

Abstract
Microtubule-targeting chemotherapies are linked to impaired cellular metabolism, which may contribute to skeletal muscle dysfunction. However, the mechanisms by which metabolic homeostasis is perturbed remains unknown. Tubulin, the fundamental unit of microtubules, has been implicated in the regulation of mitochondrial-cytosolic ADP/ATP exchange through its interaction with the outer membrane voltage-dependent anion channel (VDAC). Based on this model, we predicted that disrupting microtubule architecture with the stabilizer paclitaxel and destabilizer vinblastine would impair skeletal muscle mitochondrial bioenergetics. Here, we provide in vitro evidence of a direct interaction between both α-tubulin and βII-tubulin with VDAC2 in untreated single extensor digitorum longus (EDL) fibers. Paclitaxel increased both α- and βII-tubulin-VDAC2 interactions, whereas vinblastine had no effect. Utilizing a permeabilized muscle fiber bundle preparation that retains the cytoskeleton, paclitaxel treatment impaired the ability of ADP to attenuate H2O2 emission, resulting in greater H2O2 emission kinetics. Despite no effect on tubulin-VDAC2 binding, vinblastine still altered mitochondrial bioenergetics through a surprising increase in ADP-stimulated respiration while also impairing ADP suppression of H2O2 and increasing mitochondrial susceptibility to calcium-induced formation of the proapoptotic permeability transition pore. Collectively, these results demonstrate that altering microtubule architecture with chemotherapeutics disrupts mitochondrial bioenergetics in EDL skeletal muscle. Specifically, microtubule stabilization increases H2O2 emission by impairing ADP sensitivity in association with greater tubulin-VDAC binding. In contrast, decreasing microtubule abundance triggers a broad impairment of ADP’s governance of respiration and H2O2 emission as well as calcium retention capacity, albeit through an unknown mechanism.
Keywords: chemotherapy; fatigue; microtubule; mitochondria; paclitaxel, vinblastine
INTRODUCTION
Two common classes of chemotherapeutics are the microtubule-targeting taxanes (e.g., paclitaxel or taxol) and vinka alkaloids (e.g., vinblastine), which are used as treatments for solid-state tumors in multiple cancers. Many patients receiving these types of chemotherapies report symptoms of fatigue (31), and tumor-free rat models injected with these compounds show severe hind-limb muscle weakness and impaired cardiac contraction (11, 16). This weakness has been associated with the development of peripheral neuropathies induced by chemotherapies, where paclitaxel and vinblastine alter neuronal microtubules, causing axonal dysfunctions and transmission impairments (5, 13). These peripheral nerve dysfunctions have been associated with marked mitochondrial respiratory impairments, leading to substantial morphological changes to skeletal muscle and cellular metabolic stress (5, 10, 16). However, the degree to which this also occurs in skeletal muscle fibers remains unknown. Likewise, the mechanism by which these drugs impair oxidative phosphorylation is uncertain, as is their influence on mitochondrial reactive oxygen species emission and calcium-induced permeability transition pore (mPTP) formation. Given that all three bioenergetic functions are critical determinants of cell fate (22), understanding the relationship between altered microtubule dynamics and mitochondrial bioenergetics may provide insight into the mechanism by which these compounds disturb cellular metabolism in muscle.
Both paclitaxel and vinblastine disrupt the normal dynamics of microtubules, leading to cell death following mitotic arrest in tumors (3). An emerging model has proposed that microtubules regulate mitochondrial bioenergetics in muscle through the direct binding of tubulin to the ADP/ATP exchanger voltage-dependent anion channel (VDAC) on the outer mitochondrial membrane (Fig. 1A) (9). The available literature suggests that α- and β-tubulin likely bind VDAC in muscle, which may impede ADP import, thereby implicating microtubules as regulators of ADP-dependent mitochondrial bioenergetics (27, 28, 32). As the model posits that any change in tubulin-VDAC binding would alter ADP’s control of bioenergetics (Fig. 1A), an extended prediction is that stressors that manipulate microtubule architecture may alter ADP-dependent bioenergetics. If true, this relationship may represent a novel mechanism by which microtubule-targeting chemotherapy disrupts cellular energy homeostasis in muscle.
Fig. 1.

α-Tubulin- voltage-dependent anion channel (VDAC)2 interactions in extensor digitorum longus (EDL) single fibers increase following paclitaxel stabilization. A: model of tubulin regulation of VDAC-dependent ADP/ATP exchange (9). Confocal microscopy images of single EDL fiber. B: control experiments in the presence of either α-tubulin, βII-tubulin, or VDAC2, incubated alone or together (n = 3) C: proximity ligation assay representative images. D: graphical depiction of α-tubulin-VDAC2 protein interaction (n = 8, *P = 0.001 vs. control). E: graphical depiction of βII-tubulin-VDAC2 interaction (n = 8–9, *P = 0.0001 vs. control). Scale bar, 16 μm. Results are reported as means ± SEM.
The purpose of this study was to determine whether paclitaxel and vinblastine alter mitochondrial bioenergetics by manipulating tubulin-VDAC2 binding in skeletal muscle. VDAC2 is of particular interest, as global gene knockdown of VDAC1 (abundant on both sarcolemma and mitochondrial outer membranes) and VDAC3 (present on the mitochondrial outer membrane) isoforms permit proper embryonic and pup development, whereas VDAC2 knockdown resulted in embryonic death, signifying its importance for survival (26). We employed a proximity ligation assay with <30 nm resolution (27) to determine the extent of isoform-specific tubulin binding to VDAC2. We then determined the effect of these compounds on ADP-dependent bioenergetics, namely ADP-stimulated oxidative phosphorylation; ADP-attenuation of H2O2 emission, as occurs through lowering membrane potential; and the propensity for mPTP formation, as VDAC has been implicated in this process (33).
MATERIALS AND METHODS
Animal care.
Male and female Wistar rats (180.9 ± 7.0 g) were bred at York University (Toronto, ON, Canada) from breeders purchased at Charles River Laboratories (Toronto, ON, Canada). Housing was maintained on a 12:12-h light-dark cycle in pairs. Rats were fed standard rodent chow and had ad libitum access to food and water. A total of 35 rats, age 6–8 wk, were used for the completion of bioenergetic (n = 24) and immunohistochemical (n = 11) experiments. All procedures were in accordance with the Canadian Care Animal Committee and with approval from the York University Animal Care Committee (AUP 2016–19).
Preparation of permeabilized muscle fiber bundles and mitochondrial bioenergetic assays.
The extensor digitorum longus (EDL) muscle was removed and immediately placed in BIOPS buffer on ice for further separation of fibers into intact bundles (2–4 mg wet wt) that were incubated at 4°C with either the microtubule stabilizer, paclitaxel (Tocris, 1097, Minneapolis, MN) at 23 μM for 2 h (21), or the microtubule destabilizer vinblastine (Sigma-Aldrich, V1377, St. Louis, MO) at 3 μM for 1 h (23). Control bundles were incubated in corresponding concentrations of the vehicle DMSO. Following incubations, bundles were permeabilized (PmFB) with saponin (18, 24) and washed for 15 min at 4°C. Bundles dedicated to H2O2 emission were permeabilized in the presence of 2,5-dinitrochlorobenzene (CDNB) to deplete glutathione and allow the detection of H2O2 emission (4). Respiration assays were completed using a high-resolution respirometer (Oroboros Instruments Corp., Innsbruck, Austria) at 37°C in MiRO5 respiration media (6) supplemented with 20 mM creatine (1, 29) and 5 μM blebbistatin to prevent PmFB contraction (20, 25). Creatine was used to saturate mitochondrial creatine kinase to promote efficient cytosolic-mitochondrial exchange of creatine and phosphocreatine through VDAC, in addition to ADP/ATP, to optimize respiratory conditions (9).
Separate PmFBs were used for fluorometric detection of H2O2 emission (QuantaMaster 40, HORIBA Scientific) with Amplex UltraRed as described previously (20). Briefly, PmFBs were placed into a quartz cuvette containing Amplex UltraRed dissolved in Buffer Z. H2O2 emission was induced with the addition of 10 mM pyruvate and 4 mM malate (NADH, complex I), followed by a titration of ADP to determine the efficiency of attenuating H2O2 emission. Other PmFBs were used to determine calcium retention capacity (CRC), utilizing EGTA-coated cuvettes, loaded with 300 μl of Buffer Y supplemented with 1 μM calcium green-5N (Invitrogen), 2 µM thapsigargin, 5 mM 2-deoxyglucose, 2 U/ml hexokinase, 20 mM creatine, 5 mM glutamate, 2 mM malate, 5 µM blebbistatin, and 40 µM EGTA. Briefly, Ca2+ uptake was initiated with 8 nmol pulses of CaCl2, with subsequent 4 nmol pulses until an mPTP opening was observed (QuantaMaster 80; HORIBA Scientific) (20).
Immunohistological experiments and image acquisition.
EDL muscle was collagenase-treated and single fibers were isolated and then treated with 23 μM paclitaxel, 3 μM vinblastine, or DMSO for 2 h at room temperature. Following drug incubations, fibers were washed, fixed, blocked, and permeabilized in preparation for staining. Primary and secondary antibodies used were as follows: α-tubulin (1:1,000, Sigma-Aldrich; T6199), βII-tubulin (1:250, Abcam; ab28036; separate fibers), VDAC2 (1:250, Santa Cruz; 32059), Alexa Fluor 488 (Invitrogen/Thermo-Fisher Scientific; A21121), and Alexa Fluor 555 (Invitrogen/Thermo-Fisher Scientific; AS1431). The proximity ligation procedures were completed as previously done (12, 27). Briefly, fibers containing primary antibodies were incubated with oligonucleotide secondary probes and then subsequently with detection reagents, allowing the quantification at a specific wavelength. All fibers were coated with mounting media and secured with a coverslip. Minimal background signal during the proximity ligation assay was verified by incubating fibers with the probes, ligase, and polymerase, using each antibody above in isolation (Fig. 1B). Control experiments confirmed the specificity of the proximity ligation assay used to detect tubulin-VDAC2 interactions (Fig. 1B).
Images were acquired using a Zeiss laser scanning confocal microscope 700 (Carl Zeiss, Thornwood, NY). Images were collected with a ×40 oil immersion lens with the pinhole adjusted to 1 Airy unit capturing three fields of view per sample, with 8–12 stacks and a z-step of 0.23 μm. Image quantitation was completed using Imaris image quantifying software (Bitplane, Zurich, Switzerland) using the spot tool to identify protein-protein interactions.
Analysis and statistics.
Results are reported as means ± SEM, with significance accepted at P < 0.05. PmFBs were randomized into groups, and all analyses were completed in an unblinded fashion. Outliers were omitted in accordance with the ROUT test. The D’Agostino-Pearson omnibus or the Shapiro-Wilks normality test was first performed to determine whether data resembled a Gaussian distribution. Given that all data passed normality, differences between the means were then tested (GraphPad Prism 7, La Jolla, CA) by one-way ANOVA for proximity ligation assays and a two-way ANOVA for ADP-stimulated respiration and attenuation of H2O2 emission followed by Fisher’s least squares difference post hoc analyses when a significant F ratio was obtained. A Student’s unpaired t-test was used to test differences in CRC and all other H2O2 emission data in the absence of ADP.
RESULTS
Paclitaxel increased both α-tubulin-VDAC2 (P = 0.001) and βII-tubulin-VDAC2 interactions (P = 0.0001) (Fig. 1, C–E). This effect appeared to be associated with altered organization of α-tubulin, as has been shown previously (14), but not βII-tubulin (Fig. 2). α- (P = 0.0002) and βII-tubulin-VDAC2 (P = 0.002) interaction was also significantly higher when comparing paclitaxel and vinblastine. Vinblastine had no effect on either isoform’s interaction with VDAC2 (Fig. 1, C–E), despite an apparent reduction in α-tubulin content (Fig. 2). We next determined the degree to which ADP-dependent mitochondrial bioenergetics were affected by either drug. After stimulating complex I-supported respiration with NADH (generated by pyruvate and malate), subsequent titrations of ADP were not affected by paclitaxel (Fig. 3A, P = 0.81), yet they were surprisingly increased by vinblastine (Fig. 3B, main effect, P = 0.006). Although neither drug affected H2O2 emission in the absence of ADP [Fig. 3C, P = 0.47 (paclitaxel) and Fig. 3D, P = 0.20 (vinblastine)], paclitaxel impaired the ability of ADP to attenuate H2O2 emission (Fig. 3E; P = 0.0006, main effect), consistent with greater interactions between VDAC2 and both α- and βII-tubulin. Vinblastine, although having no effect on tubulin-VDAC2 binding, also impaired ADP’s attenuation of H2O2 (Fig. 3F, P = 0.002, main effect).
Fig. 2.

α-Tubulin but not βII-tubulin organization is altered following microtubule-targeted chemotherapy. Confocal microscopy representative images of single extensor digitorum longus fibers stained with antibodies recognizing α-tubulin and βII-tubulin following either control (DMSO), paclitaxel, or vinblastine treatment. Scale bar, 16 μm.
Fig. 3.

ADP-dependent mitochondrial bioenergetics is altered following microtubule-targeted chemotherapy incubations in extensor digitorum longus permeabilized muscle fiber bundles. Complex I-supported (NADH from 5 mM pyruvate/4 mM malate) State 3 (+ADP) respiration was tested following incubations with (n = 6–8) paclitaxel (A) and vinblastine (B). Complex I-supported (10 mM pyruvate/4 mM malate) H2O2 emission in the absence (C and D) and presence of ADP (E and F) was determined following both paclitaxel and vinblastine incubations (n = 6–8). ADP-insensitive pyruvate dehydrogenase complex (PDC)-supported (10 mM pyruvate) H2O2 emission was assessed following paclitaxel (G) and vinblastine (H) incubations (n = 6–7). **P < 0.01 and ***P < 0.001 vs. control. Results are reported as means ± SEM.
Similar impairments in the ability of ADP to attenuate H2O2 by both drugs were seen when supported by succinate (reverse electron flow to complex I, data not shown). Pyruvate dehydrogenase complex-supported H2O2 was also assessed as a control, given it is not regulated directly by ADP. No effect of either drug was observed (Fig. 3, G and H), suggesting that the drugs act specifically on ADP’s ability to attenuate H2O2, as noted above.
Given VDAC’s potential role in the formation of the mPTP and mitochondrial induction of cell death (18), a calcium-titration “stress” protocol was used to determine CRC as an index of the propensity for mPTP induction. Similar concentrations of calcium were required to open the mPTP following paclitaxel incubations (Fig. 4A), whereas vinblastine demonstrated a reduced CRC, suggesting a greater sensitivity to calcium-induced mPTP (Fig. 4B, P = 0.01).
Fig. 4.
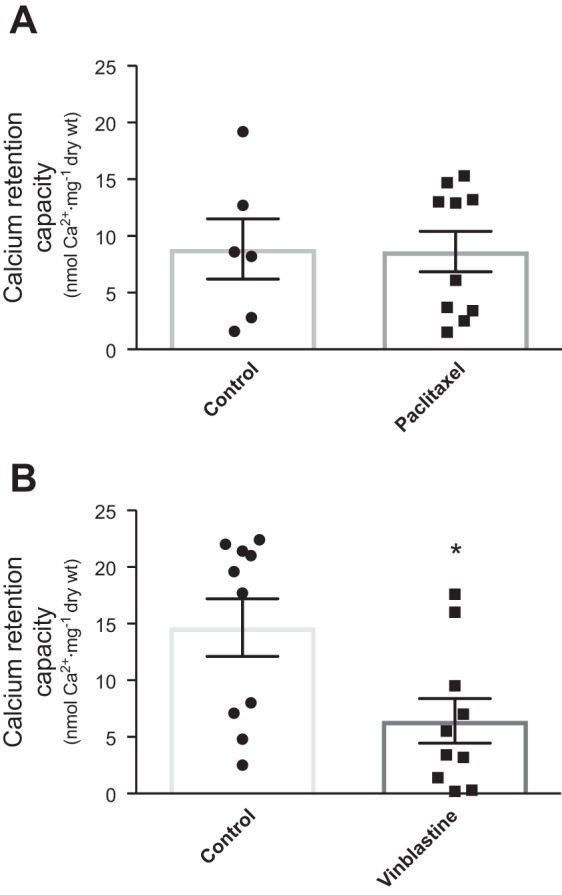
Calcium retention capacity is altered following vinblastine incubation in extensor digitorum longus permeabilized muscle fiber bundles. Mitochondrial calcium retention capacity following paclitaxel (A) and vinblastine (B). *P < 0.05 vs. control (n = 6–10). Results are reported as means ± SEM.
DISCUSSION
Using a high-resolution proximity ligation assay, we demonstrate that both α- and βII-tubulin interact with VDAC2 in EDL muscle, confirming previous suggestions in skeletal muscle based on confocal fluorescent overlay approaches. This finding aligns with the proposed model that tubulin isoforms regulate ADP/ATP exchange through VDAC on the outer mitochondrial membrane in skeletal muscle (32). We then treated muscles with the microtubule stabilizer paclitaxel and destabilizer vinblastine to determine whether altering microtubule architecture changed tubulin-VDAC2 interactions and ADP-dependent mitochondrial bioenergetics. Here, we show that paclitaxel increased α-tubulin-VDAC2 and βII-tubulin-VDAC2 interactions and impaired the ability of ADP to attenuate H2O2 emission, which supports the model. In contrast, vinblastine did not alter tubulin-VDAC2 interactions but still impaired ADP-dependent mitochondrial bioenergetics. These findings suggest that both cytotoxic drugs cause mitochondrial dysfunction but potentially through different mechanisms.
Several lines of evidence suggest that tubulin may impede ADP’s control of mitochondrial bioenergetics by directly decreasing VDAC permeability. The organization of βII-tubulin in proximity to mitochondria in rat skeletal and cardiac muscle suggests its involvement in regulating mitochondrial bioenergetics (8, 32). This interaction has also been explored in cancer cell lines where transmission electron microscopy experiments identified both α- and β-tubulin in close proximity with the mitochondria. Coimmunoprecipitation experiments have also identified α-tubulin and VDAC in mitochondrial isolations (2). In addition, trypsin removal of βII-tubulin increased ADP-stimulated respiration in cardiomyocytes, whereas the addition of tubulin to isolated mitochondria decreased ADP-stimulated respiration, further supporting the role of tubulin in VDAC permeability regulation (8, 32). The present findings align with this model by demonstrating a direct interaction between VDAC2 and both α- and βII-tubulin in skeletal muscle, and that these interactions are increased by altering microtubule architecture with paclitaxel in conjunction with impaired ADP-control of H2O2 emission.
Whereas paclitaxel increased tubulin-VDAC2 interactions and impaired ADP-attenuation of H2O2 emission, a similar impairment in ADP-stimulated respiration was not observed. This finding does not align with the proposed model that tubulin regulates ADP’s overall control of bioenergetics. The lack of effect of paclitaxel on ADP-stimulated respiration contrasts with evidence that the removal or addition of tubulin to permeabilized cardiomyocytes (7) and isolated brain mitochondria (27) increases and decreases ADP-stimulated respiration, respectively. Although the results challenge the model of tubulin’s regulation of respiration, the differences between studies may be related to the examined tissues and cellular models or sample preparations (e.g., cardiomyocytes, isolated brain mitochondria, cancer cell culture, and permeabilized skeletal muscle fibers). A previous report in skeletal muscle demonstrated an inverse association between free/nonpolymerized tubulin and mitochondrial sensitivity to ADP, suggesting free tubulin may regulate VDAC permeability to ADP (32), although the comparison was performed between muscles with differing ADP sensitivities rather than using an approach of manipulating free or polymerized tubulin. The present study suggests a refinement to the model in EDL, whereby altering tubulin-VDAC2 binding in this muscle alters ADP-control of H2O2 emission specifically. The lack of effect on ADP control of respiration is perplexing but may be due to slight changes in ADP permeability such that H2O2 emission responds with greater sensitivity than respiration, at least in muscle. In addition, it has been suggested that VDAC also facilitates the export of mitochondrial-derived phosphocreatine in addition to ADP/ATP cycling, but this is not fully established (9). Given that creatine was present in the assays during respiration and H2O2 emission, it is possible that creatine/phosphocreatine cycling may also have been modulated by paclitaxel’s effect on tubulin-VDAC2 and may serve as an area for future research. Finally, the lack of change in calcium-induced mPTP in EDL with paclitaxel contrasts with previous work in permeabilized cardiomyocytes, which reported increased propensity for mPTP formation (17). This may suggest that paclitaxel’s effects on mPTP are fiber-type dependent.
Following vinblastine incubations, we reported a surprising increase in ADP-stimulated respiration, as well as an impairment of ADP suppression of H2O2 and increased susceptibility to apoptosis. However, there were no changes in tubulin-VDAC2 interactions, which suggests off-target effects on mitochondrial bioenergetics. For example, vinblastine activates apoptosis through the c-Jun NH2-terminal protein pathway (JNK), as reported in human cervical carcinoma cells (15), and nocodazole—another microtubule destabilizer—triggers the release of cytochrome c oxidase through the formation of the mPTP (17). This is consistent with the reduced CRC seen in the present study, whereby less calcium is required to trigger mPTP. An alternative mechanism to explore is the possibility that lower tubulin content following vinblastine treatment disrupts mitochondrial morphology by impairing tethering to microtubules through a Miro GTPase-regulated docking system (30), which would occur independently of VDAC. It is also possible that vinblastine altered VDAC2 interactions with other isoforms of tubulin or tubulin interactions with mitochondrial VDAC1 or 3. These VDAC isoforms were not examined in the present study, given that VDAC1 is also on the sarcolemma and would be difficult to separate in the proximity ligation assay, whereas VDAC3 is less abundant than VDAC2 (19). Collectively, our findings suggest that either the tubulin-VDAC model is more complex than previously proposed or vinblastine’s effects on cellular stress responses linked to mitochondrial dysfunction are independent of tubulin-VDAC binding.
In conclusion, these findings demonstrate that microtubule-targeting chemotherapy impairs mitochondrial bioenergetics in EDL skeletal muscle. In the case of paclitaxel, we identified altered tubulin-VDAC2 interactions as a novel potential mechanism underlying this compound’s effect on increasing H2O2 emission through impairing control by ADP. The effects of paclitaxel also support the developing model that mitochondria are subject to regulation by cytoskeletal dynamics through microtubule influence on bioenergetic control by ADP, but the dissociation between ADP’s control of respiration and H2O2 emission should be a focus for further investigation. Moreover, it appears that microtubule disruption by vinblastine is associated with impaired mitochondrial bioenergetics through mechanisms distinct from tubulin-VDAC interactions. Ultimately, these findings serve as a foundation for exploring the mechanism by which these chemotherapeutics contribute to muscle dysfunction and suggest that microtubule dynamics may be a potential mechanism in this regard.
GRANTS
This project was funded by Natural Sciences and Engineering Research Council of Canada (NSERC) Grant 436138-2013, a James H. Cummings Foundation grant, and Canada Foundation for Innovation and Ontario Research Fund Grant 32449 to C. G. R. Perry. S. V. Ramos received an Ontario Graduate Scholarship and M. C. Hughes received a NSERC Canada Graduate Scholarships-PhD scholarship.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
S.V.R., M.C.H., and C.G.R.P. conceived and designed research; S.V.R. and M.C.H. performed experiments; S.V.R. analyzed data; S.V.R., M.C.H., and C.G.R.P. interpreted results of experiments; S.V.R., M.C.H., and C.G.R.P. prepared figures; S.V.R., M.C.H., and C.G.R.P. drafted manuscript; S.V.R., M.C.H., and C.G.R.P. edited and revised manuscript; S.V.R., M.C.H., and C.G.R.P. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Drs. Thomas Hawke and Donna D’Souza (McMaster Univ., Hamilton, ON, Canada) for initial guidance in single EDL fiber preparation.
REFERENCES
- 1.Anmann T, Guzun R, Beraud N, Pelloux S, Kuznetsov AV, Kogerman L, Kaambre T, Sikk P, Paju K, Peet N, Seppet E, Ojeda C, Tourneur Y, Saks V. Different kinetics of the regulation of respiration in permeabilized cardiomyocytes and in HL-1 cardiac cells. Importance of cell structure/organization for respiration regulation. Biochim Biophys Acta 1757: 1597–1606, 2006. doi: 10.1016/j.bbabio.2006.09.008. [DOI] [PubMed] [Google Scholar]
- 2.Carré M, André N, Carles G, Borghi H, Brichese L, Briand C, Braguer D. Tubulin is an inherent component of mitochondrial membranes that interacts with the voltage-dependent anion channel. J Biol Chem 277: 33664–33669, 2002. doi: 10.1074/jbc.M203834200. [DOI] [PubMed] [Google Scholar]
- 3.Chen JG, Horwitz SB. Differential mitotic responses to microtubule-stabilizing and -destabilizing drugs. Cancer Res 62: 1935–1938, 2002. [PubMed] [Google Scholar]
- 4.Fisher-Wellman KH, Gilliam LAA, Lin C-T, Cathey BL, Lark DS, Darrell Neufer P. Mitochondrial glutathione depletion reveals a novel role for the pyruvate dehydrogenase complex as a key H2O2-emitting source under conditions of nutrient overload. Free Radic Biol Med 65: 1201–1208, 2013. doi: 10.1016/j.freeradbiomed.2013.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ghoreishi Z, Esfahani A, Djazayeri A, Djalali M, Golestan B, Ayromlou H, Hashemzade S, Asghari Jafarabadi M, Montazeri V, Keshavarz SA, Darabi M. Omega-3 fatty acids are protective against paclitaxel-induced peripheral neuropathy: a randomized double-blind placebo controlled trial. BMC Cancer 12: 355, 2012. doi: 10.1186/1471-2407-12-355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gnaiger E, Kuznetsov AV, Schneeberger S, Seiler R, Brandacher G, Steurer W, Margreiter R. Mitochondria in the cold. In: Life in the Cold, edited by Heldmaier G, Klingenspor M. Berlin: Springer, 2000. doi: 10.1007/978-3-662-04162-8_45. [DOI] [Google Scholar]
- 7.Gonzalez-Granillo M, Grichine A, Guzun R, Usson Y, Tepp K, Chekulayev V, Shevchuk I, Karu-Varikmaa M, Kuznetsov AV, Grimm M, Saks V, Kaambre T. Studies of the role of tubulin beta II isotype in regulation of mitochondrial respiration in intracellular energetic units in cardiac cells. J Mol Cell Cardiol 52: 437–447, 2012. doi: 10.1016/j.yjmcc.2011.07.027. [DOI] [PubMed] [Google Scholar]
- 8.Guerrero K, Monge C, Brückner A, Puurand U, Kadaja L, Käämbre T, Seppet E, Saks V. Study of possible interactions of tubulin, microtubular network, and STOP protein with mitochondria in muscle cells. Mol Cell Biochem 337: 239–249, 2010. doi: 10.1007/s11010-009-0304-1. [DOI] [PubMed] [Google Scholar]
- 9.Guzun R, Gonzalez-Granillo M, Karu-Varikmaa M, Grichine A, Usson Y, Kaambre T, Guerrero-Roesch K, Kuznetsov A, Schlattner U, Saks V. Regulation of respiration in muscle cells in vivo by VDAC through interaction with the cytoskeleton and MtCK within mitochondrial interactosome. Biochim Biophys Acta 1818: 1545–1554, 2012. doi: 10.1016/j.bbamem.2011.12.034. [DOI] [PubMed] [Google Scholar]
- 10.Han Y, Smith MT. Pathobiology of cancer chemotherapy-induced peripheral neuropathy (CIPN). Front Pharmacol 4: 156, 2013. doi: 10.3389/fphar.2013.00156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Howarth FC, Calaghan SC, Boyett MR, White E. Effect of the microtubule polymerizing agent taxol on contraction, Ca2+ transient and L-type Ca2+ current in rat ventricular myocytes. J Physiol 516: 409–419, 1999. doi: 10.1111/j.1469-7793.1999.0409v.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jorquera G, Altamirano F, Contreras-Ferrat A, Almarza G, Buvinic S, Jacquemond V, Jaimovich E, Casas M. Cav1.1 controls frequency-dependent events regulating adult skeletal muscle plasticity. J Cell Sci 126: 1189–1198, 2013. doi: 10.1242/jcs.116855. [DOI] [PubMed] [Google Scholar]
- 13.Juntunen J. Effects of colchicine and vinblastine on neurotubules of the sciatic nerve and cholinesterases in the developing myoneural junction of the rat. Z Zellforsch Mikrosk Anat 142: 193–204, 1973. doi: 10.1007/BF00307032. [DOI] [PubMed] [Google Scholar]
- 14.Kerr JP, Robison P, Shi G, Bogush AI, Kempema AM, Hexum JK, Becerra N, Harki DA, Martin SS, Raiteri R, Prosser BL, Ward CW. Detyrosinated microtubules modulate mechanotransduction in heart and skeletal muscle. Nat Commun 6: 8526, 2015. doi: 10.1038/ncomms9526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kolomeichuk SN, Terrano DT, Lyle CS, Sabapathy K, Chambers TC. Distinct signaling pathways of microtubule inhibitors–vinblastine and Taxol induce JNK-dependent cell death but through AP-1-dependent and AP-1-independent mechanisms, respectively. FEBS J 275: 1889–1899, 2008. doi: 10.1111/j.1742-4658.2008.06349.x. [DOI] [PubMed] [Google Scholar]
- 16.Konishi S, Kishida S. Studies on the morphological changes of skeletal muscle induced by vincristine, vinblastine and colchicine. Bull Osaka Med Sch 30: 19–40, 1984. [PubMed] [Google Scholar]
- 17.Kumazawa A, Katoh H, Nonaka D, Watanabe T, Saotome M, Urushida T, Satoh H, Hayashi H. Microtubule disorganization affects the mitochondrial permeability transition pore in cardiac myocytes. Circ J 78: 1206–1215, 2014. doi: 10.1253/circj.CJ-13-1298. [DOI] [PubMed] [Google Scholar]
- 18.Kuznetsov AV, Tiivel T, Sikk P, Kaambre T, Kay L, Daneshrad Z, Rossi A, Kadaja L, Peet N, Seppet E, Saks VA. Striking differences between the kinetics of regulation of respiration by ADP in slow-twitch and fast-twitch muscles in vivo. Eur J Biochem 241: 909–915, 1996. doi: 10.1111/j.1432-1033.1996.00909.x. [DOI] [PubMed] [Google Scholar]
- 19.Messina A, Reina S, Guarino F, De Pinto V. VDAC isoforms in mammals. Biochim Biophys Acta 1818: 1466–1476, 2012. doi: 10.1016/j.bbamem.2011.10.005. [DOI] [PubMed] [Google Scholar]
- 20.Monaco CMF, Hughes MC, Ramos SV, Varah NE, Lamberz C, Rahman FA, McGlory C, Tarnopolsky MA, Krause MP, Laham R, Hawke TJ, Perry CGR. Altered mitochondrial bioenergetics and ultrastructure in the skeletal muscle of young adults with type 1 diabetes. Diabetologia 61: 1411–1423, 2018. doi: 10.1007/s00125-018-4602-6. [DOI] [PubMed] [Google Scholar]
- 21.Needleman DJ, Ojeda-Lopez MA, Raviv U, Ewert K, Miller HP, Wilson L, Safinya CR. Radial compression of microtubules and the mechanism of action of taxol and associated proteins. Biophys J 89: 3410–3423, 2005. doi: 10.1529/biophysj.104.057679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nicholls DG, Ferguson SJ. Bioenergetics 4. New York: Elsevier, 2013, p. 434. [Google Scholar]
- 23.Panda D, Jordan MA, Chu KC, Wilson L. Differential effects of vinblastine on polymerization and dynamics at opposite microtubule ends. J Biol Chem 271: 29807–29812, 1996. doi: 10.1074/jbc.271.47.29807. [DOI] [PubMed] [Google Scholar]
- 24.Perry CG, Kane DA, Lanza IR, Neufer PD. Methods for assessing mitochondrial function in diabetes. Diabetes 62: 1041–1053, 2013. doi: 10.2337/db12-1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Perry CGR, Kane DA, Lin C-T, Kozy R, Cathey BL, Lark DS, Kane CL, Brophy PM, Gavin TP, Anderson EJ, Neufer PD. Inhibiting myosin-ATPase reveals a dynamic range of mitochondrial respiratory control in skeletal muscle. Biochem J 437: 215–222, 2011. doi: 10.1042/BJ20110366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Raghavan A, Sheiko T, Graham BH, Craigen WJ. Voltage-dependant anion channels: novel insights into isoform function through genetic models. Biochim Biophys Acta 1818: 1477–1485, 2012. doi: 10.1016/j.bbamem.2011.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rostovtseva TK, Gurnev PA, Hoogerheide DP, Rovini A, Sirajuddin M, Bezrukov SM. Sequence diversity of tubulin isotypes in regulation of the mitochondrial voltage-dependent anion channel. J Biol Chem 293: 10949–10962, 2018. doi: 10.1074/jbc.RA117.001569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rostovtseva TK, Sheldon KL, Hassanzadeh E, Monge C, Saks V, Bezrukov SM, Sackett DL. Tubulin binding blocks mitochondrial voltage-dependent anion channel and regulates respiration. Proc Natl Acad Sci USA 105: 18746–18751, 2008. doi: 10.1073/pnas.0806303105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Saks VA, Kuznetsov AV, Khuchua ZA, Vasilyeva EV, Belikova JO, Kesvatera T, Tiivel T. Control of cellular respiration in vivo by mitochondrial outer membrane and by creatine kinase. A new speculative hypothesis: possible involvement of mitochondrial-cytoskeleton interactions. J Mol Cell Cardiol 27: 625–645, 1995. doi: 10.1016/S0022-2828(08)80056-9. [DOI] [PubMed] [Google Scholar]
- 30.Saotome M, Safiulina D, Szabadkai G, Das S, Fransson A, Aspenstrom P, Rizzuto R, Hajnóczky G. Bidirectional Ca2+-dependent control of mitochondrial dynamics by the Miro GTPase. Proc Natl Acad Sci USA 105: 20728–20733, 2008. doi: 10.1073/pnas.0808953105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tew WP, Radovich D, O’Reilly E, Schwartz G, Schrag D, Saltz LB, Kelsen DP, Kepler S, Ilson DH. Phase I trial of weekly cisplatin, irinotecan and paclitaxel in patients with advanced gastrointestinal cancer. Invest New Drugs 27: 366–373, 2009. doi: 10.1007/s10637-008-9194-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Varikmaa M, Bagur R, Kaambre T, Grichine A, Timohhina N, Tepp K, Shevchuk I, Chekulayev V, Metsis M, Boucher F, Saks V, Kuznetsov AV, Guzun R. Role of mitochondria-cytoskeleton interactions in respiration regulation and mitochondrial organization in striated muscles. Biochim Biophys Acta 1837: 232–245, 2014. doi: 10.1016/j.bbabio.2013.10.011. [DOI] [PubMed] [Google Scholar]
- 33.Vercesi AE, Castilho RF, Kowaltowski AJ, de Oliveira HCF, de Souza-Pinto NC, Figueira TR, Busanello ENB. Mitochondrial calcium transport and the redox nature of the calcium-induced membrane permeability transition. Free Radic Biol Med 129: 1–24, 2018. doi: 10.1016/j.freeradbiomed.2018.08.034. [DOI] [PubMed] [Google Scholar]