

Abstract
Objectives:
We sought to describe the phenotype for patients with P.I.G. including presentation, evaluation, cardiac co-morbidities, high resolution computed tomography findings, and outcomes.
Methods:
With institutional review board approval, we performed a retrospective review of patients with biopsy-proven P.I.G. Biopsies, high resolution chest computed tomography, and cardiac evaluations were reviewed and characterized by experts in each field.
Results:
Sixty-two percent of the patients were male. The median gestational age was 37 weeks (range 27–40). The median age at biopsy was 1.6 months (range 0.3–6 months). Structural heart disease was present in 63% of patients. Pulmonary hypertension (diagnosed by echocardiogram and/or cardiac catheterization) was noted in 38% of patients. Alveolar simplification was present in 79% of patients. Fifty percent of available biopsies revealed patchy disease. An increase in age at biopsy was associated with patchy (vs diffuse) disease. Ninety-two percent of patients were treated with systemic corticosteroids. Median age at last follow-up was 1234 days with a range of 37 days to 15 years. At the time of last follow-up, 12 patients were off all support, eight were on supplemental oxygen, two were mechanically ventilated, one underwent lung transplantation, and one died. CT findings commonly included ground glass opacities (86%) and cystic change (50%).
Conclusions:
The P.I.G. phenotype has not been comprehensively described, and poor recognition and misconceptions about P.I.G. persist. P.I.G. is a disease that presents in early infancy, requires significant medical intervention, and frequently is seen in association with alveolar simplification and/or cardiovascular disease. CT findings include ground glass opacities and cysts. Patients should be monitored for pulmonary hypertension. Without life-threatening comorbidities, many patients do well over time, although respiratory symptoms may persist into adolescence.
Keywords: biopsy, cysts, glucocorticords, infant newborn, interstitial lung diseases, oxygen, phenotype, pulmonary hypertension
1 |. INTRODUCTION
Pulmonary histologic findings of cellular interstitial space widening in infants was first described in 1992 and named as “infantile cellular interstitial pneumonitis.”1 Pulmonary Interstitial Glycogenosis (P.I.G.) was coined by Canakis et al2 in 2002 based upon the identification of glycogen in interstitial mesenchymal “clear cells” similar to those seen in infantile cellular interstitial pneumonitis. Young and Deutsch recently characterized these cells as lipid-containing mesenchymal cells in the interstitium that are consistent with lipofibroblasts and suggested that P.I.G. is best conceptualized as an abnormal developmental process.3
A large, multi-center retrospective clinical and histologic review of lung biopsies from children with diffuse lung disease classified P.I.G. as a distinct entity more common in infancy with an unknown etiology.4 Since these early descriptions, infants with P.I.G. have been reported around the world, including in twins.5 Common features to these reports include presentation in the infant period (under 9 months of age) with hypoxemia and respiratory distress. Co-associations with alveolar simplification and structural heart disease have been described.3,4,6 The diagnostic gold standard is lung biopsy.7 Many infants with P.I.G. have received therapeutic trials of glucocorticoids with anecdotal positive responses.6 Outcomes have overall been favorable, but few data on long term follow-up exist, with the longest being 6 years.2,4,8
Our aim was to more completely characterize the clinical, radiological, and histological spectrum of P.I.G. and to describe treatment approaches and long term outcomes.
2 |. MATERIALS AND METHODS
With institutional review board approval for which informed consent was waived, we performed a single-center retrospective review of patients with biopsy-proven P.I.G. evaluated at Children’s Hospital Colorado before 5/30/2015. The earliest patient included was born in 2001. Patients were identified from a rare lung disease patient registry as well as a search through Informatics for Integrating Biology and the Bedside (i2b2). Three patients in this cohort were included in a previous study.4 Chart review was performed to gather data. Comorbidities were determined by problem lists and chart review. Structural heart disease and pulmonary hypertension were identified by echocardiogram (JRD). The echocardiogram closest to lung biopsy was included. Cardiac catheterization, when available, was used to assess degree of pulmonary hypertension. Infants born at a recorded “full term” were considered to be born at 39 weeks for statistical analysis. Response to steroids was determined by clinician report and notation in the chart. Biopsies were characterized as having patchy versus diffuse findings of P.I.G., alveolar simplification, and pulmonary artery changes (arterial media thickening with increased smooth muscle cells) by a pediatric pathologist with expertise in pediatric lung pathology and chILD (MKD and CG). A pediatric radiologist with expertise in chest radiology and chILD (JPW) reviewed each high resolution chest computed tomography scan (HRCT) and applied a standardized score.9
2.1 |. Statistical analysis plan
Distributions of outcomes were evaluated using the Shapiro-Wilks test for normality. When the test suggested normality, summary statistics are presented as mean and standard deviation (±SD) otherwise outcomes are summarized by medians, interquartile ranges [IQR], maximums and minimums of the observed outcomes. Categorical outcomes are presented as frequencies (n) and percentages (%). Logistic regression was used to assess an association of age with findings on lung biopsy (patchy vs diffuse as the outcome). All P-values <0.05 were considered statistically significant. All calculations were done using SAS software version 9.4 (SAS Institute, Cary, NC).
3 |. RESULTS
Demographics are described in detail in Table 1. Fifteen of 24 patients were male (62.5%). The racial and ethnic make-up of the study cohort included 58% Caucasian with the remainder either reporting more than one race, a race other than Caucasian, or not reporting race. Eight children (33%) were Hispanic. For comparison, the Colorado demographic make-up is roughly 72% Non-Hispanic white, 21% Hispanic, 4% Black, and 3% other.10 Most patients (18 of 24) were late preterm or term (gestational age ≥38 weeks), with an overall median gestational age at birth of 37 weeks (range of 27–40 weeks). Seventeen patients (71%) required neonatal resuscitation, including non-invasive ventilation and/or invasive mechanical ventilation. Most (62.5%) patients had structural heart disease, including atrial septal defects, ventricular septal defects, patent ductus arteriosus, hypoplastic left heart syndrome, Tetralogy of Fallot, coarctation of the aorta, pulmonary vein stenosis, mitral stenosis, or a combination of the above. Non-cardiac comorbidities included midline defects (cleft lip/palate [n = 2], unilateral kidney [n = 1]), neurologic deficits (developmental delay [n = 4], autism [n = 1], hypotonia [n = 1], seizures [n = 1]), and other (aspiration [n = 1], connective tissue disease [n = 1], 22q deletion [n = 1], hypothyroidism [n = 1], airway malacia [n = 3], restless leg syndrome [n = 1], urinary retention [n = 1]). No comorbidities were identified in 12.5% of patients.
TABLE 1.
Basic demographic information
| Median [IQR] n (%) | |
|---|---|
| Age at lung biopsy (months) | 1.6 [0.3–6] |
| Sex | |
| Female | 9 (37.5) |
| Male | 15 (62.5) |
| Race | |
| More than 1 race | 1 (4.2) |
| Caucasian | 14 (58.3) |
| Other | 6 (25.0) |
| Not reported | 3 (12.5) |
| Ethnicity | |
| Latino | 8 (33.3) |
| Non-Latino | 13 (54.2) |
| Not reported | 3 (12.5) |
| Gestational age at birth (weeks)a | 37 [32, 39] range 27–40 |
| Neonatal resuscitation | |
| Yes | 17 (70.8) |
| No | 4 (16.7) |
| Missing | 3 (12.5) |
| Structural Heart Disease | |
| Yes | 15 (62.5) |
| No | 8 (33.3) |
| Undetermined | 1 (4.2) |
Three infants had a gestational age of “Full term” recorded.
3.1 |. Evaluation
Ten patients (41.6%) were tested for at least one surfactant dysfunction mutation. Five were tested for ABCA3, SFTPB, SFTPC, and NKX2.1 mutations; the remainder were tested for two or three of the mutations. No pathogenic mutations were found. Two patients had additional genetic testing, including a negative evaluation for Noonan’s syndrome and negative chromosomal microarray analysis. Seventeen of 24 patients (71%) were evaluated by flexible bronchoscopy, and many patients underwent multiple flexible bronchoscopies. Initial flexible bronchoscopies with lavage were notable for airway malacia (five patients), pale or edematous mucosa (eight patients), and/or mucus (four patients). Seven patients were noted to have bacterial growth or viruses. Infant pulmonary function tests were performed in five patients. All five patients were tachypneic and three out of five demonstrated air trapping. Infant pulmonary function testing was performed between 8 and 172 weeks. A summary of evaluations and treatments is listed in Table 2.
TABLE 2.
Evaluations and treatments
| n (%) | |
|---|---|
| Evaluation | |
| Aspiration | |
| Yes | 2 (8.3) |
| No | 13 (54.2) |
| Undetermined | 9 (37.5) |
| Genetic testing for surfactant dysfunction mutations | |
| Yes | 10 (41.6) |
| No | 14 (58.4) |
| Bronchoscopy | |
| Yes | 17 (70.8) |
| No | 6 (25.0) |
| Undetermined | 1 (4.2) |
| Infant pulmonary function tests? (Y/N) | |
| Yes | 5 (20.8) |
| No | 19 (79.2) |
| Treatment | |
| Maximal respiratory support | |
| Extracorporeal membrane oxygenation | 3 (12.5) |
| High frequency oscillatory ventilation | 7 (29.2) |
| Mechanical ventilation | 10 (41.7) |
| Non-Invasive ventilation | 1 (4.2) |
| Nasal cannula | 3 (12.5) |
| Respiratory support at hospital discharge | |
| Mechanical ventilation | 8 (33.3) |
| Nasal cannula | 14 (58.3) |
| None | 2 (8.3) |
| Gastrostomy tube and nissen fundoplication | |
| Gastrostomy tube | 3 (12.5) |
| Gastrostomy tube with nissen fundoplication | 10 (41.7) |
| Undetermined | 2 (8.3) |
| No gastrostomy tube | 9 (37.5) |
| Treatment with systemic corticosteroids | |
| Yes | 22 (91.7) |
| Enterally (PO) | 5 (23.8) |
| Intravenously (IV) | 12 (57.1) |
| Both IV and PO | 4 (19.1) |
| Timing of steroid administration relative to biopsy | |
| Before | 4 (18.2) |
| After | 10 (45.5) |
| Before and After | 6 (27.3) |
3.2 |. Imaging
Twenty-two of 24 patients had record of a single (n = 19) or multiple CT scans (n = 3) in the Children’s Hospital Colorado system. Median age at first HRCT was 9.0 weeks (IQR: 4–22). Five of 22 patients had their first recorded HRCT after 1 year of life. HRCT findings are shown in Table 3. The most common primary HRCT findings were ground glass opacities (GGO) (13 patients), followed by linear reticular opacities (three patients). The most common secondary CT findings were cysts (six patients) followed by GGO (five patients). The most common CT findings overall (primary, secondary, and tertiary) were GGO (19 patients), followed by cysts (11 patients). Typical HRCT findings of GGO and cysts are shown in Figure 1. Most of the HRCT findings were diffuse, however some were localized.
TABLE 3.
Computed tomography findings
| Median [IQR] n (%) | |
|---|---|
| Age at CT scan (days) | 64 [30 to 1575] range 10–1610 |
| Primary CT findings (n = 22) | |
| Ground glass opacities | 13 (59.1) |
| Linear reticular opacities | 3 (13.6) |
| Other | 6 (27.3) |
| Secondary CT findings (n = 16) | |
| Cysts | 6 (37.5) |
| Ground glass opacities | 5 (31.3) |
| Other | 5 (31.3) |
FIGURE 1.
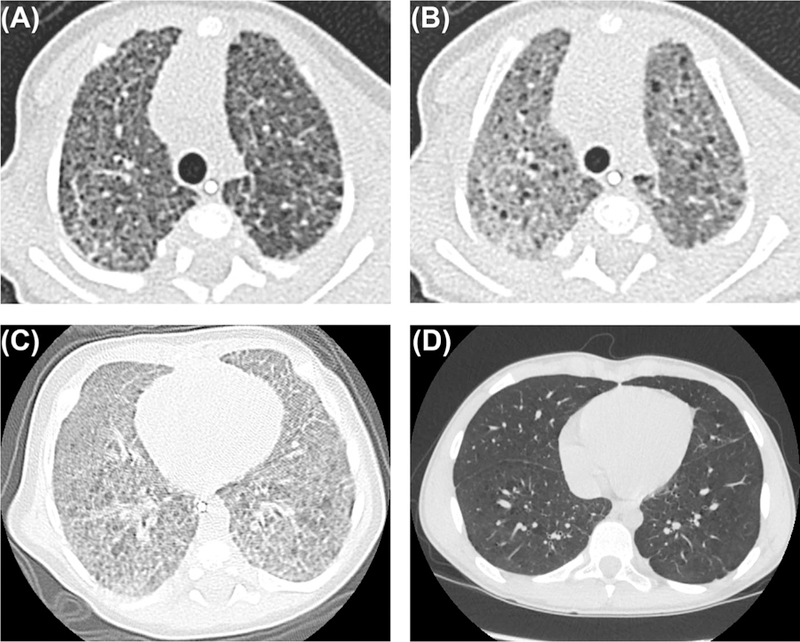
HRCT Scans in two patients with P.I.G.: Inspiratory (A) and expiratory (B) CT scans at 53 days of age demonstrating typical findings of P.I.G.: ground glass opacities and cysts, which are accentuated in expiration. CT scan from infancy (C) with ground glass opacities and cysts. Subsequent CT scan at age 14 years (D) with resolution of ground glass opacities and improvement in cysts which subtly persist
Three patients had subsequent HRCT scans. One patient had HRCTs at 18 days, 1 year, and 9 years. This patient initially had air trapping primary in the left upper lobe which evolved to linear reticular opacities in the right upper lobe and right lower lobe as well as persistent air trapping in the left upper lobe. This air trapping in the left upper lobe persisted in the third HRCT. One patient had HRCTs at 74 days and 14 years, both of which showed interlobular septal thickening. The first HRCT also demonstrated GGO and cysts that resolved by the second HRCT. One patient had HRCTs at 111 days and 180 days that initially showed cysts, GGO, and linear reticular opacities and subsequently showed cysts and decreased but persistent GGO.
3.3 |. Biopsy findings
The median age of lung biopsy was 7 weeks (IQR: 4–13) after birth. Median corrected gestational age at biopsy was 76 weeks (IQR: 65–80). Two of the biopsies were not available for review; these patients presented to our center for second opinions. Respiratory support at the time of biopsy ranged from extracorporeal membrane oxygenation to oxygen via nasal cannula. One patient required extracorporeal membrane oxygenation, one was treated with high frequency oscillatory ventilation, 10 were mechanically ventilated, one was non-invasively ventilated, three were treated with heated humidified high flow nasal cannula, and four were treated with nasal cannula oxygen. Four had unknown respiratory support at the time of the biopsy. Pathology findings are summarized in Table 4.
TABLE 4.
Pathology findings
| Median [IQR] n (%) | |
|---|---|
| Age at lung biopsy (days) | 50 [31, 93] range 10–182 |
| Distribution of pulmonary interstitial glycogenosis | |
| Patchy | 11 (45.8) |
| Diffuse | 11 (45.8) |
| Unknown | 2 (8.3) |
| Alveolar simplification | |
| Yes | 19 (79.2) |
| No | 3 (12.5) |
| Unknown | 2 (8.3) |
| Pulmonary artery thickening | |
| Yes | 19 (79.2) |
| No | 3 (12.5) |
| Undetermined | 2 (8.3) |
Forty-six percent of all biopsies revealed patchy disease (n = 11); 46% revealed diffuse disease (n = 11); two biopsies were unknown. Most (n = 19) demonstrated alveolar simplification and most (n = 19) demonstrated pulmonary artery changes. All demonstrated either alveolar simplification or pulmonary artery changes, and 16 had both. Other biopsy findings included infection (cytomegalovirus [n = 1], bronchiolitis [n = 1], pneumonia/pneumonitis [n = 3]), lipid laden macrophages (n = 1), hemorrhage (n = 3), lymphocytes (n = 1), neuroendocrine cell hyperplasia (n = 1), and pleural changes (n = 2). Classic biopsy findings are shown in Figure 2. An increase in age at biopsy was associated with an increase in the odds of patchy compared to diffuse disease (OR 1.37, 95%CI 1.03–1.82, P = 0.0332). Eight patients had electron microscopy available for review; one of these patients had lipid bodies present on electron microscopy.
FIGURE 2.

Pathology findings in P.I.G.: A, 10× H&E staining with alveolar simplification, pulmonary arterial changes, and interstitial widening. B, 40× H&E staining with bland polygonal cells with pale cytoplasm filling interstitium (arrow). C, Electron micrograph with rare organelles and abundant monoparticulate glycogen (arrow) within the cytoplasm of interstitial cells
One patient underwent lung biopsy and subsequent lung transplantation for pulmonary hypertension secondary to pulmonary veins stenosis and alveolar simplification. The explanted lung demonstrated progression of pulmonary vascular changes and similar alveolar growth abnormality.
3.4 |. Cardiology findings
Nine of 19 patients who had echocardiograms at Children’s Hospital Colorado demonstrated some degree of pulmonary hypertension. Nine patients underwent cardiac catheterization, four of whom had severe congenital heart disease: (HLHS or variant [2], pulmonary vein stenosis [1], and pulmonary valve atresia [1]). Of the remaining five who underwent catheterization, four had septal defects (ASD, VSD, PDA), one was free of CHD. No additional patients had pulmonary hypertension by cardiac catheterization. All patients were treated with oxygen. Nine patients were treated with additional pulmonary vasodilators (nitric oxide, sildenafil, tadalafil, bosentan). At the time of last echocardiogram, five patients had residual cardiac defects. All nine of the patients with pulmonary hypertension by echocardiogram had pulmonary artery changes on biopsy. An additional eight patients had pulmonary artery changes despite normal echocardiograms, and two had pulmonary artery changes with no echocardiograms performed. All four patients with severe congenital heart disease demonstrated pulmonary artery changes on biopsy (two of whom did not have pulmonary hypertension by echocardiogram/cardiac catheterization), and eight of the nine patients total who underwent cardiac catheterization demonstrated pulmonary artery changes on biopsy. Five patients did not undergo cardiac evaluation with echocardiogram and/or cardiac catheterization.
3.5 |. Treatment
Respiratory support ranged from extracorporeal membrane oxygenation (n = 3) to nasal cannula (n = 3), as shown in Table 2. Seventy-one percent of patients required either invasive or non-invasive ventilation (n = 17). By the time of hospital discharge, most patients required nasal cannula oxygen, although two weaned off all support and eight were discharged on mechanical ventilation.
Nearly all patients (92%; n = 22) were treated with systemic corticosteroids, either orally, intravenously, or both. Ten patients were treated with systemic corticosteroids before the biopsy. Sixteen were treated with systemic corticosteroids after the biopsy. Three patients were felt to have an excellent clinical response to steroids as reported in the medical records by the care teams. Quantifying the clinical response to steroids was not possible.
Two patients were found to aspirate (out of 15 evaluated) by videofluoroscopic study or upper gastrointestinal series. Thirteen patients had a gastrostomy tube, and the majority of those with gastrostomy tubes underwent fundoplications (n = 10).
3.6 |. Outcomes
Median age at last follow-up was about 3 years (1234 days) with a range of 37 days to 15 years. At the time of last follow-up, 12 patients were off all respiratory support, including ventilation and supplemental oxygen. Among eight patients receiving mechanical ventilation at discharge, three remained on ventilation, one was treated with oxygen, and four required no support at follow-up. The four individuals requiring no support at follow-up after being mechanically ventilated were off support at 34, 35, 37, and 57 months after lung biopsy. In 14 patients receiving nasal cannula oxygen at discharge, eight (57%) were still receiving oxygen by nasal cannula and six (43%) required no respiratory support at follow-up. Time to wean from nasal cannula ranged from 4 to 38 months, with a median time of 25 months after lung biopsy. Two patients in the cohort required no respiratory support both at discharge and at follow-up. One patient was transferred while mechanically ventilated and subsequently died of unknown causes and at an unknown age. One patient received a lung transplant secondary to pulmonary vein stenosis. Of the eight patients treated with home mechanical ventilation via tracheostomy, five (63%) were liberated from mechanical ventilation and decannulated. Spirometry was performed in one of these patients and demonstrated reversible airflow obstruction.
4 |. DISCUSSION
Infantile cellular interstitial pneumonitis was first described in 19921 and named P.I.G. in 2002.2 Since this initial report, case reports, and several case series P.I.G. have further defined P.I.G.4,11 However, no in depth review has been published to define a phenotype that clinicians can use to consider this diagnosis. This pathologic diagnosis is poorly understood, and has only been seen in biopsies from infants. In our experience, there is still significant under-recognition and confusion about P.I.G and we receive multiple consultations each year from other institutions regarding patients with P.I.G. This manuscript defines a clinical phenotype for P.I.G. that includes neonatal respiratory failure or distress in late preterm or term infants with comorbid chronic lung disease of prematurity, pulmonary hypertension, and/or structural heart disease. Many patients in our cohort suffered from a variety of additional co-morbidities which may be secondary to underlying genetic syndromes. We did not note any associated with congenital lung malformations, although we did not review all biopsies of patients with congenital lung malformations to determine if P.I.G. may have been present and missed as has been presented in other series.11 We do not have lung tissue on all patients with structural heart disease or at every gestational age to better understand the prevalence of P.I.G in populations such as congenital heart disease or prematurity. Understanding this phenotype will help clinicians evaluate infants with unexplained respiratory distress or failure, especially in late pre-term or term infants with comorbid cardiac disease. P.I.G. may be overlooked in patients who never undergo lung biopsy; CT can suggest P.I.G. with cysts and GGO, but clinicians should suspect P.I.G. in infants with respiratory requirements out of proportion to their gestational age.
Few lung biopsies are performed in patients born prior to 28 weeks gestational age. In the current study, younger patients had more diffuse disease on biopsy, suggesting that P.I.G. could be part of normal lung development that improves over time. Most patients in the current study had alveolar simplification seen in association with P.I.G. In a larger series of patients with diffuse lung disease, P.I.G. was recognized as the primary diagnosis in six patients and as a secondary diagnosis in 19 patients with alveolar simplification.4 It is unknown if the P.I.G. precedes or follows structural developmental defects. It is also unknown if the P.I.G. cells interfere with normal structural development or are part of normal development.
HRCT findings, which were not previously described in detail, are typically characterized by GGO with cystic changes. We believe that the cystic regions correlate with areas of alveolar simplification on biopsy and are therefore likely not true cysts. Radiographic findings differ between bronchopulmonary dysplasia and P.I.G. CT findings in bronchopulmonary dysplasia typically consist of cystic changes indicating an alveolar growth abnormality,12 however, in P.I.G., GGO were observed in the majority of our patients. As GGO and cysts on CT scans may suggest a surfactant dysfunction mutation, laboratory testing for these mutations may be appropriate prior to pursuing lung biopsy.7 Three of our patients had repeat CTs; in two patients, the ground glass opacities resolved while the cysts persisted. Though follow-up CTs were limited, this resolution of GGO corroborates our working hypothesis that P.I.G. improves over time while the changes consistent with alveolar simplification may persist. A third patient had persistent left upper lobe air trapping that persisted over time. In our experience, the CT findings of GGO and cystic changes are typical, but not pathognomonic for P.I.G., and not all patients had these findings; previous studies have not found unifying radiographic findings.11 Biopsy, therefore, is still considered the gold standard for diagnosis.
Rauch et al13 evaluated patients with persistent tachypnea of infancy; these infants had either neuroendocrine cell hyperplasia of infancy (NEHI) or P.I.G.. While we agree with Rauch et al that patients with NEHI and P.I.G. have tachypnea and good long term outcomes (in the absence of major comorbidities), we argue that NEHI is a very different disease process physiologically, pathologically, radiologically, and clinically than what we describe with P.I.G. NEHI presents with a classic clinical picture (hypoxemia, tachypnea, retractions) without respiratory failure and outside the newborn period.14 CT findings in NEHI typically demonstrate air trapping and GGO especially in the perihilar and/or right middle lobe/lingua but not cysts.15 NEHI patients rarely have pulmonary hypertension or structural heart disease. Classifying these very different diseases together prevents defining mechanisms of disease and cellular biology and confuses longitudinal clinical trials and understanding outcomes. Thus, we maintain NEHI and P.I.G. should be classified as their own disease entities with recognized distinct phenotypes, biology and outcomes. Interestingly, one patient in our series had both NEHI and P.I.G. demonstrated in her biopsy findings and the interaction between these two poorly understood disease processes is not well understood. This patient was the only one who presented outside the newborn period. Pulmonary neuroendocrine cell hyperplasia and P.I.G. has been previously reported in one case series.11
Fatalities have been described in P.I.G.2,11 One patient in our series died. Unfortunately, this patient was lost to follow-up after initial discharge, so the cause of death is unknown. This patient had a history of congenital diaphragmatic hernia, atrial septal defect, patent ductus arteriosus, and was transferred with chronic mechanical ventilation to a facility closer to home from which he was discharged. One of our patients also required lung transplant for pulmonary vein stenosis. Thus, these patients had significant comorbidities consistent with their severe disease course.
The role of corticosteroids in P.I.G. is controversial. A case report described histologic resolution of P.I.G. after treatment with systemic steroids.6 Biopsies in P.I.G. typically lack active inflammation, but corticosteroids are postulated to help with tissue maturation, possibly through acceleration of lipofibroblasts apoptosis.2,3,6 At least three patients in our series experienced dramatic clinical response to corticosteroids documented in their medical records. The risks and benefits of treatment with corticosteroids, including potential neuro-developmental concerns and negative impacts on lung growth, must be assessed prior to initiating treatment,16 especially when high dose regimens (10–30 mg/kg methylprednisolone) are recommended. If a patient is responsive to corticosteroid therapy, prolonged mechanical ventilation, hospital length of stay, and other medical complication associated with respiratory failure may be significantly reduced. Even in an organized ventilator care program, the length of stay from tracheostomy placement to discharge is 116 days and may be fraught with socio-economic challenges.17 If, however, a patient is on minimal oxygen support, observation may be the best course of therapy. The decision to use corticosteroid therapy in patients with P.I.G. should be personalized to each patient and his/her underlying disease process and level of respiratory support. In the previously reported case of resolution of P.I.G. after corticosteroids, the patient was treated with methylprednisolone 2 mg/kg every 6 h for 5 days with dramatic weaning of respiratory support6; high dose regimens may not be necessary.
A large portion of patients with P.I.G. have pulmonary hypertension. Pulmonary hypertension is a known complication of lung disease associated with early gestational age and alveolar simplification18 and has been described in P.I.G.19 There were significantly more pathologic findings consistent with pulmonary hypertension than by echocardiogram or cardiac catheterization. We suggest that this could be secondary to differences in timing between biopsy and pulmonary hypertension evaluation and interim resolution and/or adequate treatment of pulmonary hypertension. Histologic changes consistent with pulmonary hypertension compared to echocardiographic/cardiac catheterization findings have not systematically been evaluated for clinical correlation. Given the significant percentage of patients with pulmonary artery changes pathologically, patients with documented P.I.G., or the P.I.G. phenotype be evaluated and followed for pulmonary hypertension.
In conclusion, we describe a phenotype of patients with P.I.G. who presented with respiratory failure and distress out of proportion to their gestational age and not explained by cardiac disease. P.I.G. is seen in association with structural heart disease, pulmonary hypertension, and/or alveolar simplification. These comorbidities may lead to persistent symptoms outside of infancy rather than the P.I.G. itself. P.I.G. is a disease only seen in infants less than 8 months; the oldest age of pathologic findings of P.I.G. prior to the current manuscript is 5.5 months.20 Once patients are older than 12 months, continued respiratory symptoms should be attributed to other comorbidities and treated accordingly. Although there are typical CT findings of GGO and cystic regions,21 biopsy is required for diagnosis of P.I.G. Treatment with steroids remains controversial. Further study is needed to determine the indications for steroid therapy as well as optimal dosing, route, and duration of treatment. Despite very significant disease burden with most patients requiring mechanical ventilation, high frequency oscillation, or extracorporeal membrane oxygenation, there was only one death in our cohort. Long term pulmonary follow-up is important for these patients and outcomes overall are favorable.
ACKNOWLEDGMENTS
The authors wish to thank Deborah Batson for her assistance with i2b2. Robin Deterding is Co-Founder, Board member, and consultant Triple Endoscopy, Inc., Co-Founder and Board member Now Vitals, Inc., and Pediatric Interstitial Lung Disease Consultant, Boehringer Ingelheim.
Funding information
K23-HL121090–01 (PI: CDB)
Abbreviations:
- chILD
children’s interstitial and diffuse lung disease
- GGO
ground glass opacities
- NEHI
neuroendocrine cell hyperplasia of infancy
- P.I.G.
pulmonary interstitial glycogenosis
Footnotes
CONFLICTS OF INTEREST
The authors have no conflict of interest to report.
REFERENCES
- 1.Schroeder SA, Shannon DC, Mark EJ. Cellular interstitial pneumonitis in infants. A clinicopathologic study. Chest 1992;101:1065–1069. [DOI] [PubMed] [Google Scholar]
- 2.Canakis AM, Cutz E, Manson D, O’Brodovich H. Pulmonary interstitial glycogenosis: a new variant of neonatal interstitial lung disease. Am J Respir Crit Care Med 2002;165:1557–1565. [DOI] [PubMed] [Google Scholar]
- 3.Deutsch GH, Young LR. Lipofibroblast phenotype in pulmonary interstitial glycogenosis. Am J Respir Crit Care Med 2016;193:694–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Deutsch GH, Young LR, Deterding RR, et al. Diffuse lung disease in young children: application of a novel classification scheme. Am J Respir Crit Care Med 2007;176:1120–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Onland W, Molenaar JJ, Leguit RJ, et al. Pulmonary interstitial glycogenosis in identical twins. Pediatr Pulmonol 2005;40:362–366. [DOI] [PubMed] [Google Scholar]
- 6.Deutsch GH, Young LR. Histologic resolution of pulmonary interstitial glycogenosis. Pediatr Dev Pathol 2009;12:475–480. [DOI] [PubMed] [Google Scholar]
- 7.Kurland G, Deterding RR, Hagood JS, et al. An official American Thoracic Society clinical practice guideline: classification, evaluation, and management of childhood interstitial lung disease in infancy. Am J Respir Crit Care Med 2013;188:376–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ehsan Z, Montgomery GS, Tiller C, Kisling J, Chang DV, Tepper RS. An infant with pulmonary interstitial glycogenosis: clinical improvement is associated with improvement in the pulmonary diffusion capacity. Pediatr Pulmonol 2014;49:E17–E20. [DOI] [PubMed] [Google Scholar]
- 9.Brody AS, Kosorok MR, Li Z, et al. Reproducibility of a scoring system for computed tomography scanning in cystic fibrosis. J Thorac Imaging 2006;21:14–21. [DOI] [PubMed] [Google Scholar]
- 10.Children’s Hospital Colorado Community Health Assessment 2012: Priorities and opportunities for outreach and advocacy 2012. (Accessed 3/24/2017, at https://www.childrenscolorado.org/contentassets/b779a1a9e0ef47d7999ca5126233df5a/2012-chco-community-health-needs-assessment.pdf.).
- 11.Cutz E, Chami R, Dell S, Langer J, Manson D. Pulmonary interstitial glycogenosis associated with a spectrum of neonatal pulmonary disorders. Hum Pathol 2017;68:154–165. [DOI] [PubMed] [Google Scholar]
- 12.Castillo M, Vade A, Lim-Dunham JE, Masuda E, Massarani-Wafai R. Pulmonary interstitial glycogenosis in the setting of lung growth abnormality: radiographic and pathologic correlation. Pediatr Radiol 2010;40:1562–1565. [DOI] [PubMed] [Google Scholar]
- 13.Rauch D, Wetzke M, Reu S, et al. Persistent tachypnea of infancy. usual and aberrant. Am J Respir Crit Care Med 2016;193:438–447. [DOI] [PubMed] [Google Scholar]
- 14.Deterding RR, Pye C, Fan LL, Langston C. Persistent tachypnea of infancy is associated with neuroendocrine cell hyperplasia. Pediatr Pulmonol 2005;40:157–165. [DOI] [PubMed] [Google Scholar]
- 15.Brody AS, Guillerman RP, Hay TC, et al. Neuroendocrine cell hyperplasia of infancy: diagnosis with high-resolution CT. AJR Am J Roentgenol 2010;194:238–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Barrington KJ. The adverse neuro-developmental effects of postnatal steroids in the preterm infant: a systematic review of RCTs. BMC Pediatr 2001;1:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Baker CD, Martin S, Thrasher J, et al. A standardized discharge process decreases length of stay for ventilator-dependent children. Pediatrics 2016;137:e1–e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Baker CD, Abman SH, Mourani PM. Pulmonary hypertension in preterm infants with bronchopulmonary dysplasia. Pediatr Allergy Immunol Pulmonol 2014;27:8–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ivy DD, Abman SH, Barst RJ, et al. Pediatric pulmonary hypertension. J Am Coll Cardiol 2013;62:D117–D126. [DOI] [PubMed] [Google Scholar]
- 20.O’Reilly R, Kilner D, Ashworth M, Aurora P. Diffuse lung disease in infants less than 1 year of age: histopathological diagnoses and clinical outcome. Pediatr Pulmonol 2015;50:1000–1008. [DOI] [PubMed] [Google Scholar]
- 21.Weinman JP, White CJ, Liptzin DR, Deterding RR, Galambos C, Browne LP. High-resolution CT findings of pulmonary interstitial glycogenosis. Pediatr Radiol 2018;1–7. [Epub ahead of print]. [DOI] [PubMed]