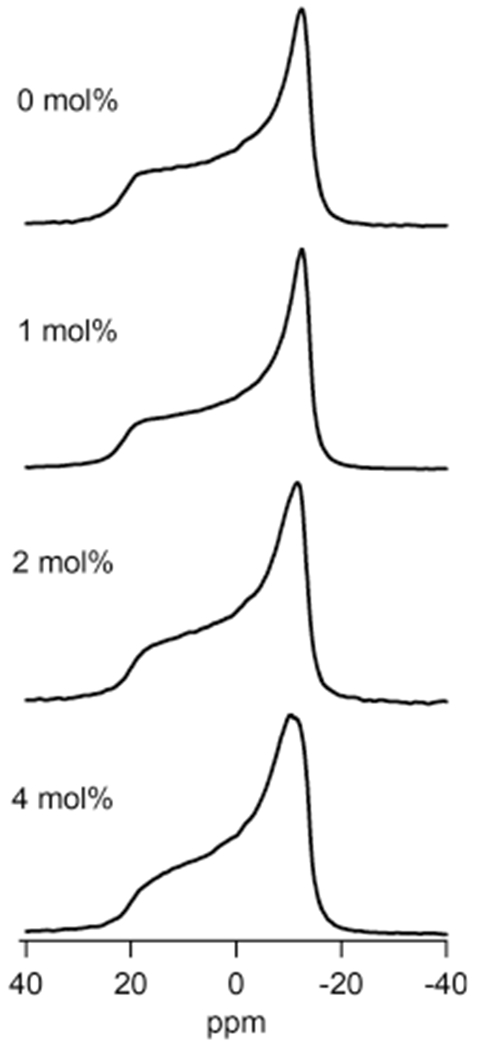
31P solid-state NMR spectra of Q1-VSD incorporated into multilamellar vesicles with varying protein:lipid molar ratios at room temperature.
CD measurements provide a reliable method for determining the overall secondary structure of a protein. Figure 2 depicts the CD data of WT Q1-VSD in 0.5% DPC micelles and in POPC/POPG vesicles at pH 7.5 and 296 K. CD experiments demonstrated a typical α-helical pattern in both micelle and vesicle environments. However, quantitative analysis of the CD data indicated an increase in the helical content from 67% in 0.5% DPC micelles to 76% for Q1-VSD reconstituted in lipid bilayer vesicles, clearly indicating that Q1-VSD adopts a more ordered structure upon incorporation into a lipid bilayer with a higher α-helical content.36–38,43–45
Figure 2.
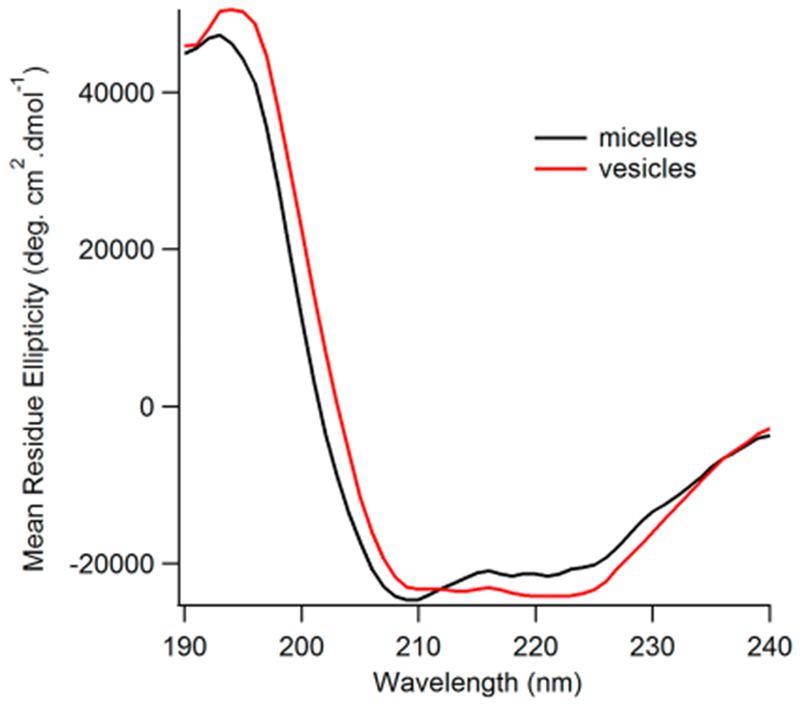
CD spectra of WT Q1-VSD in micelle and vesicle environments: (black) 0.5% DPC micelles, WT Q1-VSD, pH 7.5, and 296 K and (red) POPC/POPG vesicles, 250:1 lipid:protein ratio, WT Q1-VSD, pH 7.5, and 296 K.
On the basis of previous KCNE1 studies, the method used to incorporate the protein into a lipid bilayer is crucial in determining the type of secondary structure the protein adopts.22,23,45,46 In the case of Q1-VSD, the TMDs are very hydrophobic and have several polar residues on either end that help to anchor the protein to the polar membrane surface, while the inner nonpolar residues interact with the lipid carbon chains. Because of the amphipathic nature of Q1-VSD, it becomes very insoluble and prone to aggregation in aqueous solution. Therefore, the efficient membrane reconstitution methodology was important for this study. In this study, SM2 Bio Beads were used for selective removal of the detergent from mixed detergent/lipid micelle samples. This reconstitution method keeps Q1-VSD in contact with the detergent or lipids and prevents any aggregation. A protein:lipid molar ratio of 1:250 was found to be best for maintaining its folding pattern.
Probing the Membrane Topology of Q1-VSD.
X-Band CW-EPR spectra were collected on 20 spin-labeled Q1-VSD mutants, and the results are shown in Figures 3, 4, and 6. Our results indicate that residues F130C in helix 1 and F232C in helix 4 showed restricted mobility in a lipid bilayer environment at pH 7.5, whereas Q147C and F222C were found to be more mobile in the reconstituted phospholipid bilayer, suggesting a relatively higher level of motion of the MTSL spin-label in these mutants. The overall broadening of the EPR spectra indicates that the location of the labeled residue affects the mobility of the nitroxide spin-label and therefore illuminates the overall dynamics of the residues with respect to the membrane. It was also observed that the line broadening was significantly different between the DPC micelle and the vesicle samples for the same residue, indicating different packing in the detergent as opposed to a lipid bilayer environment.
Figure 3.
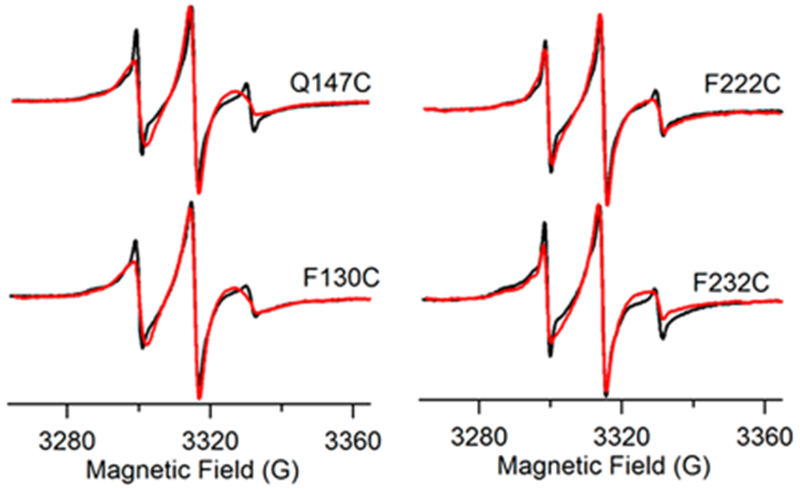
CW EPR (X-band) spectra of Q1-VSD mutants. The left panel shows Q147C (outside membrane) and F130C (inside membrane) in helix 1, and the right panel shows F222C (outside membrane) and F232C (inside membrane) in helix 4: (black) 0.5% DPC micelles and (red) 3:1 POPC/POPG proteoliposomes (1:250 protein:lipid). The pH was 7.5, and the temperature was 296 K for all samples. EPR spectra were normalized by spin concentration to show the difference in the dynamic properties between the residues in a bilayer as compared to DPC micelles.
Figure 4.
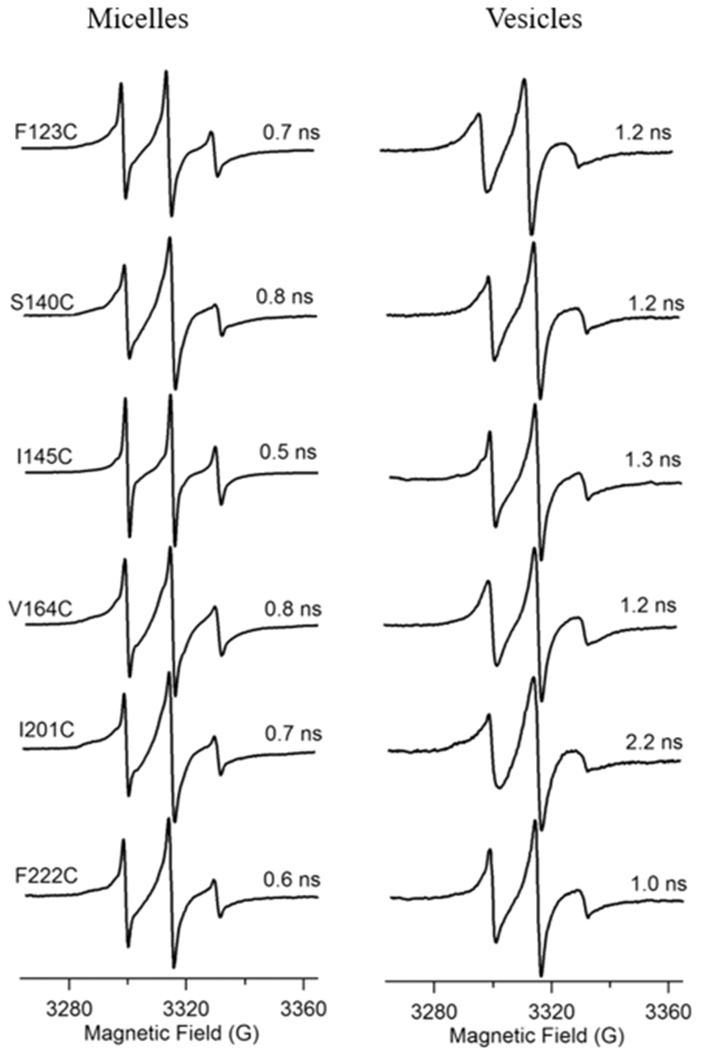
Estimated rotational correlation times (τc) of Q1-VSD (F123C, S140C, I145C, V164C, I201C, and F222C) in DPC micelles (left) and POPC/POPG vesicles (right).
Figure 6.
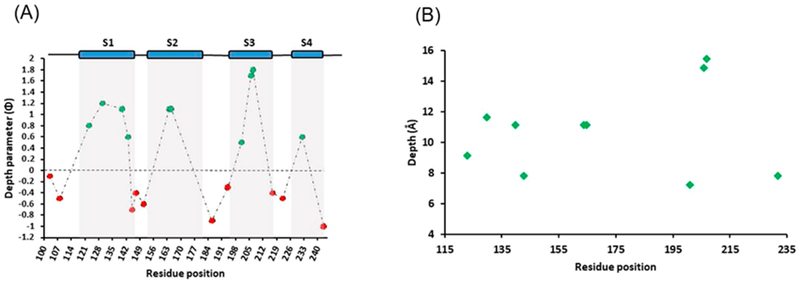
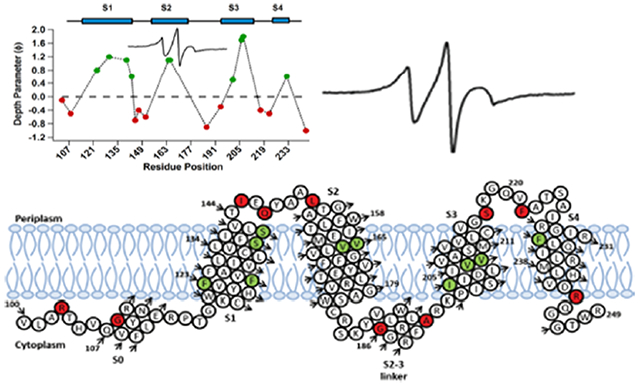
(A) Membrane depth parameter values (Φ) for different spin-labeled Q1-VSD residues in POPC/POPG vesicles. Positive Φ values (green) indicate that the residue under investigation is embedded within the membrane, and negative Φ (red) values indicate that the residue is in the aqueous environment. (B) EPR depth measurement data for the residues residing in the lipid bilayer vesicles.
Figure 4 shows the estimated motional parameter (rotational correlation time) τc values for several residues (F123C, S140C, I145C, V164C, I201C, and F222C) of Q1-VSD in detergent micelles and lipid bilayer vesicles. The τc values for Q1-VSD in detergent micelles vary between 0.5 and 0.8 ns, while the values for Q1-VSD incorporated in lipid bilayer vesicles range from 1.2 to 2.2 ns.
The overall rotational correlation time was found to be higher in lipid bilayer vesicles than in detergent micelles, suggesting an overall restricted motion of spin-labeled Q1-VSD upon incorporation into lipid bilayer vesicles.
CW-EPR data were also collected on several other mutant sites covering all four TMDs and the regions connecting S1–S4. We collected data for residues R103C and G108C around helix S0; F123C, F130C, S140C, and S143C in helix 1; V164C and V165C in helix 2; I201C, V206C, and V207C in helix 3; and F232C in helix 4. The mutants connecting these TMDs scanned for CW-EPR experiments included I145C, Q147C, L151C, G186C, A194C, S217C, F222C, and R243C. The CW-EPR data from micelles (0.5% DPC) were compared with those from lipid bilayer vesicles (Figures 3 and 4). The CW-EPR line shapes of the Q1-VSD in vesicles were found to be broader than those of the same Q1-VSD in DPC micelles.
To gain better insight into the location as well as the insertion of different spin-labeled residues of Q1-VSD in the membrane, power saturation CW-EPR experiments were conducted on Q1-VSD mutants in POPC/POPG vesicles. Representative power saturation EPR results are shown in Figure 5. POPC/POPG reconstituted Q1-VSD samples were analyzed under the conditions upon exposure to NiEDDA, N2, and air as a source of O2. The contrasting nature of NiEDDA (polar) and air (nonpolar) helps to determine the location of MTSL-labeled residues with respect to the interacting environment. Both NiEDDA and O2 act as paramagnetic relaxation agents that decrease the T1 of the unpaired electron in the nitroxide spin-label.24,28,32,33 This property can be used to probe the location of the MTSL spin-label and consequently the amino acid residue to which the spin-label is attached with respect to the membrane.
Figure 5.
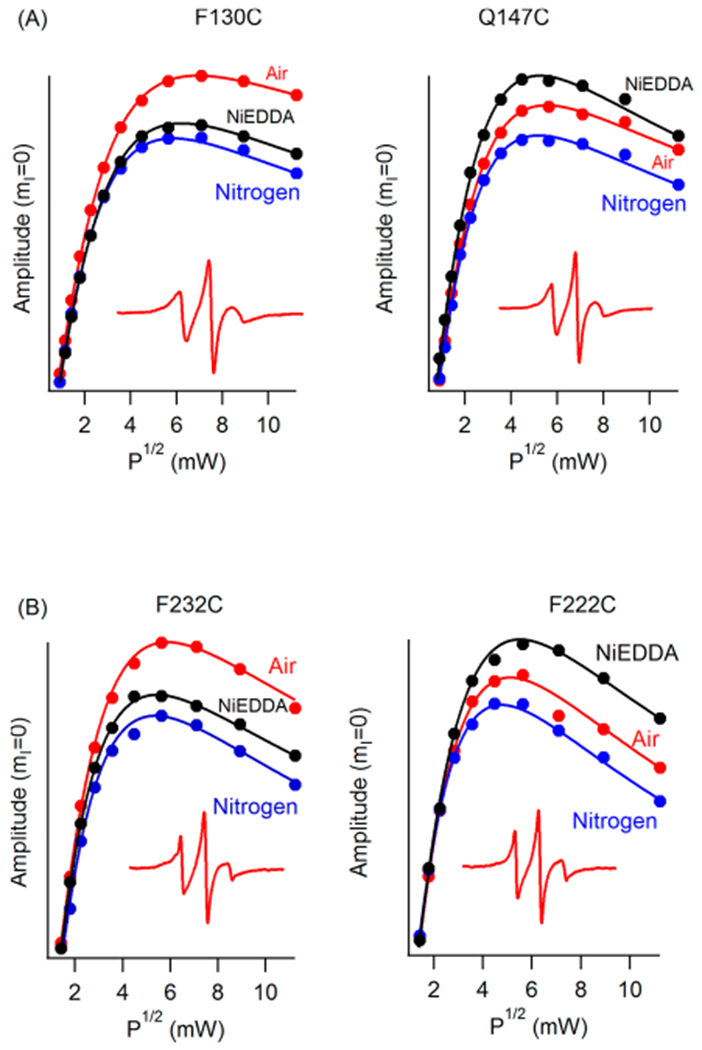
EPR power saturation curves for Q1-VSD in POPC/POPG vesicles at pH 7.5 and 296 K. (A) Mutation F130C is inside the lipid bilayer (helix 1), and mutation Q147C is outside the lipid bilayer (helix 1). (B) Mutation F232C is inside the lipid bilayer (helix 4), and mutation F222C is outside the lipid bilayer (helix 4).
Figure 5 shows that residues Q147C and F222C of Q1-VSD have greater accessibility to NiEDDA, while F130C and F232C appear to interact more with O2 in the nonpolar lipid environment. The power saturation profile of the residues with respect to nitrogen also demonstrates that both air and NiEDDA have a significant effect on the relaxation profile depending on the location of the residue with respect to the membrane when compared to nitrogen that acted as a control.
Additional CW-EPR power saturation measurements performed on mutants R103C and G108C (S0 helical region); F123C, S140C, S143C, I145C, and L151C (S1); V164C and V165C (S2); G186C and A194C (S2–S3 linker); I201C, V206C, and V207C (S3); S217C (S3–S4 loop); and R243C (S4) in each of the TMDs and the residues connecting the TMDs and the depth parameters for each were calculated to investigate their location with respect to the membrane. Figure 6A shows the graph of depth parameter (Φ) values from CW-EPR power saturation data using eq 2. The Φ values follow an increasing trend as we move from the surface toward the interior of the membrane and then decrease on the other side of the helix, indicating the correct insertion of the transmembrane segment. The negative value of Φ indicates that the residue under investigation is not interacting with the membrane and is solvent-exposed. Our data reveal that of 20 residues investigated using power saturation EPR in this study, V207C showed the highest Φ value of 1.8 (helix S3), indicating that it was exposed to the hydrophobic core and may be buried close to the center of the lipid bilayer. The residues with less positive Φ values ranging of 0.5 and 0.8 (I201C and F123C respectively) might be closer to the surface of the membrane. Mutants Q147C, G186C, A194C, F222C, and R243C showed negative Φ values (−0.3 to −1.0), indicating that these residues face the more aqueous environment outside the lipid bilayer. Overall, our data suggest that residues V206C and V207C are buried the deepest in the hydrophobic core of the lipid bilayer when compared to the other residues. Our data are consistent with the previously published studies.23,35,47–49
A calibration experiment was conducted using 5-, 7-, 10-, and 12-doxyl-labeled POPC incorporated into lipid bilayer vesicles containing WT (unlabeled) Q1-VSD. Power saturation experiments were performed under conditions similar to those used for the labeled protein samples. The Φ values obtained from calibration experiments were used to derive eq 3 from previously published depths (in angstroms) for POPC spin-labels.34 Equation 3 was then used to calculate the depth for each Q1-VSD mutant in the membrane, and the results are presented in Figure 6B. Residues in helix 1 (F123C, F130C, S140C, and S143C) showed depth values ranging from 7.8 to 11.6 Å. V164C and V165C in helix 2 had a depth value of 11.1 Å. Among the residues in helix 3, V207C showed a membrane depth of approximately 15.5 Å, indicating this mutant is buried deep in the hydrophobic core of the membrane,34 whereas I201C and V206C had values of 7.2 and 14.8 Å, respectively. The calculated depth value for F232C in helix 4 was 7.8 Å. The errors in these measurements are reported as ±2 Å.
On the basis of the data obtained from EPR measurements, we have put together a preliminary structural topology model of Q1-VSD in a lipid bilayer as shown in Figure 7. The EPR power saturation results provide insight into the location of amino acid residues spread across helices S1–S4 in the lipid bilayer environment. Helix S4 exhibits greater motional freedom than helices S1–S3 do. The smaller membrane depth parameter value for the sites in helix S4 also suggests that the helix is more flexible. This is further supported by the notion that the phenomenon of membrane thinning in the vicinity of helix S4 allows the permeability of water molecules to keep the various charged residues in this helix hydrated.8,50,51 However, when compared to other VSDs, helix S4 of Q1-VSD demonstrates an overall lower net positive charge, a feature contrasting with the charges of other voltage-gated ion channels.8–10 The motional flexibility in helix S4 is expected as it bears the charged residues acting as a voltage sensor and is involved in opening and closing the channel for voltage-dependent gating16.
Figure 7.
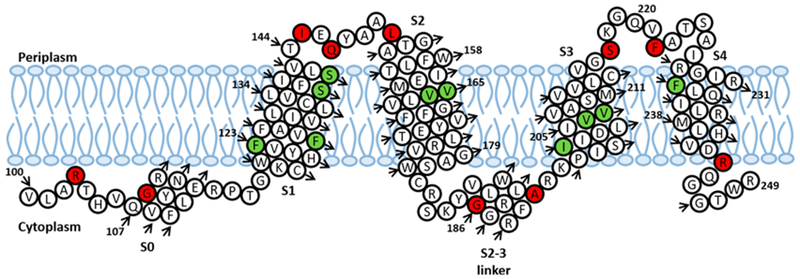
Predicted topology (residues 100–249) of the KCNQ1 voltage sensor domain (Q1-VSD) in a lipid bilayer based upon EPR studies. Green residues are buried inside the lipid bilayer, while red residues are located outside the lipid bilayer.
The findings of this study match closely with the recently published structural details of Q1-VSD.3,4,15 The range of Φ values in the S1 helical region suggests that it is significantly longer15 and has previously been described as the anchoring helix along with S2 for structural rearrangements in S4 in the case of KvAP.52 Although the S2–S3 linker is now known to be an α-helix,3 the region was found to be sensitive to most point mutations and expressed poorly for EPR studies. Similar observations were made for helix S0 where most mutants failed to express, which is consistent with previous studies.4
CONCLUSION
This study for the first time investigates the secondary structure, side chain motion, and structural topology of the human Q1-VSD protein in POPC/POPG lipid bilayer vesicles. The results are compared with those of the Q1-VSD in DPC micelles. Detailed quantitative information about the structural dynamics and topology of Q1-VSD in lipid bilayers is seminal to understanding its function during its interaction with KCNE1 during K+ channel gating. The data reported in this study will provide a better understanding of side chain motions and the locations of different segments of Q1-VSD in lipid bilayers. The findings of this work integrated with future studies will help in the structural refinement of the overall topographical orientation of Q1-VSD with respect to the membrane, laying the foundation for understanding the interaction between Q1-VSD and KCNE1 proteins in nativelike membrane environments.
Acknowledgments
Funding
This work was generously supported by National Institute of General Medical Sciences Maximizing Investigator’s Research Award (MIRA) R35 GM126935 and National Science Foundation (NSF) Grant CHE-1807131 (to G.A.L.) and by National Institutes of Health Grant RO1 HL122010 (to C.R.S.). The pulsed EPR spectrometer was purchased through funding provided by the NSF (MRI-1725502), the Ohio Board of Reagents, and Miami University. G.A.L. also acknowledges support from the John W. Steube Professorship.
Footnotes
The authors declare no competing financial interest.
REFERENCES
- (1).Jespersen T, Grunnet M, and Olesen S-P (2005) The KCNQ1 potassium channel: From gene to physiological function. Physiology 20, 408–416. [DOI] [PubMed] [Google Scholar]
- (2).Abbott GW (2014) Biology of the KCNQ1 potassium channel. New J. Sci. 2014, No. e237431. [Google Scholar]
- (3).Sun J, and MacKinnon R (2017) Cryo-EM structure of a KCNQ1/ CaM complex reveals insights into congenital long QT syndrome. Cell 169, 1042–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Huang H, Kuenze G, Smith JA, Taylor KC, Duran AM, Hadziselimovic A, Meiler J, Vanoye CG, George AL Jr., and Sanders CR (2018) Mechanisms of KCNQ1 channel dysfunction in long QT syndrome involving voltage sensor domain mutations. Sci. Adv 4, No. eaar2631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Robbins J (2001) KCNQ potassium channels: Physiology, pathophysiology, and pharmacology. Pharmacol. Ther 90, 1–19. [DOI] [PubMed] [Google Scholar]
- (6).Wang Q, Curran ME, Splawski I, Burn TC, Millholland JM, VanRaay TJ, Shen J, Timothy KW, Vincent GM, de Jager T, Schwartz PJ, Towbin JA, Moss AJ, Atkinson DL, Landes GM, Connors TD, and Keating MT (1996) Positional cloning of a novel potassium channel gene: KVLQT1 mutations cause cardiac arrythmias. Nat. Genet 12, 17–23. [DOI] [PubMed] [Google Scholar]
- (7).Hedley PL, Jorgensen P, Schlamowitz S, Wangari R, Moolman-Smook J, Brink PA, Kanters JK, Corfield VA, and Christiansen M (2009) The genetic basis of long QT and short QT syndromes: a mutation update. Hum. Mutat 30, 1486–1511. [DOI] [PubMed] [Google Scholar]
- (8).Smith JA, Vanoye CG, George AL, Meiler J, and Sanders CR (2007) Structural models for the KCNQ1 voltage-gated potassium channel. Biochemistry 46, 14141–14152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Schroeder BC, Waldegger S, Fehr S, Bleich M, Warth R, Greger R, and Jentsch TJ (2000) A constitutively open potassium channel formed by KCNQ1 and KCNE3. Nature 403, 196–199. [DOI] [PubMed] [Google Scholar]
- (10).Cui J (2016) Voltage-dependent gating: Novel insights from KCNQ1 channels. Biophys. J 110, 14–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Sanguinetti MC, Curran ME, Zou A, Shen J, Specter PS, Atkinson DL, and Keating MT (1996) Coassembly of KvLQT1 and mink (IsK) proteins to form cardiac Iks potassium channel. Nature 384, 80–83. [DOI] [PubMed] [Google Scholar]
- (12).Vanoye CG, Welch RC, Tian C, Sanders CR, and George AL Jr. (2010) KCNQ1/KCNE1 assembly, co-translation not required. Channels 4, 108–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Kapplinger JD, Tseng AS, Salisbury BA, Tester DJ, Callis TE, Alders M, Wilde AAM, and Ackerman MJ (2015) Enhancing the predictive power of mutations in the C-terminus of the KCNQ1-encoded Kv7.1 voltage-gated potassium channel. J. Cardiovasc Transl Res. 8(3) 8, 187–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Wiener R, Haitin Y, Shamgar L, Fernández-Alonso MC, Martos A, Chomsky-Hecht OC, Rivas G, Attali B, and Hirsch JA (2008) The KCNQ1 (Kv7.1) COOH terminus, a multilayered scaffold for subunit assembly and protein interaction. J. Biol. Chem 283, 5815–5830. [DOI] [PubMed] [Google Scholar]
- (15).Peng D, Kim JH, Kroncke BM, Law CL, Xia Y, Droege KD, van Horn WD, Vanoye CG, and Sanders C (2014) Purification and structural study of the voltage-sensor domain of the human KCNQ1 potassium ion channel. Biochemistry 53, 2032–2042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Chakrapani S, Cuello LG, Cortes MD, and Perozo E (2008) Structural dynamics of an isolated-voltage sensor domain in lipid bilayer. Structure 16 (3), 398–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Jiang Y, Lee A, Chen J, Ruta V, Cadene M, Chait BT, and MacKinnon R (2003) X-Ray structure of a voltage-dependent K + channel. Nature 423, 33–41. [DOI] [PubMed] [Google Scholar]
- (18).Ruta V, and Mackinnon R (2004) Localization of the voltage-sensor toxin receptor on KvAP. Biochemistry 43, 10071–10079. [DOI] [PubMed] [Google Scholar]
- (19).Lee SY, Lee A, Chen J, and MacKinnon R (2005) Structure of the KvAP voltage-dependent K+ channel and its dependence on the lipid membrane. Proc. Natl. Acad. Sci. U. S. A 102, 15441–15446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Long SB, Tao X, Campbell EB, and MacKinnon R (2007) Atomic structure of a voltage- dependent K+ channel in a lipid membrane-like environment. Nature 450, 376–382. [DOI] [PubMed] [Google Scholar]
- (21).Schmidt D, Jiang QX, and MacKinnon R (2006) Phospholipids and the origin of cationic gating charges in voltage sensors. Nature 444, 775–779. [DOI] [PubMed] [Google Scholar]
- (22).Coey AT, Sahu ID, Gunasekera TS, Troxel KR, Hawn JM, Swartz MS, Wickenheiser MR, Reid RJ, Welch RC, Vanoye CG, Kang CB, Sanders CR, and Lorigan GA (2011) Reconstitution of KCNE1 into lipid bilayers: Comparing the structural, dynamic, and activity differences in micelle and vesicle environments. Biochemistry 50, 10851–10859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Sahu ID, Craig AF, Dunagan MM, Troxel K, Zhang R, Meiberg AG, Harmon CN, McCarrick RM, Kroncke BM, Sanders CR, and Lorigan GA (2015) Probing Structural dynamics and topology of the KCNE1 membrane protein in lipid bilayers via site-directed spin labeling and electron paramagnetic resonance spectroscopy. Biochemistry 54, 6402–6412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Barrett PJ, Song Y, van Horn WD, Hustedt EJ, Schafer JM, Hadziselimovic A, Beel AJ, and Sanders CR (2012) The amyloid precursor protein has a flexible transmembrane domain and binds cholesterol. Science 336, 1168–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Yu L, Wang W, Ling S, Liu S, Xiao L, Xin Y, Lai C, Xiong Y, Zhang L, and Tian C (2015) CW-EPR studies revealed different motional properties and oligomeric states of the integrin β1a transmembrane domain in detergent micelles or liposomes. Sci. Rep 5, 7848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Alders M, Bikker H, and Christiaans I (2003) Long QT syndrome. In GeneReviews [Internet] (Adam MP, Ardinger HH, Pagon RA, et al. , Eds.) University of Washington, Seattle: https://www.ncbi.nlm.nih.gov/books/NBK1129/. [PubMed] [Google Scholar]
- (27).Altenbach C, Marti T, Khorana HG, and Hubbell WL (1990) Transmembrane protein structure: Spin labeling of bacteriorhodopsin mutants. Science 248, 1088–1092. [DOI] [PubMed] [Google Scholar]
- (28).Oh KJ, Altenbach C, Collier RJ, and Hubbell WL (2000) Site-directed spin labeling of proteins. Applications to diphtheria toxin. Methods Mol. Bio 145, 147–69. [DOI] [PubMed] [Google Scholar]
- (29).Rauch ME, Ferguson CG, Prestwich GD, and Cafiso DS (2002) Myristoylated alanine-rich C kinase substrate (MARCKS) sequesters spin-labeled phosphatidylinositol 4,5-bisphosphate in lipid bilayers. J. Biol. Chem 277, 14068–14076. [DOI] [PubMed] [Google Scholar]
- (30).Oppenheim SF, Buettner GR, and Rodgers VGJ (1996) Relationship of rotational correlation time from EPR spectroscopy and protein-membrane interaction. J. Membr. Sci 118, 133–139. [Google Scholar]
- (31).Goldman SA, Bruno GV, and Freed JH (1972) Estimating slow motional rotational correlational times for nitroxides by electron spin resonance. J. Phys. Chem 76, 1858–1860. [Google Scholar]
- (32).Altenbach C, Greenhalgh DA, Khorana HG, and Hubbell WL (1994) A collision gradient method to determine the immersion depth of nitroxides in lipid bilayers: application to spin-labeled mutants of bacteriorhodopsin. Proc. Natl. Acad. Sci. U. S. A 91, 1667–1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Hubbell WL, and Altenbach C (1994) Investigation of structure and dynamics in membrane proteins using site directed spin labeling. Curr. Opin. Struct. Biol 4, 566–573. [Google Scholar]
- (34).Dalton LA, McIntyre JO, and Fleischer S (1987) Distance estimate of the active center of D-β-hydroxybutyrate dehydrogenase from the membrane surface. Biochemistry 26, 2117–2130. [DOI] [PubMed] [Google Scholar]
- (35).Klug CS, Su W, and Feix JB (1997) Mapping the residues involved in a proposed β-strand located in the ferric enterobactin receptor FepA using site-directed spin-labeling. Biochemistry 36, 13027–13033. [DOI] [PubMed] [Google Scholar]
- (36).Findlay EH, and Booth PJ (2017) The folding, stability and function of lactose permease differ in their dependence on bilayer lipid composition. Sci. Rep 7, 13056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Whitmore L, and Wallace BA (2004) DICHROWEB, an online server for protein secondary structure analyses from circular dichroism spectroscopic data. Nucleic Acids Res. 32, W668–W673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Whitmore L, and Wallace BA (2008) Protein secondary structure analyses from circular dichroism spectroscopy: Methods and reference databases. Biopolymers 89, 392–400. [DOI] [PubMed] [Google Scholar]
- (39).Zhang R, Sahu ID, Comer RG, Maltsev S, Dabney-Smith C, and Lorigan GA (2017) Probing the interaction of the potassium channel modulating KCNE1 in lipid bilayers via solid-state NMR spectroscopy. Magn. Reson. Chem 55, 754–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Abu-Baker S, and Lorigan GA (2006) Phospholamban and its phosphorylated form interact differently with lipid bilayers: A 31P, 2H and 13C solid-state NMR spectroscopic study. Biochemistry 45, 13312–13322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Seelig J (1978) 31P Nuclear magnetic resonance and the head group structure of phospholipids in membranes. Biochim. Biophys. Acta, Rev. Biomembr. 515, 105–140. [DOI] [PubMed] [Google Scholar]
- (42).Kariev AM, and Green ME (2015) Caution is required in interpretation of mutations in the voltage sensing domain of voltage gated channels as evidence for gating mechanisms. Int. J. Mol. Sci 16, 1627–1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Park K, Perczel A, and Fasman GD (1992) Differentiation between transmembrane helices and peripheral helices by deconvolution of circular dichroism spectra of membrane proteins. Protein Sci. 1, 1032–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Mercer EAJ, Abbott GW, Brazier SP, Ramesh B, Haris PI, and Srai SKS (1997) Synthetic putative transmembrane region of minimal potassium channel protein (minK) adopts an alpha-helical conformation in phospholipid membranes. Biochem. J 325, 475–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Corbin J, Methot N, Wang HH, Baenziger JE, and Blanton MP (1998) Secondary structure analysis of individual transmembrane segments of the nicotinic acetylcholine receptor by circular dichroism and fourier transform infrared spectroscopy. J. Biol. Chem 273, 771–777. [DOI] [PubMed] [Google Scholar]
- (46).Aggeli A, Bannister ML, Bell M, Boden N, Findlay JBC, Hunter M, Knowles PF, and Yang JC (1998) Conformation and ion-channeling activity of a 27-residue peptide modeled on the single transmembrane segment of the IsK (minK) protein. Biochemistry 37, 8121–8131. [DOI] [PubMed] [Google Scholar]
- (47).Frazier AA, Wisner MA, Malmberg NJ, Victor KG, Fanucci GE, Nalefski EA, Falke JJ, and Cafiso DS (2002) Membrane orientation and position of the C2 domain from cPLA2 by site-directed spin labeling. Biochemistry 41 , 6282–6292. [DOI] [PubMed] [Google Scholar]
- (48).Fanucci GE, Cadieux N, Piedmont CA, Kadner RJ, and Cafiso DS (2002) Structure and dynamics of the beta-barrel of the membrane transporter BtuB by site directed spin-labeling. Biochemistry 41, 11543–11551. [DOI] [PubMed] [Google Scholar]
- (49).Kohout SC, Corbalan-Garcia S, Gomez-Fernandez JC, and Falke JJ (2003) C2 domain of protein kinase C alpha: elucidation of the membrane docking surface by site-directed fluorescence and spin labeling. Biochemistry 42, 1254–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Krepkiy D, Mihailescu M, Freites JA, Schow EV, Worcester DL, Gawrisch K, Tobias DJ, White SH, and Swartz KJ (2009) Structure and hydration of membranes embedded with voltage-sensing domains. Nature 462, 473–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Doherty T, Su Y, and Hong M (2010) High-resolution orientation and depth of insertion of the voltage-sensing S4 helix of a potassium channel in lipid bilayers. J. Mol. Biol 401, 642–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Li Q Wanderling S, Sompornpisut P, and Perozo E (2014) Structural basis of lipid-driven conformational transitions in the KvAP voltage-sensing domain. Nat. Struct. Mol. Biol 21, 160. [DOI] [PMC free article] [PubMed] [Google Scholar]