

Abstract
Among several available antimalarial drugs, mefloquine has proven to be effective against drug-resistant Plasmodium falciparum and remains the drug of choice for both therapy and chemoprophylaxis. However, mefloquine is known to cause adverse neurological and/or psychiatric symptoms, which offset its therapeutic advantage. The exact mechanisms leading to the adverse neurological effects of mefloquine are poorly defined. Alterations in neurotransmitter release and calcium homeostasis, the inhibition of cholinesterases and the interaction with adenosine A2A receptors have been hypothesized to play prominent roles in mediating the deleterious effects of this drug. Our recent data have established that mefloquine can also trigger oxidative damage and subsequent neurodegeneration in rat cortical primary neurons. Furthermore, we have utilized a system biology-centered approach and have constructed a pathway model of cellular responses to mefloquine, identifying non-receptor tyrosine kinase 2 (Pyk2) as a critical target in mediating mefloquine neurotoxicity. In this study, we sought to establish an experimental validation of Pyk2 using gene-silencing techniques (siRNA). We have examined whether the downregulation of Pyk2 in primary rat cortical neurons alters mefloquine neurotoxicity by evaluating cell viability, apoptosis and oxidative stress. Results from our study have confirmed that mefloquine neurotoxicity is associated with apoptotic response and oxidative injury, and we have demonstrated that mefloquine affects primary rat cortical neurons, at least in part, via Pyk2. The implication of these findings may prove beneficial in suppressing the neurological side effects of mefloquine and developing effective therapeutic modalities to offset its adverse effects.
Keywords: mefloquine, Pyk2, neuronal viability, oxidative stress, apoptosis
Introduction
Malaria is a global infectious disease with devastating human impacts. The malarial parasite infects 3.6% of the world’s population and is responsible annually for more than 800,000 deaths in over 80 nations (WHO, 2009). Approximately half of the world’s population is at risk of malaria, particularly those living in lower-income countries. Malaria also poses a continued threat to a large number of people from non-malarial regions (military and business personnel, diplomats and visitors). These populations are considerably more vulnerable to severe or even fatal forms of the disease because of their non-immune status.
Plasmodium falciparum is responsible for most malaria-related deaths. Since there are no effective antimalarial vaccines, the evolution of drug resistance in Plasmodium falciparum presents a major global health threat. Among several available antimalarial drugs (mefloquine, doxycycline, malarone, chloroquine and hydroxychloroquine sulfate), mefloquine offers an effective treatment against drug-resistant Plasmodium falciparum and maintains high concentrations in the blood for several weeks. With a half-life permitting weekly dosing, mefloquine is prescribed as the drug of choice for both chemoprophylaxis (250 mg/week) and malarial therapy (1250 mg/day). Weekly prophylactic administration of mefloquine is also optimal for military forces and has been shown to minimize non-compliance among enlisted service personnel (Dow et al, 2004).
However, the clinical utility of mefloquine has been compromised by reports of its adverse side-effects. Medical diagnoses consistent with psychiatric and neurological contraindication to mefloquine administration include depression, generalized anxiety disorder, psychoses, convulsions, seizures, extrapyramidal diseases and other movement and psychiatric disorders (Nevin et al., 2008). Adverse neurological effects also include ataxia and mood changes (Dow et al., 2006), thereby offsetting the positive prophylactic aspects of mefloquine therapy. Mefloquine is known to cause adverse neurological and/or psychiatric symptoms in as many as 25% of individuals taking the drug at prophylactic doses and 70% of individuals taking mefloquine at treatment doses (Rendi-Wagner et al., 2002). A randomized, double-blind study has also reported that neuropsychiatric-related adverse events were inherent to a similar proportion of travelers who had received mefloquine at prophylactic doses (Overbosch et al., 2001). The prevalence of mefloquine-induced adverse events requiring hospitalization (e.g. seizures and hallucinations) is low, but milder CNS events (e.g. dizziness, headache, insomnia and vivid dreams) are more frequently observed.
Limited scientific knowledge exists with regard to the exact mechanisms which lead to the adverse neurological effects associated with mefloquine exposure. Nevertheless, acethylcholinesterase and butylcholinesterase inhibition (Lim et al., 1985; McArdle et al., 2005; Zhou et al., 2006), adenosine receptor modulation (Weiss et al., 2003), the blockage of ion channels (Gribble et al., 2000; Maertens et al., 2000), the inhibition of P-glycoprotein (Pussard et al., 2007) and the destabilization of calcium (Ca2+) homeostasis (Dow et al., 2003; 2005; Toovey, 2009) have all been identified as potential mediators of mefloquine-induced neurotoxicity. Since cholinesterase inhibition, alterations in neurotransmitter release and Ca2+ homeostasis are associated with oxidative stress, neuronal oxidative injury is an expected consequence of mefloquine exposure. Our recent research supports this hypothesis by demonstrating that mefloquine induces concentration-dependent oxidative stress in primary rat cortical neurons (Hood et al., 2010). In addition, mefloquine was shown to induce a concentration-dependent decrease in the number of spines per neuron as well as a decrease in dendritic length, suggesting that mefloquine-induced oxidative stress is associated with synaptodendritic degeneration (Hood et al., 2010), reflecting morphological changes in neurons.
Furthermore, based on these and other experimental data and relevant databases, we have utilized a system biology-centered approach to construct a biological pathway model of the neuronal response to mefloquine (Jenkins et al., 2007; Jenkins, 2007). Analysis of the resultant pathway model capitalizes upon an impressive suite of computational tools, collectively known as the Boolean Network Dynamics Target Identification algorithm (BNDTI). This analysis convincingly identified Pyk2 as critical node across multiple data sets and multiple toxicity markers (Jenkins et al., 2007; Jenkins, 2007). Pyk2 encodes for a non-receptor tyrosine kinase 2 (also known as non-receptor protein tyrosine kinase-2βPTK2β or cell-adhesion kinase β, CAKβ), a member of the focal adhesion kinase family (FAK), which is involved in the Ca2+-induced regulation of ion channels and the activation of the MAP kinase signaling pathway (Avraham, 2000). Pyk2 is activated by stress stimuli that either increase intracellular Ca2+ concentrations or activate protein kinase C (PKC) (Girault et al., 1999; Nicodemo et al., 2010). Pyk2 is a mediator of several signaling pathways and has been implicated in neuronal synaptic plasticity and survival (Strappazzon et al., 2006; Faure et al., 2007; Huang et al., 2001).
In this study, we sought to establish an experimental validation of Pyk2 function by using gene-silencing techniques (siRNA). We have examined whether the downregulation of Pyk2 in primary rat cortical neurons alters mefloquine neurotoxicity by evaluating cell viability, apoptosis and oxidative stress. Understanding the role of Pyk2 and the mechanisms of mefloquine toxicity may provide new therapeutic strategies for minimizing its adverse neurological effects.
Materials and Methods
2.1. Materials
Mefloquine hydrochloride was purchased from Sigma (St. Louis, MO). All culture media and supplements were purchased from Invitrogen (Carlsbad, CA), except for Hyclone Ham’s F12 and Hyclone heat-inactivated fetal bovine serum, which were purchased from VWR (Suwanee, GA). Mouse GIPZ lentiviral shRNAmir individual clone, V2LMM_21947, for Pyk2 silencing in primary rat cortical neurons was purchased from Open Biosystems (Huntsville, AL). A Rat Neuron Nucleofector Kit was purchased from Amaxa biosystem (Gaithersburg, MD). Antibodies against PYK2 (Rabbit monoclonal, E354 and YE353) and Cy3-labeled donkey anti-chicken IgG were purchased from Abcam (Cambridge, MA) and Jackson Immunoresearch, Inc. (West Grove, PA), respectively. VECTASHIELD Mounting Medium with DAPI (4’,6-diamidino-2-phenylindole) was purchased from Vector Laboratories (Burlingame, CA), and glass coverslips for cell cultures were purchased from Carolina Biological Supply (Burlington, NC).
2.2. Primary Rat Cortical Cultures
All experiments were approved by the Institutional Animal Care and Use Committee of Vanderbilt University and were performed according to the Guidelines for Animal Experimentation as set forth by Vanderbilt University. Primary rat cortical neuron cultures were prepared from rats as previously described (McLaughlin et al., 1998). Briefly, the brains of E17 Harlan Sprague-Dawley rat embryos were removed and placed in a Petri dish filled with cold Hank’s balanced salt solution (HBSS). The cortices were dissected from the brain and transferred to another Petri dish containing HBSS, in order to remove blood vessels and meninges. The isolated cortices were then transferred to a Petri dish containing 0.6% (w/v) trypsin in HBSS and digested for 30 minutes at room temperature. Following centrifugation, dissociated cortical cells (cell pellet) were plated onto poly-L-ornithine-coated glass coverslips in 6- and/or 96-well plates using a medium of glutamine-free Dulbecco’s Modified Eagle Medium (DMEM)-Eagle’s salts (Invitrogen, Carlsbad, CA), supplemented with Ham’s F12, 10% heat-inactivated fetal bovine serum (FBS), and 200 IU/ml penicillin/streptomycin at a density of 700,000 cells/well or transfected using the nucleofection method.
2.3. Transfection of Rat Cortical Neurons
For the nucleofection procedure, we re-suspended the cell pellet into 100 μl Nucleofector solution (at room temperature) (Rat Neuronal Nucleofector Kit) and added 3 μg shRNA construct (V2LMM_21947). The cell/shRNA suspension was transferred into certified cuvettes, inserted into the Nucleofector cuvette holder, and the Nucleofector program O-03 was applied. Once the electroporation program was completed, 500 μl of the pre-equilibrated culture medium I (DMEM, supplemented with 10% fetal calf serum, 0.2 unit/ml Pen/Strep) was added, and the suspension was gently transferred into the prepared culture dish with the coated coverslips. After 3 hours, the medium was replaced with fresh culture medium I to remove cellular debris. Medium I was then replaced with fresh culture medium II (Neurobasal medium) and supplemented with 5% B27 supplement and 0.2 unit/ml pen/strep 24 hours later. Cell viability was evaluated by determining the proportion of cells attached to the coverslips at 24 hours after incubation.
For the transfection of cells with the Lipofectamine 2000 procedure, primary rat cortical neurons were re-suspended in 2 ml growth medium (DMEM, supplemented with 8% HyQ Ham’s/F-12m and 8% FBS) in the absence of antibiotics. Each well contained 4–5 ×106 cells. Separate dilutions of shRNA (V2LMM_21947, 4.0 μg in 250 μl of optimal MEM I reduced serum medium) and Lipofectamine 2000 (10 μl in 250 μl of optimal MEM I reduced serum medium for each well) were prepared. After incubation for 5 minutes at room temperature, diluted shRNA and Lipofectamine were combined, gently mixed and incubated for 20 minutes at room temperature. Complexes of shRNA and Lipofectamine were transferred into the cell suspension. Gently mixed cells were plated into the prepared culture dish with the coated coverslips. The medium was changed after 4 hours.
2.4. Evaluation of transfection efficiency
The transfection efficiency and knockdown of Pyk2 in primary rat cortical neurons were verified and quantified by fluorometric approaches with DAPI staining and an immunocytochemistry-staining method. Since shRNAmir individual clones for silencing Pyk2 protein expression were fused with green fluorescent protein (GFP), transfection efficiency was calculated as the number of GFP-positive cells (green) divided by the total number of cells (DAPI-positive cells, blue) in each microscopic field. Fluorescence images were acquired by an AxioVision Rel. 4.8, Carl Zeiss/Axoplan 2 microscope. Immunocytochemistry was performed using rabbit monoclonal primary antibody (PYK2) (Chellaiah et al., 2007). Neurons were washed in PBS three times for 5 minutes, and cells were fixed with 4% paraformaldehyde for 15 minutes on ice. After washing in PBS containing 0.05% Tween-20 (PBST), fixed neurons were permeabilized with 0.1% Triton X-100 for 15 minutes on ice and washed with PBS. Coverslips were then blocked with 10% normal goat serum in PBS for 1 hour at room temperature (RT). The primary and secondary antibodies were diluted in 0.1% NGS. Coverslips were incubated with primary antibody PYK2 (ab 32571, 1:100 dilution) for 1 hour at RT. After washing with PBS three times, the coverslips were incubated with a secondary antibody, Cy3-labeled donkey anti-chicken (1:100 dilution), for 30 minutes at RT in the dark. Then, after washing three times with PBS, the coverslips were mounted with ProLong Gold anti-fade reagent (Invitrogen, Carlsbad, CA). The edges of the coverslips were sealed with nail polish, images were acquired with a Carl Zeiss/Axoplan 2 microscope, and the numbers of PYK2 positive cells were quantified with NIS-Elements AR 3.0.
2.5. Cortical Cell Culture Treatment with Mefloquine
Two days after isolation/transfection, the cells were treated with mefloquine for 24 hours, at 37°C in a humidified atmosphere with 5% CO2 in the air. A stock solution of mefloquine (100 mM in dimethyl sulfoxide) was diluted with a treatment buffer, which consisted of minimum essential medium (MEM) supplemented with 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), 2× N2 media supplement. Control cells were exposed to the same treatment buffer containing an equal dilution of dimethyl sulfoxide. Since mefloquine highly concentrate in the brain, we have used cortical neuronal cultures treated with mefloquine concentrations up to 100 μM. Concentrations of mefloquine, at nearly 100 μM, have been measured in the central nervous system following therapeutic doses (Dow et al., 2005).
2.6. Cortical Neuron Viability and Cytotoxicity
Cell viability was assessed by measuring formazan production after the addition of 3-(4,5-dimethythiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT; Sigma Chemical Co., St. Louis, MO). The number of surviving cells following 24 hours of treatment with mefloquine (0, 1 μM, 5 μM, 10 μM, 50 μM, 100 μM) was determined by measuring the optical density (OD) of the dissolved formazan product at A570 nm after the addition of MTT for 1 hour according to the manufacturer’s instructions. Untreated negative controls were run together with the treated cells, and plates with reagent only served as background controls. After background OD subtraction, the results were expressed as a percentage of the average negative control. Each experiment with two groups on neuronal cultures was performed in at least triplicate wells and repeated three times.
Cytotoxicity, reflecting cell membrane integrity, was assessed by measuring the activity of lactate dehydrogenase (LDH) in the culture media by colorimetric detection of highly-colored formazan, using a LDH diagnostic kit (Promega, Madison, WI, USA). Following 24 hours of treatment with mefloquine (0, 1 μM, 5 μM, 10 μM, 50 μM, 100 μM), the supernatant was transferred to a 96-well plate and incubated with the reaction mixture for 30 minutes at room temperature to develop color. The optical density was measured at A492 nm using a spectrophotometer (Molecular Devices, VMax Kinetic Microplate Reader). Each experiment with different groups of neuronal cultures was performed in at least triplicate wells and repeated three times.
2.7. Apoptosis
Neuronal death following mefloquine exposure was evaluated by using the DeadEnd™ Colorimetric TUNEL System (Promega, Madison, WI), which, by labeling fragmented DNA, enables the detection and quantification of apoptotic cells within a cell population. Following the manufacturer’s instructions, previously fixed (immersed in 4% paraformaldehyde for 25 minutes) and washed (twice in PBS) cells were treated with 0.2% Triton® X-100 in PBS (for 5 minutes). Next, the cells were washed in PBS (twice) and exposed to Equilibration Buffer with Terminal Deoxynucleotidyl Transferase Recimbinant (rTdT) enzyme for 60 minutes. Following treatment with 0.3% hydrogen peroxide, washed slides were exposed to Streptavidin HRP (diluted 1:500 in PBS) for 30 minutes at room temperature. Cells washed with PBS were first exposed to 100 μl of chromogen, diaminobenzidine (DAB) and then placed in hematoxylin for 30 seconds to 1 minute followed by several rinses with water. As a result of the procedures described, apoptotic nuclei are stained dark brown, and control cells are stained blue. Using this method, primary rat cortical neurons from control and Pyk2-transfected cells, exposed to different concentrations of mefloqune (0, 1 μM, 5 μM , 10 μM, 50 μM or 100 μM) for 24 hours, were evaluated by light microscopy. Apoptotic cells were evaluated by NIS-Elements AR 3.0 software, and the data were analyzed by one-way ANOVA followed by repeated t tests with Bonferroni’s correction for multiple comparisons.
2.8. Oxidative stress
The oxidative stress response was evaluated by quantifying the formation of glutathione (GSH) and F2-isoprostanes (F2-IsoPs). Glutathione, a major antioxidant, was quantified in cortical neurons after 24 hours of treatment with mefloquine (0, 1 μM, 5 μM, 10 μM, 50 μM, 100 μM) using monochlorobimane (Molecular Probes, Eugene, OR), a fluorescent probe for glutathione (Neely et al., 2005). Cortical neurons were plated in culture medium in round-bottom 96-well plates and treated with mefloquine. Following mefloquine treatment, the cortical neurons were rinsed twice with HBSS and treated with 40 μM monochlorobimane for 15 minutes in the incubator. Fluorescence was quantified with a fluorescence microplate reader.
F2-IsoPs, products of arachidonic acid peroxidation and biomarkers of oxidative stress, were quantified in cortical neurons after 24 hours of treatment with mefloquine (0, 1 μM, 5 μM, 10 μM, 50 μM, 100 μM) using a stable isotope dilution method with detection by gas chromatography/mass spectrometry (GC/MS) and selective ion monitoring (SIM) as previously described (Milatovic et al., 2009; Morrow and Roberts, 1991; Roberts and Morrow, 1994). Following mefloquine treatment, cortical neurons were sonicated, lipids chemically hydrolyzed using KOH, and a stable isotope, 8-isoprostaglandin F2α-d4 internal standard, was added. Following extraction using C-18 and silica Sep-Pac cartridges, purification by thin layer chromatography and conversion to O-methyloxime pentafluorobenzyl ester trimethylsilyl derivatives, the compound was dissolved in undecane that is dried over a bed of calcium hydride. Negative ion chemical ionization MS was performed with Hewlett-Packard HP5989A and Agilent 5973 instruments interfaced with monitoring ions for F2-IsoPs (m/z 569) and the internal standard (m/z 573). The ion source temperature was 250°C, and the electron energy was 70 eV.
2.9. Data Analysis
Measurements of MTT, LDH, GSH and F2-IsoPs, as well as experiments for apoptosis detection were conducted in duplicate or triplicate wells/experiment, and the mean from three to four independent experiments was used for statistical analysis. The data were analyzed by one-way analysis of variance (ANOVA) followed by Bonferroni’s correction for multiple comparisons with statistical significance of p<0.05. All analyses were carried out with GraphPad Prism 4.02 for Windows (GraphPad Software, San Diego, CA).
Results
3.1. Transfection of primary rat cortical neurons
Transfection and knockdown of Pyk2 in primary rat cortical neurons was carried out using Nucleofector Kit/Nucleofection technology and lipofection. Forty-eight hours post-transfection (see Materials and Methods) 36.3% ± 3.45% of the cells expressed the GFP-transgene. Representative fluorescence images of transfected (GFP protein fused with plasmid) and control cortical neurons (blue, DAPI stained neurons) evaluated 48 hours post-transfection are shown in Figure 1a. Lower transfection efficiency (11.08% ± 1.03%) was observed when rat cortical primary neurons were evaluated 7 days after the transfection. Low transfection efficiency was also observed following Lipofectamine 2000 Reagent treatment, with transfection efficiencies in primary rat cortical neurons 48 hours and 7 days following transfection measured at 9.56% and 3.58%, respectively.
Figure 1.
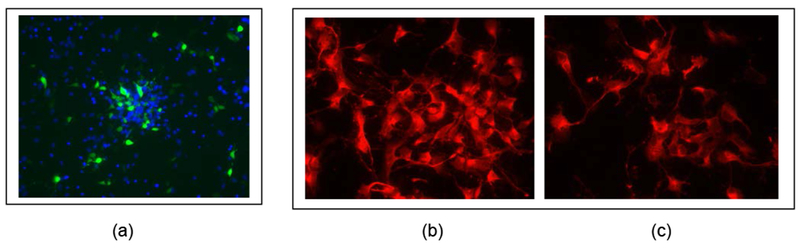
Representative fluorescence images of rat cortical neurons transfected with plasmid encoded for silencing of Pyk2 by using a Rat Neuron Nucleofector Kit/Nucleofector program O-03. (a) Rat cortical neurons transfected with plasmid (shRNA construct V2LMM_21947) fused with green fluorescence protein (GFP) and stained by DAPI (4’,6-diamidino-2-phenylindole) fluorescent DNA marker (blue). Transfection efficiency was analyzed by comparing DAPI’s blue to GFP’s green emissions. Images of immunocytochemistry/immunofluorescence–Pyk2 positive control (b) and Pyk2-transfected (c) primary rat cortical neurons. Transfection efficiency was analyzed by comparing the numbers of Pyk2 positive cells in control and Pyk2-transfected cells.
In addition to the fluorometric approach with DAPI staining, transfection efficiency and knockdown of Pyk2 in primary rat cortical neurons by nucleofection was verified by immunocytochemistry. Representative fluorescence images of Pyk2-positive control and transfected rat cortical neurons evaluated 48 hours after transfection are shown in Figure 1b and 1c, respectively. The number of Pyk2-positive cells was 34.4 % ± 3.9% less in transfected primary rat cortical neurons compared to control cells. The immunocytochemistry result indicates a transfection efficiency similar to the result obtained with DAPI staining.
Transfection and knockdown of Pyk2 in primary rat cortical neurons was verified by SDS-PAGE/Western Blot Analysis. Western blot analysis was performed using goat anti-pyk2 (1:100 in 5% milk/ PBS, R/T 3 hours, SATA CRUZ SC-1514) and was detected using an enhanced chemiluminescence technique (ECL) (Pierce, Rockford, IL). Representative western blot images with band densities from control and Pyk2- transfected primary rat cortical neurons are shown in supplemental figures 1 and 2.
3.2. Viability of Primary Rat Cortical Neurons Treated with Mefloquine
The cytotoxic effect of mefloquine in control and Pyk2-transfected primary rat cortical neurons was tested with the MTT assay. The absorbance of MTT (A570 nm) is positively correlated with cell viability. As shown in Figure 2a, after mefloquine treatment for 24 hours, the viability of non-transfected primary rat cortical neurons was indistinguishable between control and 1 μM mefloquine-treated cells. However, higher concentrations of mefloquine caused a concentration-dependent decrease in control primary rat cortical neuron viability. As shown in Figure 2b, Pyk2-transfected cells did not show decreased viability when exposed to 1 μM or 5 μM mefloquine, and a significant decrease (p<0.01) in the number of viable Pyk2-transfected primary rat cortical neurons was noted only after treatments equal to or exceeding 10 μM mefloquine (but not with 5 μM as seen in control cells). These concentrations are highly relevant to physiological exposure in humans since peak plasma levels of mefloquine are 3.8 and 2.1 to 23 μM after prophylaxis and treatment, respectively (Simpson et al., 1999; Dow et al., 2004).
Figure 2.

Mefloquine causes a concentration-dependent decrease in the number of viable control (a) and transfected (b) cells. The quantity of purple formazan product was measured at 570 nm after 24-hour incubations with increasing concentrations of mefloquine. Cytotoxicity is expressed as the percent of MTT activity in control cells (non-treated neurons). Data represent mean ± SEM values from 3 independent experiments with n ≥ 24. *p<0.05 versus control by one-way ANOVA followed by Bonferroni’s multiple comparison test.
Cell viability was also measured by the LDH assay. The absorbance of LDH (A490 nm) is inversely correlated with cell viability. As shown in Figure 3a, non-transfected primary rat cortical neurons showed a concentration-dependent increase in LDH release after treatment with 10 μM, 50 μM and 100 μM mefloquine (p < 0.001), reflecting decreased cell viability. In contrast, Pyk2-transfected primary rat cortical neurons did not show an increase in LDH release after treatment with 10 μM mefloquine. Further, a significant effect was noted only after exposure to 50 μM (p < 0.001) and 100 μM mefloquine (p < 0.001) (Figure 3b).
Figure 3.
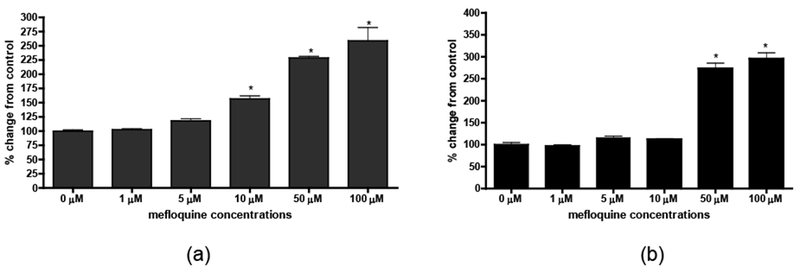
Mefloquine causes a concentration-dependent increase in the number of dead and dying control (a) and transfected (b) cells. Activity of lactate dehydrogenase (LDH) released into the media was measured spectrophotometrically according to the manufacturer’s protocol. Cytotoxicity is expressed as the percent of LDH activity in control cells (non-treated neurons). Data represent mean ± SEM (bars) values from 3 independent experiments with n ≥ 24. *p<0.05 versus control by one-way ANOVA followed by Bonferroni’s multiple comparison test.
3.3. Apoptosis
We used the DeadEnd™ Colorimetric TUNEL System and sought to determine whether the suppression of Pyk2 alters neuronal apoptosis following mefloquine exposure. This assay is based on biotinylated nucleotides, which, by labeling fragmented DNA, enable the detection and quantification of apoptotic cells within a cell population. As shown in Figure 4, mefloquine treatment was associated with an increase in the apoptotic bodies in both control and Pyk2-transfected primary rat cortical neurons. Although Pyk2-transfected neurons had a significantly lower (p<0.05) percentage of apoptotic bodies (Figure 5b) compared to control neurons (Figure 5a) following exposure to high mefloquine concentrations (50 μM and 100 μM), there was no difference in the pattern of the mefloquine-induced concentration-dependent increase in the percentage of apoptotic bodies between control and Pyk2-transfected primary rat cortical neurons.
Figure 4.
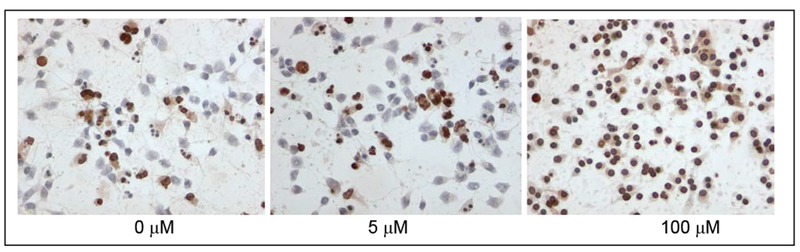
Representative images of apoptotic bodies in Pyk2-transfected cortical neurons exposed to a graded concentration of mefloquine. Apoptotic nuclei, evaluated by using the DeadEnd Colorimetric TUNEL System, are stained dark brown, and control cells are stained blue.
Figure 5.

Percent of apoptotic bodies in control (a) and Pyk2-transfected (b) primary rat cortical neurons following mefloquine exposure. The number of apoptotic neurons was evaluated after 24-hour incubations with graded concentrations of mefloquine using the DeadEnd™ Colorimetric TUNEL System. Percentages of apoptotic neurons are presented as mean±SEM (bars) values from two experiments with n=18. *p<0.05 versus control by one-way ANOVA followed by Bonferroni’s multiple comparison test.
3.4. Oxidative Stress in Rat Cortical Neurons Treated with Mefloquine
To determine whether suppression of Pyk2 in primary rat cortical neurons alters mefloquine-induced oxidative stress, levels of GSH were evaluated in control and Pyk2-transfected cells. The GSH assay was based on the quantification of cellular fluorescence resulting from the glutathione S-transferase-catalyzed conjugation of GSH to monochlorobimane (Rice et al., 1986; Neely et al., 2005). Mefloquine treatment of control cortical neurons caused a concentration-dependent decrease in GSH, with an approximate depletion of 60% following 24 hours of exposure to 100 μM of mefloquine (p<0.001) (Figure 6a). Pyk2-transfected primary cortical neurons exposed to increasing concentrations of mefloquine showed a similar pattern of concentration-dependent GSH depletion compared to the controls (Figure 6b).
Figure 6.
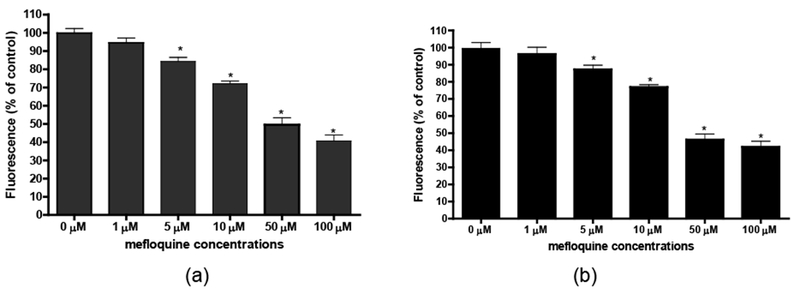
Mefloquine causes a concentration-dependent decrease in intracellular GSH levels. The GSH content of control (a) and transfected (b) primary rat cortical neurons was determined after 24-hour incubations with graded concentrations of mefloquine using monochlorobimane, a fluorescent probe for GSH. Data represent mean ± SEM (bars) values from 3 independent experiments with n ≥ 24. *Significant difference between values from control and mefloquine-treated neuronal cells (p<0.05).
Furthermore, we also utilized a more sensitive and specific biomarker of oxidative stress in neurons and evaluated levels of F2-IsoPs, free radical-catalyzed peroxidation products of arachidonic acid. Control and transfected neuronal cultures treated with mefloquine for 24 hours exhibited increased F2-IsoPs levels (data not shown). These results correlate with our previous findings (Hood et. al., 2010) and confirm the presence of oxidative stress in cortical neurons exposed to mefloquine. However, mefloquine-induced increases in F2-IsoPs levels were indistinguishable between control and Pyk2-transfected primary rat cortical neurons (data not shown).
Discussion
Corroborating a previous report (Hood et al., 2010), we have confirmed that mefloquine neurotoxicity is associated with oxidative injury, and we have shown that mefloquine’s effect on primary rat cortical neurons is mediated, at least in part, by Pyk2. Utilizing a gene-silencing technique, we have demonstrated that the suppression of Pyk2 alters the overall cytotoxic effects of mefloquine, reducing the sensitivity to this drug (Figures 2 and 3). The implication of these findings may prove beneficial in designing new therapeutic modalities that efficiently suppress the neurological side-effects of mefloquine.
The neurological side-effects reported following mefloquine treatment include depression, nausea, nightmares, fatique anxiety, dizziness and vertigo (Nevin et al., 2008; Toovey, 2009). Although the biological bases for these effects have not yet been fully determined, animal studies have suggested that mefloquine induces dose-related neurological effects (Dow et al., 2006). Our recent study with primary rat cortical neurons strongly indicates that the neurotoxic effects of mefloquine can be attributed to neuronal oxidative damage and subsequent neurodegeneration (Hood et al., 2010). Structural changes in neurons from affected areas, characterized by temporal alterations in spine density and dendritic length, may represent a biological basis for some of the clinical neurological effects associated with mefloquine neurotoxicity. Various studies and clinical reports have also indicated that cellular processes associated with mefloquine neurotoxicity include cholinesterase inhibition and neurotransmitter release (Lim and Go, 1985; McArdle et al., 2005; Zhou et al., 2006), ion channel inhibition (Gribble et al., 2000; Maertens et al., 2000), interactions with adenosine and dopamine receptors and transporters (Weiss, 2003) and the destabilization of calcium (Ca2+) homeostasis, likely due to endoplasmic reticulum stress (Dow et al., 2003; Dow et al., 2005; Gribble et al., 2000; Maertens et al., 2000). Mefloquine-induced alterations in cholinergic transmission may affect the catalysis of inositol 1,4,5-triphosphate (IP3) by phospholipase C (PLC) or the catalysis of cyclic adenosine monophosphate (cAMP) by adenylate cyclase (Siegel et al., 1999), second messengers that regulate Ca2+ homeostasis via modulation of the plasma membrane and the endoplasmic reticulum membrane ion channels. Based on these data, scientific literature and relevant databases, we have constructed a pathway model of cellular response to mefloquine by applying a novel system biology-centered approach. Pathway analysis performed using the BNDTI algorithm identified Pyk2 as critical target in mefloquine neurotoxicity (Jenkins, 2007; Jenkins et al., 2007). Pyk2 is highly enriched in the central nervous system (Girault et al., 1999). It has been implicated in the signaling of G protein-coupled receptors, nicotinic acetylcholine receptors, stress stimuli and membrane depolarization (Lev et al., 1995; Sicilianet al., 1996; Tokiwa et al., 1996; Dikic et al., 1996; Corvol et al., 2004). It has also been implicated in the Ca2+-mediated regeneration of gene expression in neuronal cells induced by NMDA receptor or voltage-sensitive calcium channels (Ghosh et al., 1994). Activation of Pyk2 in neurons appears to be a Ca2+- and/or PKC-dependent mechanism (Girault et al., 1999; Lev et al.,1995). Pyk2 activation in response to a rise in intracellular calcium can also be mediated by calmodulin or calmodulin-dependent kinase II (Zwick et al., 1999; Ginnan and Singer, 2002; Heidinger et al., 2002). Pyk2 activation and the subsequent interaction with Src family kinases lead to the formation of multiple molecular complexes that regulate several neuronal processes, such as neurite outgrowth, synaptic plasticity and differentiation (Girault et al., 1999; Xiong and Mei, 2003; Strappazzon et al., 2006).
In the present study, we have demonstrated that Pyk2 is involved in mefloquine neurotoxicity. Utilizing a gene-silencing technique, we have shown that the suppression of Pyk2 alters the overall cytotoxic effects of mefloquine. Primary rat cortical neurons were transfected by nucleofection/nucleofector program O-03 and Pyk2 transfection efficiency of ~35% corroborated by a fluorometric approach using green fluorescence GFP protein fused with plasmid (Fig. 1a) and an immunochemistry staining approach (Fig. 1b and 1c). Results from the MTT (Fig. 2) and LDH (Fig. 3) assays demonstrated that mefloquine was less toxic in Pyk2-transfected primary rat cortical neurons compared to control cells. While exposure to 5 μM mefloquine induced cell death in 19% of control neurons, no viability change was observed in Pyk2-transfected cells (Figs 2a and 2b). At the same time, exposure to 10 μM mefloquine induced a 59% increase in LDH levels in control cells, but not in Pyk2-transfected primary rat cortical neurons (Figs. 3a and 3b). Notably, the threshold for mefloquine-induced cell death in Pyk2-transfected cells was higher compared to control cells (Figures 2 and 3).
Pyk2 has been also implicated in apoptosis induced by several stress signals (Strappazzon et al., 2006). It has been shown that the overexpression of Pyk2 in rat and mouse fibroblasts leads to apoptotic cell death (Xiong and Parsons, 1977) and that a high level of chronic Pyk2 activation promotes apoptotic signaling in cardiomyocytes (Melendez et al., 2004). Therefore, we sought to determine whether the suppression of Pyk2 in primary rat cortical neurons alters apoptosis following mefloquine exposure. As shown in Figures 4 and 5, Pyk2-transfected neurons showed a significantly lower percentage of apoptotic bodies compared to control cells following exposure to high mefloquine concentrations (50 μM and 100 μM). However, the same mefloquine-induced, concentration-dependent increases in the percentage of apoptotic bodies were seen in both control and Pyk2-transfected primary rat cortical neurons.
In addition, GSH and F2-isoPs analyses confirmed that mefloquine induced a concentration-dependent oxidative stress response in primary rat cortical neurons. However, Pyk2-transfected primary rat cortical neurons exposed to increasing concentrations of mefloquine showed a concentration-dependent oxidative stress response (Fig. 6a and 6b) which was analogous to the response of the control cells. These results suggest that a greater than 35% downregulation of Pyk2 expression in primary rat cortical neurons may be required for the significant suppression of mefloquine-induced oxidative stress.
Furthermore, several lines of evidence have implicated Pyk2 in inflammatory responses. Pyk2 interacts with the adaptor protein, MyD88, and regulates lipopolysaccharide (LPS)-induced Nuclear Factor-KappaB (NF-kB) activation and signaling (Xi et al., 2010). Our previous in vivo study with activated innate immunity demonstrated that intracerebroventricular injections of LPS induced oxidative injury, an increase in cerebral F2-isoPs, F4-NeuroPs (neuroprostanes, biomarkers of neuronal oxidative damage) and inflammatory prostaglandine, PGE2 (Milatovic et al., 2003; Milatovic et al., 2004). Our results also showed that the genetic ablation of adaptor MyD88 or the p50 subunit of NF-kB completely blocked the LPS-induced cerebral oxidative damage (Milatovic et al., 2004). Since mefloquine caused oxidative stress in primary rat cortical neurons as well as a more than 2-fold increase in inflammatory prostaglandine PGE2 when exposed to 50 μM for 24 hours (not shown), it is likely that the enhanced downregulation of Pyk2 suppresses the neurotoxic effects of mefloquine. Together, suppression of Pyk2 and treatment with non-steroidal anti-inflammatory drugs and antioxidants such as N-acetylcysteine, Vitamin C and E, or their combination may be potentially used for reduction of mefloquine toxicity both in vitro and in vivo.
In conclusion, the present study elucidates the putative involvement of oxidative stress and apoptosis in mefloquine-induced neuronal injury and indicates that Pyk2 is, at least in a part, involved in mefloquine-induced neurotoxicity in primary rat cortical neurons. Further studies exploring the enhanced downregulation of Pyk2 and the suppression of oxidative injury, as well as investigations conducted in vivo, could provide beneficial therapeutic strategies to minimize the detrimental neurological effects of mefloquine treatment.
Supplementary Material
Acknowledgements
The authors wish to thank Dr. Geoffrey Dow of the Walter Reed Army Institute of Research (WRAIR) for several insightful discussions. Funding for this work was provided by the US Army Medical Research and Material Command (USAMRMC) (COL Alan Magill, Director, Division of Experimental Therapeutics, WRAIR). The views and/or opinions expressed herein are those of the authors and do not reflect the views and/or opinions of the Department of the Army and/or the Department of Defense.
References
- Avraham H, Park SY, Schinkmann K, Avraham S. RAFTK/Pyk2-mediated cellular signaling. Cell Signalling 2000; 12:123–133. [DOI] [PubMed] [Google Scholar]
- Chellaiah MA, Kuppuswamy D, Lasky L, Linder S. Phosphorylation of a Wiscott-Aldrich syndrome protein-associated signal complex is critical in osteoclast bone resorption. J Biol Chem 2007; 282:10104–16. [DOI] [PubMed] [Google Scholar]
- Corvol JC, Valjent E, Toutant M, Enslen H, Irinopoulou T, Lev S, Herve D, Girault JA. Depolarization Activates ERK and Proline-rich Tyrosine Kinase 2 (PYK2) Independently in Different Cellular Compartments in Hippocampal Slices. J Biol Chem 2005; 280:660–8. [DOI] [PubMed] [Google Scholar]
- Dikic I, Tokiwa G, Lev S, Courtneidge SA, Schlessinger J. A role for Pyk2 and Src in linking G-protein-coupled receptors with MAP kinase activation. Nature 1996; 383:547–550. [DOI] [PubMed] [Google Scholar]
- Dow G, Bauman R, Caridha D, Cabezas M, Du F, Gomez-Lobo R, Park M, Smith K, Cannard K. Mefloquine induces dose-related neurological effects in a rat model. Antimicrob Agents Chemother 2006; 50:1045–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dow GS, Caridha D, Goldberg M, Wolf L, Koenig ML, Yourick DL, Wang Z. Transcriptional profiling of mefloquine-induced disruption of calcium homeostasis in neurons in vitro. Genomics 2005; 86:539–50 [DOI] [PubMed] [Google Scholar]
- Dow GS, Hudson TH, Vahey M, Koenig ML. The acute neurotoxicity of mefloquine may be mediated through a disruption of calcium homeostasis and ER function in vitro. Malar J 2003; 2:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dow GS, Koenig ML, Wolf L, Gerena I, Lopez-Sanchez M, Hudson TH, Bhattacharjee AK. The Antimalarial Potential of 4-Quinolinecarbinolamines May Be Limited due to Neurotoxicity and Cross-Resistance in Mefloquine-Resistant Plasmodium falciparum Strains. Antimicrobial Agents and Chemotherapy 2004; 48:2624–2632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faure C, Corvol JC, Toutant M, Valjent E, Hvalby O, Jensen V, El Messari S, Corsi JM, Kadare G, Girault JA. Calcineurin is essential for depolarization-induced nuclear translocation and tyrosine phosphorylation of PYK2 in neurons J Cell Sci 2007; 120:3034–3044. [DOI] [PubMed] [Google Scholar]
- Ghosh A, Ginty DD, Bading H, Greenberg ME. Calcium regulation of gene expression in neuronal cells. J Neurobiol 1994; 25:294–303 [DOI] [PubMed] [Google Scholar]
- Ginnan R, Singer HA. CaM kinase II-dependent activation of tyrosine kinases and ERK1/2 in vascular smooth muscle. Am J Physiol: Cell Physiol 2002; 282:C754–C761. [DOI] [PubMed] [Google Scholar]
- Girault JA, Costa A, Derkinderen P, Studler JM, Toutant M. FAK and PYK2/CAKbeta in the nervous system: a link between neuronal activity, plasticity and survival? Trends Neurosci 1999; 22:257–263. [DOI] [PubMed] [Google Scholar]
- Gribble FM, Davis TM, Higham CE, Clark A, Ashcroft FM. The antimalarial agent mefloquine inhibits ATP-sensitive K-channels. Br J Pharmacol 2000; 131:756–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidinger V, Manzerra P, Wang XQ, Strasser U, Yu SP, Choi DW, Behrens MM. Metabotropic glutamate receptor 1-induced upregulation of NMDA receptor current: mediation through the Pyk2/Src-family kinase pathway in cortical neurons. J Neurosci 2002: 22:5452–5461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hood JE, Jenkins JW, Milatovic D, Rongzhu L, Aschner M. Mefloquine induces oxidative stress and neurodegeneration in primary rat cortical neurons. Neurotoxicology 2010; 31:518–523. [DOI] [PubMed] [Google Scholar]
- Huang Y, Lu W, Ali DW, Pelkey KA, Pitcher GM, Lu YM, Aoto H, Roder JC, Sasaki T, Salter MW, MacDonald JF. CAKbeta/Pyk2 kinase is a signaling link for induction of long-term potentiation in CA1 hippocampus. Neuron 2001; 29:485–496. [DOI] [PubMed] [Google Scholar]
- Jenkins J Biological Pathway-Centric Approach to Integrative Analysis of Array Data as Applied to Mefloquine Neurotoxicity. J Biomol Tech 2007; 18:83, SP12. [Google Scholar]
- Jenkins JW, Hood JE, Soni AS, Sundaram S, Aschner M, Jiang CT. Biological Pathway-Centric Approach to Integrative Analysis of Array Data as Applied to Mefloquine Neurotoxicity. The 2007 Annual Meeting Salt Lake City –UT. 657-Genomic Approaches to System Biology. 2007. [Google Scholar]
- Lev S, Moreno H, Martinez R, Canoll P, Peles E, Musacchio JM, Plowman GD, Rudy B, Schlessinger J. Protein tyrosine kinase PYK2 involved in Ca(2+)-induced regulation of ion channel and MAP kinase functions. Nature 1995: 376:737–745. [DOI] [PubMed] [Google Scholar]
- Lim LY, Go ML. The anticholinesterase activity of mefloquine. Clin Exp Pharmacol Physiol 1985; 12:527–531. [DOI] [PubMed] [Google Scholar]
- Maertens C, Wei L, Droogmans G, Nilius B. Inhibition of volume-regulated and calcium-activated chloride channels by the antimalarial mefloquine. J Pharmacol Exp Ther 2000; 295:29–36. [PubMed] [Google Scholar]
- McArdle JJ, Sellin LC, Coakley KM, Potian JG, Quinones-Lopez MC, Rosenfeld CA, Sultatos LG, Hognason K. Mefloquine inhibits cholinesterases at the mouse neuromuscular junction. Neuropharmacology 2005; 49:1132–1139. [DOI] [PubMed] [Google Scholar]
- McLaughlin BA, Nelson D, Silver IA, Erecinska M, Chesselet MF. Methylmalonate toxicity in primary neuronal cultures. Neuroscience 1998; 86:279–290. [DOI] [PubMed] [Google Scholar]
- Melendez J, Turner C, Avraham H, Steinberg SF, Schaefer E, Sussman MA. Cardiomyocyte apoptosis triggered by RAFTK/pyk2 via Src kinase is antagonized by paxillin. J Biol Chem 2004; 279:53516–53523. [DOI] [PubMed] [Google Scholar]
- Milatovic D, Ascher M. Measurement of isoprostanes as markers of oxidative stress in neuronal tissue. Curr Protoc Toxicol 2009; 39- unit12.14:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milatovic D, Zaja-Milatovic S, Montine KS, Horner PJ, Montine TJ. Pharmacologic suppression of neuronal oxidative damage and dendritic degeneration following direct activation of glial innate immunity in mouse cerebrum. J Neurochem 2003; 87:1518–1526. [DOI] [PubMed] [Google Scholar]
- Milatovic D, Zaja-Milatovic S, Montine KS, Shie FS, Montine TJ. Neuronal oxidative damage and dendritic degeneration following activation of CD14-dependent innate immune response in vivo. J Neuroinflammation 2004; 1:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrow JD, Roberts LJ 2nd. Quantification of noncyclooxygenase derived prostanoids as a marker of oxidative stress. Free Radic Biol Med 1991; 10:195–200. [DOI] [PubMed] [Google Scholar]
- Neely MD, Boutte A, Milatovic D, Montine TJ. Mechanisms of 4-hydroxynonenal-induced neuronal microtubule dysfunction. Brain Res 2005; 1037:90–98. [DOI] [PubMed] [Google Scholar]
- Nevin RL, Pietrusiack PP, Caci JB. Prevalence of contraindications to mefloquine use among USA military personnel deployed to Avganistan. Malaria J 2008; 7:1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicodemo AA, Pampillo M, Ferreira LT, Dale LB, Cregan T, Ribeiro FM, Ferguson SS. Pyk2 uncouples metabotropic glutamate receptor G protein signaling but facilitates ERK1/2 activation. Mol Brain 2010; 3:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overbosch D, Schilthuis H, Bienzle U, Behrens RH, Kain KC, Clarke PD, Toovey S, Knobloch J, Nothdurft HD, Shawn D, Roskell NS, Chulay JD. Atovaquone-proguanil versus mefloquine for malaria prophylaxis in nonimmune travelers: results from arandomized, double-blind study. Clin Infectious Dis 2001; 33:1015–1021. [DOI] [PubMed] [Google Scholar]
- Pussard E, Merzouk M, Barennes H. Increased uptake of quinine into the brain by inhibition of P-glycoprotein. Eur J Pharm Sci 2007; 32:123–7. [DOI] [PubMed] [Google Scholar]
- Rendi-Wagner P, Noedl H, Wernsdorfer WH, Wiedermann G, Mikolasek A, Kollaritsch H. Unexpected frequency, duration and spectrum of adverse events after therapeutiv dose of mefloquine in healthy adults. Acta Trop 2002; 81:167–173. [DOI] [PubMed] [Google Scholar]
- Rice GC, Bump EA, Shrieve DC, Lee W, Kovacs M. Quantitative analysis of cellular glutathione by flow cytometry utilizing monochlorobimane: some applications to radiation and drug resistance in vitro and in vivo. Cancer Res 1986; 46:6105–6110. [PubMed] [Google Scholar]
- Roberts LJ 2nd, Morrow JD. Isoprostanes. Novel markers of endogenous lipid peroxidation and potential mediators of oxidant injury. Ann N Y Acad Sci 1994; 744:237–242. [DOI] [PubMed] [Google Scholar]
- Siciliano JC, Toutant M, Derkinderen P, Sasaki T, Girault JA. Differential Regulation of Proline-rich Tyrosine Kinase 2/Cell Adhesion Kinase β (PYK2/CAKβ) and pp125FAK by Glutamate and Depolarization in Rat Hippocampus. J Biol Chem 1996; 271:28942–28946. [DOI] [PubMed] [Google Scholar]
- Simpson J, Price A, terKuile R, Teja-Isavatharm F, Nosten P, Chongsuphajaisiddhi F, Looareesuwan T, Aarons SL, White NJ. Population pharmacokinetics of mefloquine in patients with acute falciparum ma;aria. Clin Pharmacol Ther 1999; 66:472–484. [DOI] [PubMed] [Google Scholar]
- Siegel GJ, Agranoff BW, Albers RW, Fisher SK, Uhler MD. Basic Neurochemistry, Lippincott, Williams & Wilkins, Philadelphia, PA, 1999 [Google Scholar]
- Strappazzon F, Torch S, Trioulier Y, Blot B, Sadoul R, Verna JM. Survival response-linked Pyk2 activation during potassium depletion-induced apoptosis of cerebellar granule neurons Molecular and Cellular Neuroscience 2007; 34:3355–365. [DOI] [PubMed] [Google Scholar]
- Tokiwa G, Dikic I, Lev S, Schlessinger J. Activation of Pyk2 by Stress Signals and Coupling with JNK Signaling Pathway. Science 1996; 273:792–794. [DOI] [PubMed] [Google Scholar]
- Toovey S Mefloquine neurotoxicity: a literature review Travel Med Infect Dis 2009; 7:2–6. [DOI] [PubMed] [Google Scholar]
- Weiss SM, Benwell K, Cliffe IA, Gillespie RJ, Knight AR, Lerpiniere J, Misra A, Pratt RM, Revell D, Upton R, Dourish CT. Discovery of nonxanthine adenosine A2A receptor antagonists for the treatment of Parkinson’s disease. Neurology 2003; 61:S101–6. [DOI] [PubMed] [Google Scholar]
- World Health Organization. WHO Expert Committee on Malaria. World Malaria Report 2009. Geneva: World Health Organization, 2009. [Google Scholar]
- Xi CX, Xiong F, Zhou Z, Mei L, Xiong WC. PYK2 interacts with MyD88 and regulates MyD88-mediated NF-B activation in macrophages. Journal of Leukocyte Biology 2010; 87:415–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong W, Parsons JT. Induction of apoptosis after expression of PYK2, a tyrosine kinase structurally related to focal adhesion kinase. J Cell Biol 1997; 139:529–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong WC, Mei L. Roles of FAK family kinases in nervous system. Front Bio sci 2003; 8:s676–s682. [DOI] [PubMed] [Google Scholar]
- Zhou C, Xiao C, McArdle JJ, Ye JH. Mefloquine enhances nigral gamma-aminobutyric acid release via inhibition of cholinesterase. J Pharmacol Exp Ther 2006; 317:1155–60. [DOI] [PubMed] [Google Scholar]
- Zwick E, Wallasch C, Daub H, Ullrich A. Distinct calcium-dependent pathways of epidermal growth factor receptor transactivation and PYK2 tyrosine phosphorylation in PC12 cells. J Biol Chem 1999; 274:20989–20996. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.