

Abstract
Cdc42 was originally discovered as a key regulator of bud site assembly and polarity in S. cerevisiae. Recent genetic studies have shown that the function of Cdc42 in regulating cell polarity appears highly conserved from budding yeast to humans. The role of Cdc42 in hematopoietic cell transformation and leukemia progression has been studied in an acute myeloid leukemia model using the MLL-AF9 oncogene-induced transformation and a Cdc42 conditional gene-targeted mouse model. Here we describe the leukemia cell polarity and division symmetry assays in the context of leukemia cell fate determination.
Keywords: Cell fate, Cell polarity, Differentiation, Division symmetry, Self-renewal
1. Introduction
Fundamental to the definition of the cancer stem cell is the capacity to undergo cell divisions to produce progeny cells that are able to perpetuate the malignant clone [1, 2]. One potential avenue for therapy is to block stem divisions and force cell divisions that result in progeny fated for terminal differentiation and apoptosis. Thus, there is great interest in understanding the molecular and cellular underpinnings of division symmetry and cell fate in both normal and malignant stem cells [3,4]. We describe methods to assay cell polarity via tubulin localization in mitotic cells, and functional division symmetry via prospective isolation of daughter cells for tracking clonogenic potential.
The Rho GTPase family member Cdc42 plays an integral role in hematopoietic stem cell polarity and cytoskeletal organization in response to inputs from the bone marrow microenvironment to coordinate polarity-dependent cellular functions such as directional migration and adhesion [5–7]. Recent evidence also implicates proper regulation of Cdc42 activity and polarity in blood stem cell division symmetry and self-renewal [8]. Immunofluorescence confocal microscopy is used to analyze distribution of tubulin within murine acute myeloid leukemia (AML) cell lines [8, 9]. These cell lines were derived from oncogene-transformed hematopoietic stem and progenitor cells (HSC/Ps) transgenic for conditional Cdc42 alleles [9]. Treatment with tamoxifen binds to estrogen receptor (ER) and induces Cre-recombinase expression to knock out (KO) Cdc42 alleles (Cdc42KO). Since Cdc42 is a critical polarity determinant, the Cdc42KO cell line demonstrates loss of tubulin polarity. Tubulin immunofluorescence is measured for pixel intensity along a vector bisecting each cell to evaluate polar versus apolar distribution.
The AML cell lines are also used for analysis of division symmetry and cell fate upon loss of Cdc42 and tubulin polarity. Cell division produces two daughter cells that may have identical (symmetric) or disparate (asymmetric) cell fates. Cells are plated in semisolid media to allow visualization of doublets resulting from single-cell divisions. The doublets are plucked and transferred to a separate well where the separated daughter cells can be prospectively observed for viability and capacity for self-renewal versus terminal differentiation. Thus, the proportion of cell divisions that are symmetric versus asymmetric, and that result in self-renewal versus differentiation, can be quantified in correlation with the proportion of polar versus apolar cells.
2. Materials
Culture conditions and imaging parameters should be optimized for the cell lines of interest. Here we detail the conditions for myeloid leukemia cell lines, as previously described [10]. Store all stock reagents according to the manufacturer’s specifications, unless otherwise noted. Diligently follow all waste disposal regulations when disposing of waste materials.
2.1. Cell Culture
Cre+;Cdc42FL-MA9 cell line: Murine AML cells derived from knock-in of the MLL-AF9 (MA9; also known as KMT2A-MLLT3) oncogene into the ROSA26CreERt2;Cdc42FL/FL background. This murine AML cell line allows tamoxifen-inducible KO of Cdc42. Vehicle control (ethanol; EtOH) treatment of these cells does not induce Cdc42 deletion, thus maintaining wild-type (WT) control MA9 cells with intact Cdc42 alleles. Cell lines are available from the authors.
Phosphate-buffered saline (PBS) with 1 mM CaCl2 and MgCl2 (Gibco).
Fetal bovine serum (FBS), heat inactivated for 30 min at 56 °C, inverting every 10 min.
Antibiotics: 1% Penicillin-streptomycin (Pen-Strep; Gibco).
Medium: Iscove’s modified Dulbecco’s medium with L-gluta-mine and HEPES (IMDM; Corning cellgro) supplemented with 20% heat-inactivated FBS, 1% Pen-Strep, and cytokine mix as noted below (item 6).
Cytokines: 20 ng/mL Recombinant rat stem cell factor (rrSCF; PeproTech), 10 ng/mL mouse granulocyte-macrophage colony-stimulating factor (mGM-CSF; PeproTech), 10 ng/mL mouse interleukin 3 (mIL-3; Miltenyi), and 10 ng/mL human interleukin 6 (hIL-6; Miltenyi). Stock solutions are prepared to a concentration of1 mg/mL in PBS with 0.1% bovine serum albumin (BSA) and stored at –20 °C. Stock solutions are diluted 100-fold in IMDM to a working concentration of 10 μg/mL. Thus, to achieve the final concentrations noted above would require 2 μL per 1 mL of medium for rrSCF, and 1 μL per 1 mL of medium for all other cytokines.
4-Hydroxytamoxifen (4-OHT, Sigma-Aldrich): Prepare a 10 mM stock in ethanol and store at –20 °C. Dilute to 1 μM final concentration with culture medium.
Culture vessel: 50 mL Non-tissue culture-treated (suspension) flasks with a 0.22 μm filter cap (Celltreat).
2.2. Immunofluorescence Confocal Microscopy
Primary antibodies/stains: Anti-α-tubulin antibody (Abcam, ab6160), and ProLong Gold Antifade Mountant with DAPI (Thermo Fisher Scientific, P-36931).
Secondary antibodies: Cy3-conjugated donkey anti-rat antibody (Jackson, 712–005-153).
Plates: 35 mm Glass-bottom dish (#1.5) with 10 mm diameter microwell (Cellvis D35–10-1.5-N).
Coating: Recombinant human fibronectin fragment RetroNectin® (Takara Bio, Inc., T100A). Dilute to 20 μg/mL in PBS (it may be frozen at –20 °C and reused up to three times). Coat a 10 mm recessed well of 35 mm dish with 150 μL of RetroNectin® solution and incubate overnight at 4 °C or for 2 h at room temperature. Remove RetroNectin®, block using 200 μL of PBS + 2% BSA for 30 min at room temperature, and then aspirate to remove. Wash with 200 μL of PBS and the well is ready for seeding cells.
Fixative: 16% Paraformaldehyde (PFA), diluted to 4% final concentration in cell culture medium.
#1.5 Glass coverslip.
Permeabilization buffer: 0.2% (v/v) Triton X-100 in PBS.
Blocking buffer: 5% Normal donkey serum in PBS.
Confocal microscope: Nikon Ti microscope with 405 and 561 nm laser lines, Plano Apo VC 60 × oil objective, numerical aperture 1.4, with a Nikon C21 camera, or equivalent.
NIS Elements AR software (Nikon) or equivalent image analysis software.
2.3. Division Symmetry and Cell Fate Analysis
Methylcellulose medium MethoCult™ GF M3434 (STEM-CELL Technologies).
Additional cytokine: 10 ng/mL Final concentration mouse GM-CSFin 50 μL of PBS.
35 mm Gridded dishes (Sigma).
3 mL Syringe with 16-gauge 1.5 in. needle.
96-Well plate coated with 20 μg/mL final concentration RetroNectin® (see Subheading 2.2, item 4).
20 μL Pipette and tips.
Inverted microscope with 4× /10 ×/20×/40× objectives and 35 mm plate insert holder.
96-Well plate map for scoring/evaluation of individual wells over time.
3. Methods
Carry out all procedures at room temperature unless otherwise specified.
3.1. Cell Polarity
Culture Cre+;Cdc42FL-MA9 cells in culture medium.
Treat cultures with 1 μM final concentration 4-OHT to delete Cdc42 (KO) versus ethanol control (WT) and assay tubulin polarity at multiple time points post-deletion. Time points should be optimized for your model to develop the kinetics of deletion and timing of phenotype development. In our model, robust deletion of Cdc42, and depletion of the protein, was seen at 48 h, and phenotypes started to develop shortly thereafter, and increased in severity over time, with the latest time point assayed being 3 weeks post-deletion (see Note 1).
Seed 105 cells in 150 μL of culture medium and shake to completely cover the bottom of a RetroNectin®-coated well. Place the plate in incubator and allow cells to attach to the RetroNectin®-coated surface overnight.
After the cells have attached, remove the 150 μL of volume, replace with 75 μL of fresh medium, and return to the incubator for 30 min.
Add 75 μL of 8% PFA, diluted from the 16% PFA stock in culture medium, to the well (final concentration of PFA is 4%) and incubate for 20 min at room temperature.
Wash 1 × quick with 3 mL of PBS, followed by 3 × with 3 mL of PBS for 5 min each (see Note 2).
Aspirate PBS completely and add 150 μL of permeabilization buffer for 5 min at room temperature (see Note 2).
Wash 1 × quick with 3 mL of PBS (see Note 2).
Aspirate liquid and block with 150 μL of blocking buffer for 30 min at room temperature (see Note 2).
Add rat anti-a-tubulin primary antibody at 1:100 dilution (1.5 μL; 10 μg/mL final concentration) and incubate for 30 min at room temperature (see Note 3).
Wash 1 × quick with 3 mL of PBS, followed by 3 × with 3 mL of PBS for 5 min each (see Note 2).
Aspirate liquid and add 150 μL of blocking buffer (see Note 2). Add Cy3-conjugated donkey anti-rat secondary antibody at 1:130 dilution (1.154 μL; 10 μg/mL final concentration) and incubate for 30 min at room temperature (see Note 3).
Wash 1 × quick with 3 mL of PBS, followed by 3 × with 3 mL of PBS for 5 min each (see Note 2).
Aspirate liquid completely and quickly add 250 μL of Prolong gold anti-fade with DAPI to the well and seal with a dry sterilized #1.5 coverslip.
Image cells on a confocal microscope. Use the 405 nm laser line to excite DAPI and the 561 nm laser line to excite Cy3.
Use NIS Elements AR software to measure fluorescence intensity along a bisecting vector passing through the center of the nucleus. Consider a cell polarized when a clear asymmetric distribution of tubulin is seen, as described in Fig. 1a and b.
Analyze 50 to 100 MA9 cells individually per sample. Plot data as a percentage of the total number of cells scored per sample (Fig. 1c).
Fig. 1.
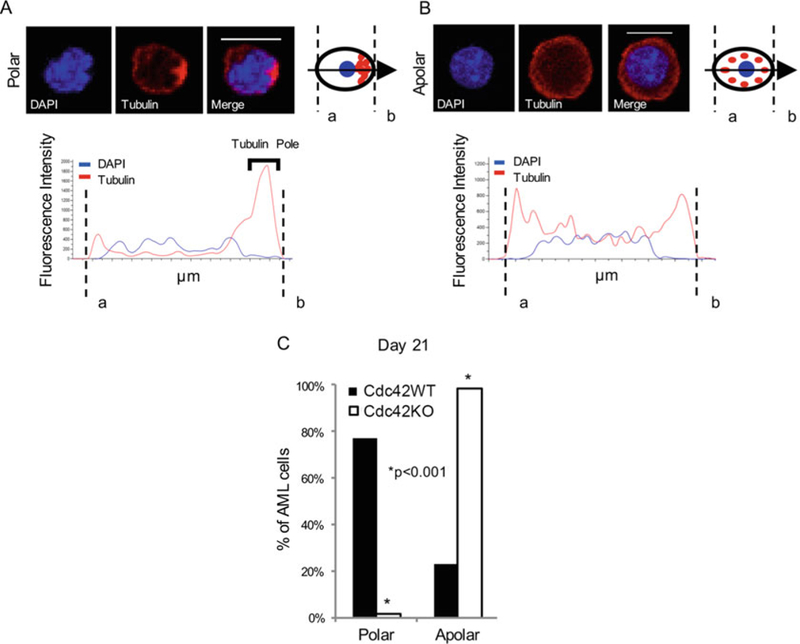
Deletion of Cdc42 caused a loss of leukemia cell polarity. MLL-AF9 leukemia cells were stained with anti-tubulin primary and rhodamine-conjugated secondary antibodies (tubulin), and DAPI (nuclear stain). Confocal images were analyzed using NIS Elements software (Nikon) to measure fluorescence intensity along a bisecting vector through individual cells. Scale bar, 10 μm. (a) Cells were considered polar when clear asymmetric distribution of tubulin was seen, or (b) apolar in the absence of tubulin asymmetry, as previously described (3). (c) Data is plotted as percentage of total number of cells scored per sample. A loss of polarity and increase in apolar cell fraction were observed following Cdc42 deletion from MA9 cells. This change became more pronounced as Cdc42KO-MA9 cells were passaged in culture, as shown at 3 weeks postdeletion of Cdc42. Functional polarity was assayed by MA9 cell adhesion and migration. Adapted from ref. 9 with permission
3.2. Isolation of Daughter Cells for Division Symmetry Assay
The procedure is depicted schematically in Fig. 2a.
Fig. 2.
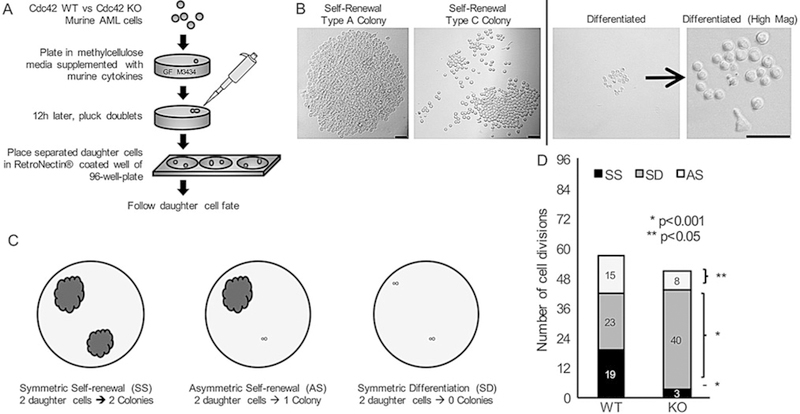
Loss of Cdc42 increased cell symmetric division and differentiation, (a) A schematic procedure measuring leukemia cell division symmetry and differentiation. Murine MLL-AF9 leukemia cells were treated with 4-OHT to induce Cre-mediated deletion of Cdc42 (K0), with ethanol-treated cells serving as vehicle control (WT). After 48 h, the cells were plated in methylcellulose medium. 16 h later, doublets were identified indicating cells at completion of first cell division. Doublets were isolated, and then transferred by pipette to a single well of RetroNectin-coated 96-well plates in IMDM +10% FBS containing 20 ng/mL rrSCF and 10 ng/mL each of murine GM-CSF, murine IL-3, and human IL-6. Daughter cells were separated by gently pipetting up and down, and then visualized 4 h later to confirm two distinct single cells adherent to the RetroNectin. Wells were visualized over the next 48 h to confirm subsequent divisions by each daughter cell indicating viability, (b and c) Wells were then visualized after 7 days and scored as having 2 colonies (symmetric self-renewal = SS), 1 colony (asymmetric self-renewal = AS), or 0 colonies (symmetric differentiation = SD). Scale bar, 50 μm. (d) Data are shown as the number of cell divisions of each type of cell division with the number of daughter cell pairs for each division type. Statistics was calculated using Student’s t-test with multiple repeats. Data are representative of six independent experiments. Adapted from ref. 9 with permission
Treat Cre+;Cdc42FL-MA9 cells for 48 h with 4-OHT or vehicle in culture medium. Seed cells at typical in vitro subculture concentrations to ensure no overgrowth of control (vehicle) cells during the 48 h (see Subheading 3, step 2). If the experimental cell group undergoes loss of viability, plate more wells to get equivalent numbers of control and treated cells. Cre+; Cdc42FL-MA9 are seeded at 105 cells/mL for in vitro culture.
Plate cells in methylcellulose. Resuspend 3300 cells in 300 μL of culture medium in a 15 mL conical tube. While vortexing, add 3 mL of MethoCult™ methylcellulose medium using a 3. mL syringe and 1.5” 16-gauge needle. After mixing is complete, leave the syringe and needle in the mixture and allow bubbles to rise to the top. Dispense 50 μL of 10 ng/mL mouse GM-CSF in PBS directly to the dish before plating of the cells. Once the mixture is degassed, dispense 1 mL aliquots to three separate 35 mm gridded dishes.
Doublets can be identified 16 h later, indicating cells at the completion of the first cell division. Isolate doublets by visualization under an inverted microscope (see Note 4) and pluck them using a P20 pipette, taking care to visualize the acquisition in real time. Each doublet plucked from the methylcellulose plate is then transferred by pipette to a single well of a RetroNectin®-coated 96-well plate with culture medium with cytokines (see Notes 5 and 6).
Separate daughter cells by pipetting gently, and then visualize 4 h later to confirm two distinct single cells (day 0). Monitor wells over the next 48 h to confirm subsequent divisions by each daughter cell, indicating viability. All visualizations are tracked using a 96-well plate map for scoring/evaluation of individual wells over time, expediting subsequent visualizations as the cell placement within the well is marked (see Notes 4 and 7).
3.2. Self-Renewal or Differentiated Cell Fate Readout
- Isolated daughter cells (Subheading 3.2, step 4) can be analyzed for colony-forming unit (CFU) potential indicating self-renewal, versus failure to form a colony indicating differentiated cell fate (see Note 6).
- (a)
-
(b)Differentiated cells are defined as daughter cells that were able to undergo at least one subsequent division, indicating initial viability, but that ultimately failed to form a colony, indicating terminal differentiation (Fig. 2b, right panel).
Evaluable wells are visualized at day 7 and scored as having 2 colonies (symmetric self-renewal, SS), 1 colony (asymmetric self-renewal, AS), or 0 colonies (symmetric differentiation, SD) as shown in Fig. 2c (see Note 4).
Plot data as the number of each type of cell division scored per 96 wells plated with WT versus KO leukemia daughter cell pairs, and calculate the statistical significance of the differences between the two (Fig. 2d).
4. Notes
In our hands protein expression of Cdc42 was shown to be absent as assessed by immunoblot by 48 h post-deletion in liquid culture. Additionally, cell cycle analysis was performed and no significant difference was observed in S/G1/G2/M populations.
During all wash steps of the staining process, add the wash solution dropwise, and never directly over the cells to prevent dislodging any adherent cells. Similarly, during the aspiration steps, tilt the plate, and aspirate from the edge of the well, leaving cells covered with a small layer of liquid to prevent dehydration and dislodgement.
Resuspend and spin all primary and secondary antibodies at full speed in a tabletop microcentrifuge for 10 min right before using for staining. Perform secondary antibody incubations and all further steps after secondary antibody addition in the dark.
Scanning methylcellulose wells or 96-well plates in a horizontal serpentine path may lead to nausea. To avoid this, it is advised to scan plates/wells in a vertical serpentine path starting at the top, scanning downwards to the bottom of the observed area, forming a column-like path, and then shifting the viewing frame over, partially overlapping the initial column, to ensure that no area is left uncovered. Repeat this process until the entire viewing area is covered.
Plucking doublets from methylcellulose and successfully transferring does require some skill, which can be improved upon with practice. Identifying the cell doublet at low magnification is recommended, and once located using higher magnification to perform the removal. The pipette should be depressed, in preparation for the subsequent aspiration. The overhead light will cast a shadow of the pipette tip, which will help in colocalization of the pipette into the doublet field of view. Typically, the pipette is inserted at an angle into the methylcellulose just to the right of the cell doublet, and the full 20 μL is brought into the pipette. Through this process you are able to see the movement of the doublet as the pipette tip enters the methylcellulose and the entrance of the doublet into the pipette tip via aspiration.
Plucking of doublets that are able to undergo cell division in the 16 h allotted in methylcellulose indicates the ability of those cells to divide in the absence of the protein. Following the fate of these individual daughter cells over time in a liquid culture colony-forming assay allows us to correlate polarity of the cells with cell fate, giving insight into the function of Cdc42. The 16 h time will likely need to be adjusted for each cell line to reflect the growth rate.
To reassure against selection bias in the division symmetry analysis, the rate of successful plucking and transfer of two distinct, viable daughter cells to wells should be determined for each group to confirm similar transfer efficiency.
Acknowledgment
The work was supported in part by NIH grants R01 CA204895, R01 CA193350, K12 HD028827, and K12 HD000850, and a St. Baldrick’s Foundation Scholar Award (BM).
References
- 1.Clarke MF, Fuller M (2006) Stem cells and cancer: two faces of eve. Cell 124:1111–1115 [DOI] [PubMed] [Google Scholar]
- 2.Dick JE (2008) Stem cell concepts renew cancer research. Blood 112:4793–4807 [DOI] [PubMed] [Google Scholar]
- 3.Hawkins ED, Russell SM (2008) Upsides and downsides to polarity and asymmetric cell division in leukemia. Oncogene 27:7003–7017 [DOI] [PubMed] [Google Scholar]
- 4.Pham K, Sacirbegovic F, Russell SM (2014) Polarized cells, polarized views: asymmetric cell division in hematopoietic cells. Front Immunol 5:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yang L, Wang L, Geiger H, Cancelas JA, Mo J, Zheng Y (2007) Rho GTpase Cdc42 coordinates hematopoietic stem cell quiescence and niche interaction in the bone marrow. Proc Natl Acad Sci U S A 104:5091–5096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yang L, Wang L, Kalfa TA, Cancelas JA, Shang X, Pushkaran S, Mo J, Williams DA, Zheng Y (2007) Cdc42 critically regulates the balance between myelopoiesis and erythropoiesis. Blood 110:3853–3861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang L, Zheng Y (2007) Cdc42: a signal coordinator in hematopoietic stem cell maintenance. Cell Cycle 6:1445–1450 [PubMed] [Google Scholar]
- 8.Florian MC, Dörr K, Niebel A, Daria D, Schrezenmeier H, Rojewski M, Filippi MD, Hasenberg A, Gunzer M, Scharfetter-Kochanek K, Zheng Y, Geiger H (2012) Cdc42 activity regulates hematopoietic stem cell aging and rejuvenation. Stem Cell 10:520–530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mizukawa B, O’Brien E, Moreira DC, Wunderlich M, Hochstetler CL, Duan X, Liu W, Orr E, Grimes HL, Mulloy JC, Zheng Y (2017) Cell polarity determinant CDC42 controls division symmetry to block leukemia cell differentiation. Blood 130:1336–1346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wei J, Wunderlich M, Fox C, Alvarez S, Cigudosa JC, Wilhem JS, Zheng Y, Cancelas JA, Gu Y, Jansen M, Dimartino JF, Mulloy JC (2008) Microenvironment determines lineage fate in a human model of MLL-AF9 leukemia. Cancer Cell 13:483–495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lavau C, Szilvassy SJ, Slany R, Cleary ML (1997) Immortalization andleukemic transformation of a myelomonocytic precursor by retrovirally transduced HRX-ENL. EMBO J 16:4226–4237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Johnson JJ, Chen W, Hudson W, Yao Q, Taylor M, Rabbitts TH, Kersey JH (2003) Prenatal and postnatal myeloid cells demonstrate stepwise progression in the pathogenesis of MLL fusion gene leukemia. Blood 101:3229–3235 [DOI] [PubMed] [Google Scholar]