

Abstract
HLA class Ι molecules can communicate a range of cellular alterations (mutations, changes in protein copy number, aberrant post-translational modifications, or pathogen proteins) to CD8+ T lymphocytes in the form of HLA peptide ligands. At any given moment, tens of thousands of different self and foreign HLA class Ι peptides may be presented on the cell surface by HLA class Ι complexes. Due to the enormous biochemical diversity and low abundance of each of these peptides, HLA ligandome analysis presents unique challenges. Even with advances in enrichment strategies and MS instrumentation and fragmentation, sufficient ligandome depth for identification of viral pathogens and immuno therapeutically important tumor neo-antigens is still not routinely achievable. In this study, we evaluated two pre-fractionation techniques, high-pH reversed-phase and strong cation exchange, for the complementary analyses of HLA class Ι peptide ligands. We observe that pre-fractionation substantially extends the detectable HLA class Ι ligandome but also creates an identification bias. We thus advocate a rational choice between high-pH reversed-phase or strong cation exchange pre-fractionation for deeper HLA class Ι ligandome analysis, depending on the HLA locus, allele, or peptide ligand modification in question.
Keywords: HLA class Ι ligandome, HLA class Ι peptide ligands, high-pH reversed-phase, strong cation exchange, pre-fractionation
Introduction
One critical function of the immune system is to counteract cancer development by identifying abnormal cells for destruction through the recognition of neo-antigens presented on these cells by human leukocyte antigen (HLA) molecules. Promising strategies in cancer immunotherapy target these neo-antigens, which arise from the proteolytic processing of tumor-specific and mutated proteins.1 While some of these cancer signatures can be picked up by next-generation sequencing at the DNA or RNA level, mass spectrometry (MS) remains the method of choice that enables the direct measurement and identification of neo-antigens that are presented on the tumor cell surface. Therefore, ultrasensitive MS profiling methodologies are needed to delve deeper into the HLA class Ι ligandome to support the rational design of immunotherapy at a personalized level.2,3
HLA class Ι peptide ligands are challenging to analyze due to enormous diversity in the repertoire and the inherently low abundance of most of these species. Through the advances in mass spectrometry over the recent decades, the HLA class Ι peptide ligandome can now be analyzed in depth, reaching to the detection of hundreds up to thousands of unique peptide ligands per cell line or tissue sample. We previously demonstrated that the use of complementary peptide fragmentation techniques, especially EThcD as an alternative “spectra-rich” fragmentation method, can further boost the identification of HLA class Ι peptide ligands, including those post-translationally modified by arginine methylation, glycosylation and phosphorylation.4−6 Sample pre-fractionation, notably by strong cation exchange (SCX), has also been shown to further increase the detection depth of the HLA class Ι ligandome.7 The outcomes of these deep HLA class Ι peptide ligand profiling experiments have prominently altered the developmental path of immunotherapeutics but also indicate that we still have plenty to gain by delving deeper into the HLA class Ι peptide ligandome.3
The analysis of HLA class Ι ligandomes benefit in general from advances in shotgun proteomics because not all but many experimental aspects of these analyses are shared. With the advent of high-pH reversed-phase (RP) fractionation in 20128−11 in shotgun proteomics, it has become widely accepted as a robust and high-performance alternative to SCX.12−14 Given the ideal compatibility of high-pH RP pre-fractionation to the second dimension, low-pH reversed-phase liquid chromatography–mass spectrometry (RP-LC–MS), without the need for additional desalting procedures, many laboratories have adopted high-pH RP for routine proteomics analyses. We reasoned that reducing purification steps between HLA class Ι peptide ligand isolation and MS analysis could further minimize the loss of low abundant HLA class Ι peptide species and extend the identification depth. To this end, we benchmarked the performance of high-pH RP pre-fractionation and SCX pre-fractionation against “no-fractionation” analysis by using an Epstein–Barr virus transformed immortalized B lymphoblastoid cell line, JY. This cell line is a routinely used model cell line in the field of immuno-peptidomics (that is, homozygous for HLA-A*02:01, HLA-B*07:02, and HLA-C*07:02).
We describe here high-pH RP pre-fractionation on HLA class Ι peptide ligands as a reliable alternative approach for detailed HLA class Ι ligandome profiling (that is, highly complementary to the conventional SCX). We observed that high-pH RP performed comparably to SCX in terms of total HLA class Ι peptide ligand identification but presents a distinct allele-specific identification bias that could be exploited for focused studies of hydrophobic peptide ligands and post-translationally modified peptide ligands, especially for charge-reducing modifications such as phosphorylation and citrullination. We conclude that the choice between these two pre-fractionation methods will depend on rational consideration of the research question surrounding a particular HLA locus, allele, or peptide ligand post-translational modification.
Materials and Methods
Cell Culture
The HLA class Ι homozygous cell line JY (HLA-A*02:01, HLA-B*07:02, and HLA-C*07:02) was cultured in RPMI 1640 supplemented with 10% fetal bovine serum, 10 mM l-glutamine, 50U/mL, penicillin and 50 μg/mL streptomycin in a humidified atmosphere at 37 °C with 5% CO2.
Immuno-Affinity Purification
Per immuno-affinity purification (IP), 5 × 108 cells were harvested by centrifugation and washed three times with cold phosphate-buffered saline (PBS). Cells were lysed for 1.5 h at 4 °C in 10 mL of lysis buffer per gram of cell pellet. The lysis buffer consisted of Pierce IP lysis buffer (Thermo Fischer Scientific) supplemented with 1× complete protease inhibitor cocktail (Roche Diagnostics), 50 μg/mL DNase Ι (Sigma-Aldrich), and 50 μg/mL RNase A (Sigma-Aldrich). Subsequently, the lysate was cleared by centrifugation for 1 h at 18000g at 4 °C. Protein concentration was determined with the Bradford assay (Bio-Rad). HLA class Ι complexes and peptide ligands were immunoprecipitated using 0.5 mg W6/32 antibody15 coupled to 125 μL of Protein A/G beads (Santa Cruz) from 25 mg of whole-cell lysate. Antibodies were cross-linked to protein A/G beads to prevent coelution. Incubation took place at 4 °C for approximately 16 h. After immunoprecipitation, the beads were washed with 40 mL of cold PBS. HLA class Ι complexes and peptide ligands were subsequently eluted with 10% acetic acid. Peptide ligands were separated from HLA class Ι complexes using 10 kDa molecular weight cutoff filters (Millipore). The flowthrough containing the HLA class Ι peptide ligands was dried by vacuum centrifugation.
Peptide Fractionation
To test the performance of high-pH RP and SCX fractionation against identification with no pre-fractionation, we pooled HLA peptide material derived from 9 IP equivalents and divided the sample into 3 equal parts for (i) the injection of 12 high-pH RP fractions, (ii) the injection of 12 SCX fractions, or (iii) 12 repeated injections of unfractionated sample. In high-pH reversed-phase fractionation, peptides were loaded on C18 STAGE-tips in 200 mM ammonium formate at pH 10 and eluted into 12 fractions with 11–100% acetonitrile. For strong cation exchange, peptides were loaded on SCX SPE cartridges (1 mg, Supelco) in 20% acetonitrile with 0.1% formic acid and eluted into 12 fractions with 50–500 mM ammonium acetate. All samples were dried by vacuum centrifugation and reconstituted in 10% formic acid prior to LC–MS/MS analyses.
LC–MS/MS
The data was acquired with an UHPLC 1290 system (Agilent) coupled to an Orbitrap Fusion Lumos Tribrid mass spectrometer (Thermo Fischer Scientific). Peptides were trapped (Dr Maisch Reprosil C18, 3 μM, 2 cm × 100 μM) for 5 min in solvent A (0.1% formic acid in water) before being separated on an analytical column (Agilent Poroshell, EC-C18, 2.7 μm, 50 cm × 75 μm). Solvent B consisted of 0.1% formic acid in 80% acetonitrile. For high-pH reversed-phase samples (fraction 1 and 2), the gradient was as follows: first 5 min of trapping, followed by 85 min of gradient from 12 to 30% solvent B and, subsequently, 10 min of washing with 100% solvent B and 10 min of re-equilibration with 100% solvent A. For fraction 3 and 4 the gradient was from 15 to 32% solvent B. For fraction 5 and 6 the gradient was from 18 to 36% solvent B. For fraction 7 to 10 the gradient was from 20 to 38% solvent B and for fraction 11 and 12 from 22 to 44% solvent B. For the SCX fractions, the gradient was as follows: first 5 min of trapping, followed by 85 min of gradient from 7 to 35% solvent B and, subsequently, 10 min of washing with 100% solvent B and 10 min re-equilibration with 100% solvent A. The mass spectrometer operated in data-dependent mode. Full scan MS spectra from m/z 400–650 were acquired at a resolution of 60 000 after accumulation to a target value or 4 × 105 or a maximum injection time of 50 ms. Up to 3 most intense precursors with a charge state of 2+ or 3+ starting at m/z 100 were chosen for fragmentation. EThcD fragmentation was performed at 35% normalized collision energy on selected precursors with 18s dynamic exclusion after accumulation of 5 × 104 ions or a maximum injection time of 250 ms. Tandem mass spectrometry (MS/MS) spectra were acquired at a resolution of 15 000.
Data Analysis
Raw files were searched using Sequest HT in Proteome Discoverer 2.2 against the Swissprot human database (20 258 entries, downloaded on Feb 2nd, 2018) appended with the 20 most abundant FBS contaminants.16 The search was set to unspecific with a minimum precursor mass of 797 Da to a maximum precursor mass of 1950 Da corresponding to peptides between 8 and 12 amino acids long. Identified peptides were filtered to a 1% false discovery rate (FDR) using the percolator algorithm, 5% peptide FDR, and Xcorr of >1. Cysteine cysteinylation and methionine oxidation were set as variable modifications. Serine phosphorylation and arginine citrullination were set as variable modifications in separate workflows. From the identified peptides, FBS contaminants were removed. The mass spectrometry peptidomics data have been deposited to the ProteomeXchange Consortium via the PRIDE17 partner repository with the data set identifier PXD011257. Binding affinity of HLA class Ι peptide ligands was predicted using the NetMHC 4.0 algorithm18 with a stringent binder cutoff of IC50 < 500 nM. The data was visualized with Graphpad PRISM 7.
Results
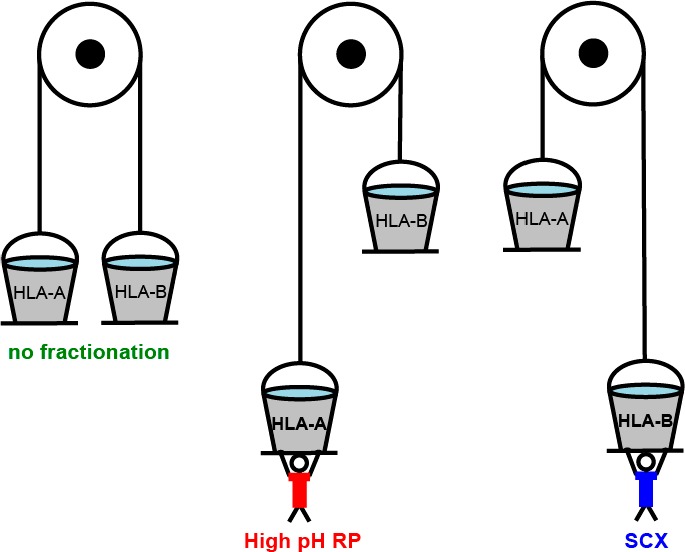
To compare the effectiveness of offline high-pH reversed-phase and strong cation exchange pre-fractionation in extending coverage of the HLA class Ι ligandome, we started with a “master pool” of immuno-affinity purified peptide ligands (nine IP equivalents). This master pool was split into three equal samples. A pair of these were subsequently either separated into 12 high-pH RP fractions or 12 SCX fractions prior to further LC–MS/MS analyses (Figure 1). The final 1/3 was analyzed as 12 repeated MS injections without pre-fractionation. Ideally, a single injection of 12 times the sample load should be used for a comparison against fractionation. This was, however, not possible because LC overloading would compromise separation. Instead, we analyzed the same sample load in 12 repeated injections using the same total MS time.
Figure 1.

Experimental workflow applied for HLA class Ι peptide ligand analysis. (A) Cell harvest and lysis steps for lysate preparation for HLA class Ι immuno-affinity purification. (B) HLA class Ι complex and peptide ligand immuno-affinity purification using W6/32 antibodies coupled to protein A/G beads. (C) HLA peptide ligand purification, fractionation, and identification.
Fractionation Reaches Deeper into the HLA Class Ι Ligandome
Without any pre-fractionation, we identified approximately 6500 ± 100 peptides per single LC–MS injection, of which 5700 ± 100 were HLA class Ι peptide ligands. Repeated injections of the same unfractionated sample did not substantially increase the cumulative identification further; with an excellent identification overlap of >95% between injection replicates, the same peptide ligand species were fragmented over 12 repeated injections (Figure 2A.B). This implied that the limit in identification was not due to stochastic sampling (Figure 2A), making it also very clear that for deeper analysis of the HLA class Ι ligandome, sample pre-fractionation is essential.
Figure 2.

HLA class Ι peptide ligand identification. (A) Cumulative unique peptides (black), predicted peptide ligands (green), and PSM counts (gray) over repeated injections of the same unfractionated sample. (B) Peptide identification overlap between repeated injections of the same unfractionated sample. (C) Cumulative unique peptides (black), predicted peptide ligands (red), and PSM (gray) counts over all analyzed high-pH RP fractions. (D) Cumulative unique peptides (black), predicted peptide ligands (blue), and PSM (gray) counts over all analyzed SCX fractions. (E) Overlap between identified peptides in high-pH RP (n = 10737, red) and SCX (n = 10511, blue). (F) Cumulative unique peptides (black), predicted peptide ligands (orange), and PSM (gray) count over combined high-pH RP and SCX fractions.
By pre-fractionation of the immuno-purified HLA class Ι ligandome into 12 fractions, with either SCX or high-pH RP, we were able to extend the number of identifications to above 10 000 peptides, of which about 8600 were HLA class Ι peptide ligands (Figure 2C,D). Considering the total number of identifications, either pre-fractionation method could increase identifications by ∼50% compared to analysis without fractionation. However, the overlap in identified peptide ligands between high-pH RP fractionation and SCX fractionation turned out to be only ∼40%, alluding to the idea that these fractionation methods are very complementary in ligandome analyses (Figure 2E). Indeed, when we combined the data from both fractionation methods, we identified 13 500 peptides, of which 11 200 HLA class Ι peptide ligands (Figure 2F), representing an increase of ∼100% in identified HLA class Ι peptide ligands compared to the analysis of the same samples but unfractionated. Therefore, we demonstrate here for the first time that high-pH reversed-phase fractionation is a good alternative to SCX for the deeper analysis of HLA class Ι peptide ligands. High-pH reversed-phase and SCX fractionation also appear to be complementary in the analysis of HLA class Ι peptide ligands, both reaching much deeper into the HLA class Ι peptide ligandome, compared to unfractionated analysis.
Boosting HLA Class Ι Peptide Ligand Extraction
While pre-fractionation boosts identification of HLA class Ι peptide ligands considerably (Figure 2), more starting material is also needed than in single-shot LC-MS analyses. This is often a limiting bottleneck, especially in the context of ligandome profiling from patient material. Because obtaining larger biopsies is not feasible, maximizing the extraction of HLA class Ι complexes and peptide ligands from limited material is then crucial to maximize ligandome coverage, for more in-depth patient-specific analyses.
It has been demonstrated that immuno-affinity purifications cannot fully deplete target protein complexes in one cycle, largely due to affinity characteristics of antibody-based capture and equilibrium dissociation.19,20 Therefore, we further tested if recycling the flowthrough from immuno-affinity purification for sequential enrichments could still increase the yield of HLA class Ι peptide ligands. We performed sequential immuno-affinity purifications using the same starting cell lysate, injected these samples in separate LC–MS runs with the same parameters, and then analyzed the purity of HLA class Ι peptide ligands measured. As shown in panels A and B of Figure 3, approximately 5000 peptides were identified from the first and second IP, of which about 4500 were HLA class Ι peptide ligands. In the third IP, the purity was still good, although the total identification number was about 10% lower than in the first two IPs. By the fourth reuse of the same lysate, peptide ligand identification declined to approximately 2600, with a marginal increase of unassigned peptides (15%), suggesting the lysate was starting to become depleted of HLA class Ι complexes. Collectively, these data demonstrate that the same starting material for HLA class Ι peptide ligand immuno-affinity purification may be reused for several times for repeated extraction of the same HLA class Ι complexes. We further verified that the HLA class Ι peptide ligands identified in sequential IPs share a large overlap of ∼80%, validating that repeated use of the same starting material does not introduce profiling bias or sampling artifacts associated with extended incubation times (Figure 3C). Because we performed these experiments in “bulk incubation” mode with free agarose beads, it remains to be verified empirically if these observations are transferrable to immunoaffinity capture in resin-filled tip format.
Figure 3.

HLA class Ι peptide ligand identification in sequential immuno-affinity purifications using single LC–MS/MS runs. (A) Unique peptide (black) and predicted peptide ligand (gray) identifications. (B) Proportion of HLA-A*02:01, HLA-B*07:02, and HLA-C*07:02 peptide ligands. (C) Identification overlap of the first three sequential IPs.
Peptide Characteristics of HLA Class Ι Peptide Ligands Detected with High-pH Reversed-Phase or SCX Pre-fractionation
By pooling HLA class Ι peptide ligands obtained from two sets of immuno-affinity purifications, each performed for three sequential times, we created a common pool of HLA class Ι peptide ligands that was then used to compare and contrast the properties of peptide ligands identified by either high-pH RP or SCX fractionation. The goal was to rationalize the substantial nonoverlap in the HLA peptide ligand identifications between these two complementary pre-fractionation approaches (Figure 2E).
Specifically, we examined peptide characteristics within predicted ligands for each HLA allele (HLA-A*02:01, HLA-B*07:02, and HLA-C*07:02) to check for potential bias in peptide lengths (Figure 4A) and sequence motifs (Figure 4B). HLA class Ι peptide ligands are typically between 8 and 12 amino acids long, with 9-residue ligands being the most frequent. By comparing the peptide ligands identified with and without pre-fractionation, we found no significant difference in the peptide length distribution (Figure 4A). Among HLA-A*02:01 peptide ligands, ∼70% were 9-mers, ∼20% 10-mers, and <10% were 11-mers regardless of pre-fractionation approach. Comparable trends were also observed in HLA-B*07:02 and HLA-C*07:02.
Figure 4.

Comparable HLA class Ι peptide ligand characteristics in high-pH RP, SCX, and without fractionation. (A) Length distribution of HLA-A*02:01 peptide ligands (high-pH RP, n = 4705; SCX, n = 3419; no fractionation, n = 2921), HLA-B*07:02 peptide ligands (high-pH RP, n = 4075;, SCX, n = 4825; no fractionation, n = 2804), and HLA-C*07:02 peptide ligands (high-pH RP, n = 389; SCX, n = 290; no fractionation, n = 252). (B) Gibbs clustering21 sequence motifs for HLA-A*02:01, HLA-B*07:02, and HLA-C*07:02 peptides identified with high-pH RP, SCX, or without pre-fractionation.
Regardless of the fractionation approach used, HLA-A*02:01 ligands anchor with leucine at position 2 and a C-terminal leucine/valine; HLA-B*07:02 ligands show enrichment for proline at position 2 and a C-terminal leucine/valine; whereas HLA-C*07:02 ligands over-represent arginine at position 2 and a C-terminal leucine/phenylalanine. Taken together, our data here suggest that within ligands of the same HLA allele, increased identification through pre-fractionation appears not to correlate with unique peptide properties that induce preferential binding and/or separation in either high-pH RP or SCX. This could be tested further with more focused HLA peptide ligandome profiling using monoallelic cell lines, allele-specific monoclonal antibodies, or both.
High-pH Reversed-Phase or SCX Fractionation can Introduce an Allele-Specific Ligand Identification Bias
We next sought to explain the substantial non-overlap in HLA class Ι peptide ligand identification in high-pH RP or SCX by calculating the contribution of peptide ligands, from each allele, to total ligand identification in the JY cell line (Figure 5). Specifically, we examined separately the proportion of HLA-A*02:01, HLA-B*07:02, and HLA-C*07:02 peptide ligands in high-pH RP or SCX fractionation to check for any allele-specific bias. Using binding affinity of peptides, predicted by NetMHC 4.0 with a stringent cutoff of IC50 < 500 nM, we binned identified peptide ligands by allele specificity. As shown in Figure 5, except for a small percentage of HLA-C*07:02 ligands and 11% non-assigned peptides, HLA-A*02:01 and HLA-B*07:02 ligands were represented almost evenly without pre-fractionation (41.0% and 44.1%, respectively) using the data from 12 repeated injections of the same sample (Figure 5A). However, with pre-fractionation, HLA-A*02:01 ligands appear to be over-represented in high-pH RP, while HLA-B*07:02 ligands appear to be identified more frequently with SCX. Because HLA-C*07:02 ligands represent only a small and consistent proportion in either fractionation approach, we visualized only the binary distribution of HLA-A*02:01 and HLA-B*07:02 ligands in Figure 5B, where a clear bias in HLA allele specificity was observed compared to analysis without pre-fractionation.
Figure 5.

Bias in allele-specific HLA class Ι peptide ligand identification in high-pH RP and SCX compared to analysis without fractionation. (A) Identification of HLA-A*02:01, HLA-B*07:02, and HLA-C*07:02 peptide ligands per fractionation method. (B) Identification bias in HLA-A*02:01 and HLA-B*07:02 peptide ligands when using high-pH RP fractionation, SCX fractionation, or no fractionation. (C) Cumulative unique HLA-A*02:01 peptide ligand count in high-pH RP (red), SCX (blue), or without fractionation (green). (D) Isoelectric point distribution of all identified HLA-A*02:01 peptide ligands. (E) Cumulative unique HLA-B*07:02 peptide ligand count in high-pH RP (red), SCX (blue), or without fractionation (green). (F) Isoelectric point distribution of all identified HLA-B*07:02 peptide ligands.
By following cumulative peptide ligand identification over either 12 high-pH RP or 12 SCX fractions, we further rationalized the lower identification of HLA-A*02:01 ligands in SCX. As shown in Figure 5C, the cumulative increase in unique HLA-A*02:01 ligands was restricted to the first 3 SCX fraction in a linear salt gradient, while HLA class Ι peptide ligands continue to be fragmented over the 12 fractions (Figure 2D, cumulative PSM count). This indicates that HLA-A*02:01 binders are not optimally separated by using the charge selective SCX gradient, likely due to higher peptide hydrophobicity attributed to the leucine anchor and prevalent leucine/valine C-terminus. We assessed the isoelectric point distribution of HLA-A*02:01 peptides, which revealed a striking majority of peptides with an isoelectric point between 3 and 7. These peptides would be negatively charged at pH 3 and therefore not optimally separated on a positive-charge selective SCX gradient (Figure 5D). However, HLA-B*07:02 ligands feature prominently arginine residues flanking the proline anchor at position 2. This makes HLA-B*07:02 ligands more charged in low-pH SCX and therefore better separated and consequently better identified in SCX across all 12 fractions compared to high-pH RP (Figure 5E). Assessment of the isoelectric point distribution of these peptides again confirms that more HLA-B*07:02 peptide ligands have isoelectric points between 8 and 12 (Figure 5F). Based on these observations we therefore conclude that in high-pH RP there is preferential identification of HLA-A*02:01 peptide ligands, whereas SCX performs better in the analysis of HLA-B*07:02 peptide ligands.
High-pH Reversed-Phase Fractionation and Enabling of In-Depth Profiling of HLA Class Ι Peptide Ligands Containing Serine Phosphorylations
Serine phosphorylation on HLA class Ι peptide ligands have been reported previously and may function as neo-antigens arising from aberrant phosphorylation in tumors.6,22,23 Because serine phosphorylation reduces the charge of peptides at low pH, we reasoned serine phosphorylated HLA class Ι peptide ligands would be better separated and identified using high-pH RP pre-fractionation compared to SCX.24 We therefore attempted to detect and localize serine phosphorylation on HLA class Ι peptide ligands with either high-pH RP or SCX fractionation (Figure 6A–C). In total, we detected 45% more phosphopeptides using the high-pH RP workflow than SCX (90 phospho-HLA peptides versus 62, respectively; Table S1), in strong agreement with our theoretical understanding. Remarkably, the SCX workflow even provided a lower number of phosphorylated peptide ligands than the unfractionated workflow (62 phospho-HLA peptides versus 74, respectively; Table S1). As also shown in Figure 6A, serine phosphorylation occurs predominantly on HLA-B*07:02 peptide ligands and is indeed most frequently detected in high-pH RP fractionation.
Figure 6.

Serine phosphorylation and arginine citrullination on JY HLA class Ι peptide ligands preferred on HLA-B*07:02. (A) HLA-assigned peptides containing phosphoserine distributed per allele per fractionation method. (B) Serine phosphorylation per position in HLA-B*07:02 peptide ligands identified with high-pH RP compared to overall serine percentage per position in HLA-B*07:02 peptide ligands. (C) Gibbs cluster sequence logo from HLA-B*07:02 phosphorylated peptides (n = 66) and motifs specific for proline directed kinases (n = 32) and basophilic kinases (n = 34). (D) HLA-assigned peptides containing citrullinated arginine distributed per allele per fractionation method. (E) Arginine citrullination per position in HLA-B*07:02 peptide ligands identified with high-pH RP compared to overall arginine percentage per position in HLA-B*07:02 peptide ligands. (F) Gibbs cluster sequence logo from HLA-B*07:02 citrullinated peptides (n = 39).
We next focused on the characteristics of the phosphorylated HLA-B*07:02 peptide ligands detected with high-pH RP. Interestingly, although positions 1 and 8 of HLA-B*07:02 peptide ligands are most often occupied by serine residues, almost no phosphorylations are observed at these sites (Figure 6B). On the contrary, phosphorylated serines are most frequently localized to position 4 in ∼50% of all of the serine phosphorylated HLA-B*07:02 peptides. Deeper analysis of serine phosphorylated peptides revealed the presence of two distinct kinase motifs (Figure 6C). Hence, it seems that the majority of phosphorylated peptide ligands for loading on HLA-B*07:02 may have been provided by proline-directed kinases with SP consensus and basophilic kinases with an RxxS consensus motif.25
Pre-fractionation is Essential for the Detection of Citrullinated HLA Class Ι Peptide Ligands
Extending the depth in analysis of the HLA class Ι ligandome by pre-fractionation not only leads to increased identification of phosphorylated HLA class Ι peptide ligands but has also been shown to improve detection of HLA class Ι peptide ligands containing other post-translational modifications, e.g., OGlcNAcylation4 or arginine (di)methylation.5 In our data, we next looked for HLA class Ι peptide ligands harboring arginine citrullination, another important modification linked to rheumatoid arthritis.26−30 In contrast to serine phosphorylation, arginine citrullination is detected rather poorly without pre-fractionation. In our analysis, we were able to detect 48 and 34 citrullinated HLA-B*07:02 peptide ligands with high-pH RP and SCX, respectively, compared to 16 in unfractionated samples (Figure 6D and Table S2). This implies that pre-fractionation is almost a necessity for detailed study of this modification. In our data, arginine citrullination is mostly detected on HLA-B*07:02 peptide ligands. This is not surprising because binders of HLA- B*07:02 contain more arginine residues on average.
In contrast to the preference for serine phosphorylation at position 4, the distribution of arginine citrullination per position in the peptide ligands followed largely the overall arginine frequency per position in HLA-B*07:02 peptide ligands (Figure 6E,F). Cumulatively, we detected here many citrullinated HLA peptide ligands (Table S2), suggesting that high-pH RP could potentially be a preferred fractionation method to study this relatively scarcely studied modification in more detail. Notably, some of these citrullinated peptides originate from the filaggrin source protein, which has been reported to harbor several citrullinated rheumatoid arthritis-specific epitopes.31 Some of citrullinated peptides detected in this work overlap with the rheumatoid arthritis epitopes reported earlier, while we also detect novel arginine citrullination sites on filaggrin.
Discussion
Here, we profiled HLA class Ι peptide ligands from the HLA homozygous cell line JY to benchmark the utility of two complementary fractionation approaches, namely high-pH RP and SCX. Compared to analysis without fractionation, each of these strategies expanded the HLA class Ι ligandome coverage by about 50%, but significant non-overlap between these two strategies also provides a cumulative gain in peptide identification of >100% compared to analysis without fractionation. This significant boost in ligandome coverage is, however, highly dependent on intrinsic properties of peptide ligands. Hence, we clearly demonstrate that the choice of pre-fractionation approach can introduce an allele specific analytical bias. We showed experimentally that HLA class Ι peptide ligands with largely hydrophobic residues [e.g., HLA-A*02:01, motif xLxxxLLx(V/L)] are better pre-fractionated on a less charge selective high-pH gradient, whereas charged HLA class Ι peptide ligands containing arginines [HLA-B*07:02, motif RPRxxRxx(L/V)] are better pre-fractionated and thus detected with SCX. Because this bias is strongly influenced by conserved properties of the HLA class Ι peptide ligands, we think there is a critical need to rationalize which fractionation approach to use, depending on both the HLA locus to be investigated and the allele specificity within the locus.
However, this apparent bias presents an opportunity to further improve allele-specific ligandome coverage. For instance, a large majority of HLA-A*02 alleles have hydrophobic ligands that feature predominantly in leucine and valine residues [e.g., HLA-A*02:03, motif xLAxx(L/V)xx(L/V); HLA-A*02:06, motif (xLLxxLxx(V/L)]. This implies that high-pH RP pre-fractionation would also be the better choice for most HLA-A*02 peptide ligands to maximize specific coverage in the respective ligandomes (Figure S1). Conversely, just like for HLA-B*07:02, ligands of other HLA-B alleles (e.g., HLA-B*27:02, motif RRLxxxxxL; HLA-B*27:20, motif RRxxxxxRL) or HLA-A alleles with affinity for charged peptides [e.g., HLA-A*30:01, motif RPRxxRxx(L/V); HLA-A*31:01, motif RTRxxxxxR] would also be better analyzed by using SCX. In view of these considerations, we put forth high-pH RP as a valuable alternative analytical strategy to choose from to further expand the coverage of hydrophobic HLA class Ι peptide ligands. It is key to note that employing high-pH RP alone instead of SCX already profiles the ligandome to similar depth with comparable total identifications but in addition with a bigger proportion of hydrophobic peptide ligands. This further implies that by choosing the appropriate strategy between high-pH RP and SCX, the allele-specific ligandome space could be expanded.
In addition, post-translational modifications on HLA class Ι peptide ligands can also alter the biophysical and electrostatic properties of these peptides to further impact the ideal choice of analytical strategy. For instance, phospho-modifications will reduce the net charge of peptide ligands and theoretically makes these peptides better retained and separated on a non-charge-selective high-pH RP gradient. We validated this experimentally with indeed more phosphorylated peptides detected using the high-pH RP workflow. In fact, fractionating phosphorylated peptide ligands on a suboptimal charge-selective gradient results in even fewer identifications compared to analysis without any pre-fractionation. This further affirms the importance of a rational choice of pre-fractionation method and shows that an inappropriate strategy will defeat the purpose of fractionation altogether. With the same rational thought, modifications that involve removal of a charge, such as citrullination, would also be better analyzed on high-pH RP, as we documented here experimentally.
While pre-fractionation offers distinct advantages in allele-specific and PTM-specific HLA peptide ligand identification, more starting material is inherently needed to harness these benefits compared to the unfractionated workflow. To tackle this, we show here that it is possible to obtain more HLA peptide ligands by reusing the flowthrough from immuno-affinity purifications without increasing the initial input material. This can also further relieve the specimen bottleneck on patient-specific HLA peptide ligand analyses. We verify here that repeated use of the lysate does not compromise the quality and purity of the immuno-affinity purification, even to the point of HLA complex depletion, and that a large overlap of >80% in MS identification is still possible between sequential reuse.
Utilizing all strategies and considerations described above, we deeply profiled the phosphorylated HLA-B*07:02 ligandome by high-pH RP to examine the preferred site of serine phosphorylation against serine occupancy in the peptide ligand sequence. We found a strong preference for phospho-serine at position 4 but not at positions 1 and 8 despite higher occurrence of serine in the latter. This, we further rationalized against the loading model proposed previously; HLA-B antigens are collectively stabilized at position 1 by π–π stacking at position 1 with the R62 guanidinium group, hydrophobic interaction with the W167 indole group, and the salt bridge with the N163 carboxyl group on the HLA-B backbone.6 Phosphorylation at serine in position 1 is likely to critically destabilize these docking interactions, such that peptides serine-phosphorylated at position 1 can no longer be loaded, whereas serine phosphorylations at position 4 can be stabilized through contact with R62 and a water-mediated reaction with the carboxyl group of E163 in the HLA-B backbone6 and, thus, are more prominently observed. Thus, our data strongly supports the loading preference of HLA-B peptide ligands reported previously, where a phospho-serine neo-antigen at P4 can extend out of the binding groove, to bind putatively to T-cells in a PTM-dependent manner.22,23,32−34
Taken together, we show in this work that the detection of HLA class Ι peptide ligands can be improved tremendously by a carefully deliberated choice, or complementary use of high-pH RP and SCX fractionation. We demonstrate here that the physical and chemical properties of HLA class Ι peptide ligands can strongly influence the choice of analytical strategy and that with a research question in mind surrounding a particular HLA locus, allele, or peptide ligand PTM, a rational consideration of which pre-fractionation to adopt will meaningfully expand coverage in the ligandome space of interest and potentially boost the identification of the much-sought-after tumor neo-antigens.
Acknowledgments
We acknowledge The Netherlands Organization for Scientific Research (NWO) for supporting this research through funding of the large-scale proteomics facility Proteins@Work (project no. 184.032.201) embedded in The Netherlands Proteomics Centre. L.C.D. and A.J.R.H. are further supported by the NWO Gravitation Program Institute for Chemical Immunology (ICI00003). We acknowledge additional funding through the European Union’s Horizon 2020 research and innovation program under grant agreement no. 686547 (MSMed).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jproteome.8b00821.
A figure showing Gibbs clustering motifs for nine different HLA types; tables showing a list of identified phosphopeptides and a list of identified citrullinated peptides (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Vinay D. S.; Ryan E. P.; Pawelec G.; Talib W. H.; Stagg J.; Elkord E.; Lichtor T.; Decker W. K.; Whelan R. L.; Kumara H. M. C. S.; et al. Immune evasion in cancer: Mechanistic basis and therapeutic strategies. Semin. Cancer Biol. 2015, 35, S185–S198. 10.1016/j.semcancer.2015.03.004. [DOI] [PubMed] [Google Scholar]
- Schumacher T. N.; Schreiber R. D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. 10.1126/science.aaa4971. [DOI] [PubMed] [Google Scholar]
- Bassani-Sternberg M.; Coukos G. Mass spectrometry-based antigen discovery for cancer immunotherapy. Curr. Opin. Immunol. 2016, 41, 9–17. 10.1016/j.coi.2016.04.005. [DOI] [PubMed] [Google Scholar]
- Marino F.; et al. Extended O-GlcNAc on HLA Class-I-Bound Peptides. J. Am. Chem. Soc. 2015, 137, 10922–10925. 10.1021/jacs.5b06586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marino F.; et al. Arginine (Di)methylated Human Leukocyte Antigen Class I Peptides Are Favorably Presented by HLA-B*07. J. Proteome Res. 2017, 16, 34–44. 10.1021/acs.jproteome.6b00528. [DOI] [PubMed] [Google Scholar]
- Alpizar A.; et al. A Molecular Basis for the Presentation of Phosphorylated Peptides by HLA-B Antigens. Mol. Cell. Proteomics 2017, 16, 181–193. 10.1074/mcp.M116.063800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mommen G. P.; et al. Expanding the detectable HLA peptide repertoire using electron-transfer/higher-energy collision dissociation (EThcD). Proc. Natl. Acad. Sci. U. S. A. 2014, 111, 4507–4512. 10.1073/pnas.1321458111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilar M.; Olivova P.; Daly A. E.; Gebler J. C. Two-dimensional separation of peptides using RP-RP-HPLC system with different pH in first and second separation dimensions. J. Sep. Sci. 2005, 28, 1694–1703. 10.1002/jssc.200500116. [DOI] [PubMed] [Google Scholar]
- Gilar M.; Olivova P.; Daly A. E.; Gebler J. C. Orthogonality of separation in two-dimensional liquid chromatography. Anal. Chem. 2005, 77, 6426–6434. 10.1021/ac050923i. [DOI] [PubMed] [Google Scholar]
- Essader A. S.; Cargile B. J.; Bundy J. L.; Stephenson J. L. Jr. A comparison of immobilized pH gradient isoelectric focusing and strong-cation-exchange chromatography as a first dimension in shotgun proteomics. Proteomics 2005, 5, 24–34. 10.1002/pmic.200400888. [DOI] [PubMed] [Google Scholar]
- Delmotte N.; Lasaosa M.; Tholey A.; Heinzle E.; Huber C. G. Two-dimensional reversed-phase x ion-pair reversed-phase HPLC: an alternative approach to high-resolution peptide separation for shotgun proteome analysis. J. Proteome Res. 2007, 6, 4363–4373. 10.1021/pr070424t. [DOI] [PubMed] [Google Scholar]
- Yang F.; Shen Y.; Camp D. G. 2nd; Smith R. D. High-pH reversed-phase chromatography with fraction concatenation for 2D proteomic analysis. Expert Rev. Proteomics 2012, 9, 129–134. 10.1586/epr.12.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H.; et al. An off-line high pH reversed-phase fractionation and nano-liquid chromatography-mass spectrometry method for global proteomic profiling of cell lines. J. Chromatogr. B: Anal. Technol. Biomed. Life Sci. 2015, 974, 90–95. 10.1016/j.jchromb.2014.10.031. [DOI] [PubMed] [Google Scholar]
- Batth T. S.; Francavilla C.; Olsen J. V. Off-line high-pH reversed-phase fractionation for in-depth phosphoproteomics. J. Proteome Res. 2014, 13, 6176–6186. 10.1021/pr500893m. [DOI] [PubMed] [Google Scholar]
- Barnstable C. J.; et al. Production of monoclonal antibodies to group A erythrocytes, HLA and other human cell surface antigens-new tools for genetic analysis. Cell 1978, 14, 9–20. 10.1016/0092-8674(78)90296-9. [DOI] [PubMed] [Google Scholar]
- Zheng X.; et al. Proteomic analysis for the assessment of different lots of fetal bovine serum as a raw material for cell culture. Part IV. Application of proteomics to the manufacture of biological drugs. Biotechnol. Prog. 2006, 22, 1294–1300. 10.1021/bp060121o. [DOI] [PubMed] [Google Scholar]
- Vizcaino J. A.; et al. 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 2016, 44, D447–456. 10.1093/nar/gkv1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreatta M.; Nielsen M. Gapped sequence alignment using artificial neural networks: application to the MHC class I system. Bioinformatics 2016, 32, 511–517. 10.1093/bioinformatics/btv639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talmage D. W. The primary equilibrium between antigen and antibody. Ann. N. Y. Acad. Sci. 1957, 70, 82–93. 10.1111/j.1749-6632.1957.tb35380.x. [DOI] [PubMed] [Google Scholar]
- Reverberi R.; Reverberi L. Factors affecting the antigen-antibody reaction. Blood Transfus 2007, 5, 227–240. 10.2450/2007.0047-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreatta M.; Alvarez B.; Nielsen M. GibbsCluster: unsupervised clustering and alignment of peptide sequences. Nucleic Acids Res. 2017, 45, W458–W463. 10.1093/nar/gkx248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarling A. L.; et al. Phosphorylated peptides are naturally processed and presented by major histocompatibility complex class I molecules in vivo. J. Exp. Med. 2000, 192, 1755–1762. 10.1084/jem.192.12.1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarling A. L.; et al. Identification of class I MHC-associated phosphopeptides as targets for cancer immunotherapy. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 14889–14894. 10.1073/pnas.0604045103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beausoleil S. A.; et al. Large-scale characterization of HeLa cell nuclear phosphoproteins. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 12130–12135. 10.1073/pnas.0404720101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amanchy R.; et al. A curated compendium of phosphorylation motifs. Nat. Biotechnol. 2007, 25, 285–286. 10.1038/nbt0307-285. [DOI] [PubMed] [Google Scholar]
- James E. A.; et al. HLA-DR1001 presents ″altered-self″ peptides derived from joint-associated proteins by accepting citrulline in three of its binding pockets. Arthritis Rheum. 2010, 62, 2909–2918. 10.1002/art.27594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen H.; James E. A. Immune recognition of citrullinated epitopes. Immunology 2016, 149, 131–138. 10.1111/imm.12640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scally S. W.; et al. A molecular basis for the association of the HLA-DRB1 locus, citrullination, and rheumatoid arthritis. J. Exp. Med. 2013, 210, 2569–2582. 10.1084/jem.20131241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seward R. J.; Drouin E. E.; Steere A. C.; Costello C. E. Peptides presented by HLA-DR molecules in synovia of patients with rheumatoid arthritis or antibiotic-refractory Lyme arthritis. Mol. Cell. Proteomics 2011, 10, M110002477. 10.1074/mcp.M110.002477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ting Y. T.; et al. The interplay between citrullination and HLA-DRB1 polymorphism in shaping peptide binding hierarchies in rheumatoid arthritis. J. Biol. Chem. 2018, 293, 3236–3251. 10.1074/jbc.RA117.001013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Union A.; et al. Identification of citrullinated rheumatoid arthritis-specific epitopes in natural filaggrin relevant for antifilaggrin autoantibody detection by line immunoassay. Arthritis Rheum. 2002, 46, 1185–1195. 10.1002/art.10229. [DOI] [PubMed] [Google Scholar]
- Andersen M. H.; et al. Phosphorylated peptides can be transported by TAP molecules, presented by class I MHC molecules, and recognized by phosphopeptide-specific CTL. J. Immunol 1999, 163, 3812–3818. [PubMed] [Google Scholar]
- Mohammed F.; et al. Phosphorylation-dependent interaction between antigenic peptides and MHC class I: a molecular basis for the presentation of transformed self. Nat. Immunol. 2008, 9, 1236–1243. 10.1038/ni.1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen J.; et al. Phosphorylated self-peptides alter human leukocyte antigen class I-restricted antigen presentation and generate tumor-specific epitopes. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 2776–2781. 10.1073/pnas.0812901106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.