

Abstract
We present a succession of structural changes involved in hormone peptide activation of a prototypical GPCR. Microsecond molecular dynamics simulation generated conformational ensembles reveal propagation of structural changes through key “microswitches” within human AT1R bound to native hormone. The endocrine octa-peptide angiotensin II (AngII) activates AT1R signaling in our bodies which maintains physiological blood pressure, electrolyte balance, and cardiovascular homeostasis. Excessive AT1R activation is associated with pathogenesis of hypertension and cardiovascular diseases which are treated by sartan drugs. The mechanism of AT1R inhibition by sartans has been elucidated by 2.8 Å X-ray structures, mutagenesis, and computational analyses. Yet, the mechanism of AT1R activation by AngII is unclear. The current study delineates an activation scheme initiated by AngII binding. A van der Waals “grasp” interaction between Phe8AngII with Ile2887.39 in AT1R induced mechanical strain pulling Tyr2927.43 and breakage of critical interhelical H-bonds, first between Tyr2927.43 and Val1083.32 and second between Asn1113.35 and Asn2957.46. Subsequently changes are observed in conserved microswitches DRYTM3, Yx7K(R)TM5, CWxPTM6, and NPxxYTM7 in AT1R. Activating the microswitches in the intracellular region of AT1R may trigger formation of the G-protein binding pocket as well as exposure of helix-8 to cytoplasm. Thus, the active-like conformation of AT1R is initiated by the van der Waals interaction of Phe8AngII with Ile2887.39, followed by systematic reorganization of critical interhelical H-bonds and activation of microswitches.
Graphical Abstract
INTRODUCTION
Nearly 5% of the human genome encodes G-protein coupled receptors (GPCRs), which are targets of ~34% of therapeutic drugs currently in use.1 GPCRs are seven-transmembrane helical proteins that modulate vital cell-signaling responses to a variety of natural and pharmaceutical ligands.1–5 Extracellular agonists bind and initiate receptor conformational changes which propagate across the trans-membrane (TM) α-helical domain to intracellular “microswitches” formed by highly conserved sequence motifs in GPCRs. These events result in the transient formation of a cytosolic cleft for binding and activating a cognate G protein. By comparing the antagonist-bound inactive and [agonist + G protein/antibody] bound active state structures of GPCRs, the activating structural changes were identified.6–11 Structure−function studies have shown that mutating a microswitch may still allow the activating motion of TM-helices but impair cell signaling.12 Intramolecular events that form the electromechanical basis of signal transduction are predominantly documented for amine activated GPCR prototypes. Although three agonist-peptide bound GPCR structures are available (PDB ids: 6DDF, 5UNF, 5T04), an ensemble view of the conformational dynamics of peptide hormone activation of a GPCR is absent to date.13–16
The angiotensin type 1 receptor (AT1R) is a clinically important GPCR that binds the renin−angiotensin system hormone angiotensin II (AngII). Cell signaling by activated AT1R involves stimulation of the Gq11/12 leading to intra-cellular production of calcium, lipid messengers, and activation of protein kinases.17–20 In addition, G protein-independent signaling pathways are also activated.21–25 AngII-activated AT1R is responsible for homeostatic regulation of blood pressure and water−electrolyte balance in the body.26,27 AT1R is widely expressed in vascular and myocardial tissue, the liver, the kidney, the adrenal cortex, and specific brain centers. Chronic AT1R activation produces disease states including hypertension, inflammation, endothelial dysfunction, athero-sclerosis, congestive heart failure, and end organ damage. AT1R blocking drugs (ARBs) such as olmesartan are used as a first line of therapy in fighting these diseases. Previous studies have suggested a transition scheme for functional states of AT1R [Figure 1A] upon antagonist and agonist binding.28,29 Two high-resolution inactive state structures of AT1R are reported but attempts made to crystallize the AngII bound AT1R did not succeed.13,14 To bridge the knowledge gap with regard to the AngII-activated state of AT1R, we performed microsecond molecular dynamics simulation (MDS) analysis in a membrane bilayer environment.
Figure 1.
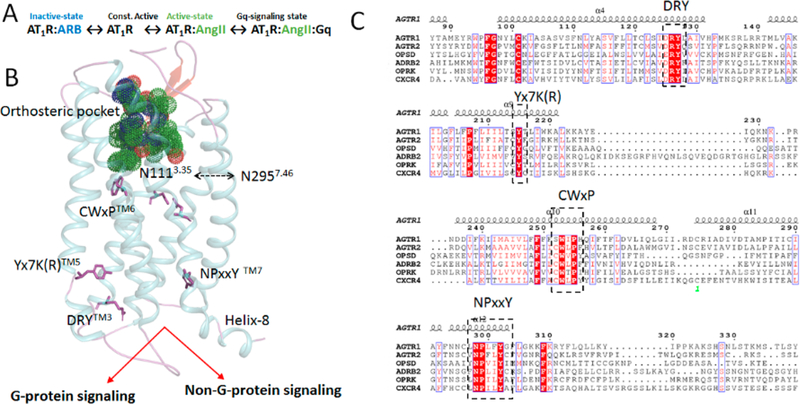
(A) Scheme of proposed conformational states governing AT1R function. (B) 3D structure of AT1R showing orthosteric pocket and important motifs in G-protein and non-G-protein signaling. (C) Multiple sequence alignment of AT1R with Class A GPCRs highlighting conserved motifs.
In this first report delineating conformational ensemble of a clinically relevant peptide hormone—GPCR—we carried out MDS starting with the 2.8 Å X-ray crystallographic structure of human AT1R.13,14 We corroborate the MDS results [Figure 1B] by (i) comparing to active-state structures of established GPCRs;30–34 (ii) using stereochemical features of AngII-analogs defined by structure activity relationship (SAR),35–38 and (iii) by combining with site-directed mutagenesis findings on pharmacology of AT1R.37,39–49 As in all GPCRs, several conserved residues form functional motifs within AT1R [Figure 1C]. During AT1R-AngII signaling, these motifs may act as microswitches that change from an inactive to an active conformation. Hence, inspecting perturbation in CWxPTM6, DRYTM3, Yx7K(R) TM5, and NPxxYTM7 motifs in comparison to different established activated GPCRs39–42 will further validate our MDS. SAR analysis of the AngII peptide, Asp1-Arg2-Val3-Tyr4-Ile5-His6-Pro7-Phe8 has established that the charged property of Asp1, Arg2, Phe8-COO− and bulk-hydrophobicity of Tyr4 determine binding affinity and specificity for AT1R [Figure S1A]. The aromatic Phe8 side chain and cumulative influence of Tyr4, His6, and Pro7 are responsible for agonist potency of AngII in AT1R activation.50–52 Mutagenesis analysis showed that AngII binding requires residues, Tyr351.39, Trp842.60, Arg162ECL2, Lys1995.42, and Asp2817.32 [Figure S1B]. In addition, the critical role of Asp742.50, Asn1113.35, Arg1263.50, Trp2536.48, His2566.51, Ile2887.39, Tyr2927.43, and Asn2957.46 in receptor activation are documented.37,44,46,48,49,53,54 How these features of the receptor and the hormone cooperatively produce an AngII-activated state of AT1R is unknown.
Our working hypothesis assumes that microswitches constrain spontaneous activation in the basal state of AT1R. Distinct interactions of AngII with the AT1R binding pocket break the prime constraint instigating the microswitches to change from inactive to active conformation in the course of receptor activation. Understanding the details of AngII activation of AT1R could further help in designing more potent ARBs to prevent AngII associated pathogenesis.
RESULTS
Global Conformational Changes.
In the 1 μS MDS following flexible docking of AngII to membrane embedded AT1R (PDB id: 4ZUD),13 the trajectory plot for AngII and the receptor backbone reached equilibrium (Figure S2A). Significant changes in root-mean-square fluctuation (RMSF) was observed in DRY, ECL2, and ICL3 motifs as shown in Figure S2B in the AT1R-AngII compared to AT1R-Olmesartan. High fluctuation in these motif may correlate with the activation of the receptor. The RMSD plot indicated significant movement in TM-helices, ECL2, ICL3, and helix-8 (Figure S3). The AT1R conformation ensembles produced from MDS closely match the crystal structure of peptide-agonist bound ETBR (PDB id: 5GLH)16 (Figure 2A and B). The volume of the AngII bound pocket of AT1R is ~1203.11 ± 335.83 Å3, comparable to the pocket of ETBR which has a volume of 895.92 Å3 (PDB id: 5GLH).16 The extracellular portions of TM1, TM3, TM4, TM5, and TM7 move in the AngII-bound state (Figure S4). These structural changes are associated with the orthosteric pocket adopting a compact “closed” configuration enabling strong interactions with AngII. Together the N-terminal tail (NT) and EC-loops (ECL) including the β-strands of ECL2 form a lid-like architecture that covers the orthosteric pocket (Figure 2C and D). Mutagenesis and accessibility mapping studies have predicted ligand-specific movements in NT and ECL regions of AT1R, especially in the ECL2.55–57 The water accessible entrance in the apo-AT1R might facilitate the entry of ligands to the orthosteric pocket.
Figure 2.
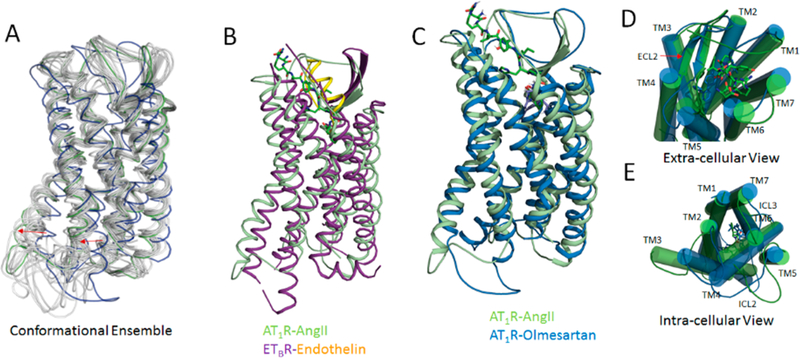
(A) Representative conformational ensembles from 1 μs MDS of AngII bound AT1R. (B) Superimposed structure of AT1R-AngII and ETBR-Endothelin. Superimposed structure of AT1R-AngII (from MDS) and AT1R-Olmesartan (crystal structure) bound states showing transmembrane view (C), extra-cellular view (D), and intra-cellular view (E).
By contrast, in the intracellular region, the TM1, TM3, TM5, TM6, and TM7 move outward (Figure S4). The movement of ICL3 associated with TM3, TM6, and TM7 opens a crevice that is supposed to bind the G protein (Figure 2C and E), but the opening is not complete as seen in the Gs protein bound β2AR structure.40 The outward movement of TM6 is comparatively less than observed in the active state crystal structures of GPCRs.40 Functional studies in AT1R have indicated that exposure of the DRYTM3 motif, the membrane proximal segments of ICL3, and the NPxxYTM7 motif are necessary and sufficient to recruit the G protein.20 A well-defined helix-8 observed in the inactive state AT1R shows ~15 Å shift toward the cytoplasmic space with decreased helical stability in a small number of the ensemble frames (Figure S4). Observed conformational change in helix-8 is relevant because functional studies have shown that helix-8 of AT1R is a hotspot for recruitment of non-G protein signaling molecules such as Jak2, Cam kinase, and Filamin A.20,58 These AngII-induced receptor conformational changes are not observed when olmesartan is used as the ligand for MDS and the ensemble frames are indicative of agonist bound structures of β2AR, ETBR, NT1R, and μ-opioid receptor in which structural changes linking the orthosteric pocket and the cytoplasmic G-protein binding pocket is observed.
AngII Binding Interactions.
Supporting the mutagenesis findings, AngII residues 3−8 are embed within the orthosteric pocket (Figure 3A). Asp1AngII and Arg2AngII extend to the NT and ECL2 and ECL3.55,56,59–61 Binding of AngII involves ~17 AT1R residues forming ionic, H-bond, van der Waals, and π−π stacking interactions (Figure 3B). The conformation of bound AngII observed in our MDS is similar to NMR,62 flexible docking,38 or modeling studies.35 Superimposition of AngII and Olmesartan indicates overlap of their aromatic−acidic pharmacophore groups surrounded by shared residues, Tyr353.91, Trp842.60, Tyr872.63, Lys1995.42, Arg167ECL2, Ile2887.39, and Tyr2927.43 which validate competitive antagonism of AngII and ARBs (Figure S5). Mutagenesis studies indicate that these are critical residues for binding both AngII and ARBs (see Table S1).
Figure 3.
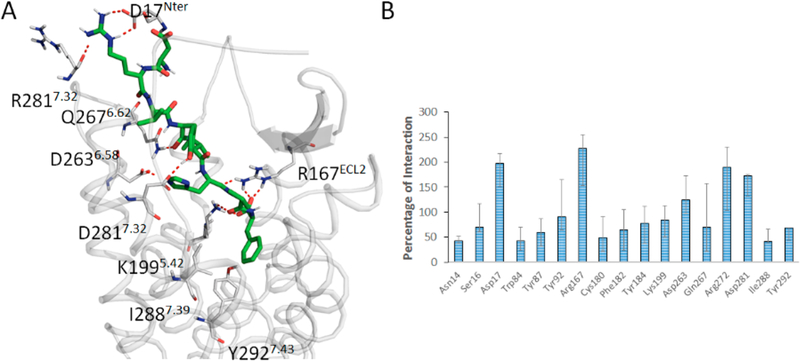
(A) Binding mode of AngII in the orthosteric pocket of AT1R. (B) Interaction of AngII with AT1R residues >25% in at least 3 out of 6 independent MDS experiments are shown. A single residue can have >100% value due to multiple interactions (e.g., weak, ionic, hydrophobic, etc.).
The octapeptide is oriented so that the Phe8AngII α-carboxyl interaction with Lys1995.42 is robust (Figure 3). MDS trajectory analysis of Phe8AngII α-carboxyl H-bond and ionic interaction with Lys1995.42 estimated to be >75% in multiple trials. It is known that the Phe8AngII α-carboxyl, if blocked by methylation or amination causes tachphylaxis in vivo. Lys1995.42 mutation to Ala/Gln in AT1R completely abolished binding of AngII and its analog [Sar1, Ile8]AngII. Noda et al.45 suggesting that Lys1995.42 interaction with Phe8AngII α-carboxyl may be important for the orientation of His6-Pro7-Phe8AngII in mediating receptor agonism. After 200 ns the guanidinium group of Arg167ECL2 forms multiple interactions including water mediated bonding with backbone carbonyl groups of Phe8AngII, His6AngII, and Ile5AngII amounting to 250% of interaction. When Arg167ECL2 was mutated, AngII binding is abolished. Therefore, projecting Arg167ECL2 into the depth of the orthosteric pocket to support interaction with AngII backbone carbonyls might be critical for shaping the receptor-bound conformation of AngII. We have explored every critical binding pocket residue of AT1R involved in AngII binding. We did not find significant change in the conformation of Arg167ECL2 in AT1R bound with AngII or olmesartan. Supporting result was observed in our mutagenesis and crystal structure experiments (Table S1).
The NH2-backbone amide of Asp1AngII interacts with Asp263ECL3 and Ser186ECL2 and the Asp1AngII carboxyl group maintains interaction with Lys20NT. Deletion of Asp1AngII reduces the binding affinity slightly and substitution with sarcosine enhances the binding affinity. The Arg2AngII interacts with Asp2817.32 validating the SAR studies, which established its importance for the hormone’s binding affinity. The Asp2817.32 mutation was shown to reduce AngII affinity as well as efficacy.60 Previous modeling studies also indicate that additional stabilization of this interaction may involve Asp2787.29. During simulation, Arg2AngII appears to form a strong interaction with Asp1AngII as well as Asp17NT. These interactions contributed ≈130% each, implicating their role in binding Arg2AngII.
Phe8AngII reaches deep toward the core of the receptor, while Tyr4 AngII is positioned near the extracellular entrance of the orthosteric binding-cavity.Tyr4AngII and Phe8AngII are important for agonistic potency, [Ile4, Ile8]-AngII functions as a low affinity antagonist and [Ile4]AngII and [Ile8]AngII are low efficacy partial agonists for AT1R. Hence, Tyr4AngII and Phe8AngII interactions are critical for activation of the AT1R (Figure 3). In MDS we observed that dynamic state of Tyr4AngII precludes a stable interaction of this side chain with the receptor.
Phe8AngII is oriented toward Trp842.60 forming a strong π−π stacking interaction. Also Trp842.60 forms intramolecular interactions with Leu812.57, Tyr872.63, Thr882.64, Ala1043.28, and Arg167ECL2. Trp842.60 mutation abolished AngII binding because the smaller aliphatic residues introduced alter conformational geometry and disrupt interactions with Tyr872.63 and Arg167ECL2. Tyr92ECL1 mutation to Ala completely abolished AngII binding. Tyr92ECL1 forms hydrophobic interaction with Pro7AngII, which is important for the active conformation of the AngII. Pro7AngII substitution with a positively or uncharged residue completely abolishes binding affinity of AngII.
The Ile2887.39 side chain is oriented toward Phe8AngII making a van der Waals interaction. Mutation with Ala in this position abolished AngII binding, since the shorter side chain of Ala could not reach Phe8AngII to establish a van der Waals interaction. Mutagenesis further showed that a bulkier Phe2887.39 side chain does not significantly affect AngII binding. This observation is consistent with modifications of Phe8AngII such as [Sar1, Ile8]AngII and des-Phe8-heptapeptide, which are devoid of AT1R agonism.
AngII-Induced Conformational Changes.
Systematic changes observed in AT1R conformation upon AngII binding in our MDS mimic structures of the active-like state GPCRs. In the following sections, we describe the time sequence of structural changes connecting AngII binding site to different cytoplasmic microswitches (Table 1).
Table 1.
Time Course of Molecular Events Observed in MD Simulationa
| Molecular Events |
||
|---|---|---|
| time | AT1R-AngII | AT1R-olmesartan |
| 0 ns | strong interaction of Phe8AngII-Ile2887.39 | strong interaction of alkyl tail of omesartan-Ile2887.39 |
| ~200 ns | Phe8AngII pulls up Ile2887.39 | position of Ile2887.39 remain same, strong interaction with alkyl tail of Omesartan |
| ~400 ns | breakage of Val1083.35 Cα-OH-Tyr2927.34 H-bond (TM3-TM7) | Val1083.32 Cα-OH-Tyr2927.34 (TM3-TM7) H-bond is consistent |
| ~600 ns | breakage of Asn1113.32 N2957.46 H-bond (TM3-TM7) | Asn1113.32 N2957.46 H-bond (TM3-TM7) is consistent |
| ~700 | opening of intracellular domain for Gprotein binding | intracellular domain are closed for Gprotein binding |
| ~1000 ns | open intracellular domain with active like conformation | closed intracellular domain with inactive conformation |
All these events are captured and shown in MDS trajectory videos1−4.
Breaking Primary Constraint (Asn1113.35-Asn2957.46 H-Bond).
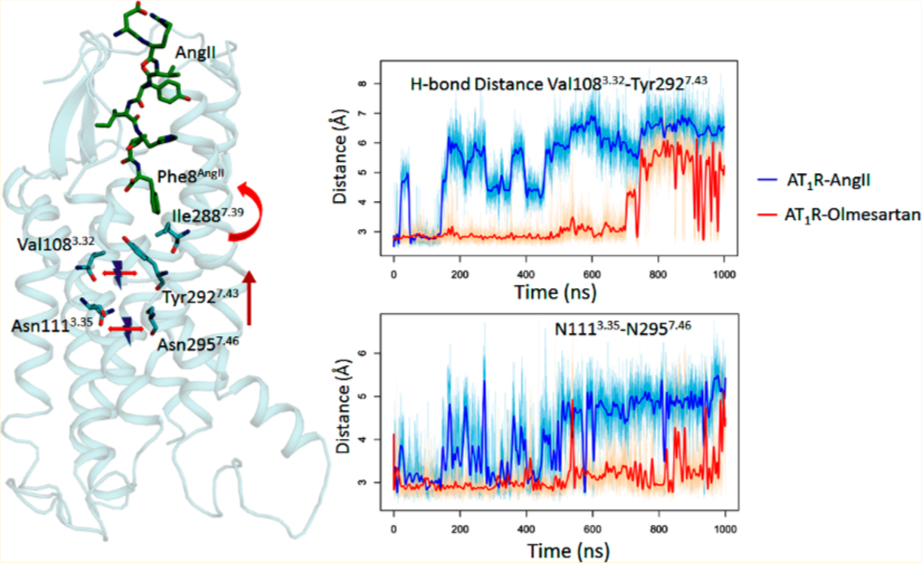
The most important agonism determinant is Phe8AngII side chain which orientates toward a cluster formed by Tyr351.39, Trp842.60, Pro2857.36, Ile2887.39, and Tyr2927.43 throughout the 1000 ns MDS. The Phe8AngII aromatic side chain “grasps” Ile2887.39 in van der Waals attraction within ~100 ns (Figure 4), and this “grasp” pulls the aromatic side chain of Tyr2927.43, resulting in breakage of the interhelical H-bond between Tyr2927.43−OH and the backbone carbonyl of Val1083.32 in ~200 ns which in turn breaks interhelical H-bonds between Asn1113.35-Asn2957.46 in ~500 ns. Interestingly, equivalent intrahelical H-bonds between TM3-TM7 are absent in AT2R.63The AT2R structure (PDB id: 5unf) resembles active like GPCRs and it has a Phe residue at TM7.43 position that is equivalent to Tyr2927.43 in AT1R. This evidence further supports the conclusion that MDS produced an active-like state of AT1R.
Figure 4.
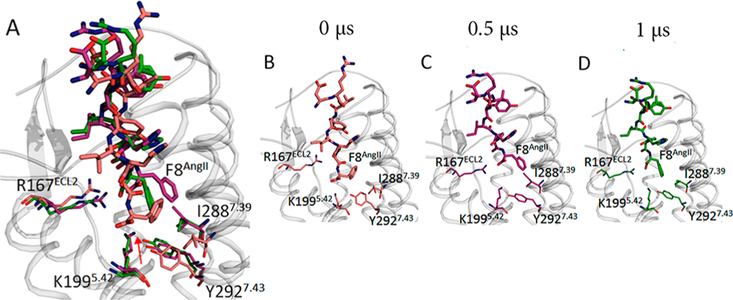
(A) Superimposed Phe8AngII orientation changes at time intervals indicated. Orientation of Phe8AngII captured at 0 (B), 0.5 (C), and 1 μs (D).
In contrast, binding olmesartan establishes independent interaction with both Ile2887.39 and Tyr2927.43 and consequently stabilizes the TM7-TM3 interhelical H-bonds between Tyr2927.43-OH-Val1083.32 and Asn1113.35-Asn2957.46 (Figures 5, S6, and S7). In support of this finding, independent studies have shown that nonpeptide AT1R antagonists show reduced binding affinity to active like state of AT1R.63,64 MD simulations with -Ile8AngII, -Ala8AngII, and -Gly8AngII indicated that the aliphatic side chains replacing Phe8AngII systematically decrease interaction with Ile2887.39 which is also validated by the agonist-efficacy experimental data shown in Figure S8.
Figure 5.
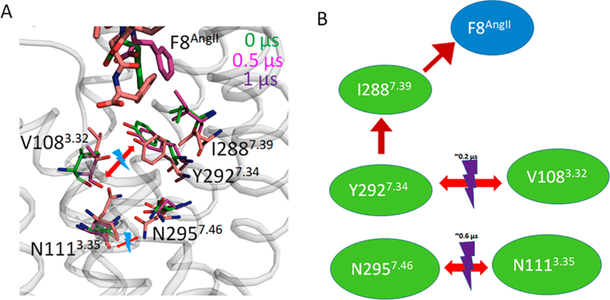
Representation of Phe8AngII induced motion of side chains leading to breakage of N1113.35-N2957.46 H-bond.
The disruption of the Asn1113.35-Asn2957.46 bond being central to AT1R activation is well-documented: (i) stable interhelical H-bonds between Asn1113.35-Asn2957.46 are confirmed in ARB-bound AT1R crystal structures;13,14 (ii) Asn1113.35 mutation to smaller residues Gly, Ala, Ser, and Cys constitutively activate AT1R;47,60,65–67 (iii) Asn2957.46 to Ser mutation mimics this effect;64 and (iv) long-range effects of mutations destabilizing Asn1113.35-Asn2957.46 bond also cause constitutive activation.57 To further examine, we modeled Asn111Gly3.35 mutant of AT1R and found that the model coordinates matched the active-like-state AT2R structure better than inactive state AT1R structure. Substitution of larger side chains, such as the Trp1113.35 produced the most inactive AT1R mutant.49 The residues involved are conserved in AT2R, suggesting that the same mechanism may be involved in AngII activation of AT2R.
Mechanism of TM-Bundle Entropy.
Interhelical polar interaction networks involving conserved residues, Asn461.50, Asp742.60, Asn2957.46, and Asn2987.49 are rearranged. The Tyr2927.43 side chain forms π−π stacking with Phe772.53 and the Asn1113.35-Asn2957.46 H-bond interacts with Asp742.50 in the AngII bound state. When the Asn1113.35-Asn2957.46 H-bond breaks, the Asn2957.46 interacts strongly with Asp742.50 and Asn461.50. The weakening of TM7-TM3 interaction sways the side chain of Leu1123.36 toward Lys1995.42. In a previous MDS study, Cabana et al.49 compared constitutively active Gly1113.35 mutant with the inactive Trp1113.35 mutant. Although the Asn1113.35-Asn2957.46 H-bond is broken in both of these mutants, the activation switch to Asn2957.46-Asp742.50-Asn461.50 H-bonds was not observed in the Trp1113.35 mutant. Instead, Asp742.50 stably interacted with Trp1113.35. In the constitutively active Gly1113.35 mutant, they observed H-bond between Asp742.50-Asn461.50. Therefore, orderly rearrangement of interhelical network coordinated with Asn1113.35-Asn2957.46 interaction in the AT1R are critical steps in activation. AngII binding also perturbed another important hydrogen bonding network involving Asp742.50, with Ser1093.33, Tyr1133.37 and Cys2967.47 that is conserved among class A GPCRs (Figure S9). Biochemical experiments showed that agonist-activation of AT1R alters the orientation of TM2, reducing its interaction with TM7. The Asp742.50 and Phe772.53 side chains buried in AT1R become solvent exposed when AngII-binds.61,68,69
In addition to the networks in the AngII binding pocket, extensive H-bond network linking the orthosteric ligand pocket and the G-protein-binding cleft also rearrange in the active-like state. In this network, polar residues are conserved as documented in family A GPCR structures and may involve water molecules and a sodium ion as discovered in the high-resolution inactive-state structure of a few GPCRs.10,11,40 Since involvement of allosteric sodium is not experimentally confirmed in AT1R, we did not include a sodium ion in our MDS analysis. However, the cytoplasmic H-bond networks can be traced to the side chains of Asp742.50, Asn1113.35, Trp2536.48, and Asn2957.46 that could be involved in coordination of a sodium ion. As discussed below the polar networks extend from CWxPTM6 motif to the cytoplasmic surface involving microswitches, the DRYTM3, Yx7K(R)TM5, and NPxxYTM7 sequences.
Motion of Trp2536.48.
Trp2536.48 is part of the CWxP motif, a rotamer toggle switch which leads to the opening of G protein cleft in class A GPCRs. This highly conserved amino acid residue is located on the floor of the ligand pocket and adjacent to the conserved Pro2556.50. No significant change in orientation of Pro2556.50 is observed in our MD simulation. In the AngII activated state the orientation of Trp2536.48 side chain tilts intracellular toward His2566.51 and Phe2496.44, which are in close interactions with the cluster of aromatic residues Phe2045.47, Phe2085.51, and Tyr2927.43 (Figure 6). Of these, His2566.51 is a key coupling residue as mutations in this residue uncouple AT1R agonist binding and G protein activation.44,45 While the Cα torsion remained constant, the Trp2536.48 side chain torsion was stabilized in ~200 ns in the AngII-bound state and in 500 ns in the Olmesartan-bound states (Figure S10). The nature of CWxP conformational changes is not identical in eight agonist or G-protein bound active state GPCR structures in PDB (PDB ids: 2YDV, 3SN6, 5GLH, 4MQS, 5C1M, 3PQR, 5YNF). However, the motion observed in our simulation best matched with ETBR and A2ARs (Figure S10B). The hydrophobic interaction between Trp2536.48 and Phe2496.44 is also observed in the active-like states of NTSR1 and μ-opioid receptor.
Figure 6.
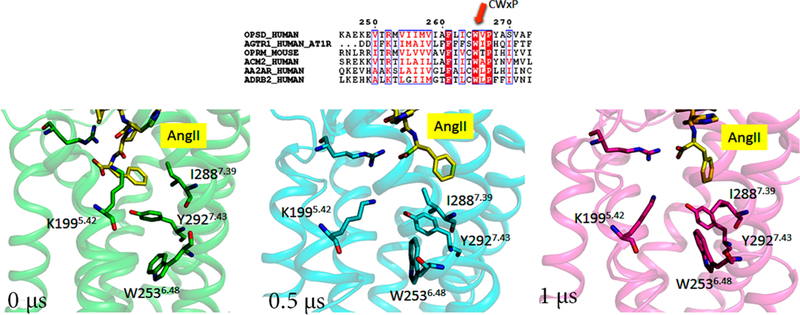
Conformation of toggle switch Trp2536.48 of AT1R at different time intervals.
Motion of Tyr2155.58.
Tyr2155.58 is located closer to the cytoplasmic end of TM5 and is part of the Yx7KTM5 motif. Highly conserved throughout the GPCR family, the Tyr side chain at this location may serve the need for the polarity and the bulky hydrophobic ring system to ensure requirements for an efficient stabilizing motif in the active states of GPCRs.70,71 In crystal structures of GPCRs, Tyr2155.58 forms an H-bond with Arg1263.50 side chain of the DRY motif in the active state. In AT1R, this bonding is not observed throughout the simulation. Instead we observed a strong and continuous interaction of Tyr2155.58 with Leu1193.43 which helps TM3 and TM5 remain closer. But in the active state, the Tyr2155.58 side chain moves toward the surface of the protein and forms π−π interaction with Trp2195.62 which makes the intracellular region of TM5 less flexible (Figure 7 and S11).
Figure 7.
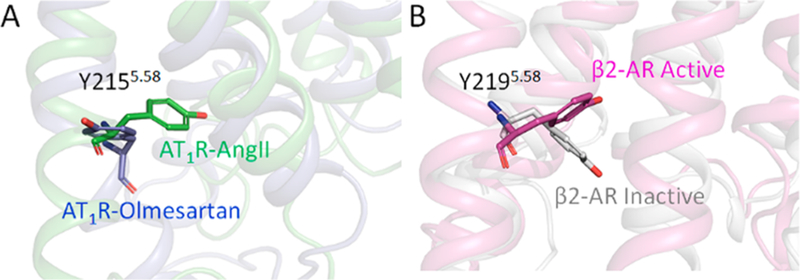
Conformation of Tyr2155.58 projecting inward in AngII-bound AT1R and outward in olmesartan-bound AT1R (A). The same pattern is also observed in active and inactive states of β2AR (B).
DRY3.51 Switch.
The Asp1253.49-Arg1263.50-Tyr1273.51 located at the cytoplasmic end of TM3 in AT1R is a highly conserved motif in GPCRs and often called an “ionic-lock”, restricting G protein interaction. In the current simulation we observed a distinct difference in orientation and H-bond networking of Arg1263.50. In the inactive state, Arg1263.50 is involved in stable intrahelical salt bridge with Asp1253.49 which stabilizes the intracellular portion of TM3 to remain compact. This observation was also reported by Matsoukas et al.38 Figure 8 depicts the conformation of this microswitch in the active state. The side chain of Arg1263.50 moves toward TM6 and forms an H-bond with Asp2376.32 (Figure S12). These rearrangements could be the beginning of opening of the groove for binding G-protein.
Figure 8.
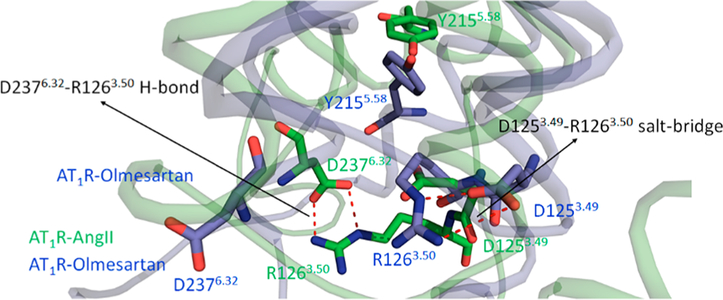
Breakage of H-bond between D1253.49-R1263.50 and formation of H-bond between R1263.50-D2376.32 in AngII bound AT1R. The D1253.49-R1263.50 H-bond is retained in the olmesartan-bound state.
NPxxYTM7.53 Motif and Helix-8.
The Asn2987.49-Pro2997.50-Leu7.51-Glu7.52-Tyr3027.53 motif is also highly conserved in GPCRs. Mutations targeting this motif reduce G protein activation by AT1R.61,72 During AngII activation, the NPxxYTM7 moves toward the conserved Tyr2155.58 forming a hydrogen-bond network with Asn2987.51, Tyr3027.53, and Leu1193.43 (Figure 9). A similar hydrogen-bond network was reported in activated structures of rhodopsin, β2AR, and M2R. The movements associated with NPxxY also contribute to opening the groove for G-protein binding.
Figure 9.
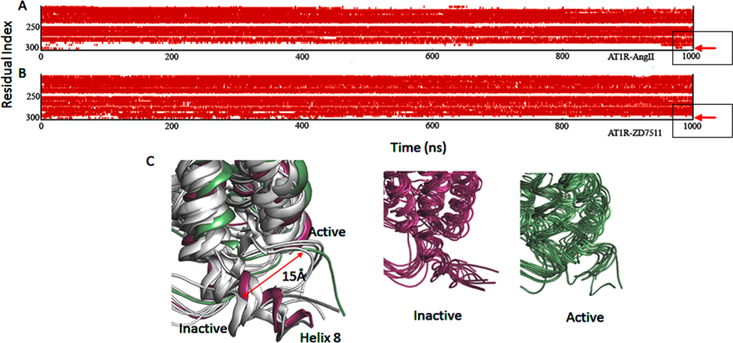
Secondary structure of H8 region of AT1R-AngII (A) and AT1R-ZD7511 (B). Loss of α helix (red color) from residue number 302 to 316 in panel A. Ensembles of structures of AT1R-AngII and AT1R-ZD7511 (C).
A ~15 Å movement of helix-8 toward the membrane is observed in AngII-bound AT1R, but not in the olmesartan-bound AT1R. The secondary structure of helix-8 changes from α helix to unstructured in AngII bound state (Figure 9). Another dynamic feature observed in most activated GPCRs including AT1R is the increase of the Tyr3027.53-Tyr1128.53 distance, indicating TM7-helix-8 conformational rearrangement. This distance increased gradually in MD simulations after the first 20 ns and remained stable for the rest of the trajectory. Large changes in helix-8 conformation are reported in agonist-bound μ-opioid receptor.73
DISCUSSION
Delineating the molecular mechanics involved in the opening of G protein binding cleft when AT1R engages AngII is an important advancement. Without MDS analysis, visualizing the dynamics of receptor activation would be difficult while accounting for AngII-analog SAR data, molecular pharmacology data, receptor mutagenesis data, and 3D-structure data. MDS analysis indicated that AT1R conforms to the general paradigm of conformational modulation by ligands. The agonists promote accumulation of the activated state and the inverse agonists depleted the basal state with minor structural changes (Figure 1). The activated state accumulated spontaneously in the constitutively active Asn111Gly3.35 mutant suggesting that the wild-type AT1R activation must involve agonist-dependent repositioning of Asn1113.35 side chain. The Asn1113.35-Asn2957.46 bond is the prime constraining interaction in AT1R. When AngII binds this bond is broken indirectly through strain induced by the van der Waals attraction of Ile2887.39 toward the well-known agonism-specifying Phe8AngII (Figures 3−5). This is a novel GPCR activation mechanism unlike the agonist trigger mechanisms in well-known GPCRs. For instance, rhodopsin photoactivation which serves as a prototype includes two triggering events, protonation breakage of the buried salt-bridge linking TM7 (retinal-Lys Schiff-base) to TM3 (counterion) and the resulting mechanical motion of functional microdomains.12,74 In other ligand activated GPCRs different variations of a Lewis acid/base interaction chemical mechanisms operate. For instance, in β-adrenergic receptor, TM3-TM7 lock is a hydrogen bond betweenTyr7.35-Asp3.29 which interacts with protonated quaternary nitrogen in adrenergic ligands and networked with an Asn7.32 residue.75,76 In activating S1P1R, the lyso-phospholipid sphingosine 1-phosphate receptor, the zwitterionic phosphonate and primary amine groups respectively bond with Arg7.35 and Glu3.29.77 Thus, release of the TM3-TM7 ionic-lock is suggested to be the first agonist activation step in GPCRs.74 However, a general GPCR mechanism for breaking prime constraint remains unclear especially for peptide GPCRs until now.
The chemical nature of interactions of AngII and ARBs with the orthosteric pocket is fundamental to pharmacological outcomes, i.e., agonism and inverse-agonism. MDS analysis indicated that inverse-agonist (olmesartan) binding stabilizes multiple TM3-TM7 interactions and dampens intrinsic motion of helices and motifs of AT1R (Table 1, Figures 6−9 and S6− S12). In addition, insights regarding the differing roles of critical residues are revealed. For example, in the activated state Arg167ECL2 establishes multiple interactions with the backbone carbonyl groups of AngII, morphing its receptor bound conformation. However, Arg167ECL2 binds the acidic pharmacophore groups at either ends of inverse-agonists, inhibiting residue rearrangements locally in the orthosteric pocket as well as globally in TM and cytoplasmic domains in the inactive-state (Figures 3 and S5).
Receptor bound conformation or the preferred conformation of a hormone peptide provides critical structural information for designing selective pharmaceutical scaffolds for a GPCR. The AngII structure in solution shows a bioactive γ-turn. Cyclic AngII analogs which restrain this conformation increase potency.78,79 Specific spatial alignment of aromatic residues in AngII, Tyr-hydroxyl (Y4), His-imidazole (H6), and Phe-benzyl (F8) in this conformation maybe important.80,81 Hormone peptides, AngII (DRVY4IH6PF8), AngIII (RVY4IH6PF8), and AngIV (VY4IH6PF8) which preserve the turn-motif are capable of modulating blood pressure via activating AT1R. The pose of AngII in our MDS analysis is consistent with spatial alignment of Tyr (Y4), His (H6), and Phe(F8). Additionally, Phe8AngII substitution with Ile and Gly, as well as its deletion (des-Phe8AngII) affect binding and activation. As shown in Figure S7, Ang1–7(DRVY4IH6P) with either unstable or absence of the bioactive γ-turn conformation binds poorly and may activate AT1R at nonphysiological concentrations which is also consistent with its poor MM/GBSA scores from IFD and MDS. Aromatic side chain of Phe8AngII helps to project toward the Ile2887.39. Substitution with nonaromatic residues (Ile, Gly, or Ala) produce a different conformation in which the side chain moves toward the ECL2 region.
The design of currently used ARBs to mimic the Y4IH6PF8 motif in AngII is confirmed by MDS analysis as well as in previous studies.19,28,82 This knowledge will be useful in designing better AT1R-selective scaffolds. Development of next generation AT1R agonists and antagonists in terms of differences in functional selectivity and tissue tropism has important clinical implications. Given that a significant number of patients are currently prescribed ARBs for indications beyond blood pressure reduction, it is important to know the mechanism of action of these drugs at the receptor level.
To our knowledge, this is the first report detailing activating structural dynamics of a clinically important peptide hormone GPCR. There are ~135 human GPCRs responding to endogenous peptide/protein ligands. Static structures of peptide-activated NTR1, ETBR, GLPR1 and opioid receptors2,83,84 are documented. Despite this progress the specific interactions that trigger activation of peptide GPCRs are not easy to decipher. Hence MDS analysis starting with human AT1R crystal structure described here should prove a useful model for gaining insights into the activation mechanism of peptide GPCRs and drug action. Energy-based models allow characterization of side-chain and backbone shift in the pocket as well as provide a 3D framework of determinants of agonism or inverse agonism essential for rational drug discovery. Important insights into agonist induced changes, establishing the path from ligand binding to G-protein activation can be gained.
In functioning GPCRs, thermodynamic motions within the protein produce a conformational landscape with discernible functional substates.85,86 The MDS ensemble frames provide an integrated view of substate dynamics and time evolution of receptors from inactive to basal to activated states (Figures 2−9). We observed that the orthosteric pocket and cytoplasmic domain where signal transducer molecules converge are semi-independent. The cytoplasmic domain harbors conserved motifs DRYTM3, Yx7KTM5, NPxxYTM7, and helix-8. These microswitches were originally described in rhodopsin,71 and functional studies in AT1R demonstrate that a high degree of stability of functional motifs in the inactive state prevents spontaneous receptor activation. Mutagenesis of microswitches either constitutively activate the receptor or interferes with overall signaling. The impetus for activation of cytoplasmic microswitches is transmitted through a network of hydrogen bonds from the conserved CWxPTM6 toggle-switch motif. Our MDS study shows critical positioning of this TM6-motif, sharing a hydrogen-bonded network between TM1-TM2-TM3-TM7 which includes the highly conserved Asp742.50 in AT1R. The CWxP motif is part of the proposed sodium-ion binding site formed with Asn1113.35 and Asn2957.46.12,13 Further, it interacts with a cluster of residues, His2566.51, Phe2045.47, Phe2085.51, and Tyr2927.43 which are associated with TM6 movement.36,37 Because of high level of conservation of residues at TM6.44, TM6.48, and TM2.50, the role of the CWxP motif in AT1R may be same as in other GPCRs. Recent experimental dynamics study in adenosine A2R found that the interplay between TM2.50 and TM6.48 regulated structural dynamics localized at cytoplasmic motifs associated with receptor activation87 which mimics similar observations made in rhodopsin activation.88,89
Time-evolution of changes involving residues, Ile2887.39, Tyr2927.43, V1083.32, Asn1113.35, Asn2957.46, Asn461.50, Asp742.50, and Trp2536.48 suggests a sequential conformational transition model in which disruption of the TM3-TM7 interaction leads to movement of TM6 followed by reorientation of the TM1-TM2-TM3-TM7 interhelical network. Alternative nonlinear transition model as proposed earlier27 with multiple kinetic scheme to account for native cellular and system-wide conditions is more realistic. Nonlinear transition models do include elements of a linear transition model as has been shown for rhodopsin.90–92
CONCLUSION
Phe8AngII engages Ile2887.39 of AT1R in van der Waals interaction which by pulling Tyr2927.43 and breaking H-bond with Val1083.32 initiates AT1R activation. This is a novel mechanism unlike those described in prototypical GPCR activation models. Breaking the Asn1113.35-Asn2957.46 bond relaxes the prime constraint; subsequently Asn2957.46 interacts with Asp742.50 and Asn461.50 leading to AngII activation of AT1R. This change in the molecular network leads to systematic change in the intracellular domain, and these changes may be a necessary for creation of a pocket for G-protein binding. In the olmesartan-bound inactive state, we observed a stable Asn1113.35-Asn2957.46 H-bond interaction and in addition Asn2957.46 interacts with Asp742.50. Capturing and understanding the molecular details of activation of AT1R by AngII and inhibition by ARBs could lead to better design of novel and more potent drugs targeting AT1R which could effectively prevent AngII pathogenesis.
MATERIALS AND METHODS
Protein Preparation.
The starting coordinates of AT1R PDB IDs: 4ZUD, 4YAY] were retrieved from Protein Data Bank (www.rcsb.org) and further modified for Glide docking calculations (Glide, Schrodinger, LLC, NY, USA).93 For these calculations, energy minimization was applied to the protein using the Protein Preparation Wizard by applying an OPLS3 (Optimized Potentials for Liquid Simulations) force field.94 Missing segments of loops were filled using Prime, Schrodinger, LLC, during the protein preparation.95 Progressively weaker restraints applied to only the non-hydrogen atoms and structure refinement was done based on the default protocol of Schrodinger, LLC, NY, USA, as Glide uses the full OPLS3 force field at an intermediate docking stage and is claimed to be more sensitive to geometrical details than other docking tools. The most likely positions of hydroxyl and thiol hydrogen atoms, protonation states, and tautomers of His residues, and Chi “flip” assignments for Asn, Gln, and His residues were selected. Finally, energy minimizations of protein were performed until the average root-mean-square deviation of the non-hydrogen atoms reached 0.3 Å.
Ligand Preparation.
We retrieved AngII crystal structure from Protein Data Bank [PDB ID: 1N9V] and the structure was prepared using LigPrep package from Schrodinger, LLC, NY, USA (LigPrep, 2018)96 by assigning appropriate bond order. AngII PDB structure was converted to mae format (Maestro, Schrodinger, LLC, NY, USA), geometrically optimized and partial atomic charges were computed. Then, at most, 32 poses were generated with different steric features for the subsequent docking study. Olmesartan structure was also prepared in Ligprep using the protocol described earlier.28
Induced Fit Docking (IFD).
Induced fit docking (IFD) (Schrodinger, LLC, NY, USA) was used to dock AngII and olmesartan inside the ligand binding pocket of AT1R. First, the ligand was docked into a rigid receptor model with scaled-down vdW radii. A vdW scaling of 0.5 was used for both the protein and ligand nonpolar atoms. A constrained energy minimization was carried out on the protein structure, keeping it close to the original crystal structure while removing bad steric contacts. Energy minimization was carried out using the OPLS3 force field with implicit solvation model. The Glide XP mode was used for the initial docking and ligand poses were retained for protein structural refinements. Prime (Schrodinger, LLC, NY, USA) was then used to generate the induced-fit protein−ligand complexes (Prime, Schrodinger, LLC, NY, USA).97 Each of the structures from the previous step were subjected to sidechain and backbone refinements.98 All residues with at least one atom located within 4.0 Å of each corresponding ligand pose were included in the Prime refinement. The refined complexes were ranked by Prime energy, and the 20 receptor structures within 30 kcal/mol of the minimum energy structure were passed through for a final round of Glide docking and scoring. In the final step, each ligand was redocked into the top 20 refined structures using Glide XP.
MM/GBSA.
Prime/MM-GBSA was used to predict the free energy of binding between the receptor and the AngII or olmesartan. The binding free energy (ΔGbind) was calculated using Prime.99 The binding free energy (Gbind) is then estimated using the following equation:
where ER:L is energy of the complex, ER + EL is the sum of the energies of the ligand and the apo protein, using the OPLS3 force field, and ΔGsolv (ΔGSA) is the difference between GBSA solvation energy (surface area energy) of complex and sum of the corresponding energies for the ligand and apoprotein.100–102
MD Simulation.
Molecular dynamics (MD) simulations were carried out for AT1R-AngII and AT1R-olmesartan complexes using Desmond MD code and the OPLS3 force field92 for minimization of the system. We ran the MD simulation for 1 μS for each of the complexes. For AT1R-AngII complex, we repeated the MD simulation six times for both the crystal structures solved for AT1R (PDB id’s: 4ZUD and 4YAY) and two times for the AT1R-olmesartan complex. Using the Desmond system builder, a 10 Å buffered orthorhombic system with periodic boundary conditions was constructed using a POPC lipid membrane103–106 and an SPC explicit water solvent. The overall charge was neutralized by 0.15 mol/L NaCl. The simulations were performed in the NPT ensemble with number of elements, pressure, and temperature controlled. The temperature of 300 K and pressure of 1.013 bar were kept constant by coupling the system to a Berendsen thermostat and barostat, which are normal temperature and pressure mimicking the real life environment. An integration step of 2.0 was used, Coulombic interactions were calculated using a cutoff radius of 9.0 Å and long-range electrostatic interactions were calculated using the smooth particle mesh Ewald method.107 Before each MD simulation, a default Desmond membrane protein relaxation protocol was applied.108 To monitor protein stability and conformational fluctuations throughout the simulations, we computed backbone RMSDs after least-squares fitting to the starting structure. This was done for the entire protein and transmembrane α-helices. The trajectory was selected when the system is fully equilibrated and the RMSD reached its plateau. Statistical analyses are based on the structural ensemble from each cluster. Clustering of trajectory was performed using RMSD clustering tool of Schrodinger software package. Five clusters were generated and average structures were generated from each cluster. Additional structural analyses were further performed using these average structures. All the figures were generated using PyMol (The PyMOL Molecular Graphics System, Version 2.0 Schrödinger, LLC). The volume of the binding pocket was calculated using SiteMap, Schrödinger Release 2018–2: SiteMap, Schrödinger, LLC, New York, NY, 2018. Five representative structure of AT1R-AngII were generated from the MDS trajectory and average value was reported.
125I-AngII Competition Binding Analysis.
The detailed procedure is described in previous reports from our laboratory.12,55 Ligand binding was analyzed using membranes prepared from HEK293T cells expressing wild-type HA-AT1R as described earlier and suspended in membrane binding buffer (140 mMNaCl, 5.4 mMKCl, 1 mM EDTA, 0.006% bovine serum albumin, 25 mMHEPES, pH 7.4). A 10 μg of homogeneous cell membrane was used per well. Competition binding assays were performed under equilibrium conditions, with 2 nM radioligand (125I-AngII purchased from Dr. Robert Speth, University of Mississippi) and concentrations of the competing ligand ranging between 0.04 and 1000 nM. Binding reaction was terminated by filtering the binding mixture through Whatman GF/C glass fiber filters, washed with buffer (20 mMsodium phosphate, 100 mMNaCl, 10 mM MgCl2, 1 mMEGTA, pH 7.2). The bound ligand concentration was determined as the counts/min (MicroBeta2 Plate Counter, PerkinElmer Life Sciences). Nonspecific binding was measured in the presence of 105 M 127I-AngII (Bachem). The binding kinetics were analyzed by the nonlinear curve-fitting program GraphPad Prism 5. The means ± SE for the IC50 values were calculated.
Comparative Agonism by Intracellular Calcium Levels.
Calcium levels were measured using FLIPR_ calcium 5 assay kit (Molecular Devices) as described previously.12,13,55 The HA-AT1R expressing cells were seeded at a density of 100 000 cells/well in a 96-well clear bottom black plate. Following serum starvation, calcium-sensitive dye was added to the cells. The Flipper instrument was programmed in FLEX mode to add ligands (0.04 and 1000 nM concentration) to the cells and to monitor the fluorescence before and after adding the ligands. The dose−response curves were plotted and percent agonism was calculated assuming 100% stimulation by AngII.
Supplementary Material
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jcim.8b00583.
Tables showing the effect of amino acid mutation on AngII binding, structure of AngII, snake plot of AT1R showing important residues for AngII, RMSD of TM helices throughout MDS, superimposition of TM helices with active and inactive structures, superimposition of AngII and olmesartan in the orthosteric site of AT1R, time scale of breakage of V1083.32-Y2927.43 H-bonding, time scale of breakage of N1113.35-N2957.46 H-bonds, experimental binding affinity of Ile8-AngII, Ala8-AngII, Gly8-AngII and its binding pose, comparison of W2536.48 torsion angle of AT1R-AngII and AT1R-olmesartan, movement of Tyr2155.58 in the AT1R activation and movement of Arg1263.50 in the AT1R activation (PDF)
2D display of interactions of AngII with AT1R(PDF) MDS trajectory videos 1, 2, 3, and 4 (MP4) PDB file of AT1R bound with AngII (PDB)
Acknowledgments
Funding
This work was supported by National Institutes of Health Grants HL132351 and R01HL142091 and LRI Chair’s Innovative Research Award to S.S.K.
Footnotes
Notes
The authors declare no competing financial interest.
REFERENCES
- (1).Hauser AS; Chavali S; Masuho I; Jahn LJ; Martemyanov KA; Gloriam DE; Babu MM Pharmacogenomics of GPCR Drug Targets. Cell 2018, 172, 41–54.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Takeda S; Kadowaki S; Haga T; Takaesu H; Mitaku S Identification of G Protein-Coupled Receptor Genes From The Human Genome Sequence. FEBS Lett 2002, 520, 97–101. [DOI] [PubMed] [Google Scholar]
- (3).Fredriksson R; Lagerstrom MC; Lundin LG; Schioth HB The G-protein-Coupled Receptors in the Human Genome form Five Main Families. Phylogenetic analysis, paralogon groups, and finger prints. Mol. Pharmacol 2003, 63, 1256–1272. [DOI] [PubMed] [Google Scholar]
- (4).Salon JA; Lodowski DT; Palczewski K The Significance of G Protein-Coupled Receptor Crystallography for Drug Discovery. Pharmacol Rev 2011, 63, 901–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Hauser AS; Attwood MM; Rask-Andersen M; Schioth HB; Gloriam DE Trends In GPCR Drug Discovery: New Agents, Targets and Indications. Nat. Rev. Drug Discovery 2017, 16, 829–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Wang W; Qiao Y; Li Z New Insights into Modes of GPCR Activation. Trends Pharmacol. Sci 2018, 39, 367–386. [DOI] [PubMed] [Google Scholar]
- (7).Kobilka BK Gprotein Coupled Receptor Structure and Activation. Biochim. Biophys. Acta, Biomembr 2007, 1768, 794–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Rosenbaum DM; Rasmussen SG; Kobilka BK The Structure and Function of G-Protein-Coupled Receptors. Nature 2009, 459, 356–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Park PS; Lodowski DT; Palczewski K Activation of G Protein-Coupled Receptors: Beyond Two-State Models and Tertiary Conformational Changes. Annu. Rev. Pharmacol. Toxicol 2008, 48, 107–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Cherezov V; Rosenbaum DM; Hanson MA; Rasmussen SG; Thian FS; Kobilka TS; Choi HJ; Kuhn P; Weis WI; Kobilka BK; Stevens RC High-Resolution Crystal Structure of An Engineered Human Beta2-Adrenergic G Protein-Coupled Receptor. Science 2007, 318, 1258–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Ring AM; Manglik A; Kruse AC; Enos MD; Weis WI; Garcia KC; Kobilka BK Adrenaline-Activated Structure of Beta2-Adrenoceptor Stabilized By An Engineered Nanobody. Nature 2013, 502, 575–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Lin SW; Han M; Sakmar TP Analysis of Functional Micro Domains of Rhodopsin. Methods Enzymol 2000, 315, 116–130. [DOI] [PubMed] [Google Scholar]
- (13).Zhang H; Unal H; Desnoyer R; Han GW; Patel N; Katritch V; Karnik SS; Cherezov V; Stevens RC Structural Basis for Ligand Recognition and Functional Selectivity at Angiotensin Receptor. J. Biol. Chem 2015, 290, 29127–29139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Zhang H; Unal H; Gati C; Han GW; Liu W; Zatsepin NA; James D; Wang D; Nelson G; Weierstall U; Sawaya MR; Xu Q; Messerschmidt M; Williams GJ; Boutet S; Yefanov OM; White TA; Wang C; Ishchenko A; Tirupula KC; Desnoyer R; Coe J; Conrad CE; Fromme P; Stevens RC; Katritch V; Karnik SS; Cherezov V Structure of The Angiotensin Receptor Revealed By Serial Femtosecond Crystallography. Cell 2015, 161, 833–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Zhang X; Perez-Sanchez H; Lightstone FC A Comprehensive Docking and MM/GBSA Rescoring Study of Ligand Recognition upon Binding Antithrombin. Curr. Top. Med. Chem 2017, 17, 1631–1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Shihoya W; Nishizawa T; Okuta A; Tani K; Dohmae N; Fujiyoshi Y; Nureki O; Doi T Activation Mechanism of Endothelin ETB Receptor by Endothelin-1. Nature 2016, 537, 363–368. [DOI] [PubMed] [Google Scholar]
- (17).Unger T The Role Of The Renin-Angiotensin System in the Development of Cardiovascular Disease. Am. J. Cardiol 2002, 89, 3A–9A discussion 10A. [DOI] [PubMed] [Google Scholar]
- (18).Duprez DA Role of the Renin-Angiotensin-Aldosterone System in Vascular Remodeling And Inflammation: A Clinical Review. J. Hypertens 2006, 24, 983–991. [DOI] [PubMed] [Google Scholar]
- (19).Pacurari M; Kafoury R; Tchounwou PB; Ndebele K The Renin-Angiotensin-Aldosterone System in Vascular Inflammation and Remodeling. Int. J. Inflammation 2014, 2014, 689360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Karnik SS; Unal H; Kemp JR; Tirupula KC; Eguchi S; Vanderheyden PM; Thomas WG International Union of Basic and Clinical Pharmacology. XCIX. Angiotensin Receptors: Interpreters of Pathophysiological Angiotensinergic Stimuli [corrected]. Pharmacol. Rev 2015, 67, 754–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Shenoy SK; Lefkowitz RJ Angiotensin II-Stimulated Signaling Through G Proteins and Beta-Arrestin. Sci. Signaling 2005, 2005, cm14. [DOI] [PubMed]
- (22).Balla T; Varnai P; Tian Y; Smith RD Signaling Events Activated by Angiotensin II Receptors: What Goes Before and After the Calcium Signals. Endocr. Res 1998, 24, 335–344. [DOI] [PubMed] [Google Scholar]
- (23).Rodrigues-Ferreira S; Nahmias C G-Protein Coupled Receptors of the Renin-Angiotensin System: New Targets Against Breast Cancer? Front. Pharmacol 2015, 6, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Yee DK; Suzuki A; Luo L; Fluharty SJ Identification of Structural Determinants for G Protein-Independent Activation of Mitogen-Activated Protein Kinases in the Seventh Transmembrane Domain of the Angiotensin II Type 1 Receptor. Mol. Endocrinol 2006, 20, 1924–1934. [DOI] [PubMed] [Google Scholar]
- (25).Doan TN; Ali MS; Bernstein KE Tyrosine Kinase Activation by the Angiotensin II Receptor in the Absence of Calcium Signaling. J. Biol. Chem 2001, 276, 20954–20958. [DOI] [PubMed] [Google Scholar]
- (26).Mehta PK; Griendling KK Angiotensin II Cell Signaling: Physiological and Pathological Effects in the Cardiovascular System. Am. J. Physiol Cell Physiol 2007, 292, C82–97. [DOI] [PubMed] [Google Scholar]
- (27).Higuchi S; Ohtsu H; Suzuki H; Shirai H; Frank GD; Eguchi S Angiotensin Ii Signal Transduction Through The AT1 Receptor: Novel Insights into Mechanisms and Pathophysiology. Clin. Sci 2007, 112, 417–428. [DOI] [PubMed] [Google Scholar]
- (28).Unal H; Karnik SS Domain Coupling in GPCRS: The Engine for Induced Conformational Changes. Trends Pharmacol. Sci 2012, 33, 79–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Singh KD; Unal H; Desnoyer R; Karnik SS Divergent Spatiotemporal Interaction of Angiotensin Receptor Blocking Drugs with Angiotensin Type 1 Receptor. J. Chem. Inf. Model 2018, 58, 182–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Lu M; Wu B Structural Studies of G Protein-Coupled Receptors. IUBMB Life 2016, 68, 894–903. [DOI] [PubMed] [Google Scholar]
- (31).Sanchez-Reyes OB; Cooke ALG; Tranter DB; Rashid D; Eilers M; Reeves PJ; Smith SO G Protein-Coupled Receptors Contain Two Conserved Packing Clusters. Biophys. J 2017, 112, 2315–2326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Fredriksson R; Hoglund PJ; Gloriam DE; Lagerstrom MC; Schioth HB Seven Evolutionarily Conserved Human Rhodopsin G Protein-Coupled Receptors Lacking Close Relatives. FEBS Lett 2003, 554, 381–388. [DOI] [PubMed] [Google Scholar]
- (33).Venkatakrishnan AJ; Deupi X; Lebon G; Heydenreich FM; Flock T; Miljus T; Balaji S; Bouvier M; Veprintsev DB; Tate CG; Schertler GF; Babu MM Diverse Activation Pathways in Class A GPCRS Converge Near the G-Protein-Coupling Region. Nature 2016, 536, 484–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Venkatakrishnan AJ; Deupi X; Lebon G; Tate CG; Schertler GF; Babu MM Molecular Signatures of G-Protein-Coupled Receptors. Nature 2013, 494, 185–194. [DOI] [PubMed] [Google Scholar]
- (35).Fillion D; Cabana J; Guillemette G; Leduc R; Lavigne P; Escher E Structure of The Human Angiotensin II Type 1 (AT1) Receptor Bound to Angiotensin II From Multiple Chemo-selective photo probe Contacts Reveals A Unique Peptide Binding Mode. J. Biol. Chem 2013, 288, 8187–8197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Clement M; Martin SS; Beaulieu ME; Chamberland C; Lavigne P; Leduc R; Guillemette G; Escher E Determining the Environment of The Ligand Binding Pocket of the Human Angiotensin II Type I (hAT1) Receptor Using the Methionine Proximity Assay. J. Biol. Chem 2005, 280, 27121–27129. [DOI] [PubMed] [Google Scholar]
- (37).Cabana J; Holleran B; Leduc R; Escher E; Guillemette G; Lavigne P Identification of Distinct Conformations of the Angiotensin-II Type 1 Receptor Associated with the Gq/11 Protein Pathway and the beta-Arrestin Pathway Using Molecular Dynamics Simulations. J. Biol. Chem 2015, 290, 15835–15854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Matsoukas MT; Potamitis C; Plotas P; Androutsou ME; Agelis G; Matsoukas J; Zoumpoulakis P Insights into AT1 Receptor Activation Through AngII Binding Studies. J. Chem. Inf. Model 2013, 53, 2798–2811. [DOI] [PubMed] [Google Scholar]
- (39).Lebon G; Warne T; Edwards PC; Bennett K; Langmead CJ; Leslie AG; Tate CG Agonist-Bound Adenosine A2a Receptor Structures Reveal Common Features of GPCR Activation. Nature 2011, 474, 521–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Rasmussen SG; DeVree BT; Zou Y; Kruse AC; Chung KY; Kobilka TS; Thian FS; Chae PS; Pardon E; Calinski D; Mathiesen JM; Shah ST; Lyons JA; Caffrey M; Gellman SH; Steyaert J; Skiniotis G; Weis WI; Sunahara RK; Kobilka BK Crystal Structure of the Beta2 Adrenergic Receptor-Gs Protein Complex. Nature 2011, 477, 549–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Kruse AC; Ring AM; Manglik A; Hu J; Hu K; Eitel K; Hubner H; Pardon E; Valant C; Sexton PM; Christopoulos A; Felder CC; Gmeiner P; Steyaert J; Weis WI; Garcia KC; Wess J; Kobilka BK Activation and Allosteric Modulation of a Muscarinic Acetylcholine Receptor. Nature 2013, 504, 101–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Huang W; Manglik A; Venkatakrishnan AJ; Laeremans T; Feinberg EN; Sanborn AL; Kato HE; Livingston KE; Thorsen TS; Kling RC; Granier S; Gmeiner P; Husbands SM; Traynor JR; Weis WI; Steyaert J; Dror RO; Kobilka BK Structural Insights into Micro-Opioid Receptor Activation. Nature 2015, 524, 315–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Choe HW; Kim YJ; Park JH; Morizumi T; Pai EF; Krauss N; Hofmann KP; Scheerer P; Ernst OP Crystal Structure of Metarhodopsin II. Nature 2011, 471, 651–655. [DOI] [PubMed] [Google Scholar]
- (44).Miura S; Feng YH; Husain A; Karnik SS Role of Aromaticity of Agonist Switches of Angiotensin II in the Activation of the AT1 Receptor. J. Biol. Chem 1999, 274, 7103–7110. [DOI] [PubMed] [Google Scholar]
- (45).Noda K; Feng YH; Liu XP; Saad Y; Husain A; Karnik SS The Active State of the At1 Angiotensin Receptor is Generated by Angiotensin II Induction. Biochemistry 1996, 35, 16435–16442. [DOI] [PubMed] [Google Scholar]
- (46).Nikiforovich GV; Zhang M; Yang Q; Jagadeesh G; Chen HC; Hunyady L; Marshall GR; Catt KJ Interactions Between Conserved Residues in Transmembrane Helices 2 and 7 During Angiotensin AT1 Receptor Activation. Chem. Biol. Drug Des 2006, 68, 239–249. [DOI] [PubMed] [Google Scholar]
- (47).Auger-Messier M; Turgeon ES; Leduc R; Escher E; Guillemette G The Constitutively Active N111G-AT1 Receptor for Angiotensin II Modifies the Morphology and Cytoskeletal Organization of Hek-293 Cells. Exp. Cell Res 2005, 308, 188–195. [DOI] [PubMed] [Google Scholar]
- (48).Bhuiyan MA; Hossain M; Miura S; Nakamura T; Ozaki M; Nagatomo T Constitutively Active Mutant N111G of Angiotensin II Type 1 (AT(1)) Receptor Induces Homologous Internalization Through Mediation of AT(1)-receptor Antagonist. J. Pharmacol. Sci 2009, 111, 227–234. [DOI] [PubMed] [Google Scholar]
- (49).Cabana J; Holleran B; Beaulieu ME; Leduc R; Escher E; Guillemette G; Lavigne P Critical Hydrogen Bond Formation for Activation of the Angiotensin II Type 1 Receptor. J. Biol. Chem 2013, 288, 2593–2604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Wilkes BC; Masaro L; Schiller PW; Carpenter KA Angiotensin II vs its Type I Antagonists: Conformational Requirements for Receptor Binding Assessed From NMR Spectroscopic and Receptor Docking Experiments. J. Med. Chem 2002, 45, 4410–4418. [DOI] [PubMed] [Google Scholar]
- (51).Holloway AC; Qian H; Pipolo L; Ziogas J; Miura S; Karnik S; Southwell BR; Lew MJ; Thomas WG Side-Chain Substitutions Within Angiotensin II Reveal Different Requirements for Signaling, Internalization, and Phosphorylation of Type 1A Angiotensin Receptors. Mol. Pharmacol 2002, 61, 768–777. [DOI] [PubMed] [Google Scholar]
- (52).Zimmerman B; Beautrait A; Aguila B; Charles R; Escher E; Claing A; Bouvier M; Laporte SA Differential Beta-Arrestin-Dependent Conformational Signaling and Cellular Responses Revealed by Angiotensin Analogs. Sci. Signaling 2012, 5, ra33. [DOI] [PubMed] [Google Scholar]
- (53).Noda K; Saad Y; Karnik SS Interaction of Phe8 of Angiotensin II with Lys199 and His256 of AT1 Receptor in Agonist Activation. J. Biol. Chem 1995, 270, 28511–28514. [DOI] [PubMed] [Google Scholar]
- (54).Auger-Messier M; Clement M; Lanctot PM; Leclerc PC; Leduc R; Escher E; Guillemette G The Constitutively Active N111G-AT1 Receptor for Angiotensin II Maintains a High Affinity Conformation Despite Being Uncoupled From Its Cognate G Protein Gq/11alpha. Endocrinology 2003, 144, 5277–5284. [DOI] [PubMed] [Google Scholar]
- (55).Schambye HT; Hjorth SA; Bergsma DJ; Sathe G; Schwartz TW Differentiation Between Binding Sites for Angiotensin II and Nonpeptide Antagonists on the Angiotensin II Type 1 Receptors. Proc. Natl. Acad. Sci. U. S. A 1994, 91, 7046–7050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Unal H; Jagannathan R; Bhat MB; Karnik SS Ligand-Specific Conformation of Extracellular Loop-2 in the Angiotensin II Type 1 Receptor. J. Biol. Chem 2010, 285, 16341–16350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Unal H; Jagannathan R; Bhatnagar A; Tirupula K; Desnoyer R; Karnik SS Long Range Effect of Mutations on Specific Conformational Changes in the Extracellular Loop 2 of Angiotensin II Type 1 Receptor. J. Biol. Chem 2013, 288, 540–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Tirupula KC; Ithychanda SS; Mohan ML; Naga Prasad SV; Qin J; Karnik SS G Protein-Coupled Receptors Directly Bind Filamin A With High Affinity and Promote Filamin Phosphorylation. Biochemistry 2015, 54, 6673–6683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Bihoreau C; Monnot C; Davies E; Teutsch B; Bernstein KE; Corvol P; Clauser E Mutation of Asp74 of the Rat Angiotensin II Receptor Confers Changes in Antagonist Affinities and Abolishes G-Protein Coupling. Proc. Natl. Acad. Sci. U. S. A 1993, 90, 5133–5137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Feng YH; Noda K; Saad Y; Liu XP; Husain A; Karnik SS The Docking of Arg2 of Angiotensin II with Asp281 of AT1 Receptor is Essential for Full Agonism. J. Biol. Chem 1995, 270, 12846–12850. [DOI] [PubMed] [Google Scholar]
- (61).Boucard AA; Roy M; Beaulieu ME; Lavigne P; Escher E; Guillemette G; Leduc R Constitutive Activation of the Angiotensin II Type 1 Receptor Alters the Spatial Proximity of Transmembrane 7 to the Ligand-Binding Pocket. J. Biol. Chem 2003, 278, 36628–36636. [DOI] [PubMed] [Google Scholar]
- (62).Kellici TF; Ntountaniotis D; Kritsi E; Zervou M; Zoumpoulakis P; Potamitis C; Durdagi S; Salmas RE; Ergun G; Gokdemir E; Halabalaki M; Gerothanassis IP; Liapakis G; Tzakos A; Mavromoustakos T Leveraging NMR and X-ray Data of the Free Ligands to Build Better Drugs Targeting Angiotensin II Type 1 G-Protein Coupled Receptor. Curr. Med. Chem 2015, 23, 36–59. [DOI] [PubMed] [Google Scholar]
- (63).Zhang H; Han GW; Batyuk A; Ishchenko A; White KL; Patel N; Sadybekov A; Zamlynny B; Rudd MT; Hollenstein K; Tolstikova A; White TA; Hunter MS; Weierstall U; Liu W; Babaoglu K; Moore EL; Katz RD; Shipman JM; Garcia-Calvo M; Sharma S; Sheth P; Soisson SM; Stevens RC; Katritch V; Cherezov V Structural Basis for Selectivity and Diversity in Angiotensin II Receptors. Nature 2017, 544, 327–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Takezako T; Unal H; Karnik SS; Node K Structure-Function Basis of Attenuated Inverse Agonism of Angiotensin II Type 1 Receptor Blockers for Active-State Angiotensin II Type 1 Receptor. Mol. Pharmacol 2015, 88, 488–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Unal H; Karnik SS Constitutive Activity in the Angiotensin II Type 1 Receptor: Discovery and Applications. Adv. Pharmacol 2014, 70, 155–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Noda K; Feng YH; Liu XP; Saad Y; Husain A; Karnik SS The Active State of the AT1 Angiotensin Receptor is Generated by Angiotensin II Induction. Biochemistry 1996, 35, 16435–16442. [DOI] [PubMed] [Google Scholar]
- (67).Groblewski T; Maigret B; Larguier R; Lombard C; Bonnafous JC; Marie J Mutation of Asn111 in the Third Transmembrane Domain of the AT1A Angiotensin II Receptor Induces its Constitutive Activation. J. Biol. Chem 1997, 272, 1822–1826. [DOI] [PubMed] [Google Scholar]
- (68).Miura S; Karnik SS Constitutive Activation of Angiotensin II Type 1 Receptor Alters the Orientation of Transmembrane Helix-2. J. Biol. Chem 2002, 277, 24299–24305. [DOI] [PubMed] [Google Scholar]
- (69).Miura S; Zhang J; Boros J; Karnik SS TM2-TM7 Interaction in Coupling Movement of Transmembrane Helices to Activation of the Angiotensin II Type-1 Receptor. J. Biol. Chem 2003, 278, 3720–3725. [DOI] [PubMed] [Google Scholar]
- (70).Goncalves JA; South K; Ahuja S; Zaitseva E; Opefi CA; Eilers M; Vogel R; Reeves PJ; Smith SO Highly Conserved Tyrosine Stabilizes the Active State of Rhodopsin. Proc. Natl. Acad. Sci. U. S. A 2010, 107, 19861–19866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Elgeti M; Kazmin R; Heck M; Morizumi T; Ritter E; Scheerer P; Ernst OP; Siebert F; Hofmann KP; Bartl FJ Conserved Tyr223(5.58) plays different roles in the activation and G-protein interaction of rhodopsin. J. Am. Chem. Soc 2011, 133, 7159–7165. [DOI] [PubMed] [Google Scholar]
- (72).Laporte SA; Servant G; Richard DE; Escher E; Guillemette G; Leduc R The tyrosine within the NPXnY motif of the human angiotensin II type 1 receptor is involved in mediating signal transduction but is not essential for internalization. Mol. Pharmacol 1996, 49, 89–95. [PubMed] [Google Scholar]
- (73).Sounier R; Mas C; Steyaert J; Laeremans T; Manglik A; Huang W; Kobilka BK; Demene H; Granier S Propagation of Conformational Changes During Mu-Opioid Receptor Activation. Nature 2015, 524, 375–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Kim JM; Altenbach C; Kono M; Oprian DD; Hubbell WL; Khorana HG Structural Origins of Constitutive Activation in Rhodopsin: Role of the K296/E113 Salt Bridge. Proc. Natl. Acad. Sci. U. S. A 2004, 101, 12508–12513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Dror RO; Arlow DH; Maragakis P; Mildorf TJ; Pan AC; Xu H; Borhani DW; Shaw DE Activation Mechanism of the Beta2-Adrenergic Receptor. Proc. Natl. Acad. Sci. U. S. A 2011, 108, 18684–18689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Trzaskowski B; Latek D; Yuan S; Ghoshdastider U; Debinski A; Filipek S Action of Molecular Switches IN GPCRS-Theoretical and Experimental Studies. Curr. Med. Chem 2012, 19, 1090–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Hanson MA; Roth CB; Jo E; Griffith MT; Scott FL; Reinhart G; Desale H; Clemons B; Cahalan SM; Schuerer SC; Sanna MG; Han GW; Kuhn P; Rosen H; Stevens RC Crystal Structure of a Lipid G Protein-Coupled Receptor. Science 2012, 335, 851–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (78).Murphy KP; Xie D; Garcia KC; Amzel LM; Freire E Structural Energetics of Peptide Recognition: Angiotensin II/Antibody Binding. Proteins: Struct., Funct., Genet 1993, 15, 113–20. [DOI] [PubMed] [Google Scholar]
- (79).Nikiforovich GV; Kao JL; Plucinska K; Zhang WJ; Marshall GR Conformational Analysis of Two Cyclic Analogs of Angiotensin: Implications For the Biologically Active Conformation. Biochemistry 1994, 33, 3591–3598. [DOI] [PubMed] [Google Scholar]
- (80).Nikiforovich GV; Marshall GR Three-Dimensional Recognition Requirements for Angiotensin Agonists: A Novel Solution for an Old Problem. Biochem. Biophys. Res. Commun 1993, 195, 222–228. [DOI] [PubMed] [Google Scholar]
- (81).Rosenstrom U; Skold C; Lindeberg G; Botros M; Nyberg F; Karlen A; Hallberg A A Selective AT2 Receptor Ligand with A Gamma-Turn-Like Mimetic Replacing The Amino Acid Residues 4− 5 Of Angiotensin II. J. Med. Chem 2004, 47, 859–870. [DOI] [PubMed] [Google Scholar]
- (82).Potamitis C; Zervou M; Katsiaras V; Zoumpoulakis P; Durdagi S; Papadopoulos MG; Hayes JM; Grdadolnik SG; Kyrikou I; Argyropoulos D; Vatougia G; Mavromoustakos T Antihypertensive Drug Valsartan in Solution and at the AT1 Receptor: Conformational Analysis, Dynamic NMR Spectroscopy, In Silico Docking, and Molecular Dynamics Simulations. J. Chem. Inf. Model 2009, 49, 726–739. [DOI] [PubMed] [Google Scholar]
- (83).Foord SM Receptor Classification: Post Genome. Curr. Opin. Pharmacol 2002, 2, 561–566. [DOI] [PubMed] [Google Scholar]
- (84).Ghosh E; Kumari P; Jaiman D; Shukla AK Methodological Advances: The Unsung Heroes of the GPCR Structural Revolution. Nat. Rev. Mol. Cell Biol 2015, 16, 69–81. [DOI] [PubMed] [Google Scholar]
- (85).Ramanathan A; Savol A; Burger V; Chennubhotla CS; Agarwal PK Protein Conformational Populations and Functionally Relevant Sub States. Acc. Chem. Res 2014, 47, 149–156. [DOI] [PubMed] [Google Scholar]
- (86).Shukla D; Hernandez CX; Weber JK; Pande VS Markov State Models Provide Insights into Dynamic Modulation of Protein Function. Acc. Chem. Res 2015, 48, 414–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (87).Eddy MT; Lee MY; Gao ZG; White KL; Didenko T; Horst R; Audet M; Stanczak P; McClary KM; Han GW; Jacobson KA; Stevens RC; Wuthrich K Allosteric Coupling of Drug Binding and Intracellular Signaling in the A2A Adenosine Receptor. Cell 2018, 172, 68–80.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (88).Lin SW; Sakmar TP Specific Tryptophan UV-Absorbance Changes are Probes of the Transition of Rhodopsin to its Active State. Biochemistry 1996, 35, 11149–11159. [DOI] [PubMed] [Google Scholar]
- (89).Crocker E; Eilers M; Ahuja S; Hornak V; Hirshfeld A; heves M; Smith SO Location of Trp265 in Meta rhodopsin II: Implications for the Activation Mechanism of the Visual Receptor Rhodopsin. J. Mol. Biol 2006, 357, 163–172. [DOI] [PubMed] [Google Scholar]
- (90).Szundi I; Lewis JW; Kliger DS Deriving Reaction Mechanisms from Kinetic Spectroscopy. Application to Late Rhodopsin Intermediates. Biophys. J 1997, 73, 688–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (91).Zaitseva E; Brown MF; Vogel R Sequential Rearrangement of Interhelical Networks Upon Rhodopsin Activation in Membranes: The Meta II(A) Conformational Sub state. J. Am. Chem. Soc 2010, 132, 4815–4821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (92).Altenbach C; Kusnetzow AK; Ernst OP; Hofmann KP; Hubbell WL High-Resolution Distance Mapping in Rhodopsin Reveals the Pattern of Helix Movement Due to Activation. Proc. Natl. Acad. Sci. U. S. A 2008, 105, 7439–7444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (93).Glide; Schrödinger Release 2018–1; Schrödinger, LLC: New York, NY, 2018. [Google Scholar]
- (94).Harder E; Damm W; Maple J; Wu C; Reboul M; Xiang JY; Wang L; Lupyan D; Dahlgren MK; Knight JL; Kaus JW; Cerutti DS; Krilov G; Jorgensen WL; Abel R; Friesner RA OPLS3: A Force Field Providing Broad Coverage of Drug-like Small Molecules and Proteins. J. Chem. Theory Comput 2016, 12, 281–296. [DOI] [PubMed] [Google Scholar]
- (95).Jacobson MP; Pincus DL; Rapp CS; Day TJ; Honig B; Shaw DE; Friesner RA A Hierarchical Approach to All-Atom Protein Loop Prediction. Proteins: Struct., Funct., Genet 2004, 55, 351–367. [DOI] [PubMed] [Google Scholar]
- (96).Schrödinger Release 2018–1: LigPrep; Schrödinger, LLC, New York, NY, 2018. [Google Scholar]
- (97).Schrödinger Release 2018–1: Prime; Schrödinger, LLC, New York, NY, 2018. [Google Scholar]
- (98).Friesner RA; Banks JL; Murphy RB; Halgren TA; Klicic JJ; Mainz DT; Repasky MP; Knoll EH; Shelley M; Perry JK; Shaw DE; Francis P; Shenkin PS Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem 2004, 47, 1739–1749. [DOI] [PubMed] [Google Scholar]
- (99).Dhanachandra Singh K; Karthikeyan M; Kirubakaran P; Nagamani S Pharmacophore Filtering and 3D-QSAR in the Discovery of New JAK2 inhibitors. J. Mol. Graphics Modell 2011, 30, 186–197. [DOI] [PubMed] [Google Scholar]
- (100).Osajima T; Suzuki M; Neya S; Hoshino T Computational and Statistical Study on the Molecular Interaction between Antigen and Antibody. J. Mol. Graphics Modell 2014, 53, 128–139. [DOI] [PubMed] [Google Scholar]
- (101).Sun H; Tian S; Zhou S; Li Y; Li D; Xu L; Shen M; Pan P; Hou T Revealing the Favorable Dissociation Pathway of Type II Kinase Inhibitors via Enhanced Sampling Simulations and Two-End-State Calculations. Sci. Rep 2015, 5, 8457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (102).Zhang X; Perez-Sanchez H; Lightstone FC A Comprehensive Docking and MM/GBSA Rescoring Study of Ligand Recognition upon Binding Antithrombin. Curr. Top. Med. Chem 2017, 17, 1631–1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (103).Braganza LF; Worcester DL Hydrostatic Pressure Induces Hydrocarbon Chain Inter digitation in Single-Component Phospholipid Bilayers. Biochemistry 1986, 25, 2591–2596. [DOI] [PubMed] [Google Scholar]
- (104).Nath Chakraborty SN; Gelb LD A Monte Carlo Simulation Study of Methane Clathrate Hydrates Confined in Slit-Shaped Pores. J. Phys. Chem. B 2012, 116, 2183–2197. [DOI] [PubMed] [Google Scholar]
- (105).Faraldo-Gomez JD; Smith GR; Sansom MS Setting Up and Optimization of Membrane Protein Simulations. Eur. Biophys. J 2002, 31, 217–227. [DOI] [PubMed] [Google Scholar]
- (106).Schlegel B; Sippl W; Holtje HD Molecular Dynamics Simulations of Bovine Rhodopsin: Influence of Protonation States and Different Membrane-Mimicking Environments. J. Mol. Model 2005, 12, 49–64. [DOI] [PubMed] [Google Scholar]
- (107).Darden T; York D; Pedersen L Particle Mesh Ewald: an N log (N) Method for Ewald Sums in Large Systems. J. Chem. Phys 1993, 98, 10089–10092. [Google Scholar]
- (108).Isberg V; Balle T; Sander T; Jorgensen FS; Gloriam DE G Protein- and Agonist-Bound Serotonin 5-HT2A Receptor Model Activated by Steered Molecular Dynamics Simulations. J. Chem. Inf. Model 2011, 51, 315–325. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jcim.8b00583.
Tables showing the effect of amino acid mutation on AngII binding, structure of AngII, snake plot of AT1R showing important residues for AngII, RMSD of TM helices throughout MDS, superimposition of TM helices with active and inactive structures, superimposition of AngII and olmesartan in the orthosteric site of AT1R, time scale of breakage of V1083.32-Y2927.43 H-bonding, time scale of breakage of N1113.35-N2957.46 H-bonds, experimental binding affinity of Ile8-AngII, Ala8-AngII, Gly8-AngII and its binding pose, comparison of W2536.48 torsion angle of AT1R-AngII and AT1R-olmesartan, movement of Tyr2155.58 in the AT1R activation and movement of Arg1263.50 in the AT1R activation (PDF)
2D display of interactions of AngII with AT1R(PDF) MDS trajectory videos 1, 2, 3, and 4 (MP4) PDB file of AT1R bound with AngII (PDB)