

Production of polyP in bacteria is required for virulence and stress response, but little is known about how bacteria regulate polyP levels in response to changes in their environments. Understanding this regulation is important for understanding how pathogenic microbes resist killing by disinfectants, antibiotics, and the immune system. In this work, I have clarified the connections between polyP regulation and the stringent response to starvation stress in Escherichia coli and demonstrated an important and previously unknown role for the transcription factor DksA in controlling polyP levels.
KEYWORDS: (p)ppGpp, DksA, inorganic polyphosphate, stringent response
ABSTRACT
Production of inorganic polyphosphate (polyP) by bacteria is triggered by a variety of different stress conditions. polyP is required for stress survival and virulence in diverse pathogenic microbes. Previous studies have hypothesized a model for regulation of polyP synthesis in which production of the stringent-response second messenger (p)ppGpp directly stimulates polyP accumulation. In this work, I have now shown that this model is incorrect, and (p)ppGpp is not required for polyP synthesis in Escherichia coli. However, stringent mutations of RNA polymerase that frequently arise spontaneously in strains defective in (p)ppGpp synthesis and null mutations of the stringent-response-associated transcription factor DksA both strongly inhibit polyP accumulation. The loss of polyP synthesis in a mutant lacking DksA was reversed by deletion of the transcription elongation factor GreA, suggesting that competition between these proteins for binding to the secondary channel of RNA polymerase plays an important role in controlling polyP activation. These results provide new insights into the poorly understood regulation of polyP synthesis in bacteria and indicate that the relationship between polyP and the stringent response is more complex than previously suspected.
IMPORTANCE Production of polyP in bacteria is required for virulence and stress response, but little is known about how bacteria regulate polyP levels in response to changes in their environments. Understanding this regulation is important for understanding how pathogenic microbes resist killing by disinfectants, antibiotics, and the immune system. In this work, I have clarified the connections between polyP regulation and the stringent response to starvation stress in Escherichia coli and demonstrated an important and previously unknown role for the transcription factor DksA in controlling polyP levels.
INTRODUCTION
Bacteria in nature are rarely found under conditions that are ideal for their growth, and they regulate their gene expression and metabolism to optimize growth and survival under a variety of nonoptimal conditions (1–5). This is known as “stress response” and is key to understanding, for example, how pathogenic bacteria resist attack by antibiotics or the immune system. The intricate regulatory mechanisms that bacteria use to sense and respond to various stresses have been the subject of intense study for many years. While different bacteria have a wide variety of transcription factors and other regulators that sense and respond to particular stressors, general stress response mechanisms, which are triggered by a broad range of conditions that adversely affect bacterial growth, are among the most widely conserved pathways across the bacterial domain.
One such highly conserved bacterial general stress response pathway, the stringent response, controls how bacteria respond to a variety of starvation stresses, and it has been studied for nearly 50 years (reviewed in references 6 to 8). When bacteria are starved for amino acids, carbon, or phosphate, they accumulate the GDP- and GTP-derived signaling molecules guanosine-5′,3′-tetraphosphate (ppGpp) and guanosine-5′,3′-pentaphosphate (pppGpp), referred to collectively as (p)ppGpp (8). Homologs of the RelA and SpoT enzymes, which are responsible for (p)ppGpp synthesis or synthesis and breakdown, respectively, are found throughout the tree of life (9). In the model gammaproteobacterium Escherichia coli, where the stringent response is best understood, RelA synthesizes (p)ppGpp in response to amino acid starvation, which it senses by recognizing uncharged tRNAs at the ribosome (10). SpoT synthesizes (p)ppGpp in response to a variety of other stresses, including osmotic shock, detergents, and carbon, phosphate, iron, or fatty acid starvation (6–8), although the mechanisms by which it senses these stresses have not yet been clearly established. Levels of (p)ppGpp also increase under other stress conditions, including heat shock, ethanol treatment, and exposure to heavy metals (11). The consequences of (p)ppGpp accumulation include genome-wide changes in gene expression, notably a drastic reduction in rRNA transcription and an increase in amino acid biosynthesis gene expression, both of which are driven by allosteric effects of (p)ppGpp on RNA polymerase (6, 12, 13). The ability of RNA polymerase to control gene expression in response to (p)ppGpp is greatly enhanced by the transcription factor DksA, which binds directly to the secondary channel of RNA polymerase, where it interacts with both (p)ppGpp and the active site of the polymerase (6, 14–16). There are also direct inhibitory or activating effects of (p)ppGpp binding on a variety of proteins other than RNA polymerase that affect translation, replication, and metabolism (17–20). The increase in (p)ppGpp under stress has the overall effect of temporarily halting growth and limiting protein translation while the bacteria adapt their physiology to allow them to grow under the new, more stressful conditions (8, 21).
Inorganic polyphosphate (polyP), a linear biopolymer composed of up to a thousand phosphoanhydride bond-linked phosphate monomers, is the key component of another, much less well understood general stress response pathway in bacteria (22–26). Bacteria from diverse phyla synthesize polyP in response to multiple stressors, including amino acid starvation, oxidative stress, heat shock, salt stress, and heavy metal exposure, and mutants lacking the ppk gene, which encodes the polyP-synthesizing kinase PPK, are highly sensitive to these and other stresses (25, 27–30). The mechanisms by which polyP mediates bacterial stress resistance are not completely characterized, but in E. coli, polyP is known to act as a protein-stabilizing chaperone, as a metal chelator, and as a regulator of RNA polymerase and DNA polymerase IV activity, ribosomal translation fidelity, and transcription, including that of rpoS, which encodes the general stress response sigma factor σS (3, 25, 27, 28, 31–37). Surprisingly, however, very little is currently known about the molecular details of how polyP accumulation is itself regulated in response to different stress conditions.
There are several pieces of information that suggest a link between the stringent response and polyP production, and these have led to the wide acceptance of a simple model for polyP regulation that proposes that (p)ppGpp stimulates polyP accumulation (7, 22, 38–43). First, polyP and (p)ppGpp accumulate concurrently during a variety of different stresses in multiple species of bacteria (43–46). Second, ppk and relA spoT mutants of E. coli, which are unable to synthesize either polyP or (p)ppGpp, respectively, have similar pleiotropic phenotypes, including sensitivity to amino acid starvation and osmotic shock, poor survival in long-term stationary phase, and loss of virulence and biofilm formation (7, 8, 22, 47, 48). Third, (p)ppGpp is an efficient inhibitor of the polyP-degrading exopolyphosphatase PPX (encoded by the ppx gene) (41). Finally, there have been reports that mutants lacking relA and spoT have defects in polyP synthesis (38, 43). This model cannot, however, explain the phenotype of ppx mutants, which retain stress regulation of polyP (28, 49), nor can it account for reports that polyP accumulation in response to stresses other than amino acid starvation does not depend on RelA and SpoT but may require other regulators, including transcription factors like σS, the phosphate starvation sensor PhoB, or the nitrogen starvation sensor GlnG (38, 43). The ppk and ppx genes of E. coli are cotranscribed (50). The transcription of the ppk-ppx operon is not regulated by PhoB, GlnG, or σS (51–55) and does not change in response to stresses that stimulate polyP production (13, 28, 49, 56, 57).
In this work, I set out to explore the relationship between the stringent response and polyP accumulation in more detail. I found that, in contrast to the generally accepted model, production of (p)ppGpp was not required for polyP synthesis in E. coli but that suppressor mutations in RNA polymerase that frequently arise in relA spoT mutant strains can have dramatic inhibitory effects on polyP accumulation. Unusually, while (p)ppGpp had no influence on polyP levels, I found that DksA and other RNA polymerase secondary channel binding proteins did have strong effects on polyP synthesis. These effects were not correlated with changes in the expression of ppk itself, and they suggest a more complicated relationship between polyP and the stringent response than has been previously suspected.
RESULTS
(p)ppGpp is neither necessary nor sufficient to activate polyP synthesis in E. coli.
polyP accumulation can be stimulated in E. coli growing in rich medium (LB) by shifting them to minimal medium lacking amino acids (a nutrient limitation stress) (38, 43, 49). As expected (49, 58, 59), after nutrient limitation a Δppk mutant made no polyP, and a Δppx mutant made slightly more than the wild type (Fig. 1A). As we have previously observed (49), the fact that polyP levels are still stress regulated in a Δppx mutant indicates that regulation of PPX activity cannot be the primary means by which polyP accumulation is controlled. I was very surprised to find, however, that ΔrelA, ΔrelA spoT::cat+, and Δppx ΔrelA spoT::cat+ mutants made at least as much polyP as the wild-type strain (Fig. 1A). (It was not possible to examine a null mutation of spoT in a relA+ background because this genetic combination is lethal [48].) This contradicted the common model that (p)ppGpp is the primary signal leading to polyP accumulation in stressed bacteria and previous reports that relA spoT mutants of E. coli cannot synthesize polyP (7, 22, 38–43). I therefore did a series of experiments to validate these results.
FIG 1.

(p)ppGpp is neither necessary nor sufficient to activate polyP synthesis in E. coli. (A to C) E. coli MG1655 wild-type and isogenic Δppk-749, Δppx-750, ΔrelA782, ΔrelA782 spoT207::cat+, and Δppx-750 ΔrelA782 spoT207::cat+ strains (A), MG1655-derived strains RNAP(1+2+) (RLG14535; rpoZ+-kan+ rpoC+-tetAR+) and RNAP(1−2−) [(p)ppGpp-insensitive mutant RLG14538]; rpoZΔ2-5-kan+ rpoC R362A R417A K615A N680A K681A-tetAR+ (B), or E. coli Nissle 1917 wild-type and isogenic ΔrelA1917 spoT1000::kan+ mutant strains (C) were grown at 37°C to an A600 of 0.2 to 0.4 in rich medium (LB) (black circles) and then shifted to minimal medium (MOPS with no amino acids, 4 g liter−1 glucose, and 0.1 mM K2HPO4) for 2 h (white circles) (n = 3 or 4; means ± standard deviations [SD] are shown). polyP concentrations are in terms of individual phosphate monomers. Asterisks indicate polyP levels significantly different from those of the wild-type control for a given experiment (two-way repeated-measures ANOVA with Holm-Sidak’s multiple-comparison test; *, P < 0.05). (D) E. coli MG1655 containing plasmid pRelA′-His or pRelA′-His(D275G) was inoculated into LB containing ampicillin and incubated at 37°C with shaking. IPTG (isopropyl-β-d-thiogalactopyranoside) (100 µM) was added at the time indicated by the dashed lines (A600 of 0.3). Culture density (A600, closed symbols) and polyP concentrations (open symbols) were measured at the indicated times (n = 3; means ± SD are shown).
First, I tested polyP accumulation in an E. coli RNAP(1−2−) strain, which has mutations eliminating both (p)ppGpp-binding sites in RNA polymerase (60), and in its isogenic RNAP(1+2+) parent strain. The RNAP(1−2−) strain makes (p)ppGpp but cannot regulate transcription in response to its production. I observed no difference in polyP accumulation between the RNAP(1+2+) and RNAP(1−2−) strains (Fig. 1B), indicating that (p)ppGpp-dependent transcriptional regulation was not necessary for stress regulation of polyP levels. Second, to confirm that polyP synthesis by the ΔrelA spoT::cat+ mutant was not specific to the E. coli K-12 MG1655 strain background I used (61–63), I constructed a ΔrelA spoT::kan+ mutant of the distantly related E. coli strain Nissle 1917 (64). This mutant also had no defect in polyP production (Fig. 1C). Finally, to directly test whether (p)ppGpp production stimulated polyP synthesis, I grew wild-type E. coli MG1655 to mid-log phase in LB medium and induced expression of a plasmid-borne active fragment of RelA (RelA′) (20, 65) to artificially stimulate (p)ppGpp production under nonstress conditions. Production of (p)ppGpp slowed growth, as expected (21), but did not stimulate polyP accumulation (Fig. 1D). Expression of a catalytically inactive RelA′(D275G) variant (66) had no effect on growth and also did not lead to polyP accumulation. These results showed that, under the conditions of these experiments, (p)ppGpp does not play a role in stimulating polyP accumulation.
Stringent alleles of RNA polymerase can reduce polyP synthesis.
To resolve this apparent contradiction with previously published results, I analyzed a strain descended from the CF1693 relA spoT mutant (48, 67) that was used in the original experiments testing the role of (p)ppGpp in polyP production (41, 43). As shown in Fig. 2A, this strain, which I refer to as CF1693(M+) for reasons described below, did have a very significant defect in polyP synthesis, indicating that there was some important difference between this strain and the other relA spoT mutants I had constructed (Fig. 1).
FIG 2.
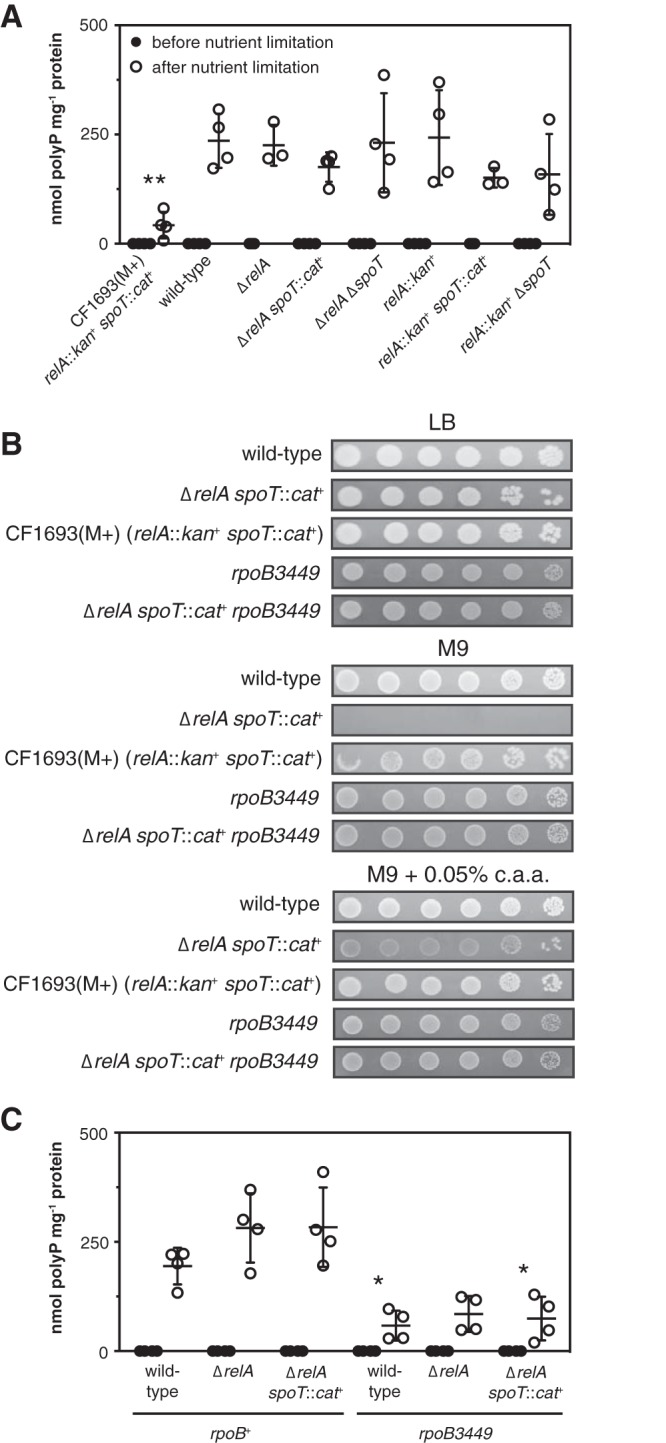
Stringent alleles of RNA polymerase can reduce polyP synthesis. (A) E. coli CF1693(M+) (ϕ80+ λ+ relA251::kan+ spoT205::cat+ rpoB1693 rpoD1693), wild-type MG1655, and isogenic MG1655 ΔrelA782, ΔrelA782 spoT207::cat+, ΔrelA782 ΔspoT1000, relA251::kan+, relA251::kan+ spoT205::cat+, and relA251::kan+ ΔspoT1000 strains were grown to an A600 of 0.2 to 0.4 in LB and then shifted to minimal medium for 2 h (n = 3 or 4; means ± SD are shown). (B) Overnight cultures of E. coli MG1655, CF1693(M+), and MG1655 isogenic ΔrelA782 spoT207::cat+, rpoB3449, and ΔrelA782 spoT207::cat+ rpoB3449 mutant strains were resuspended to an A600 of 1 in PBS, serially diluted and spotted on LB, M9 glucose, or M9 glucose plates containing 0.5 g liter−1 Casamino Acids (c.a.a.), and incubated at 37°C. The results shown are representative of at least 3 independent experiments. (C) E. coli MG1655 wild-type and isogenic ΔrelA782, ΔrelA782 spoT207::cat+, rpoB3449, ΔrelA782 rpoB3449, and ΔrelA782 spoT207::cat+ rpoB3449 strains were grown at 37°C to an A600 of 0.2 to 0.4 in rich medium (LB) (black circles) and then shifted to minimal medium (MOPS with no amino acids, 4 g liter−1 glucose, and 0.1 mM K2HPO4) for 2 h (white circles) (n = 4; means ± SD are shown). polyP concentrations are in terms of individual phosphate monomers. Asterisks indicate polyP levels significantly different from those of the wild type (rpoB+) for a given experiment (two-way repeated-measures ANOVA with Holm-Sidak’s multiple-comparison test; *, P < 0.05; **, P < 0.01).
First I tested whether differences in relA and spoT mutant alleles affected polyP accumulation. All of the alleles of these genes tested in this work are described in Table 1. CF1693 contains the relA251::kan+ and spoT207::cat+ alleles, which are insertions of antibiotic resistance cassettes associated with partial deletions of the relA and spoT genes and, in the case of relA251::kan+, also a partial deletion of the upstream rumA gene (48, 68). I made all of the possible relA and relA spoT mutants containing combinations of relA251::kan+, spoT207::cat+, and clean in-frame deletions of relA and spoT derived from the Keio gene knockout collection (28) and tested their ability to synthesize polyP after nutrient limitation. None of the freshly constructed relA or relA spoT mutants had a significant defect in polyP synthesis (Fig. 2A), indicating that the defect in CF1693(M+) was not due to differences in the nature of the relA and spoT null mutations.
TABLE 1.
Alleles of relA and spoT used in this study
| Allele | Structure of mutationa | Reference |
|---|---|---|
| relA251::kan+ | Δ−71–2023, with pUC-4K-derived Knr insertion | 68 |
| relA782::kan+ | Δ4–2214, with pKD4-derived Knr insertion | 103 |
| ΔrelA782 | Δ4–2214, with FRT scar sequence | 103 |
| relA1917::kan+ | Δ4–2232, with pKD4-derived Knr insertion | This work |
| ΔrelA1917 | Δ4–2232, with FRT scar sequence | This work |
| spoT251::cat+ | Δ407–2062, with pBR327-derived Cmr insertion | 48 |
| spoT1000::kan+ | Δ4–2106, with pKD4-derived Knr insertion | This work |
| ΔspoT1000 | Δ4–2106, with FRT scar sequence | This work |
Numbers indicate which base pairs of the coding domain sequence of the indicated gene have been deleted.
I received CF1693(M+) as a gift from Kenn Gerdes (University of Copenhagen), who recently reported that many strains distributed from his laboratory, including this one, are infected with lysogenic bacteriophages, including λ and ϕ80 (69). I confirmed that CF1693(M+) was lysogenic for both λ and ϕ80 (see Fig. S1A and B in the supplemental material) but found no defect in polyP synthesis in wild-type MG1655 infected either with ϕ80 alone or with both λ and ϕ80 (Fig. S1C, D, and E), indicating that phage infection did not affect polyP synthesis. I checked all of the strains used in this work for phage contamination, and none of the strains used for any other experiment contained either λ or ϕ80.
One of the best-known phenotypes of relA spoT mutants is their complex amino acid requirement, which is due to their inability to induce expression of amino acid synthesis genes upon amino acid limitation (6, 12, 13, 48). As shown in Fig. 2B, wild-type E. coli grows well on glucose minimal medium in the absence of amino acids, but a freshly constructed ΔrelA spoT::cat+ mutant does not grow at all. There is, however, a very common class of revertants that arise in relA spoT strains and allow these strains to grow on minimal media. These are point mutations in the β, β′, or σ70 subunit of RNA polymerase (encoded by rpoB, rpoC, and rpoD, respectively), encoded by alleles called stringent alleles, that are thought to mimic the effect of (p)ppGpp binding to RNA polymerase (14, 70–73). CF1693(M+) grew robustly in the absence of amino acids (Fig. 2B). This is what is indicated by the (M+) designation (73), and its growth strongly suggested that this strain contains a stringent mutation of RNA polymerase. A ΔrelA spoT::cat+ mutant containing the well-characterized rpoB3449 stringent allele (a deletion of RpoB alanine 532) (71, 74–76) also grew well without amino acid supplementation under these conditions (Fig. 2B).
To identify the stringent mutation(s) present in CF1693(M+), I sequenced its rpoB, rpoC, and rpoD genes and found point mutations in both rpoB and rpoD. The rpoB1693 mutation is a C3387A transversion, encoding an RpoB N1129K variant protein, and the rpoD1693 mutation is an A191G transition, encoding an RpoD D64G variant protein. As far as I can tell, neither of these mutations has been previously reported as a stringent allele (14, 70, 72–74). I transduced CF1693(M+) to either rpoB+ or rpoD+ using linked markers (77, 78) and found that the rpoD1693 mutation was primarily responsible for the amino acid prototrophy of CF1693(M+) but that the single rpoD1693 mutant did not grow as well without amino acids as the rpoD1693 rpoB1693 double mutant (see Fig. S2 in the supplemental material).
Strikingly, like CF1693(M+), rpoB3449 stringent mutants had substantial defects in polyP accumulation, regardless of the presence of relA and spoT (Fig. 2C). This showed that different, independently isolated M+ stringent mutations of RNA polymerase could have similar inhibitory effects on polyP synthesis. Given the very high frequency at which stringent mutations arise in relA spoT strains (1 in 105, with as many as a third of overnight cultures containing substantial proportions of M+ revertants [21]), the unsuspected accumulation of RNA polymerase stringent alleles in relA spoT stock cultures probably explains previous reports that (p)ppGpp is necessary for polyP synthesis. Since these are mutations of RNA polymerase, it also strongly suggested that transcription plays an important role in regulation of polyP synthesis, despite reports that the ppk-ppx operon itself is not transcriptionally regulated in E. coli (49, 56, 57).
Transcription is necessary for induction of polyP synthesis, but ppk gene expression does not correlate with polyP accumulation.
Supporting this conclusion, polyP accumulation was strongly inhibited in wild-type E. coli when transcription was blocked with rifampin (Fig. 3A). As noted above, three transcription factors have been reported to play roles in polyP accumulation under different stress conditions in E. coli: σS (also known as RpoS), GlnG (also known as NtrC), and PhoB (38). Neither ΔrpoS nor ΔglnG mutations had any significant effect on polyP accumulation under the stress conditions tested here (Fig. 3B), and while the phosphate transport gene activator PhoB (51, 79) is required for polyP accumulation (43, 49), possibly due to defects in phosphate transport in phoB mutants (80), the nutrient limitation conditions used in these experiments did not activate the PhoB regulon (see Fig. S3 in the supplemental material), suggesting that regulation by PhoB was unlikely to be a major contributor to activation of polyP synthesis under these conditions.
FIG 3.

Transcription is necessary for induction of polyP synthesis, but ppk gene expression does not correlate with polyP accumulation. (A) E. coli strain MG1655 was grown at 37°C to an A600 of 0.2 to 0.4 in rich medium (LB) (black circles) and then shifted to minimal medium (MOPS with no amino acids, 4 g liter−1 glucose, and 0.1 mM K2HPO4) with or without rifampin (Rif) (50 µg ml−1) for 2 h (white circles) (n = 3; means ± SD are shown). (B) ΔrpoS746 and ΔglnG730 strains were grown at 37°C to an A600 of 0.2 to 0.4 in rich medium (LB) (black circles) and then shifted to minimal medium (MOPS with no amino acids, 4 g liter−1 glucose, and 0.1 mM K2HPO4) for 2 h (white circles) (n = 3 or 4; means ± SD are shown). (C to E) E. coli MG1655 wild-type and isogenic ΔrelA782 spoT207::cat+, dksA761::kan+, ΔrelA782 spoT207::cat+ dksA761::kan+, greA::cat+, and dksA761::kan+ greA::cat+ strains (C and D) or an rpoB3449 mutant containing plasmid pGUSA5 (Pppk-uidA+) (E) were grown at 37°C to an A600 of 0.2 to 0.4 in rich medium (LB) (black symbols) and then shifted to minimal medium (MOPS with no amino acids, 4 g liter−1 glucose, and 0.1 mM K2HPO4) containing 0.1 mM uracil for 2 h (white symbols). polyP concentrations are in terms of individual phosphate monomers. (D and E) Expression from the Pppk promoter before and after nutrient limitation was determined using β-glucuronidase (GUS) activity assays (n = 3 or 4; means ± SD are shown). Dashed lines indicate mean expression before nutrient limitation in the wild-type strain. Asterisks indicate polyP or Pppk expression levels significantly different from those of the wild type for a given experiment (two-way repeated-measures ANOVA with Holm-Sidak’s multiple-comparison test; *, P < 0.05; **, P < 0.01; ***, P < 0.001). (For panel B, the wild type for comparison is MG1655 from panel A without rifampin treatment.)
polyP synthesis is regulated by DksA.
The RNA polymerase secondary channel binding transcriptional regulator DksA normally works in concert with (p)ppGpp to regulate expression of genes during the stringent response (6, 16). In most cases reported previously, relA spoT and dksA mutants have very similar phenotypes, due to the cooperative effects of (p)ppGpp and DksA on RNA polymerase activity (6, 81, 82). Surprisingly, unlike relA spoT mutants, both dksA and dksA relA spoT mutants were extremely defective in polyP accumulation (Fig. 3C), indicating that (p)ppGpp and DksA play very different roles in controlling polyP levels. Because dksA mutants of MG1655 are uracil auxotrophs (21, 83), I included uracil in the nutrient limitation medium used in these and all subsequent experiments.
There are a few reports in the literature of genes that are differentially regulated by (p)ppGpp and DksA in E. coli (81, 82, 84). For some of these genes (e.g., fimB and fliC), deletion of GreA, a transcription elongation factor that also binds in the secondary channel of RNA polymerase (85–87), is able to suppress the effects of a DksA knockout (82, 84). I therefore tested the effect of knocking out greA on polyP synthesis. The greA knockout had no significant effect on polyP synthesis on its own, but knocking out greA in a dksA mutant restored polyP synthesis to wild-type levels (Fig. 3C), suggesting that the interplay between transcription factors in the secondary channel (74, 88) is critical for polyP regulation.
ppk expression does not correlate with polyP production.
I used a translational fusion between the ppk promoter and β-glucuronidase (49) as a reporter to measure ppk gene expression in relA spoT, dksA, and greA mutants (Fig. 3D). In these experiments, both wild-type and greA mutant E. coli showed a slight, but not statistically significant (multiple t test with Holm-Sidak’s multiple-comparison correction, P > 0.05), increase in ppk expression after stress. Mutants lacking either relA spoT or dksA had significantly lower ppk expression both before and after stress (roughly half the expression seen in the wild-type strain). Deletion of greA did not restore ppk expression to wild-type levels in the dksA mutant. Nutrient limitation did lead to a 2.4-fold increase in ppk expression after stress in an rpoB3449 mutant (Fig. 3E). While these results showed that (p)ppGpp, DksA, and stringent alleles of RNA polymerase do affect ppk gene expression in various ways, the most important conclusion I drew from the results of this experiment was that there was no correlation between ppk gene expression (Fig. 3D and E) and polyP accumulation (Fig. 3C and 2C) and therefore that transcription of a gene or genes other than ppk is probably responsible for the transcription- and DksA-dependent activation of polyP synthesis after stress (Fig. 3A and C).
The DksA dependence of polyP synthesis is relieved by deletion of greA but not by that of greB.
To confirm the importance of secondary channel binding transcription factors in polyP regulation, I first complemented a dksA mutant strain with dksA expressed from a plasmid, which restored polyP synthesis (Fig. 4C), confirming that the loss of DksA was responsible for the observed loss of polyP. Next, I constructed dksA mutants of the (p)ppGpp-insensitive RNAP(1−2−) strain and its isogenic RNAP(1+2+) parent (60) and observed that knocking out dksA eliminated polyP synthesis in these strains as well (Fig. 4B). This reinforced the conclusion that the role of DksA in polyP regulation is independent of its interaction with (p)ppGpp in the secondary channel of RNA polymerase. Finally, I constructed all of the possible single, double, and triple combinations of dksA, greA, and greB mutations and tested the ability of these mutant strains to synthesize polyP after stress (Fig. 4C). GreB is a transcription elongation factor closely related to GreA (87, 89). Only dksA and dksA greB mutants accumulated significantly less polyP than the wild type, indicating that greB is not involved in polyP regulation. A dksA greA mutant made as much polyP as the wild type, confirming that eliminating greA restores polyP synthesis in a dksA mutant. The dksA greA greB triple mutant, which grew very poorly, made on average substantially less polyP than the wild type, but the difference was not statistically significant in this experiment (two-way repeated measures analysis of variance [ANOVA] with Holm-Sidak’s multiple-comparison test, P > 0.05).
FIG 4.
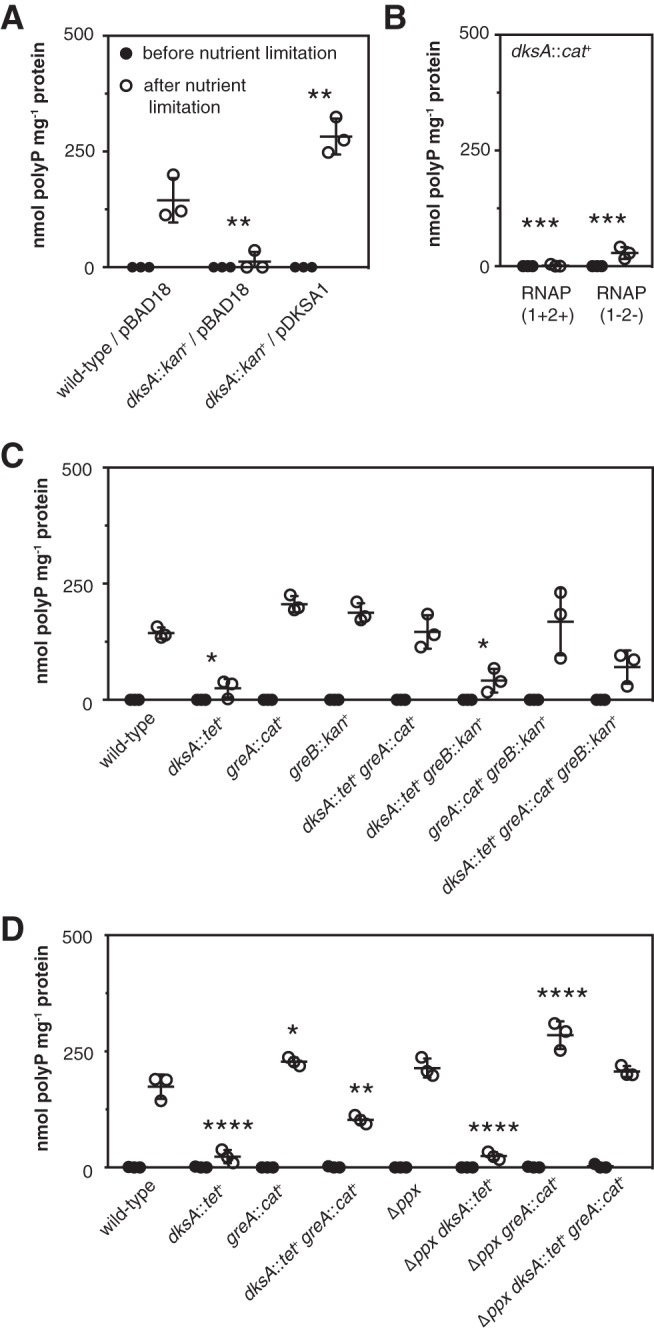
The DksA dependence of polyP synthesis is relieved by deletion of greA but not by that of greB. (A) MG1655 wild-type and dksA761::kan+ strains containing empty vector pBAD18 or the pBAD18-derived dksA+ plasmid pDKSA1 were grown at 37°C to an A600 of 0.2 to 0.4 in rich medium (LB) containing ampicillin and 2 g liter−1 arabinose (black circles) and then shifted to minimal medium (MOPS with no amino acids, 4 g liter−1 glucose, and 0.1 mM K2HPO4) containing 0.1 mM uracil and 2 g liter−1 arabinose for 2 h (white circles) (n = 3; means ± SD are shown). (B) MG1655-derived strains RNAP(1+2+) (rpoZ+-kan+ rpoC+-tetAR+) dksA1000::cat+ and RNAP(1−2−) [(p)ppGpp-insensitive mutant; rpoZΔ2-5-kan+ rpoC R362A R417A K615A N680A K681A-tetAR+] dksA1000::cat+ were grown at 37°C to an A600 of 0.2 to 0.4 in rich medium (LB) (black circles) and then shifted to minimal medium (MOPS with no amino acids, 4 g liter−1 glucose, and 0.1 mM K2HPO4) containing 0.1 mM uracil for 2 h (white circles) (n = 3; means ± SD are shown). (C and D) E. coli MG1655 wild-type and isogenic dksA::tet+, greA::cat+, greB::kan+, dksA::tet+ greA::cat+, dksA::tet+ greB::kan+, greA::cat+ greB::kan+, dksA::tet+ greA::cat+ greB::kan+, Δppx-750, Δppx-750 dksA::tet+, Δppx-750 greA::cat+, and Δppx-750 dksA::tet+ greA::cat+ strains were grown at 37°C to an A600 of 0.2 to 0.4 in rich medium (LB) (black circles) and then shifted to minimal medium (MOPS with no amino acids, 4 g liter−1 glucose, and 0.1 mM K2HPO4) containing 0.1 mM uracil for 2 h (white circles) (n = 3; means ± SD are shown). polyP concentrations are in terms of individual phosphate monomers. Asterisks indicate polyP levels significantly different from those of the wild type for a given experiment (two-way repeated-measures ANOVA with Holm-Sidak’s multiple-comparison test; *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.001). [For panel B, the wild type for comparison is RNAP(1+2+) from Fig. 1B.]
Since both ppx and greA mutants produced somewhat more polyP than the wild type (Fig. 1A, 3C, and 4C), I tested polyP accumulation in strains lacking both genes to see whether these small effects might be additive (Fig. 4D). In this experiment, the amount of polyP accumulated by a ppx greA mutant was significantly larger than that seen in the wild type, and deletion of ppx also increased the amount of polyP accumulated by a dksA greA mutant. A ppx dksA mutant, like the dksA single mutant, accumulated very little polyP.
Effects of relA, spoT, ppx, and dksA mutations on polyP accumulation over time.
PolyP accumulation was measured 2 h after nutrient limitation in all of the experiments described above. To test whether the differences in polyP levels I observed in those experiments were due to changes in the kinetics of polyP synthesis, I measured polyP levels in various mutant strains at 1-h intervals for 4 h after nutrient limitation (Fig. 5). In wild-type and ΔrelA spoT::cat+ strains, polyP accumulation peaked at 3 h, followed by a slight decrease at hour 4 (Fig. 5A). This correlated with the resumption of growth by the wild-type strain between 3 and 4 h after nutrient limitation (data not shown). As expected, mutants lacking relA, spoT, or dksA did not grow in minimal medium. A ΔrelA mutant accumulated polyP at the same rate as the wild type but continued accumulating even more at the last time point (Fig. 5A), consistent with a previous report of excess polyP accumulation in relA mutants at later time points after nutrient starvation stress (43). Deletion of ppx generally resulted in slightly higher average polyP levels at all time points in wild-type, ΔrelA, and ΔrelA spoT::cat+ backgrounds, with the Δppx ΔrelA strain containing the largest amounts of polyP (Fig. 5B). These differences were, however, relatively modest in this set of experiments (compared to the difference in polyP accumulation after 2 h between the wild-type and Δppx strains in Fig. 1A, for example). Deletion of dksA in both wild-type and ΔrelA spoT::cat+ backgrounds resulted in a strong defect in polyP synthesis at all time points (Fig. 5C). These results showed that the changes in polyP levels observed in previous experiments with dksA mutants were not due to changes in the timing of polyP synthesis but rather were due to changes in the absolute amount of polyP accumulating in stressed cells.
FIG 5.
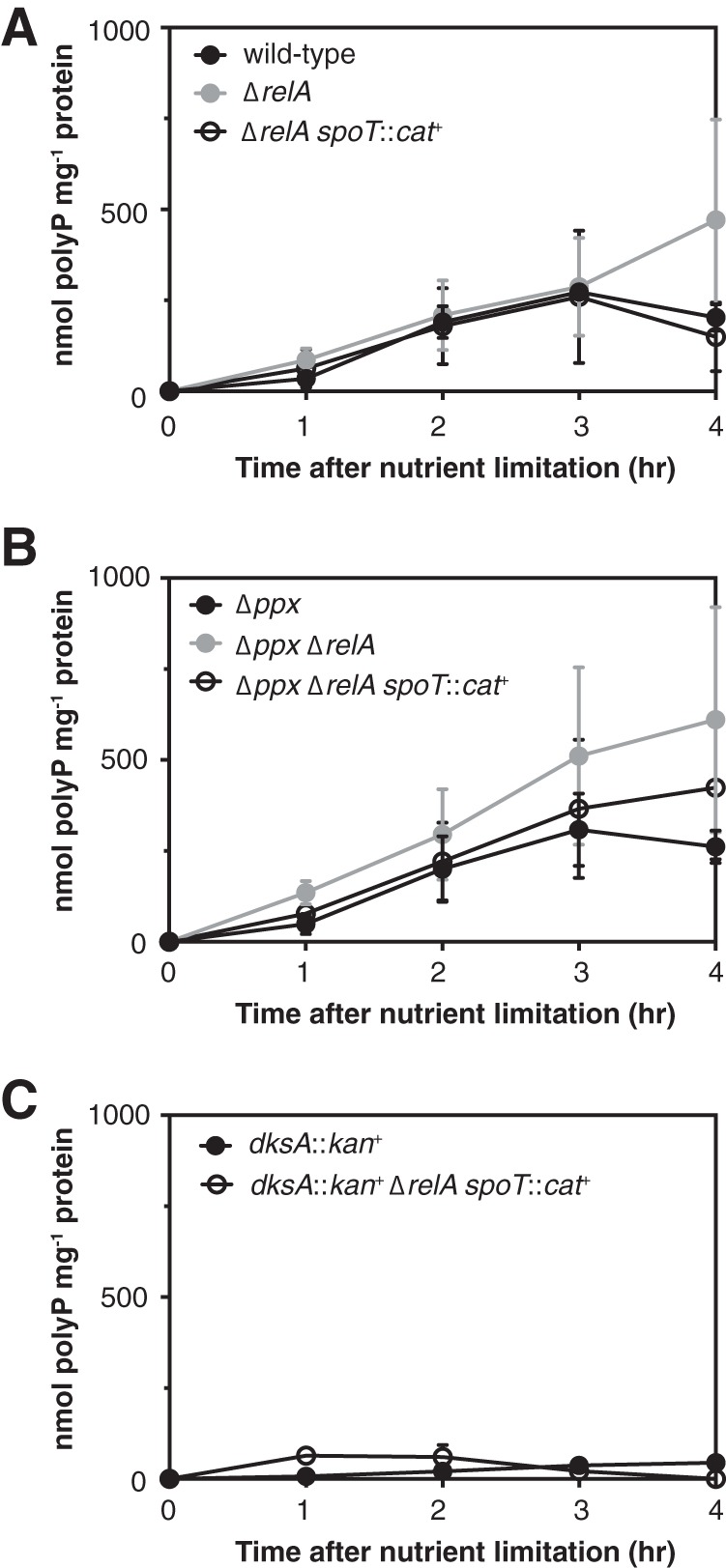
Effects of relA, spoT, ppx, and dksA mutations on polyP accumulation over time. E. coli strain MG1655 and isogenic ΔrelA782, ΔrelA782 spoT207::cat+ strains (A), Δppx-750, Δppx-750 ΔrelA782, and Δppx-750 ΔrelA782 spoT207::cat+ strains (B), and dksA761::kan+ and dksA761::kan+ ΔrelA782 spoT207::cat+ strains (C) were grown at 37°C to an A600 of 0.2 to 0.4 in rich medium (LB) and then shifted to minimal medium (MOPS with no amino acids, 4 g liter−1 glucose, and 0.1 mM K2HPO4) containing 0.1 mM uracil for the indicated times (n = 3 or 4; means ± SD are shown). polyP concentrations are in terms of individual phosphate monomers. The experiments shown in panels A, B, and C were done concurrently, and the data were divided into three panels to improve legibility.
Deletion of dksA reduces but does not eliminate the overproduction of polyP in a ppk* mutant.
We recently identified a set of point mutations in ppk that dramatically increase polyP accumulation in E. coli, although we do not yet know the mechanism by which they do so (49). To test whether polyP synthesis in these mutants (designated ppk* mutants) is affected by the transcriptional regulation identified here, I measured polyP accumulation before and after nutrient limitation in Δppk and Δppk dksA::tet+ strains containing low-copy-number plasmids encoding either wild-type PPK or the hyperactive PPKE245K variant under the control of the native ppk promoter (Fig. 6). Deletion of dksA significantly reduced the amount of polyP produced in the ppk* strain but did not reduce it to the levels seen with wild-type ppk. In strains expressing PPKE245K, nutrient limitation induced a 4.4-fold increase in polyP levels in the presence of dksA and a similar 5.6-fold increase in the dksA knockout strain. Since no polyP is produced by strains encoding wild-type PPK in the absence of stress, it was not possible to calculate the fold change in those strains. These results suggested that ppk* and dksA regulate polyP accumulation by independent mechanisms.
FIG 6.
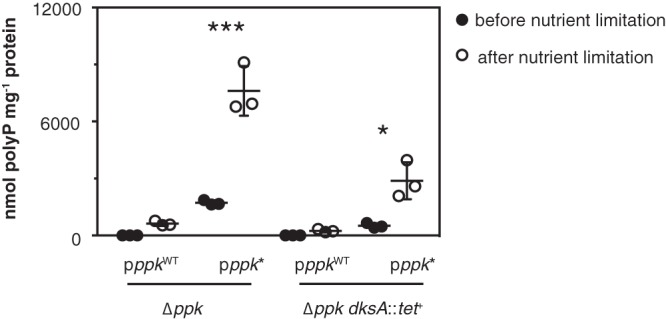
Deletion of dksA reduces but does not eliminate the overproduction of polyP by a ppk* mutant. E. coli Δppk-749 and Δppk-749 dksA::tet+ strains, containing low-copy-number plasmids encoding wild-type PPK (pppkWT; pPPK10) or hyperactive PPKE245K (pppk*; pPPK10b), were grown at 37°C to an A600 of 0.2 to 0.4 in rich medium (LB) containing kanamycin and 1 mM MgCl2 (black circles) and then shifted to minimal medium (MOPS with no amino acids, 4 g liter−1 glucose, and 0.1 mM K2HPO4) containing kanamycin and 0.1 mM uracil for 2 h (white circles) (n = 3; means ± SD are shown). polyP concentrations are in terms of individual phosphate monomers. Asterisks indicate polyP levels significantly different from those of Δppk/pppkWT (two-way repeated-measures ANOVA with Holm-Sidak’s multiple-comparison test; *, P < 0.05; ***, P < 0.001).
DISCUSSION
The most important result of this study is that (p)ppGpp is not required for polyP synthesis in E. coli, overturning a model that has been widely accepted for more than 20 years (7, 22, 38–43). The discovery that stringent mutations of RNA polymerase can dramatically reduce polyP accumulation (Fig. 2) almost certainly explains previous incorrect reports that relA spoT mutants of E. coli are defective in polyP synthesis (38, 43). I doubt that the specific mutations present in the CF1693(M+) strain used here were present in the strains used in those studies, but stringent alleles arise very commonly in relA spoT strains (21, 70). In other bacterial species, including Caulobacter crescentus and Pseudomonas aeruginosa, mutants lacking (p)ppGpp are reported to make wild-type amounts of polyP (90, 91), supporting the conclusion that, in general, (p)ppGpp production is not necessary for polyP production in bacteria. The physiological relevance of the significant activation of ppk expression by nutrient limitation in an rpoB3449 mutant (Fig. 3E) but not in the wild-type strain (Fig. 3D) is uncertain. However, my results certainly highlight the need to be extremely careful when working with relA spoT mutants of E. coli (21).
Another striking result from these studies is the fact that polyP synthesis is nearly entirely lost in a dksA mutant regardless of the presence or absence of (p)ppGpp (Fig. 3C, 4, and 5C). For most genes in E. coli, (p)ppGpp and DksA exert concerted regulatory effects, either positive or negative (6, 16, 81, 82, 84). However, a small subset of genes have been previously reported to be differentially regulated in relA spoT and dksA mutants, including genes encoding flagella, chemotaxis proteins, fimbriae, and others, including several genes of unknown function (81, 82, 84). The mechanisms by which (p)ppGpp and DksA differentially regulate expression of these genes are not entirely clear, but for some of them, notably the fimB and fliC genes (82, 84), mutations of greA have been reported to suppress the effect of dksA mutations, similar to the effect on polyP levels seen here (Fig. 3C and 4C). In the case of polyP, deletion of greA alone had a small stimulatory effect on polyP levels (Fig. 3C, 4C, and 4D), but this was greatly enhanced by deletion of ppx (Fig. 4D). One possible explanation for this result is that greA has some influence on ppx expression or activity. Another possibility is that polyP synthesis is more strongly activated in a greA mutant but wild-type PPX acts to degrade the polyP produced to maintain a normal, presumably optimal, cytoplasmic polyP level.
GreA and GreB are transcript cleavage factors that suppress RNA polymerase backtracking, arrest, and pausing, generally acting to enhance transcription elongation (85), and they have a very complex relationship with DksA in genome-wide regulation of gene expression (74, 82, 84, 88). All three proteins compete for binding to the secondary channel of RNA polymerase, such that in the absence of DksA, GreA occupies the channel more often (88). This has been proposed as a model to explain why greA mutations suppress some dksA mutant phenotypes (74, 82, 84), but the details of these interactions and the identity of all of the genes that might be controlled in such a way are not yet known. Consistent with the lack of effect on polyP synthesis seen with a greB mutant (Fig. 4C), GreB has been previously reported to play a less important role than GreA in the regulatory interactions between DksA and RNA polymerase (74, 88).
DksA itself has been reported to be able to directly sense certain stresses, including pH changes (92) and, by means of a cysteine-containing zinc finger motif, both oxidative and nitrosative redox stress (93–95). It will be interesting to determine whether these play any role in the regulation of polyP synthesis, although since a dksA greA mutant does correctly regulate polyP accumulation in response to nutrient limitation (Fig. 3C), DksA is clearly not essential for detecting this specific stress condition. polyP synthesis is activated by a variety of stresses other than amino acid starvation (25, 27–29), and we are currently exploring the roles of (p)ppGpp and DksA in polyP synthesis in response to these other stress conditions.
The role of the stringent response in regulating polyP levels in E. coli is more complicated than previously thought. In addition to the newfound importance of DksA, it is clear from the results of this study that (p)ppGpp, rather than being necessary to stimulate polyP production, has several more subtle effects on polyP regulation. Mutants lacking either (p)ppGpp or DksA have significantly lower expression of ppk (Fig. 3D), although there is no obvious relationship between ppk gene expression and the capacity to synthesize polyP in response to stress (Fig. 3C, D, and E). It is worth noting, however, that expression of the reporter fusion construct used in these experiments may not entirely reflect expression of the native ppk transcript or PPK protein, and experiments are under way in my lab to test these directly. The previously reported inhibition of PPX by (p)ppGpp (41) appears likely to play a role in tuning the maximum accumulation of polyP, not in activating its synthesis (Fig. 1A, 4D, and 5B). Surprisingly, relA, but not relA spoT, mutants appear to be unable to correctly reverse the accumulation of polyP during recovery from stress (Fig. 5A and B) (43). Since the abilities of PPX and RelA to limit polyP accumulation appear to be additive (Fig. 5A and B), this suggests that there may be an additional, currently unknown, PPX-independent mechanism by which (p)ppGpp can limit the amount of polyP accumulated by cells or control degradation of polyP once adaptation to stress has occurred.
The results presented here strongly suggest that transcription of an unknown gene or genes is required for upregulation of polyP synthesis in response to nutrient limitation stress and that DksA and GreA affect the expression of this gene(s), while (p)ppGpp does not. Future work in my laboratory is focused on identifying this gene(s) and determining how it leads to activation of polyP synthesis by stress. Since changes in ppk gene expression do not appear to account for activation of polyP synthesis, we are currently testing whether exposure to stress results in changes in PPK protein levels or PPK enzyme activity. The activity of PPK is not directly affected by (p)ppGpp (41). However, the existence of hyperactive ppk* mutations (49) (Fig. 6) suggests that it is possible for PPK activity to be upregulated in vivo, and we are actively pursuing experiments to identify the mechanism that explains the phenotype of these mutants. It will also be interesting to explore the connections between regulation of polyP and other general stress responses. polyP, DksA, and (p)ppGpp all affect the induction of the σS regulon (3, 34, 83, 96), for example, and a complete accounting of how E. coli responds to stressful changes in its environment will require a detailed understanding of how polyP, σS, and the stringent response work together to promote bacterial survival. Since both the stringent response and polyP are widely conserved among both Gram-negative and Gram-positive bacteria (9, 23), I hypothesize that the interactions between these general stress response pathways may also be conserved, and it will be intriguing to explore those connections in other species.
MATERIALS AND METHODS
Bacterial strains, growth conditions, and molecular methods.
All strains and plasmids used in this study are listed in Table 2. DNA manipulations were carried out by standard methods (97, 98) in the E. coli cloning strain DH5α (Invitrogen). Gene and protein sequences were obtained from the Integrated Microbial Genomes database (99). PCR and sequencing primers were designed with Web Primer (www.candidagenome.org/cgi-bin/compute/web-primer). E. coli was grown at the indicated temperatures in lysogeny broth (LB) (100) containing 5 g liter−1 NaCl and (where indicated) 1 mM MgCl2, MOPS (morpholinepropanesulfonic acid) minimal medium (Teknova) (101) containing 4 g liter−1 glucose, 0.1 mM K2HPO4, and (where indicated) 0.1 mM uracil, or M9 minimal medium (97) containing 4 g liter−1 glucose and (where indicated) 0.5 g liter−1 Casamino Acids or 20 µg ml−1 l-arginine. Antibiotics were added when appropriate: ampicillin (100 µg ml−1), chloramphenicol (17 or 34 µg ml−1), gentamicin (30 µg ml−1), kanamycin (12.5 or 50 µg ml−1), rifampin (50 µg ml−1), or tetracycline (10 µg ml−1).
TABLE 2.
Strains and plasmids used in this studya
| Strain or plasmid | Marker(s)b | Relevant genotype | Source or reference(s) |
|---|---|---|---|
| E. coli strains | |||
| DH5α | F− λ− ϕ80lacZΔM15 Δ(lacZYA-argF)U169 recA1 endA1 hsdR17(rK− mK+) phoA supE44 thi-1 gyrA96 relA1 | Invitrogen | |
| MG1655 | F− λ− rph-1 ilvG rfb-50 | 61 | |
| CF1693(M+) | Cmr Knr | MG1655 λ+ ϕ80+ relA251::kan+ spoT207::cat+ rpoB1693 (encoding RpoBN1129K) rpoD1693 (encoding RpoDD64G) | 67, 69 |
| CAG12152 | Tcr | MG1655 aer-3075::Tn10(tet+) | 77, 78 |
| CAG12185 | Tcr | MG1655 argE86::Tn10(tet+) | 77 |
| RLG8066 | Cmr Tcr | MG1655 dksA::tet+ greA::cat+ | 88 |
| RLG8067 | Knr Tcr | MG1655 dksA::tet+ greB::kan+ | 88 |
| RLG14535 | Knr Tcr | MG1655 rpoZ+-kan+ rpoC+-tetAR+ | 60 |
| RLG14538 | Knr Tcr | MG1655 rpoZΔ2-5-kan+ rpoC R362A R417A K615A N680A K681A-tetAR+ | 60 |
| Nissle 1917 | 106 | ||
| MJG210 | Knr | MG1655 relA782::kan+ | |
| MJG224 | MG1655 Δppk-749 | 28 | |
| MJG226 | MG1655 ΔrelA782 | ||
| MJG227 | MG1655 ΔphoB763 | 49 | |
| MJG315 | MG1655 Δppx-750 | 28 | |
| MJG341 | Knr | MG1655 glnG730::kan+ | |
| MJG342 | Knr | MG1655 rpoS746::kan+ | |
| MJG344 | MG1655 ΔrpoS746 | ||
| MJG400 | MG1655 ΔglnG730 | ||
| MJG428 | Knr | MG1655 Δppx-750 relA782::kan+ | |
| MJG432 | Knr | MG1655 phoU755::kan+ | |
| MJG443 | MG1655 Δppx-750 ΔrelA782 | ||
| MJG1136 | Cmr | MG1655 ΔrelA782 spoT205::cat+ | |
| MJG1205 | Rifr | MG1655 rpoB3449 (encoding RpoBΔAla532) | |
| MJG1235 | Rifr | MG1655 ΔrelA782 rpoB3449 | |
| MJG1237 | Cmr Rifr | MG1655 ΔrelA782 spoT205::cat+ rpoB3449 | |
| MJG1247 | Cmr | MG1655 Δppx-750 ΔrelA782 spoT205::cat+ | |
| MJG1257 | Knr | Nissle 1917 relA1917::kan+ | |
| MJG1259 | Nissle 1917 ΔrelA1917 | ||
| MJG1267 | Knr | Nissle 1917 ΔrelA1917 spoT1000::kan+ | |
| MJG1287 | Knr | MG1655 ΔrelA782 spoT1000::kan+ | |
| MJG1288 | MG1655 ΔrelA782 ΔspoT1000 | ||
| MJG1289 | Knr | MG1655 relA251::kan+ | |
| MJG1290 | Cmr Knr | MG1655 relA251::kan+ spoT205::cat+ | |
| MJG1291 | Knr | MG1655 relA251::kan+ ΔspoT1000 | |
| MJG1302 | MG1655 ϕ80+ | ||
| MJG1303 | MG1655 λ+ ϕ80+ | ||
| MJG1346 | Knr | MG1655 dksA761::kan+ | |
| MJG1378 | Cmr Knr Tcr | MG1655 λ+ ϕ80+ relA251::kan+ spoT205::cat+ argE86::Tn10(tet+) rpoB+ rpoD1693 | |
| MJG1390 | Cmr Knr | MG1655 ΔrelA782 spoT205::cat+ dksA761::kan+ | |
| MJG1398 | Cmr Knr Tcr | MG1655 λ+ ϕ80+ relA251::kan+ spoT205::cat+ argE86::Tn10(tet+) rpoB1693 rpoD+ | |
| MJG1409 | Cmr | MG1655 greA::cat+ | |
| MJG1410 | Cmr Knr | MG1655 dksA761::kan+ greA::cat+ | |
| MJG1419 | Cmr | MG1655 dksA1000::cat+ | |
| MJG1424 | Knr | MG1655 greB::kan+ | |
| MJG1425 | Tcr | MG1655 dksA::tet+ | |
| MJG1426 | Tcr | MG1655 Δppk-749 dksA::tet+ | |
| MJG1427 | Cmr Knr Tcr | MG1655 rpoZ+-kan+ rpoC+-tetAR+ dksA1000::cat+ | |
| MJG1428 | Cmr Knr Tcr | MG1655 rpoZΔ2-5-kan+ rpoC R362A R417A K615A N680A K681A-tetAR+ dksA1000::cat+ | |
| MJG1434 | Cmr Knr | MG1655 greA::cat+ greB::kan+ | |
| MJG1435 | Cmr Tcr | MG1655 dksA::tet+ greA::cat+ | |
| MJG1436 | Cmr Knr Tcr | MG1655 dksA::tet+ greA::cat+ greB::kan+ | |
| MJG1437 | Knr Tcr | MG1655 dksA::tet+ greB::kan+ | |
| MJG1511 | Tcr | MG1655 Δppx-750 dksA::tet+ | |
| MJG1512 | Cmr | MG1655 Δppx-750 greA::cat+ | |
| MJG1517 | Cmr Tcr | MG1655 Δppx-750 dksA::tet+ greA::cat+ | |
| Plasmids | |||
| pBAD18 | Apr | bla+ | 112 |
| pKD46 | Apr | λ Red+ bla+ | 104 |
| pKD3 | Cmr | cat+ | 104 |
| pKD4 | Knr | kan+ | 104 |
| pRelA′-His | Apr | RelA′(amino acids 1–455)-His8 bla+ | 20 |
| pRelA′-His(D275G) | Apr | RelA′(amino acids 1–455)D275G-His8 bla+ | 20 |
| pGUSA5 | Gmr | Pppk-uidA+ gnt+ | 49 |
| pPPK10 | Knr | ppk+ kan+ | 49 |
| pPPK10b | Knr | ppkG733A (encoding PPKE245K) kan+ | 49 |
| pDKSA1 | Apr | dksA+ bla+ |
Unless otherwise indicated, all strains and plasmids were generated in the course of this work.
Abbreviations: Apr, ampicillin resistance; Cmr, chloramphenicol resistance; Gmr, gentamicin resistance; Knr, kanamycin resistance; Rifr, rifampin resistance; Tcr, tetracycline resistance.
To induce polyP synthesis by nutrient limitation (38, 49), E. coli strains were grown in 10 ml LB at 37°C with shaking (200 rpm) to an A600 of 0.2 to 0.4. Samples (1 ml) were harvested for polyP measurements or enzyme activity assays (see below), and then 5 ml of each culture was harvested by centrifugation (5 min at 4,696 × g at room temperature), resuspended in 5 ml phosphate-buffered saline (PBS) to rinse, and then recentrifuged and resuspended in 5 ml MOPS minimal medium containing 4 g liter−1 glucose, 0.1 mM K2HPO4, and, where indicated, 0.1 mM uracil. These cultures were incubated at 37°C with shaking for the indicated times (usually 2 h) before further samples were collected for polyP measurements. Where indicated, rifampin was added to both the PBS rinse and the MOPS medium.
For assessment of in vivo growth phenotypes on solid media (1.5% agar), strains of interest were grown overnight in LB broth at 37°C, harvested by centrifugation, rinsed once with PBS, and then resuspended to an A600 of 0.1 or 1 in PBS. Tenfold serial dilutions to 10−5 were made in PBS, and 5-µl aliquots of each dilution were spotted on solid medium, allowed to dry, and incubated at 37°C for 24 or 48 h.
Strain construction.
Unless otherwise indicated, all E. coli strains are derivatives of wild-type strain MG1655 (F− rph-1 ilvG rfb-50) (61). All chromosomal mutations were confirmed by PCR. Plasmids pRelA′-His and pRelA′-His(D275G) (20) were gifts from Mike Laub (Massachusetts Institute of Technology). Strain CF1693(M+) (ϕ80+ λ+ relA251::kan+ spoT207::cat+ rpoB1693 rpoD1693) (67, 69) was a gift from Kenn Gerdes (University of Copenhagen). Strains RLG8066 (dksA::tet+ greA::cat+), RLG8067 (dksA::tet+ greB::kan+) (88), RLG14535 (rpoZ+-kan+ rpoC+-tetAR+), and RLG14538 (rpoZΔ2-5-kan+ rpoC R362A R417A K615A N680A K681A-tetAR+) (60) were gifts from Rick Gourse (University of Wisconsin—Madison). Strains CAG12152 [aer-3075::Tn10(tet+)] and CAG12185 [argE86::Tn10(tet+)] (77, 78) were gifts from Chuck Turnbough (University of Alabama at Birmingham).
P1vir transductions were carried out by a modification of a previously described protocol (102). To prepare P1vir lysates, 50 µl of an overnight culture of donor cells was subcultured to 5 ml LB medium containing 2 g liter−1 glucose and 5 mM CaCl2 and grown for 30 min at 37°C with shaking before addition of 100 µl of a P1vir lysate prepared from wild-type MG1655 cells. These cultures were incubated at 37°C with shaking until complete lysis occurred (typically 2 to 3 h), chloroform was added to kill vegetative cells, cell debris was removed by centrifugation (15 min at 3,000 × g at room temperature), and supernatants were stored at 4°C with approximately 200 µl chloroform. Transductions were carried out by harvesting 200-µl aliquots of overnight cultures of recipient cells by centrifugation (10 min at 1,500 × g at room temperature) and then gently resuspending the pellets in 100 µl 5 mM CaCl2–10 mM MgSO4. These suspensions were mixed gently with 100, 50, or 10 µl of donor phage lysate. After incubating for 30 min at 30°C, 100 µl of 1 M sodium citrate and 1 ml of LB medium were added, and cultures were incubated for 1 h at 37°C without shaking before plating on LB agar containing antibiotics and 10 mM sodium citrate. Transductants were restreaked for isolation twice on LB agar containing antibiotics and 10 mM sodium citrate to ensure that no phage remained in the resulting strains.
P1vir transduction was used to move the relA782::kan+ allele from the Keio collection (103) into MG1655 and MJG315 (Δppx-750) (28), generating strains MJG210 (relA782::kan+) and MJG428 (Δppx-750 relA782::kan+). The kanamycin resistance cassettes were resolved as described previously (104) to give strains MJG226 (ΔrelA782) and MJG443 (Δppx-750 ΔrelA782). P1vir transduction was used to move the spoT207::cat+ allele from strain CF1693(M+) into strains MJG226 and MJG443, yielding strains MJG1136 (ΔrelA782 spoT207::cat+) and MJG1247 (Δppx-750 ΔrelA782 spoT207::cat+). The spoT gene of strain MJG226 (ΔrelA782) was replaced with a pKD4-derived kanamycin resistance cassette by recombineering (104) using primers 5′ GTT ACC GCT ATT GCT GAA GGT CGT CGT TAA TCA CAA AGC GGG TCG CCC TTG GTG TAG GCT GGA GCT GCT TC 3′ and 5′ GGC GAG CAT TTC GCA GAT GCG TGC ATA ACG TGT TGG GTT CAT AAA ACA TTA CAT ATG AAT ATC CTC CTT AG 3′, yielding strain MJG1287 (ΔrelA782 spoT1000::kan+). The kanamycin resistance cassette was resolved, yielding strain MJG1288 (ΔrelA782 ΔspoT1000). P1vir transduction was used to move the relA251::kan+ allele from CF1693(M+) into MG1655, MJG1136 (ΔrelA782 spoT205::cat+), and MJG1288 (ΔrelA782 ΔspoT1000), generating strains MJG1289 (relA251::kan+), MJG1290 (relA251::kan+ spoT205::cat+), and MJG1291 (relA251::kan+ ΔspoT1000). All MG1655-derived relA and relA spoT mutants were tested for infection with ϕ80 and λ bacteriophages by phage infection assay (see below) and by PCR with primer pairs 5′ GAA GTC TTT GCG CAG TAT CAG 3′/5′ CTT TGT GTC GGT GAT CAT TG 3′ and 5′ CCG ACA GAA TCG GGC GAG AAG A 3′/5′CGC GCA CGA AAA GCA TCA GGT 3′, as previously described (69). Despite reports that ϕ80 can be transmitted during P1vir transductions (105), I did not observe any instances of this when using the transduction protocol described above. I also regularly checked relA spoT mutant strains to confirm that they could not grow on M9 minimal medium without amino acids to ensure that no M+ suppressors had arisen in those strains (21, 70).
The relA gene of E. coli strain Nissle 1917 (locus tag ECOLIN_15000) (106) was replaced with a kanamycin resistance cassette by recombineering using primers 5′ GTA TGG ACA TTG ACA CGC TGC GGG CGG CGC TGC TGT TCC CTC TGG CTG ATG GTG TAG GCT GGA GCT GCT TC 3′ and 5′ AAA AGA TAT CGC TGG ATA CGG CTG ACG CCA TAT CCA GCT ATG TAA GAT TAA CAT ATG AAT ATC CTC CTT AG 3′, yielding strain MJG1257 (Nissle 1917 relA1917::kan+). The resulting insertion was then resolved, yielding strain MJG1259 (Nissle 1917 ΔrelA1917). The spoT gene of this strain (locus tag ECOLIN_20110), which is identical to that of MG1655, was replaced by recombineering with a kanamycin resistance cassette using primers 5′ GTT ACC GCT ATT GCT GAA GGT CGT CGT TAA TCA CAA AGC GGG TCG CCC TTG GTG TAG GCT GGA GCT GCT TC 3′ and 5′ GGC GAG CAT TTC GCA GAT GCG TGC ATA ACG TGT TGG GTT CAT AAA ACA TTA CAT ATG AAT ATC CTC CTT AG 3′, yielding strain MJG1267 (Nissle 1917 ΔrelA1917 spoT1000::kan+).
Strain MG1655 was made lysogenic for ϕ80 and λ bacteriophages by a slight modification of a previously described procedure (69). An overnight culture of CF1693(M+) was grown in LB at 37°C, cells were removed by centrifugation (15 min at 3,000 × g at room temperature), and the supernatant was diluted 1:10 in 0.1 M NaCl–10 mM MgSO4–50 mM Tris-HCl (pH 7.5)–0.01% gelatin, with several drops of chloroform added to kill vegetative cells. MG1655 was grown in 5 ml LB at 30°C with shaking to an A600 of 0.2, and then 100 µl of diluted CF1693(M+) supernatant was added and incubation continued overnight at 30°C. Survivors were streaked for isolation on LB, and individual colonies were repeatedly restreaked for isolation and grown overnight at 30°C until no plaques were observed in the streak. Isolates were screened for lysogeny by PCR, as described above, and plaque assay, as described below.
P1vir transduction was used to move the argE86::Tn10(tet+) allele (required for arginine synthesis; 53% linked to rpoB) from strain CAG12185 (77) into strain CF1693(M+), selecting for growth in the presence of tetracycline and checking for arginine auxotrophy. The sequence of rpoB alleles in the resulting colonies was screened by PCR amplification of a fragment of rpoB with primers 5′ GGG TCT GTT CAG CCG TAT 3′ and 5′ ATT TGC TCC CGT CGG AGT 3′ and Sanger sequencing of the resulting products (UAB Heflin Center for Genomic Science Sequencing Core). This yielded strain MJG1378 [ϕ80+ λ+ relA251::kan+ spoT207::cat+ argE86::Tn10(tet+) rpoB+ rpoD1693].
P1vir transduction was used to move the aer-3075::Tn10(tet+) allele (90% linked to rpoD) from strain CAG12152 (77, 78) into strain CF1693(M+), selecting for growth in the presence of tetracycline. The sequence of rpoD alleles in the resulting colonies was screened by PCR amplification of rpoD with primers 5′ CGC CCT CGT AAT TAT CGT TGG 3′ and 5′ TGG AGA AAC TGG TTG AAG CG 3′ and Sanger sequencing of the resulting products. This yielded strain MJG1398 [ϕ80+ λ+ relA251::kan+ spoT207::cat+ aer-3075::Tn10(tet+) rpoB1693 rpoD+].
Oligonucleotide-directed recombineering (107) was used to construct chromosomal rpoB3449 alleles (71, 108–111) using the mutagenic primer 5′ CCT GCA CGT TCA CGG GTC AGA CCG CCT GGA CCA AGA GAA ATA CGA CGT TTG TGC GTA ATC TCA GAC AGC G 3′, which contained four 5′ phosphorothiorate linkages. This primer deletes nucleotides G1593 through A1596 of rpoB, removing the codon for alanine 532 of RpoB, and also incorporates silent mutations in four adjacent codons (C1590T, C1593T, C1599T, and C1602T) to avoid mismatch repair. Strains MG1655, MJG226 (ΔrelA782), and MJG1136 (ΔrelA782 spoT207::cat+) were transformed with pKD46 (104), induced to express the λ Red recombinase, and electroporated with 250 pmol of mutagenic primer. Recombinant colonies were selected at 37°C on LB plates containing rifampin. The sequence of rpoB alleles was confirmed by PCR amplification of a fragment of rpoB with primers 5′ GAT GTT ATG AAA AAG CTC 3′ and 5′ CTG GGT GGA TAC GTC CAT 3′ and Sanger sequencing of the resulting product. After curing pKD46 by growth at 37°C, this yielded strains MJG1205 (rpoB3449), MJG1235 (ΔrelA782 rpoB3449), and MJG1237 (ΔrelA782 spoT207::cat+ rpoB3449).
P1vir transduction was used to move the rpoS746::kan+, glnG730::kan+, phoU755::kan+, and dksA761::kan+ alleles from the Keio collection into MG1655, generating strains MJG341 (rpoS746::kan+), MJG342 (glnG730::kan+), MJG432 (phoU755::kan+), and MJG1346 (dksA761::kan+). The kanamycin resistance cassettes in MJG341 and MJG342 were resolved to give strains MJG344 (ΔrpoS746) and MJG400 (ΔglnG730). P1vir transduction was used to move the dksA761::kan+ allele from the Keio collection into MJG1136 (ΔrelA782 spoT205::cat+), generating strain MJG1390 (ΔrelA782 spoT205::cat+ dksA761::kan+). P1vir transduction was used to individually or sequentially move the dksA::tet+, greA::cat+, and greB::kan+ alleles from RLG8066 (dksA::tet+ greA::cat+) or RLG8067 (dksA::tet+ greB::kan+) (88) (which have, as far as I can determine, no assigned allele numbers) into strains MG1655, MJG224 (Δppk-749), MJG1346 (dksA761::kan+), and MJG315 (Δppx-750), generating isogenic strains MJG1409 (greA::cat+), MJG1410 (dksA761::kan+ greA::cat+), MJG1424 (greB::kan+), MJG1425 (dksA::tet+), MJG1426 (Δppk-749 dksA::tet+), MJG1434 (greA::cat+ greB::kan+), MJG1435 (dksA::tet+ greA::cat+), MJG1436 (dksA::tet+ greA::cat+ greB::kan+), MJG1437 (dksA::tet+ greB::kan+), MJG1511 (Δppx-750 dksA::tet+), MJG1512 (Δppx-750 greA::cat+), and MJG1517 (Δppx-750 dksA::tet+ greA::cat+).
The dksA gene of MG1655 was replaced with a chloramphenicol resistance cassette by recombineering (104) using primers 5′ TTT TCC CCC GAA CAT GGG GAT CGA TAG TGC GTG TTA AGG AGA AGC AAC ATG GTG TAG GCT GGA GCT GCT TC 3′ and 5′ AGG CGG GAG CAT TTC CCG CCT GTG GTA AAC GTG ATG GAA CGG CTG TAA TTA CAT ATG AAT ATC CTC CTT AG 3′, yielding strain MJG1419 (dksA1000::cat+). P1vir transduction was used to move the dksA1000::cat+ allele into strains RLG14535 and RLG14538 to yield strains MJG1427 (rpoZ+ kan+ rpoC+ tetAR+ dksA1000::cat+) and MJG1428 (rpoZΔ2-5-kan+ rpoC R362A R417A K615A N680A K681A tetAR+ dksA1000::cat+).
Sequencing rpoB, rpoC, and rpoD of CF1693(M+).
Sections of the rpoB and rpoC genes of E. coli strain CF1693(M+) were PCR amplified and Sanger sequenced with combinations of primers 5′ GAT GTT ATG AAA AAG CTC 3′, 5′ CTG GGT GGA TAC GTC CAT 3′, 5′ TTG CGG AAA GTG TTC CAT 3′, 5′ AGA GCA GAT CCT CGA CCT 3′, 5′ CTG GTA CGT GTA GAG CGT 3′, 5′ AGC TAA ACG TGG TGG TGT 3′, 5′ GGG TCT GTT CAG CCG TAT 3′, 5′ TGG TGA ACA GTT CGA GCG 3′, 5′ ATT TGC TCC CGT CGG AGT 3′, 5′ TAC CCA GGT GGG TTT CGA 3′, 5′ CGG ATT CAT CCA GTT TGG 3′, 5′ CCT GGG CGA TAA CGT AGT 3′, 5′ TGC GGT AGA TTT CTA CCA 3′, 5′ CTT TAA TGT CTT TTA CGG 3′, 5′ TTC TCG CTC AAA CAG GTC 3′, 5′ AGC GTT CGT TCA GTC TGG 3′, 5′ CGT GCG CTG ATG ATG TCT 3′, 5′ TCC GGT GCG CGT GGT TCT GC 3′, 5′ CAC TTC CCG TAA TAC TGA 3′, 5′ TGT TCA TGC TGT TAC TCG 3′, 5′ ATA TTC TGA CAA ATG CTC 3′, 5′ TCG GAA TCA TCT CTT CGT 3′, 5′ ACC ACC GAT GTG GAA CGT 3′, 5′ CGC AGC CCA GAT ATC GAT 3′, 5′ TCA GTA CCG GTT CAA ATG 3′, 5′ TGC TCT TCA GCA GAG CCT 3′, 5′ GAA CAT CGG TCT GAT CAA 3′, 5′ TAA GGC ATA TCT TCG ATC 3′, 5′ TTG GTA TCG ATG ACA TGG 3′, and 5′ TGC CGG ACG CAG ATC TTT 3′ until complete 2-fold sequence coverage of both genes and flanking intergenic sequences (a total of 8,259 bp) was obtained. The rpoB C3387A mutation found in CF1693(M+), encoding an RpoB N1129K variant protein, is referred to, for simplicity’s sake, as rpoB1693. Despite an extensive literature search, I can find no previously published or reported allele of rpoB using this allele number.
The rpoD gene and flanking sequences from CF1693(M+) (2,098 bp) were amplified with primers 5′ CGC CCT CGT AAT TAT CGT TGG 3′ and 5′ TGG AGA AAC TGG TTG AAG CG 3′, and the resulting PCR product was Sanger sequenced with those primers and with primers 5′ TCG GTT CTG AGC TTT CCC AG 3′ and 5′ TTG AGC TTG TTG ATG GTC TCA 3′. As with rpoB1693, I refer to the rpoD A191G mutation found in CF1693(M+), encoding an RpoD D64G variant protein, as rpoD1693.
Plasmid construction.
The dksA coding domain sequence (471 bp) plus 15 bp of 5′ sequence was amplified from E. coli MG1655 genomic DNA with primers 5′ TTC gaa ttc TTA AGG AGA AGC AAC ATG CAA GA 3′ and 5′ CTT aag ctt TTA GCC AGC CAT CTG TTT TTC G 3′ (lowercase letters indicate restriction sites inserted for cloning) and cloned into the EcoRI and HindIII sites of plasmid pBAD18 (112).
Phage infection assay.
To test for production of infective phage particles by E. coli strains, LB-grown overnight cultures were centrifuged for 15 min at 3,000 × g, and 3 µl of the supernatant was spotted on LB plates overlaid with wild-type MG1655 in LB containing 0.7% agar. These plates were incubated overnight at 30°C to allow plaque development.
In vivo polyphosphate assay.
Intracellular polyP levels were measured as previously described (49). Briefly, samples of bacterial cultures (50 to 100 µg of total protein; 1 ml of a culture at an A600 of 0.2 to 0.4) were harvested by centrifugation, resuspended in 250 µl of 4 M guanidine isothiocyanate–50 mM Tris-HCl (pH 7), and lysed by incubation for 10 min at 95°C. The protein concentration was determined by Bradford assay (Bio-Rad) of a 5-µl aliquot, and then 15 µl of 10% SDS and 250 µl of 95% ethanol were added to the remaining sample. The resulting mixture was applied to an EconoSpin silica spin column (Epoch Life Science) and rinsed with 1.5 ml 5 mM Tris-HCl (pH 7.5)–50 mM NaCl–5 mM EDTA–50% ethanol, and polyP was eluted with 150 µl 50 mM Tris-HCl, pH 8. The eluate was brought to 20 mM Tris-HCl (pH 7.5), 5 mM MgCl2, and 50 mM ammonium acetate with 1 µg of PPX1 exopolyphosphatase from Saccharomyces cerevisiae (113) in a final volume of 200 µl and incubated for 15 min at 37°C. The resulting polyP-derived free phosphate was measured using a malachite green colorimetric assay (114) and normalized to total protein. polyP concentrations were expressed in terms of individual phosphate monomers.
β–Glucuronidase assays.
Expression from a plasmid-encoded Pppk-uidA β-glucuronidase translational fusion before and 2 h after nutrient limitation stress was measured as previously described (49, 115). Cell pellets from 1 ml of culture were resuspended in 500 µl of 50 mM sodium phosphate (pH 7)–10 mM β-mercaptoethanol–1 mM EDTA–0.1% Triton X-100, permeabilized with 12.5 µl 0.1% sodium dodecyl sulfate and 25 µl chloroform, and stored on ice for no more than 30 min. Reaction mixtures were prewarmed to 37°C, and then reactions were started by addition of 12.5 µl 4 mg ml−1 p-nitrophenyl-β-d-glucuronide in 50 mM sodium phosphate (pH 7). Reactions were stopped after 15 min by addition of 250 µl 1 M Na2CO3, and then the mixtures were centrifuged for 10 min at 21,100 × g at room temperature to remove light-scattering particles. The p-nitrophenol product of the β-glucuronidase reaction was measured as A405 in a Genesys 10S UV-visible spectrophotometer (Thermo Fisher), blanked to a control reaction mixture containing no cells. Miller units were calculated as 1,000 × [A405/(A600 of culture × milliliters of culture added to the reaction mixture × minutes of reaction time)].
Alkaline phosphatase assays.
The activity of PhoA before and 2 h after nutrient limitation stress, as a readout for activation of the PhoB regulon (79), was measured using a slight modification of a previously described method (116). Cell pellets from 1 ml of culture were resuspended in 1 ml 1 M Tris-HCl (pH 8) and prewarmed to 37°C. Reactions were started by addition of 100 µl p-nitrophenyl-phosphate (4 mg ml−1 in 1 M Tris-HCl, pH 8) and stopped at appropriate time points (up to 1 h) by addition of 100 µl of 1 M K2HPO4. Cells were removed by centrifugation (1 min at 16,000 × g at room temperature). The absorbance of the resulting supernatant was measured at 420 and 550 nm in a Genesys 10S UV-visible spectrophotometer (Thermo Fisher), blanked to a control reaction mixture containing no cells. Units of PhoA phosphatase activity were calculated as 1,000 × [A420 – (1.75 × A550)]/(A600 of culture × minutes of reaction time).
Statistical analyses.
Two-way repeated-measures ANOVA and multiple t-test analyses with Holm-Sidak’s multiple-comparison corrections were performed using Prism version 7.00e for Macintosh (GraphPad Software, La Jolla, CA).
Data availability.
All strains and plasmids generated in the course of this work are available from the author upon request.
Supplementary Material
ACKNOWLEDGMENTS
I thank the members of the Gray lab, Charles Turnbough (University of Alabama at Birmingham), Michael Laub (Massachusetts Institute of Technology), Michael Cashel (National Institutes of Health), Kenn Gerdes (University of Copenhagen), and Wilma Ross, Richard Gourse, and Saumya Gopalkrishnan (University of Wisconsin—Madison) for helpful discussions and strains. I thank Nicole Arroyo Diaz for construction of pDKSA1.
This project was supported by University of Alabama at Birmingham Department of Microbiology startup funds and NIH grant R35GM124590.
I have no conflicts of interest to declare.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JB.00664-18.
For a commentary on this article, see https://doi.org/10.1128/JB.00070-19.
REFERENCES
- 1.Hecker M, Pané-Farré J, Völker U. 2007. SigB-dependent general stress response in Bacillus subtilis and related gram-positive bacteria. Annu Rev Microbiol 61:215–236. doi: 10.1146/annurev.micro.61.080706.093445. [DOI] [PubMed] [Google Scholar]
- 2.Fiebig A, Herrou J, Willett J, Crosson S. 2015. General stress signaling in the alphaproteobacteria. Annu Rev Genet 49:603–625. doi: 10.1146/annurev-genet-112414-054813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Battesti A, Majdalani N, Gottesman S. 2011. The RpoS-mediated general stress response in Escherichia coli. Annu Rev Microbiol 65:189–213. doi: 10.1146/annurev-micro-090110-102946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bergkessel M, Basta DW, Newman DK. 2016. The physiology of growth arrest: uniting molecular and environmental microbiology. Nat Rev Microbiol 14:549–562. doi: 10.1038/nrmicro.2016.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gottesman S. 2017. Stress reduction, bacterial style. J Bacteriol 199:e00433-17. doi: 10.1128/JB.00433-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gourse RL, Chen AY, Gopalkrishnan S, Sanchez-Vazquez P, Myers A, Ross W. 2018. Transcriptional responses to ppGpp and DksA. Annu Rev Microbiol 72:163–184. doi: 10.1146/annurev-micro-090817-062444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hauryliuk V, Atkinson GC, Murakami KS, Tenson T, Gerdes K. 2015. Recent functional insights into the role of (p)ppGpp in bacterial physiology. Nat Rev Microbiol 13:298–309. doi: 10.1038/nrmicro3448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Potrykus K, Cashel M. 2008. (p)ppGpp: still magical? Annu Rev Microbiol 62:35–51. doi: 10.1146/annurev.micro.62.081307.162903. [DOI] [PubMed] [Google Scholar]
- 9.Atkinson GC, Tenson T, Hauryliuk V. 2011. The RelA/SpoT homolog (RSH) superfamily: distribution and functional evolution of ppGpp synthetases and hydrolases across the tree of life. PLoS One 6:e23479. doi: 10.1371/journal.pone.0023479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Winther KS, Roghanian M, Gerdes K. 2018. Activation of the stringent response by loading of RelA-tRNA complexes at the ribosomal A-site. Mol Cell 70:95–105 e4. doi: 10.1016/j.molcel.2018.02.033. [DOI] [PubMed] [Google Scholar]
- 11.VanBogelen RA, Kelley PM, Neidhardt FC. 1987. Differential induction of heat shock, SOS, and oxidation stress regulons and accumulation of nucleotides in Escherichia coli. J Bacteriol 169:26–32. doi: 10.1128/jb.169.1.26-32.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Durfee T, Hansen AM, Zhi H, Blattner FR, Jin DJ. 2008. Transcription profiling of the stringent response in Escherichia coli. J Bacteriol 190:1084–1096. doi: 10.1128/JB.01092-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Traxler MF, Summers SM, Nguyen HT, Zacharia VM, Hightower GA, Smith JT, Conway T. 2008. The global, ppGpp-mediated stringent response to amino acid starvation in Escherichia coli. Mol Microbiol 68:1128–1148. doi: 10.1111/j.1365-2958.2008.06229.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rutherford ST, Villers CL, Lee JH, Ross W, Gourse RL. 2009. Allosteric control of Escherichia coli rRNA promoter complexes by DksA. Genes Dev 23:236–248. doi: 10.1101/gad.1745409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Paul BJ, Barker MM, Ross W, Schneider DA, Webb C, Foster JW, Gourse RL. 2004. DksA: a critical component of the transcription initiation machinery that potentiates the regulation of rRNA promoters by ppGpp and the initiating NTP. Cell 118:311–322. doi: 10.1016/j.cell.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 16.Molodtsov V, Sineva E, Zhang L, Huang X, Cashel M, Ades SE, Murakami KS. 2018. Allosteric effector ppGpp potentiates the inhibition of transcript initiation by DksA. Mol Cell 69:828–839. doi: 10.1016/j.molcel.2018.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dalebroux ZD, Swanson MS. 2012. ppGpp: magic beyond RNA polymerase. Nat Rev Microbiol 10:203–212. doi: 10.1038/nrmicro2720. [DOI] [PubMed] [Google Scholar]
- 18.Kanjee U, Ogata K, Houry WA. 2012. Direct binding targets of the stringent response alarmone (p)ppGpp. Mol Microbiol 85:1029–1043. doi: 10.1111/j.1365-2958.2012.08177.x. [DOI] [PubMed] [Google Scholar]
- 19.Srivatsan A, Wang JD. 2008. Control of bacterial transcription, translation and replication by (p)ppGpp. Curr Opin Microbiol 11:100–105. doi: 10.1016/j.mib.2008.02.001. [DOI] [PubMed] [Google Scholar]
- 20.Wang B, Dai P, Ding D, Del Rosario A, Grant RA, Pentelute BL, Laub MT. 2018. Affinity-based capture and identification of protein effectors of the growth regulator ppGpp. Nat Chem Biol doi: 10.1038/s41589-018-0183-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Potrykus K, Murphy H, Philippe N, Cashel M. 2011. ppGpp is the major source of growth rate control in E. coli. Environ Microbiol 13:563–575. doi: 10.1111/j.1462-2920.2010.02357.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rao NN, Gómez-García MR, Kornberg A. 2009. Inorganic polyphosphate: essential for growth and survival. Annu Rev Biochem 78:605–647. doi: 10.1146/annurev.biochem.77.083007.093039. [DOI] [PubMed] [Google Scholar]
- 23.Wang L, Yan J, Wise MJ, Liu Q, Asenso J, Huang Y, Dai S, Liu Z, Du Y, Tang D. 2018. Distribution patterns of polyphosphate metabolism pathway and its relationships with bacterial durability and virulence. Front Microbiol 9:782. doi: 10.3389/fmicb.2018.00782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Achbergerova L, Nahalka J. 2011. Polyphosphatean—ancient energy source and active metabolic regulator. Microb Cell Fact 10:63. doi: 10.1186/1475-2859-10-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gray MJ, Jakob U. 2015. Oxidative stress protection by polyphosphatenew—roles for an old player. Curr Opin Microbiol 24:1–6. doi: 10.1016/j.mib.2014.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Albi T, Serrano A. 2016. Inorganic polyphosphate in the microbial world. Emerging roles for a multifaceted biopolymer. World J Microbiol Biotechnol 32:27. doi: 10.1007/s11274-015-1983-2. [DOI] [PubMed] [Google Scholar]
- 27.Yoo NG, Dogra S, Meinen BA, Tse E, Haefliger J, Southworth DR, Gray MJ, Dahl JU, Jakob U. 2018. Polyphosphate stabilizes protein unfolding intermediates as soluble amyloid-like oligomers. J Mol Biol 430:4195–4208. doi: 10.1016/j.jmb.2018.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gray MJ, Wholey WY, Wagner NO, Cremers CM, Mueller-Schickert A, Hock NT, Krieger AG, Smith EM, Bender RA, Bardwell JC, Jakob U. 2014. Polyphosphate is a primordial chaperone. Mol Cell 53:689–699. doi: 10.1016/j.molcel.2014.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Crooke E, Akiyama M, Rao NN, Kornberg A. 1994. Genetically altered levels of inorganic polyphosphate in Escherichia coli. J Biol Chem 269:6290–6295. [PubMed] [Google Scholar]
- 30.Dahl JU, Gray MJ, Bazopoulou D, Beaufay F, Lempart J, Koenigsknecht MJ, Wang Y, Baker JR, Hasler WL, Young VB, Sun D, Jakob U. 2017. The anti-inflammatory drug mesalamine targets bacterial polyphosphate accumulation. Nat Microbiol 2:16267. doi: 10.1038/nmicrobiol.2016.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cremers CM, Knoefler D, Gates S, Martin N, Dahl JU, Lempart J, Xie L, Chapman MR, Galvan V, Southworth DR, Jakob U. 2016. Polyphosphate: a conserved modifier of amyloidogenic processes. Mol Cell 63:768–780. doi: 10.1016/j.molcel.2016.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kusano S, Ishihama A. 1997. Functional interaction of Escherichia coli RNA polymerase with inorganic polyphosphate. Genes Cells 2:433–441. doi: 10.1046/j.1365-2443.1997.13203301320330.x. [DOI] [PubMed] [Google Scholar]
- 33.McInerney P, Mizutani T, Shiba T. 2006. Inorganic polyphosphate interacts with ribosomes and promotes translation fidelity in vitro and in vivo. Mol Microbiol 60:438–447. doi: 10.1111/j.1365-2958.2006.05103.x. [DOI] [PubMed] [Google Scholar]
- 34.Shiba T, Tsutsumi K, Yano H, Ihara Y, Kameda A, Tanaka K, Takahashi H, Munekata M, Rao NN, Kornberg A. 1997. Inorganic polyphosphate and the induction of rpoS expression. Proc Natl Acad Sci U S A 94:11210–11215. doi: 10.1073/pnas.94.21.11210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stumpf JD, Foster PL. 2005. Polyphosphate kinase regulates error-prone replication by DNA polymerase IV in Escherichia coli. Mol Microbiol 57:751–761. doi: 10.1111/j.1365-2958.2005.04724.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Grillo-Puertas M, Schurig-Briccio LA, Rodríguez-Montelongo L, Rintoul MR, Rapisarda VA. 2014. Copper tolerance mediated by polyphosphate degradation and low-affinity inorganic phosphate transport system in Escherichia coli. BMC Microbiol 14:72. doi: 10.1186/1471-2180-14-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Varas M, Valdivieso C, Mauriaca C, Ortíz-Severín J, Paradela A, Poblete-Castro I, Cabrera R, Chávez FP. 2017. Multi-level evaluation of Escherichia coli polyphosphate related mutants using global transcriptomic, proteomic and phenomic analyses. Biochim Biophys Acta 1861:871–883. doi: 10.1016/j.bbagen.2017.01.007. [DOI] [PubMed] [Google Scholar]
- 38.Ault-Riche D, Fraley CD, Tzeng CM, Kornberg A. 1998. Novel assay reveals multiple pathways regulating stress-induced accumulations of inorganic polyphosphate in Escherichia coli. J Bacteriol 180:1841–1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kornberg A, Rao NN, Ault RD. 1999. Inorganic polyphosphate: a molecule of many functions. Annu Rev Biochem 68:89–125. doi: 10.1146/annurev.biochem.68.1.89. [DOI] [PubMed] [Google Scholar]
- 40.Kuroda A. 2006. A polyphosphate-lon protease complex in the adaptation of Escherichia coli to amino acid starvation. Biosci Biotechnol Biochem 70:325–331. doi: 10.1271/bbb.70.325. [DOI] [PubMed] [Google Scholar]
- 41.Kuroda A, Murphy H, Cashel M, Kornberg A. 1997. Guanosine tetra- and pentaphosphate promote accumulation of inorganic polyphosphate in Escherichia coli. J Biol Chem 272:21240–21243. doi: 10.1074/jbc.272.34.21240. [DOI] [PubMed] [Google Scholar]
- 42.Moreno SN, Docampo R. 2013. Polyphosphate and its diverse functions in host cells and pathogens. PLoS Pathog 9:e1003230. doi: 10.1371/journal.ppat.1003230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rao NN, Liu S, Kornberg A. 1998. Inorganic polyphosphate in Escherichia coli: the phosphate regulon and the stringent response. J Bacteriol 180:2186–2193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.de Almeida LG, Ortiz JH, Schneider RP, Spira B. 2015. phoU inactivation in Pseudomonas aeruginosa enhances accumulation of ppGpp and polyphosphate. Appl Environ Microbiol 81:3006–3015. doi: 10.1128/AEM.04168-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim HY, Schlictman D, Shankar S, Xie Z, Chakrabarty AM, Kornberg A. 1998. Alginate, inorganic polyphosphate, GTP and ppGpp synthesis co-regulated in Pseudomonas aeruginosa: implications for stationary phase survival and synthesis of RNA/DNA precursors. Mol Microbiol 27:717–725. doi: 10.1046/j.1365-2958.1998.00702.x. [DOI] [PubMed] [Google Scholar]
- 46.Sureka K, Dey S, Datta P, Singh AK, Dasgupta A, Rodrigue S, Basu J, Kundu M. 2007. Polyphosphate kinase is involved in stress-induced mprAB-sigE-rel signalling in mycobacteria. Mol Microbiol 65:261–276. doi: 10.1111/j.1365-2958.2007.05814.x. [DOI] [PubMed] [Google Scholar]
- 47.Rao NN, Kornberg A. 1999. Inorganic polyphosphate regulates responses of Escherichia coli to nutritional stringencies, environmental stresses and survival in the stationary phase. Prog Mol Subcell Biol 23:183–195. doi: 10.1007/978-3-642-58444-2_9. [DOI] [PubMed] [Google Scholar]
- 48.Xiao H, Kalman M, Ikehara K, Zemel S, Glaser G, Cashel M. 1991. Residual guanosine 3',5'-bispyrophosphate synthetic activity of relA null mutants can be eliminated by spoT null mutations. J Biol Chem 266:5980–5990. [PubMed] [Google Scholar]
- 49.Rudat AK, Pokhrel A, Green TJ, Gray MJ. 2018. Mutations in Escherichia coli polyphosphate kinase that lead to dramatically increased in vivo polyphosphate levels. J Bacteriol 200:e00697-17. doi: 10.1128/JB.00697-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Akiyama M, Crooke E, Kornberg A. 1993. An exopolyphosphatase of Escherichia coli. The enzyme and its ppx gene in a polyphosphate operon. J Biol Chem 268:633–639. [PubMed] [Google Scholar]
- 51.Yang C, Huang TW, Wen SY, Chang CY, Tsai SF, Wu WF, Chang CH. 2012. Genome-wide PhoB binding and gene expression profiles reveal the hierarchical gene regulatory network of phosphate starvation in Escherichia coli. PLoS One 7:e47314. doi: 10.1371/journal.pone.0047314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yuan ZC, Zaheer R, Morton R, Finan TM. 2006. Genome prediction of PhoB regulated promoters in Sinorhizobium meliloti and twelve proteobacteria. Nucleic Acids Res 34:2686–2697. doi: 10.1093/nar/gkl365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zimmer DP, Soupene E, Lee HL, Wendisch VF, Khodursky AB, Peter BJ, Bender RA, Kustu S. 2000. Nitrogen regulatory protein C-controlled genes of Escherichia coli: scavenging as a defense against nitrogen limitation. Proc Natl Acad Sci U S A 97:14674–14679. doi: 10.1073/pnas.97.26.14674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dong T, Schellhorn HE. 2009. Control of RpoS in global gene expression of Escherichia coli in minimal media. Mol Genet Genomics 281:19–33. doi: 10.1007/s00438-008-0389-3. [DOI] [PubMed] [Google Scholar]
- 55.Dong T, Kirchhof MG, Schellhorn HE. 2008. RpoS regulation of gene expression during exponential growth of Escherichia coli K12. Mol Genet Genomics 279:267–277. doi: 10.1007/s00438-007-0311-4. [DOI] [PubMed] [Google Scholar]
- 56.Gray MJ, Wholey WY, Parker BW, Kim M, Jakob U. 2013. NemR is a bleach-sensing transcription factor. J Biol Chem 288:13789–13798. doi: 10.1074/jbc.M113.454421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang S, Deng K, Zaremba S, Deng X, Lin C, Wang Q, Tortorello ML, Zhang W. 2009. Transcriptomic response of Escherichia coli O157:H7 to oxidative stress. Appl Environ Microbiol 75:6110–6123. doi: 10.1128/AEM.00914-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ahn K, Kornberg A. 1990. Polyphosphate kinase from Escherichia coli. Purification and demonstration of a phosphoenzyme intermediate. J Biol Chem 265:11734–11739. [PubMed] [Google Scholar]
- 59.Akiyama M, Crooke E, Kornberg A. 1992. The polyphosphate kinase gene of Escherichia coli. Isolation and sequence of the ppk gene and membrane location of the protein. J Biol Chem 267:22556–22561. [PubMed] [Google Scholar]
- 60.Ross W, Sanchez-Vazquez P, Chen AY, Lee JH, Burgos HL, Gourse RL. 2016. ppGpp binding to a site at the RNAP-DksA interface accounts for its dramatic effects on transcription initiation during the stringent response. Mol Cell 62:811–823. doi: 10.1016/j.molcel.2016.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Blattner FR, Plunkett G 3rd, Bloch CA, Perna NT, Burland V, Riley M, Collado-Vides J, Glasner JD, Rode CK, Mayhew GF, Gregor J, Davis NW, Kirkpatrick HA, Goeden MA, Rose DJ, Mau B, Shao Y. 1997. The complete genome sequence of Escherichia coli K-12. Science 277:1453–1462. doi: 10.1126/science.277.5331.1453. [DOI] [PubMed] [Google Scholar]
- 62.Freddolino PL, Amini S, Tavazoie S. 2012. Newly identified genetic variations in common Escherichia coli MG1655 stock cultures. J Bacteriol 194:303–306. doi: 10.1128/JB.06087-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Soupene E, van Heeswijk WC, Plumbridge J, Stewart V, Bertenthal D, Lee H, Prasad G, Paliy O, Charernnoppakul P, Kustu S. 2003. Physiological studies of Escherichia coli strain MG1655: growth defects and apparent cross-regulation of gene expression. J Bacteriol 185:5611–5626. doi: 10.1128/JB.185.18.5611-5626.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sonnenborn U. 2016. Escherichia coli strain Nissle 1917-from bench to bedside and back: history of a special Escherichia coli strain with probiotic properties. FEMS Microbiol Lett 363:fnw212. doi: 10.1093/femsle/fnw212. [DOI] [PubMed] [Google Scholar]
- 65.Schreiber G, Metzger S, Aizenman E, Roza S, Cashel M, Glaser G. 1991. Overexpression of the relA gene in Escherichia coli. J Biol Chem 266:3760–3767. [PubMed] [Google Scholar]
- 66.Li W, Bouveret E, Zhang Y, Liu K, Wang JD, Weisshaar JC. 2016. Effects of amino acid starvation on RelA diffusive behavior in live Escherichia coli. Mol Microbiol 99:571–585. doi: 10.1111/mmi.13252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hernandez VJ, Bremer H. 1993. Characterization of RNA and DNA synthesis in Escherichia coli strains devoid of ppGpp. J Biol Chem 268:10851–10862. [PubMed] [Google Scholar]
- 68.Metzger S, Dror IB, Aizenman E, Schreiber G, Toone M, Friesen JD, Cashel M, Glaser G. 1988. The nucleotide sequence and characterization of the relA gene of Escherichia coli. J Biol Chem 263:15699–15704. [PubMed] [Google Scholar]
- 69.Harms A, Fino C, Sorensen MA, Semsey S, Gerdes K. 2017. Prophages and growth dynamics confound experimental results with antibiotic-tolerant persister cells. mBio 8:e01964-17. doi: 10.1128/mBio.01964-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Murphy H, Cashel M. 2003. Isolation of RNA polymerase suppressors of a (p)ppGpp deficiency. Methods Enzymol 371:596–601. doi: 10.1016/S0076-6879(03)71044-1. [DOI] [PubMed] [Google Scholar]
- 71.Zhou YN, Jin DJ. 1998. The rpoB mutants destabilizing initiation complexes at stringently controlled promoters behave like “stringent” RNA polymerases in Escherichia coli. Proc Natl Acad Sci U S A 95:2908–2913. doi: 10.1073/pnas.95.6.2908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bartlett MS, Gaal T, Ross W, Gourse RL. 1998. RNA polymerase mutants that destabilize RNA polymerase-promoter complexes alter NTP-sensing by rrn P1 promoters. J Mol Biol 279:331–345. doi: 10.1006/jmbi.1998.1779. [DOI] [PubMed] [Google Scholar]
- 73.Hernandez VJ, Cashel M. 1995. Changes in conserved region 3 of Escherichia coli sigma 70 mediate ppGpp-dependent functions in vivo. J Mol Biol 252:536–549. doi: 10.1006/jmbi.1995.0518. [DOI] [PubMed] [Google Scholar]
- 74.Vinella D, Potrykus K, Murphy H, Cashel M. 2012. Effects on growth by changes of the balance between GreA, GreB, and DksA suggest mutual competition and functional redundancy in Escherichia coli. J Bacteriol 194:261–273. doi: 10.1128/JB.06238-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kvint K, Hosbond C, Farewell A, Nybroe O, Nystrom T. 2000. Emergency derepression: stringency allows RNA polymerase to override negative control by an active repressor. Mol Microbiol 35:435–443. doi: 10.1046/j.1365-2958.2000.01714.x. [DOI] [PubMed] [Google Scholar]
- 76.Jin DJ. 1996. A mutant RNA polymerase reveals a kinetic mechanisms for the switch between nonproductive stuttering synthesis and productive initiation during promoter clearance. J Biol Chem 271:11659–11667. [PubMed] [Google Scholar]
- 77.Singer M, Baker TA, Schnitzler G, Deischel SM, Goel M, Dove W, Jaacks KJ, Grossman AD, Erickson JW, Gross CA. 1989. A collection of strains containing genetically linked alternating antibiotic resistance elements for genetic mapping of Escherichia coli. Microbiol Rev 53:1–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Nichols BP, Shafiq O, Meiners V. 1998. Sequence analysis of Tn10 insertion sites in a collection of Escherichia coli strains used for genetic mapping and strain construction. J Bacteriol 180:6408–6411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hsieh YJ, Wanner BL. 2010. Global regulation by the seven-component Pi signaling system. Curr Opin Microbiol 13:198–203. doi: 10.1016/j.mib.2010.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kimura S, Makino K, Shinagawa H, Amemura M, Nakata A. 1989. Regulation of the phosphate regulon of Escherichia coli: characterization of the promoter of the pstS gene. Mol Gen Genet 215:374–380. doi: 10.1007/BF00427032. [DOI] [PubMed] [Google Scholar]
- 81.Magnusson LU, Gummesson B, Joksimovic P, Farewell A, Nystrom T. 2007. Identical, independent, and opposing roles of ppGpp and DksA in Escherichia coli. J Bacteriol 189:5193–5202. doi: 10.1128/JB.00330-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Aberg A, Fernández-Vázquez J, Cabrer-Panes JD, Sánchez A, Balsalobre C. 2009. Similar and divergent effects of ppGpp and DksA deficiencies on transcription in Escherichia coli. J Bacteriol 191:3226–3236. doi: 10.1128/JB.01410-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Brown L, Gentry D, Elliott T, Cashel M. 2002. DksA affects ppGpp induction of RpoS at a translational level. J Bacteriol 184:4455–4465. doi: 10.1128/JB.184.16.4455-4465.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Aberg A, Shingler V, Balsalobre C. 2008. Regulation of the fimB promoter: a case of differential regulation by ppGpp and DksA in vivo. Mol Microbiol 67:1223–1241. doi: 10.1111/j.1365-2958.2008.06115.x. [DOI] [PubMed] [Google Scholar]
- 85.Borukhov S, Laptenko O, Lee J. 2001. Escherichia coli transcript cleavage factors GreA and GreB: functions and mechanisms of action. Methods Enzymol 342:64–76. doi: 10.1016/S0076-6879(01)42536-5. [DOI] [PubMed] [Google Scholar]
- 86.Perederina A, Svetlov V, Vassylyeva MN, Tahirov TH, Yokoyama S, Artsimovitch I, Vassylyev DG. 2004. Regulation through the secondary channelstructural—framework for ppGpp-DksA synergism during transcription. Cell 118:297–309. doi: 10.1016/j.cell.2004.06.030. [DOI] [PubMed] [Google Scholar]
- 87.Borukhov S, Sagitov V, Goldfarb A. 1993. Transcript cleavage factors from E. coli. Cell 72:459–466. doi: 10.1016/0092-8674(93)90121-6. [DOI] [PubMed] [Google Scholar]
- 88.Rutherford ST, Lemke JJ, Vrentas CE, Gaal T, Ross W, Gourse RL. 2007. Effects of DksA, GreA, and GreB on transcription initiation: insights into the mechanisms of factors that bind in the secondary channel of RNA polymerase. J Mol Biol 366:1243–1257. doi: 10.1016/j.jmb.2006.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Opalka N, Chlenov M, Chacon P, Rice WJ, Wriggers W, Darst SA. 2003. Structure and function of the transcription elongation factor GreB bound to bacterial RNA polymerase. Cell 114:335–345. doi: 10.1016/S0092-8674(03)00600-7. [DOI] [PubMed] [Google Scholar]
- 90.Boutte CC, Henry JT, Crosson S. 2012. ppGpp and polyphosphate modulate cell cycle progression in Caulobacter crescentus. J Bacteriol 194:28–35. doi: 10.1128/JB.05932-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Racki LR, Tocheva EI, Dieterle MG, Sullivan MC, Jensen GJ, Newman DK. 2017. Polyphosphate granule biogenesis is temporally and functionally tied to cell cycle exit during starvation in Pseudomonas aeruginosa. Proc Natl Acad Sci U S A 114:E2440–E2449. doi: 10.1073/pnas.1615575114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Furman R, Danhart EM, NandyMazumdar M, Yuan C, Foster MP, Artsimovitch I. 2015. pH dependence of the stress regulator DksA. PLoS One 10:e0120746. doi: 10.1371/journal.pone.0120746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Crawford MA, Tapscott T, Fitzsimmons LF, Liu L, Reyes AM, Libby SJ, Trujillo M, Fang FC, Radi R, Vazquez-Torres A. 2016. Redox-active sensing by bacterial DksA transcription factors is determined by cysteine and zinc content. mBio 7:e02161. doi: 10.1128/mBio.02161-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Crawford MA, Henard CA, Tapscott T, Porwollik S, McClelland M, Vazquez-Torres A. 2016. DksA-dependent transcriptional regulation in Salmonella experiencing nitrosative stress. Front Microbiol 7:444. doi: 10.3389/fmicb.2016.00444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Henard CA, Tapscott T, Crawford MA, Husain M, Doulias PT, Porwollik S, Liu L, McClelland M, Ischiropoulos H, Vazquez-Torres A. 2014. The 4-cysteine zinc-finger motif of the RNA polymerase regulator DksA serves as a thiol switch for sensing oxidative and nitrosative stress. Mol Microbiol 91:790–804. doi: 10.1111/mmi.12498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Girard ME, Gopalkrishnan S, Grace ED, Halliday JA, Gourse RL, Herman C. 2017. DksA and ppGpp regulate the sigma(S) stress response by activating promoters for the small RNA DsrA and the anti-adapter protein IraP. J Bacteriol 200:e00463-17. doi: 10.1128/JB.00463-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Sambrook J, Fritsch EF, Maniatis T. 1989. Molecular cloning: a laboratory manual, 2nd ed Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 98.Guo B, Bi Y. 2002. Cloning PCR products. An overview. Methods Mol Biol 192:111–119. doi: 10.1385/1-59259-177-9:111. [DOI] [PubMed] [Google Scholar]
- 99.Markowitz VM, Chen IM, Palaniappan K, Chu K, Szeto E, Grechkin Y, Ratner A, Jacob B, Huang J, Williams P, Huntemann M, Anderson I, Mavromatis K, Ivanova NN, Kyrpides NC. 2012. IMG: the Integrated Microbial Genomes database and comparative analysis system. Nucleic Acids Res 40:D115–D122. doi: 10.1093/nar/gkr1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Bertani G. 1951. Studies on lysogenesis. I. The mode of phage liberation by lysogenic Escherichia coli. J Bacteriol 62:293–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Neidhardt FC, Bloch PL, Smith DF. 1974. Culture medium for enterobacteria. J Bacteriol 119:736–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Silhavy TJ, Berman ML, Enquist LW (ed). 1984. Experiments with gene fusions. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY. [Google Scholar]
- 103.Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H. 2006. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol 2:2006.0008. doi: 10.1038/msb4100050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A 97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Rotman E, Amado L, Kuzminov A. 2010. Unauthorized horizontal spread in the laboratory environment: the tactics of Lula, a temperate lambdoid bacteriophage of Escherichia coli. PLoS One 5:e11106. doi: 10.1371/journal.pone.0011106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Vejborg RM, Friis C, Hancock V, Schembri MA, Klemm P. 2010. A virulent parent with probiotic progeny: comparative genomics of Escherichia coli strains CFT073, Nissle 1917 and ABU 83972. Mol Genet Genomics 283:469–484. doi: 10.1007/s00438-010-0532-9. [DOI] [PubMed] [Google Scholar]
- 107.Sawitzke JA, Thomason LC, Costantino N, Bubunenko M, Datta S, Court DL. 2007. Recombineering: in vivo genetic engineering in E. coli, S. enterica, and beyond. Methods Enzymol 421:171–199. doi: 10.1016/S0076-6879(06)21015-2. [DOI] [PubMed] [Google Scholar]
- 108.Jin DJ, Gross CA. 1988. Mapping and sequencing of mutations in the Escherichia coli rpoB gene that lead to rifampicin resistance. J Mol Biol 202:45–58. doi: 10.1016/0022-2836(88)90517-7. [DOI] [PubMed] [Google Scholar]
- 109.Jin DJ, Gross CA. 1989. Characterization of the pleiotropic phenotypes of rifampin-resistant rpoB mutants of Escherichia coli. J Bacteriol 171:5229–5231. doi: 10.1128/jb.171.9.5229-5231.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Jin DJ, Cashel M, Friedman DI, Nakamura Y, Walter WA, Gross CA. 1988. Effects of rifampicin resistant rpoB mutations on antitermination and interaction with nusA in Escherichia coli. J Mol Biol 204:247–261. doi: 10.1016/0022-2836(88)90573-6. [DOI] [PubMed] [Google Scholar]
- 111.Jin DJ, Walter WA, Gross CA. 1988. Characterization of the termination phenotypes of rifampicin-resistant mutants. J Mol Biol 202:245–253. doi: 10.1016/0022-2836(88)90455-X. [DOI] [PubMed] [Google Scholar]
- 112.Guzman LM, Belin D, Carson MJ, Beckwith J. 1995. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J Bacteriol 177:4121–4130. doi: 10.1128/jb.177.14.4121-4130.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Wurst H, Kornberg A. 1994. A soluble exopolyphosphatase of Saccharomyces cerevisiae. Purification and characterization. J Biol Chem 269:10996–11001. [PubMed] [Google Scholar]
- 114.Carter SG, Karl DW. 1982. Inorganic phosphate assay with malachite green: an improvement and evaluation. J Biochem Biophys Methods 7:7–13. doi: 10.1016/0165-022X(82)90031-8. [DOI] [PubMed] [Google Scholar]
- 115.Wilson KJ, Hughes SG, Jefferson RA. 1992. The Escherichia coli gus operon: induction and expression of the gus operon in E. coli and the occurence and use of GUS in other bacteria, p 7–22. In Gallagher SR.(ed), GUS protocols using the GUS gene as a reporter of gene expression. Academic Press, Inc., San Diego, CA. [Google Scholar]
- 116.Brickman E, Beckwith J. 1975. Analysis of the regulation of Escherichia coli alkaline phosphatase synthesis using deletions and phi80 transducing phages. J Mol Biol 96:307–316. doi: 10.1016/0022-2836(75)90350-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All strains and plasmids generated in the course of this work are available from the author upon request.