

Abstract
The anti-inflammatory, pro-resolving annexin-A1 protein acts as an endogenous brake against exaggerated cardiac necrosis, inflammation, and fibrosis following myocardial infarction (MI) in vivo. Little is known, however, regarding the cardioprotective actions of the N-terminal-derived peptide of annexin A1, Ac2-26, particularly beyond its anti-necrotic actions in the first few hours after an ischemic insult. In this study, we tested the hypothesis that exogenous Ac2-26 limits cardiac injury in vitro and in vivo. Firstly, we demonstrated that Ac2-26 limits cardiomyocyte death both in vitro and in mice subjected to ischemia-reperfusion (I-R) injury in vivo (Ac2-26, 1 mg/kg, i.v. just prior to post-ischemic reperfusion). Further, Ac2-26 (1 mg/kg i.v.) reduced cardiac inflammation (after 48 h reperfusion), as well as both cardiac fibrosis and apoptosis (after 7-days reperfusion). Lastly, we investigated whether Ac2-26 preserved cardiac function after MI. Ac2-26 (1 mg/kg/day s.c., osmotic pump) delayed early cardiac dysfunction 1 week post MI, but elicited no further improvement 4 weeks after MI. Taken together, our data demonstrate the first evidence that Ac2-26 not only preserves cardiomyocyte survival in vitro, but also offers cardioprotection beyond the first few hours after an ischemic insult in vivo. Annexin-A1 mimetics thus represent a potential new therapy to improve cardiac outcomes after MI.
Keywords: myocardial ischemia, inflammation, cardiac remodeling, annexin-A1, formyl peptide receptors
Introduction
Myocardial infarction (MI or ‘heart attack”) and its resultant heart failure (HF) remains a major cause of death and disability, despite current thrombolytic and interventional coronary revascularization procedures (Hausenloy et al., 2013; Heusch, 2013). Development of novel strategies is essential to overcome the limitations of these approaches and the increasing morbidity and mortality due to MI. Given that MI represents a severe inflammatory insult to the myocardium, pro-resolving mediators may represent an exciting therapeutic target in this regard.
The therapeutic potential of the glucocorticoid-regulated anti-inflammatory mediator, annexin-A1 (ANX-A1), has been recognized in a range of systemic inflammatory disorders (Sugimoto et al., 2016; Ansari et al., 2018). ANX-A1 belongs to the annexin superfamily, which has ≥ 13 protein members; these differ most commonly only at their N-terminal tails (Flower and Blackwell, 1979; Gavins et al., 2003a). The N-terminal annexin-A1 peptide Ac2-26 was initially reported to elicit similar biological effects (such as inhibition of neutrophil adhesion and infiltration) as the full ANX-A1 protein, suggesting that at least part of the anti-inflammatory actions of ANX-A1 can result from its N-terminal proteolytic cleavage products (Harris et al., 1995; La et al., 2001a,b). Further, both ANX-A1 and Ac2-26 exhibit pro-resolving properties in several animal models, such as a pleurisy model of acute inflammation (Corminboeuf and Leroy, 2015; Perretti et al., 2015; Lima et al., 2017).
ANX-A1 and its mimetic peptide, Ac2-26 bind to the formyl peptide receptor (FPR) family of 7 transmembrane G-protein coupled receptors (GPCR), to inhibit neutrophil activation, migration and infiltration (Perretti and Flower, 1995; Perretti et al., 1995; Chatterjee et al., 2005; Dalli et al., 2008). There are three FPR subtypes in humans, FPR1, FPR2, and FPR3 (Ye et al., 2009; Qin et al., 2015). Encoded by the FPR1 gene, FPR1 is predominately located on neutrophils, epithelial cells and endothelial cells in multiple organs (Becker et al., 1998). Although it has a similar distribution pattern to FPR1, FPR2 has largely been considered to mediate the majority of the anti-inflammatory effects of ANX-A1 (Gavins et al., 2003b, 2005; Ye et al., 2009; Dufton et al., 2010; Maderna et al., 2010). For example, ANX-A1 fails to inhibit migration of macrophages isolated from FPR2 knockout mice in vitro (Dufton et al., 2010), suggesting that this anti-inflammatory action of ANX-A1 is mediated by FPR2 (Dufton and Perretti, 2010). In contrast to FPR1 and FPR2, FPR3 are highly expressed on dendritic cells and monocytes, with much lower levels detected in heart, and less information regarding the role of FPR3 is presently available (Ye et al., 2009). Interestingly, it has been suggested that Anx-A1 and Ac2-26 could promote an anti-inflammatory and pro-resolving signature, via interleukin (IL)-10 release secondary to induction of FPR1/2 heterodimers (Cooray et al., 2013).
Several lines of evidence indicate that endogenous ANX-A1 protects the myocardium from acute ischemic injury (Ansari et al., 2018). In isolated mouse hearts, deficiency of ANX-A1 further impairs recovery of left ventricular (LV) function, and markedly reduces the activity of the pro-cell survival kinase, Akt, following ischemia-reperfusion (I-R) in vitro (Qin et al., 2013). Excitingly, we have recently revealed the importance of endogenous ANX-A1 in the setting of I-R injury (Qin et al., 2017a). Our evidence suggests ANX-A1 is likely acting as an anti-inflammatory and pro-resolving meditator, dampening both I-R induced inflammation and myocardial damage. Mice deficient in ANX-A1 exhibited increased LV necrosis, inflammation and adverse remodeling compared with wildtype mice, after myocardial ischemic insult in vivo (Qin et al., 2017a). Given that macrophages and neutrophils invade myocardial tissue in this setting and contribute to injury, activation of FPRs may be a novel pharmacological intervention for treating I-R injury.
The protective effects of ANX-A1 in the cardiovascular system are further supported by evidence obtained using exogenous ANX-A1 and Ac2-26 in vitro and in vivo (Qin et al., 2015). ANX-A1 has been shown to attenuate neutrophil recruitment and adhesion to endothelial cells (Dalli et al., 2008). In addition, Ac2-26 has also been demonstrated to elicit direct cardioprotective actions (i.e., in the absence of circulating neutrophils and other pro-inflammatory cells). These include preservation of cardiac contractile function and cardiomyocyte viability, in both isolated cardiomyocytes obtained from the post I-R rat hearts and other settings of myocardial stress in vitro (Ritchie et al., 1999, 2003, 2005; Qin et al., 2013). The anti-inflammatory actions of ANX-A1 and Ac2-26 have also been investigated in short-term (<2h) models of I-R in vivo. The anti-inflammatory actions of ANX-A1 are evidenced by reductions in infarct size and reduced neutrophil infiltration (Gavins et al., 2003a; La et al., 2001a,b). To date, the majority of studies which have examined the cardioprotective roles of exogenous ANX-A1 have, however, utilized relatively short periods of ischemia and reperfusion (<2 h). Thus, elucidating the cardioprotective potential of the ANX-A1 mimetic, Ac2-26, over longer periods of ischemia, and at later timepoints following reperfusion in vivo, is clearly warranted. This would address the translational potential and clinical relevance of targeted ANX-A1 therapy in the management of ischemic heart disease, in both the immediate (24 h) and longer-term.
Materials and Methods
Animals and Materials
All animal research was conducted in accordance with the National Health and Medical Research of Australian Code and Practice and use of animals for scientific purposes, and the study and protocol were reviewed and approved by the Alfred Medical Education Precinct Animal Ethics Committee (E/1154/2011/B), and Directive 2010/63/EU of the European Parliament in the protection of animals used for scientific purpose. Neonatal (1–2 days old) Sprague-Dawley rats (mixed sex) and male C57/BL6 mice (11–14 weeks-of-age) were bred and housed in the Alfred Medical Research and Education Precinct (AMREP) Animal Centre and maintained under a 12 h light/dark cycle. All adult animal experiments were reported in the form of the “Animal Research Reporting of in vivo experiments (ARRIVE) Guidelines” (Drucker, 2016), as outlined in Supplementary Figure S1.
All reagents were purchased from Sigma-Aldrich (St Louis, MO, United States) except where indicated, and were of analytical grade or higher. Dulbecco’s-modified Eagle Medium (DMEM) and fetal bovine serum (FBS) were purchased from Gibco (Gaithersburg, MD, United States) and JRH Biosciences (Lenexa, KS, United States). All materials used for cardiomyocyte isolation were of tissue culture grade. Ac2-26 was synthesized by Chemieliva (Chongqing, China).
Isolation and Culture of Primary Ventricular Cardiomyocytes and Cardiofibroblasts
Cardiomyocyte isolation from neonatal rats was performed by serial enzymatic digestion, as previously described (Irvine et al., 2012, 2013; Qin et al., 2017b). Cardiomyocytes were suspended in sterile DMEM, supplemented with 100 U/mL penicillin, 100 μg/mL streptomycin, and 10% FCS. The myocyte-rich cell suspension was pre-plated twice (45 min at 37°C) to reduce fibroblast contamination. Cardiomyocytes were then plated on 60-mm dishes for gene expression analysis, and on 12-well plates at 1.3 × 105 cells per cm2 to assess cardiomyocyte injury, in the presence of 1% 5-bromo-2′-deoxyuridine (BrdU, to limit proliferation of any remaining fibroblasts) (Qin et al., 2017b). Neonatal mouse cardiac fibroblasts were obtained as a by-product of cardiomyocyte isolation (Irvine et al., 2012). Cardiofibroblasts (passage #3) were seeded on 60-mm tissue culture dishes at 9.5 × 104 cells per cm2 and allowed to grow to 80% confluence before study.
RNA Extraction and Quantitative-Real Time PCR (qPCR)
RNA was extracted from cardiomyocytes, cardiac fibroblasts or LV tissues using TRIzol® (Invitrogen, Life Technologies, Mulgrave, VIC, Australia) as previously described (Huynh et al., 2012). Briefly, tissue were homogenized using sterilized zirconium oxide beads in TRIzol® with a tissue lyser (Tissue Lyser II Qiagen®). Homogenates were then centrifuged and chloroform was added to separate the aqueous and organic layers. RNA in the aqueous layer was precipitated in isopropanol overnight at -20°C. The RNA pellets were washed in ethanol and resuspended in RNAase-free water. RNA quality was assessed by Nanodrop2000 spectrophotometer (Thermo Fisher Scientific; wavelength 260 nm) and treated with DNAse treated using commercial Ambion DNase I Kit (Ambion®; Thermo Fisher Scientific) according to the manufacturer’s instructions. Taqman reverse-transcription reagents (Applied Biosystems, Mulgrave, VIC, Australia) were used to generate approximately 20 ng/μl cDNA from 1 μg of DNase-treated RNA via transcription. Quantitative-real time PCR (qPCR) was conducted using SYBR green chemistry (Life Technologies, Victoria, Australia) to measure the expression of genes of interest using the Applied Biosystems ABI Prism 7700 Sequence Detection System described previously using sequences in Table 1 (Qin et al., 2017a). The comparative 2-ΔΔCt method was used to analyze changes in gene expression as a fold change relative to sham mice, with ribosomal 18S as the housekeeping gene (Qin et al., 2017a). If cycle threshold (Ct) for the gene of interested yield a result of “undetermined.” i.e., Ct > 40, expression was reported as zero (i.e., not detected).
Table 1.
5′-3′ primer sequences used for gene expression by qPCR.
| Forward primer | Reverse primer | |
|---|---|---|
| 18S | TGT TCA CCA TGA GGC TGA GAT C | TGG TTG CCT GGG AAA ATC C |
| rFpr1 | CAC CTC CAC TTT GCC ATT TT | TGC ACA TGA ACC AAC CAA AT |
| rFpr2 | GCT CAG AAC CAC CGC ACT | CAT AAA TCC AGG GCC CAA C |
| rFpf3 | TGT TCA CCA TGA GGC TGA GAT C | TGG TTG CCT GGG AAA ATC |
| mFpr1 | CCT TGG CTT TCT TCA ACA GC | GCC CGT TCT TTA CAT TGC AT |
| mFpr2 | ACA GCA GTT GTG GCT TCCTT | CCT GGC CCA TGA AAA CAT AG |
| mCtgf | TGA CCC CTG CGA CCC ACA | TAC ACC GAC CCA CCG AAG ACA CAG |
| mTgf-β | TGG AGC AAC ATG TGG AAC TC | GTC AGC AGC CGG TTA CCA |
| mSerca-2a | TCT GGA GTT TTC ACG GGA TAG AA | TGT CCG GCT TGG CTT GTT |
| mmyh7 | TCT CCT GCT GTT TCC TTA CTT GCT A | GTA CTC CTC TGC TGA GGC TTC CT |
| mCd68 | CCA ATT CAG GGT GGA AGA AA | CTC GGG CTC TGA TGT AGG TC |
| mS100a8 | CCA TGC CCT CTA CAA GAA TGA | TAT CAC CAT CGC AAG GAA CTC |
| mS100a9 | TCA TCG ACA CCT TCC ATC AA | GTC CAG GTC CTC CAT GAT GT |
| mArg-1 | TCA GAA GCT GTT CTT GGT CTG AAC | GTT CAT GGG GAT CCC AGT GA |
| mCd206 | GGC TAC CAG GAA GTC CAT CA | TGT AGC AGT GGC CTG CAT AG |
FPR Expression in Cardiac Cells in vitro
The optimal concentrations of Ac2-26 (over 0.3–3 μM) in hypoxic cardiomyocytes was determined initially in pilot studies (see Supplementary Table S1); 1 μM was demonstrated as the optimum concentration and was used in all subsequent cardiomyocyte studies. Cardiomyocytes plated on 60-mm dishes were incubated for 48 h at 37°C in serum-free DMEM, in the presence or absence of Ac2-26 (1 μM) or vehicle (0.1% DMSO). At the end of 48 h, RNA was extracted and qPCR performed) to determine the relative gene expression of rat Fpr subtypes (rFpr1, rFpr2, and rFpr3, primer sequences detailed in Table 1) relative to the housekeeping gene 18S, as described previously (Qin et al., 2017b). Cardiac fibroblasts were starved overnight in serum-free DMEM, before incubation for 24 h at 37°C with Ac2-26 (10 μM) or vehicle, prior to RNA extraction for qPCR determination of the relative gene expression of mFpr1 and mFpr2 (refer to primer sequences in Table 1), again relative to the housekeeping gene 18S, as described previously (Qin et al., 2017b).
Cardiomyocyte Injury Responses in vitro
Following 48 h in serum-free DMEM, cardiomyocytes plated on 12-well tissue plates were subjected to simulated ischemia, induced by replacement of culture media with sterile-filtered Krebs buffer (118 mM NaCl, 4.8 mM KCl, 1.2 mM KH2PO4, 1.2 mM MgSO4, 25 mM NaHCO3, 50 mM EDTA. 11.0 mM glucose, 1.75 mM CaCl2), before incubation for 6 h at 37°C under hypoxia (95% N2-5% CO2, using a hypoxia chamber, QNA International, Melbourne, VIC, Australia), with subsequent 48 h reoxygenation as described previously (Qin et al., 2017b). Agonists were dissolved in DMEM or Krebs buffer, and were present for the full duration of hypoxia (H) and reoxygenation (R). At the end of 48 h reoxygenation, culture supernatant aliquots were collected on ice and stored at -80°C for assessment of cardiomyocyte injury via levels of lactate dehydrogenase (LDH) released (Cyto-Tox-OneTM Homogeneous Membrane Integrity Assay, Promega Inc, Dane County, WI, United States). Levels of cardiac troponin (cTnI) released from cardiomyocytes were also determined using a high-sensitivity rat cTnI ELISA kit (Life Diagnostics Inc, West Chester, PA, United States) as per manufacturer’s instructions.
Cardiac Injury Responses in vivo
Adult male C57BL/6 mice (22–30 g) were randomly assigned to either myocardial I-R injury (cohort 1–3), permanent coronary artery ligation (cohort 4) or sham in vivo (Supplementary Figure S1). Briefly, mice were anesthetized, ketamine/xylazine/atropine (KXA, 100/20/1.2 mg/kg, i.p.), mechanically ventilated and a left thoracotomy performed to expose the left anterior descending (LAD) coronary artery, as previously described (Gao et al., 2005, 2010, 2011). For Cohorts 1, 2, and 3, the LAD was ligated using 7.0 silk suture with a slip-knot enclosing two releasing rings. Regional ischemia was induced for 40–60 min, then blood flow through the LAD was re-established by releasing the slip-knot, as we have described previously (Gao et al., 2005, 2010, 2011). Mice in cohorts 1–3 were then subjected to reperfusion for 24, 48 h, and 7-days respectively, as these are considered optimal timepoints for the assessment of cardiac necrosis, inflammation and early remodeling in vivo respectively, as reported previously (Qin et al., 2017b). Sham animals underwent identical surgical procedures, but without ligation. Mice were randomly assigned to receive either vehicle (10% dimethyl sulfoxide, DMSO) in saline, i.v.) or Ac2-26 peptide (1 mg/kg, i.v.) every 24 h, for cohorts 1–3, with the first dose administered immediately before reperfusion (until day 7).
An additional cohort of mice, cohort 4, was subjected to a more severe ischemic insult, permanent LAD ligation; late cardiac remodeling and cardiac dysfunction were assessed in these mice after 4 weeks in sham and MI mice ± vehicle (10% DMSO in saline, s.c., osmotic pump) or Ac2-26 (1 mg/kg/day, s.c., osmotic pump) from the time of cardiac surgery until experimental endpoint. The initial osmotic pumps (Alzet, Model 1002, Cupertino, CA, United States) were implanted under anesthesia at the time of LAD occlusion surgery. Two weeks later, the first pumps were removed and a second pump placement was performed under isoflurane anesthesia (induction at 3–4%, followed by 1–2% isoflurane, to maintain anesthesia during surgery). For all cohorts, infarcted or sham-operated mice were killed under KXA anesthesia at the end of study, and heparinised blood collected by cardiac puncture for later analysis as indicated.
Assessment of Cardiac Necrosis in vivo
Plasma levels of cTnI were determined in cohort 1 (following 40 min ischemia and 24 h reperfusion) using ELISA as for cardiac myocytes. To further evaluate the extent of cardiac necrosis, infarct size (IS) in relation to the area-at-risk (AAR) was also determined, using 2,3,5-triphenyltetrazolium chloride (TTC) staining of cardiac sections, as described previously (Qin et al., 2017a,b). The images were analyzed in a blinded fashion using Image J (Version 1.45S, National Institute of Health, United States). The non-ischemic zone (blue area), area-at-risk zone (AAR, red and white-yellow areas), infarct zone (white-yellow areas) and total LV were outlined, and infarct size was calculated as the percentage of the infarct zone/AAR zone (Qin et al., 2017a,b).
Assessment of Cardiac Inflammation in vivo
Lung, atria, left and right ventricles from mice in cohort 2 (following 60 min ischemia and 48 h reperfusion) were dissected, blotted dry and weighed. LVs were cut in two at the occlusion site, and the apical half placed in Tissue-Tek optimal cutting temperature compound (OTC, Tissue-Tek, Torrance, CA, United States) for storage at -80°C. The apical half of LVs were sectioned at 6 μm for immunofluorescent analysis. Sections were pre-incubated with 4% paraformaldehyde for 20 min, and 10% normal goat serum (NGS) for a further 30 min. Sections were then incubated at room temperature with either CD68+ or Ly-6B.2+ primary antibody (1:200, ABD Serotec, Raleigh, NC, United States) for 1 h, followed by 30 min incubation with the Alexa Fluor 546 secondary antibody (1:200, Invitrogen, Carlsbad, CA, United States), to detect cardiac inflammatory cells infiltration, respectively. Finally, sections were incubated with 0.001% Hoechst 33342 (Invitrogen, Melbourne, VIC, Australia) for 30 min, to elicit nuclear staining. Images were photographed and analyzed as described previously (Qin et al., 2017b). For detection of neutrophil infiltration into the heart, sections were incubated overnight at 4°C with Ly-6G+ primary antibody (1:400 in 5% NGS and 0.01% PBS-T, BD Pharm, Scoresby, Australia), followed by incubation at room temperature for 2 h with Alexa Fluor 546 secondary antibody (1:1000 in 5% NGS and 0.01% PBS-T, Invitrogen, Carlsbad, CA, United States). Finally, sections were incubated with 1 mg/ml 4′,6-diamidino-2-phenyldole (DAPI, 1:1000 in PBS) for 10 min and quenched with Sudan Black (0.1% in 70% ethanol), to elicit nuclear staining and reduce auto-fluorescence, respectively. Images were photographed and analyzed as described previously (Qin et al., 2017b).
Left ventricular tissues collected from mice in cohort 3 (following 40 min ischemia and 7-days reperfusion) were fixed in neutral-buffered formalin, embedded in paraffin (Alfred Pathology Service, Melbourne, VIC, Australia) and then sectioned at 4 μm. Sections were deparaffinised, rinsed and antigen retrieval was performed by incubating the slides for 20 min at 95°C in 0.01% citrate buffer. Sections were blocked in 5% NGS in 0.01% PBS/Tween 20 for 1 h. For detection of neutrophil and inflammatory monocyte infiltration into the heart, sections were then incubated overnight at 4°C with either Ly-6G+ (1:400 in 5% NGS and 0.01% PBS-T, BD Pharm, Scoresby, VIC, Australia) or Ly-6C+ primary antibody (1:400 in 5% NGS and 0.01% PBS-T, Bio-Rad, Hercules, CA, United States). Images were photographed and analyzed as described previously (Qin et al., 2017b).
Whole blood and plasma were collected from mice in cohort 2 to assess total and differential circulating white blood cells (WBC) numbers, as an assessment of systemic inflammation, as described previously (Qin et al., 2017a,b).
Assessment of Cardiac Fibrosis and Apoptosis in vivo
Left ventricular tissues collected from mice in cohort 3 (following 40 min ischemia and 7-days reperfusion) were fixed in neutral-buffered formalin, embedded in paraffin (Alfred Pathology Service, Melbourne, VIC, Australia), sectioned at 4 μm and stained with picrosirius red (0.1%, Fluka, Bucks, Switzerland; pH2) for cardiac collagen deposition (Qin et al., 2017a,b). In addition, the level of apoptosis in LV was examined using CardiacTAC In Situ Apoptosis Detection kit (Trevigen, Gaithersburg, MD, United States). Images were photographed and analyzed as described previously (Qin et al., 2017a,b).
Assessment of Cardiac Function by Echocardiography
M-mode echocardiography was performed in anesthetized mice (KXA: 80/8/0.96 mg/kg, i.p.) allocated to cohort 4, to obtain measures of LV function at 1 and 4 weeks post permanent ligation, utilizing a Philips iE33 ultrasound machine (North Ryde, NSW, Australia) with a 15 MHz linear transducer. Heart rate, left ventricular end-systolic dimension (LVESD), LV end-diastolic dimension (LVEDD), LV posterior wall thickness (LVPW) and fractional shortening (FS, %) were assessed from M-mode echocardiography as per the published guidelines for echocardiography in mice (Donner et al., 2018).
Left ventricular tissues were also collected from anesthetized mice in cohort 4 at study end point (4 weeks post MI). LV RNA was extracted from both the infarct and the non-infarcted AAR, and gene expression determined via qPCR as described previously (Qin et al., 2017a,b).
Data Analysis
GraphPad Prism software (Version 7.00, La Jolla, CA, United States) was used to perform statistical analyses. Data are presented as expressed as mean ± SEM, unless otherwise stated. Statistical analyses utilized Student’s t-test or one-way ANOVA followed by Dunnett’s or Tukey’s post hoc test, as appropriate. Values of P < 0.05 were considered statistically significant. The Kaplan–Meier survival curve was analyzed by the log-rank (Mantel-Cox) test. Values of P < 0.05 were considered statistically significant for all analyses.
Results
Impact of Ac2-26 on Primary Cardiomyocytes and Cardiofibroblasts in vitro
After 48 h incubation of cardiomyocytes with Ac2-26, the expression of rFpr1, rFpr2, and rFPR3 were assessed. Low levels of constitutive expression of both rFpr1 (Ct∼33) and rFpr2 (Ct∼34) were detected in untreated cardiomyocytes. Ac2-26 tended to increase rFpr1 (Figure 1A, p = 0.05, overall on one-way ANOVA with Dunnett’s post hoc test) and significantly increased rFpr2 expression (Figure 1B, p = 0.04, one-way ANOVA with Dunnett’s post hoc test) compared to vehicle-treated cardiomyocytes. Expression of rFpr3 was only detectable in 2 out of 5 cardiomyocytes preparations, the average of rFpr3 expression was 0.3- and 3.0-fold untreated cardiomycytes after vehicle or Ac2-26 in these two preparations.
Figure 1.
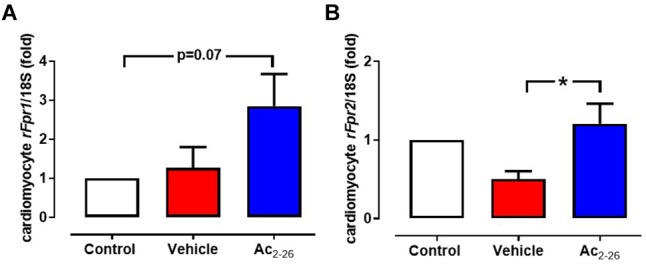
Ac2-26 increases cardiomyocyte expression of rFprs. Ac2-26 tends to increase expression of (A) rFPR1 and (B) significantly increases rFPR2 expression. ∗P < 0.05 versus paired vehicle-treated mice, one-way repeated measures ANOVA with Dunnett’s post hoc test. n (number of different cardiomyocyte preparations) = 5. Data were presented as mean ± SEM.
The direct effects of Ac2-26 in primary cardiomyocytes and cardiofibroblasts were determined in vitro. Hypoxia-reoxygenation (H-R) significantly increased LDH release (Figure 2A, p = 0.01 overall on one-way ANOVA) from cardiomyocytes by approximately 70%, while cTnI (a more sensitive measure of cardiomyocyte death) (Figure 2B, p = 0.01 overall on one-way ANOVA, and on Dunnett’s post hoc test) was almost tripled, compared to control cardiomyocytes. Ac2-26 (1 μM) prevented cardiomyocyte LDH and cTnI release whether present for the full duration of H-R (Figure 2A,B), or when only added at the start of reoxygenation (Figure 2C, p = 0.005 overall on one-way ANOVA and on Dunnett’s post hoc test; Figure 2D, p = 0.0009 overall on one-way ANOVA). These results confirm that Ac2-26 elicits direct protective actions on cardiomyocytes subjected to H-R, and that these are comparable whether treatment is commenced concomitantly with the insult, or at the onset of post-insult recovery. We also sought the impact of Ac2-26 on cardiac fibroblast FPR gene expression in vitro, and observed that Ac2-26 tends to upregulate both mFpr1 and mFpr2 (Figure 3A,B); mFpr2 was significantly upregulated > 10-fold (Figure 3B, p = 0.04 overall on one-way ANOVA and on Dunnett’s post hoc test), with a moderate but non-significant tendency for increased mFpr1 (Figure 3A, p = 0.09 overall on one-way ANOVA, and on Dunnett’s post hoc test).
Figure 2.

Protective actions of Ac2-26 against cardiomyocyte injury responses in vitro. Ac2-26 (1 μM) prevents neonatal rat cardiomyocyte hypoxia-reoxygenation (H-R) injury in vitro (assessed by measuring LDH and cTnI release), whether present for the full duration of H-R (A,B, n = 7 cardiomyocyte preparation) or only post H-R (C,D, n = 5 cardiomyocyte preparation). #P < 0.05, ##P < 0.01, ###P < 0.001 versus normoxia, ∗P < 0.05 ∗∗P < 0.01 versus vehicle-treated mice from the same cardiomyocyte preparation. One-way ANOVA with Dunnett’s post hoc test. n (number of different cardiomyocyte preparations) = 5. Data were presented as mean ± SEM.
Figure 3.

Effect of Ac2-26 on TGF-β stimulation of cardiofibroblasts in vitro. TGF-β (10 μM) downregulates (A) mFpr1 and (B) mFpf2 expression, which is elevated by Ac2-26 (10 μM) within 48 h. ####P < 0.001 versus control, ∗P < 0.05 versus TGF-β-treated cells from the same cardiofibroblast preparation. One-way ANOVA with Dunnett’s post-hoc test. n (number of different cardiofibroblasts preparations) = 7. Data were presented as mean ± SEM.
Ac2-26 Significantly Reduces Myocardial Necrosis in vivo
To assess the impact of Ac2-26 in an animal model of myocardial I-R in vivo, C57BL/6 mice underwent reversible ligation of the LAD coronary artery followed by reperfusion in the absence or presence of treatment for 24 h (cohort 1), 48 h (cohort 2), or 7-days (cohort 3).
The effect of Ac2-26 (1 mg/kg, i.v.) on cardiac necrosis was assessed in mice subjected to 40 min ischemia-24 h reperfusion (cohort 1). There were no differences between I-R groups in the identified size of the LV AAR (∼60%) after 24 h reperfusion, as measured by Evans blue staining (Figure 4A,B, p < 0.0001 overall on one-way ANOVA). Of the AAR in vehicle-treated I-R mice, 30–40% was infarcted (Figure 4C, p < 0.0001 overall on one-way ANOVA), and plasma levels of cTnI were markedly elevated (Figure 4D, p < 0.0001 overall on one-way ANOVA). Administration of Ac2-26 peptide immediately before LAD reperfusion significantly attenuated infarct size by ∼25%, and tended to lower cTnI levels, relative to vehicle-treated I-R injured mice (p = 0.05, one-way ANOVA with Dunnett’s post hoc test, Figure 4D).
Figure 4.
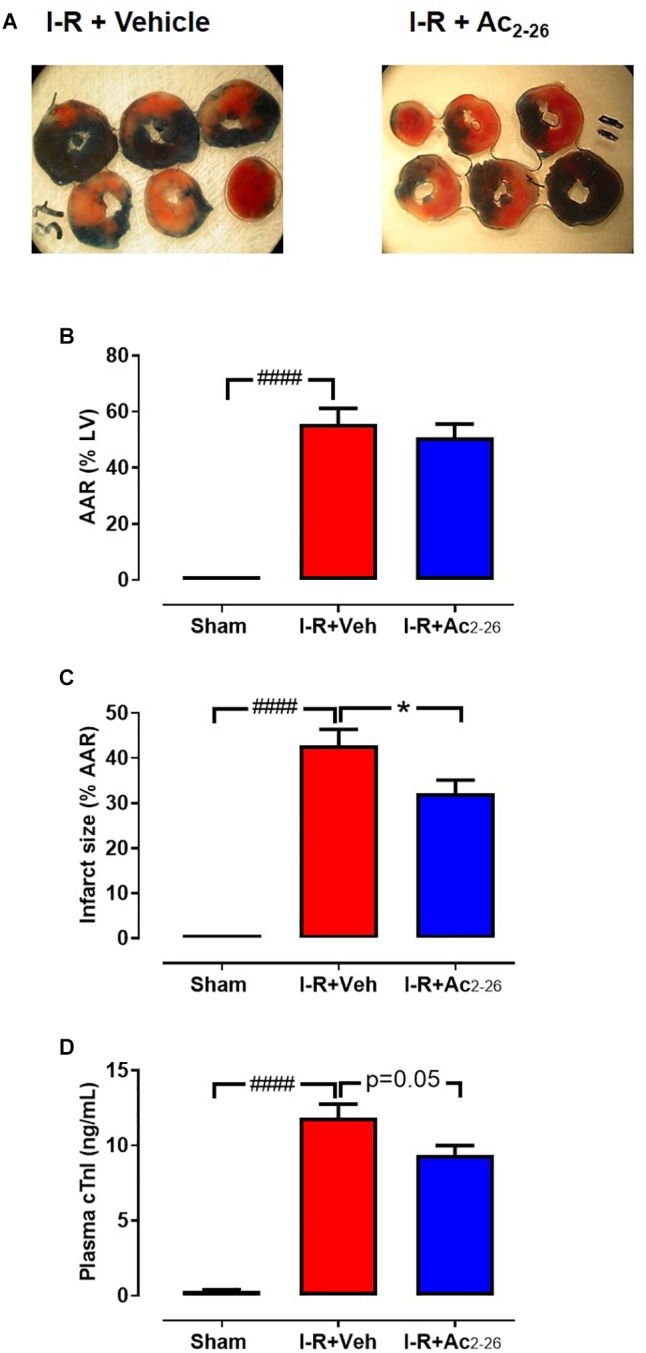
Ac2-26 reduces cardiac necrosis 24 h post I-R injury in vivo. (A) LV transverse slices from representative mice subjected to 40 min ischemia followed by 24 h reperfusion following either vehicle or Ac2-26 (1 mg/kg i.p. administered at time of reperfusion). Three different zones are visible after staining with Evans blue and 2,3,5-triphenyltetrazolium chloride. Areas stained dark blue, white and red represented non-risk, infarcted and ischemic but non-infarcted zones, respectively. The risk zone includes red and white areas. (B) Area-at-risk (AAR) in mice subjected to myocardial I-R, (C) Infarct size, and (D) Plasma levels of cardiac troponin (cTnI). ####P < 0.001 versus sham, ∗P < 0.05 versus vehicle-treated mice. One-way ANOVA with Tukey post hoc test. Data were presented as mean ± SEM; with number of mice per group. n = 3 (sham), n = 5 (I-R + vehicle), and n = 12 (I-R + Ac2-26).
Ac2-26 Attenuates Cardiac and Systemic Inflammation in vivo
The extent of I-R-induced cardiac injury and systemic inflammation was assessed after 48 h and 7 days reperfusion in vivo, as well as the impact of Ac2-26 on these changes (cohort 2 and cohort 3). As shown via immunofluorescent detection of LV inflammatory cells (anti-Ly-6B.2), I-R injury elicited a significant increase in inflammatory cells signals per section, in both vehicle- and Ac2-26-treated I-R mice, compared to sham (Figure 5A,B, p < 0.0001 overall on one-way ANOVA). Administration of Ac2-26 reduced cardiac neutrophil numbers by approximately 35% compared to vehicle-treated I-R (Figure 5A). Macrophage numbers in LV sections, detected using anti-CD68, were also increased in mice subjected to I-R (Figure 5C,D, p < 0.0001 on one-way ANOVA); Ac2-26 tended to blunt the increase in LV macrophage density (p = 0.10, one-way ANOVA with Dunnett’s post hoc test, Figure 5D). Consistent with the Ly6B results, accumulation of Ly6G (indicative of neutrophils) was also significantly increased post ischemic insult (Figure 5E,F). Ac2-26 failed to exert impact on LV neutrophil content at this 48 h timepoint. Interestingly, levels of Ly6G-positive cells appear to have returned to baseline levels by 7 days post I-R (Figure 6A,B). Levels of Ly6C-positive cells (inflammatory monocytes) is significantly elevated 7 days post-ischemic insult (Figure 6C,D). Administration of Ac2-26, however, exerted no impact on the total LV content of Ly6G- and Ly6C-positive cells 7 days post I-R (Figure 6).
Figure 5.

Ac2-26 reduces cardiac inflammation 48 h post I-R injury in vivo. Representative immunofluorescent images of (A) LV inflammatory cell content (determined using anti-Ly-6B.2 antibody, (C) LV macrophage content (determined using anti-CD68 antibody, E) LV neutrophil content (determined using anti-Ly6G+ antibody) from sham, vehicle- and Ac2-26-(1 mg/kg/day, i.v.)-treated mice, 48 h post I-R (X400 magnification, scale-bar in top panels = 100 μm; in bottom panels = 50 μm). Pooled data for (B) LV Ly6B.2 positive, (D) C68+ positive, and (F) Ly6G+ positive immunofluorescence. Higher magnification images reveals overlay of dark blue (DAPI; detecting nuclei) and red (inflammatory cells) indicating positive staining (scale-bar in top panels = 100 μm; in bottom panels = 50 μm). #P < 0.05, ##P < 0.01, ####P < 0.0001 versus sham, ∗P < 0.05 versus vehicle-treated mice. One-way ANOVA with Tukey’s post hoc test. Data were presented as mean ± SEM, with number of mice per group. n = 6 (sham), n = 7 (I-R + vehicle), and n = 8 (I-R + Ac2-26).
Figure 6.

Ac2-26 has no impact on myocardial neutrophil content 7 days post I-R injury in vivo. Representative immunofluorescent images of (A) LV neutrophil content (determined using anti-Ly6G+ antibody, and C) inflammatory monocytes (determined using anti-Ly6C+ antibody). Pooled data for both are shown in B) LV LyG+ and (D) LV Ly6C+ immunofluorescence. Images reveals overlay of dark blue (DAPI; detecting nuclei) and red (inflammatory cells) indicating positive staining (scale-bar = 50 μm). #P < 0.05 versus sham. One-way ANOVA with Tukey’s post hoc test. Data were presented as mean ± SEM, with number of mice per group. n = 7 (sham), n = 4 (I-R + vehicle), and n = 6 (I-R + Ac2-26).
Total and differential WBC quantification were performed, to investigate the effects of Ac2-26 on systemic inflammation (Figure 7). Circulating total WBCs (p = 0.009 overall on one-way ANOVA), lymphocytes (p = 0.04 overall on one-way ANOVA), neutrophils (p = 0.0005 overall on one-way ANOVA) and monocytes (p = 0.03 overall on one-way ANOVA) were all significantly increased in I-R vehicle-treated mice compared with sham (Figure 7). Ac2-26 significantly blunted the 3-fold increase in levels of circulating neutrophils (Figure 7B), without reducing the I-R induced increases in circulating lymphocytes and monocytes (Figure 7C,D).
Figure 7.

Ac2-26 reduces systemic inflammation 48 h post I-R injury in vivo. Circulating levels of (A) total white blood cells (WBC), (B) neutrophils, (C) lymphocytes and (D) monocytes in blood collected from sham, vehicle- and Ac2-26-(1 mg/kg/day, i.v.)-treated mice, 48 h post I-R. #P < 0.05, ##P < 0.01 versus sham, ∗P < 0.05 versus vehicle-treated mice. One-way ANOVA with Dunnett’s post hoc test. Data were presented as mean ± SEM, with number of mice per group. n = 6 (sham), n = 7 (I-R + vehicle), and n = 8 (I-R + Ac2-26).
Ac2-26 Limits Cardiac Remodeling in vivo
Next, the impact of Ac2-26 on I-R-induced cardiac fibrosis (by picrosirius red, Figure 8A–D) and cardiac apoptotic cell death (by CardioTACS, Figure 8E,F) after 40 min ischemia with 7-days reperfusion was determined (cohort 3). Myocardial I-R significantly increased collagen deposition (Figure 8A, p < 0.001 overall on one-way ANOVA). Our data shows that cardiac collagen (% LV area) is significantly increased in the LV of vehicle-treated I-R mice compared to sham; this was attenuated by Ac2-26 treatment (Figure 8B). Quantitative analysis revealed that the interstitial staining (% area) in the remote zone is more than doubled in vehicle-treated I-R mice compared to sham; this increase was completely abolished by Ac2-26 treatment (Figure 8C,D, p = 0.04 overall on one-way ANOVA). Apoptosis was not evident in cardiac sections from sham mice. Myocardial I-R significantly increased LV cardiac cell death, which was significantly attenuated by Ac2-26 treatment (Figure 8C,D, p < 0.0001 overall on one-way ANOVA).
Figure 8.
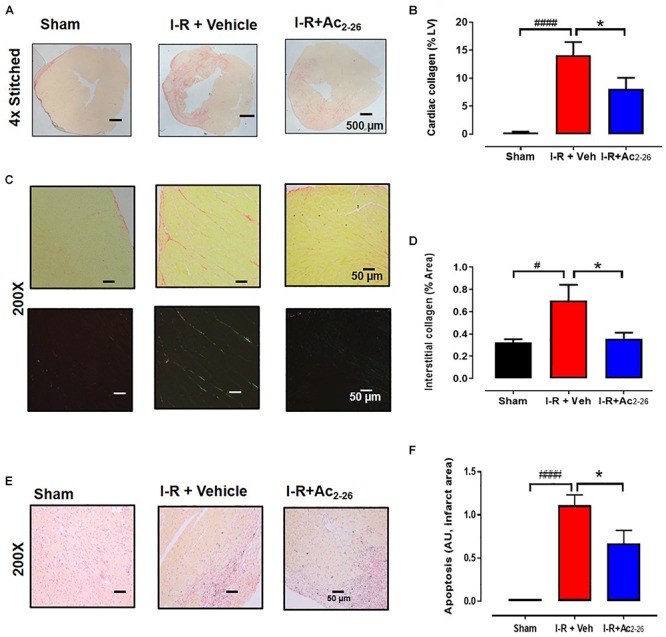
Ac2-26 reduces cardiac fibrosis at 7 days post I-R injury in vivo. (A) Representative picrosirius red-stained LV cross-sections from sham, vehicle- and Ac2-26-(1mg/kg/day, i.v.)-treated mice, 7-days post I-R (collagen appears red); (B) quantification of cardiac fibrosis. (C) Representative picrosirius red-stained remote area (bright field and polarized light) from sham, vehicle- and Ac2-26-(1mg/kg/day, i.v.)-treated mice, 7-days post I-R, (D) quantification of interstitial fibrosis under polarized light, magnification x200. (E) Representative CardioTAC-stained LV cross-sections from sham, vehicle- and Ac2-26-(1 mg/kg/day, i.v.)-treated mice, 7-days post I-R (magnification x200). (F) Quantification of dead:viable cells (expressed as fold change versus sham; magnification x200. #P < 0.05, ####P < 0.0001 versus sham, ∗P < 0.05 versus vehicle-treated I-R mice. One-way ANOVA with Tukey’s post hoc test. Data were presented as mean ± SEM, with number of mice per group. n = 4 (sham), n = 6 (I-R + vehicle), and n = 6 (I-R + Ac2-26).
Impact of Ac2-26 on Organ Weights After I-R Injury in vivo
Body and organ weights measured after 48 h (cohort 2) and 7-day (cohort 3) reperfusion are shown in Table 2. Following 48 h post-ischemic reperfusion, no differences in body weight (BW), atria or lung weights were observed, but heart weight (HW) was significantly increased in both vehicle and Ac2-26-treated hearts compared to sham (cardiac weights were normalized to BW). The increase in LV:BW ratio after 7-day reperfusion with vehicle-treated I-R compared with sham mice was not prevented by Ac2-26 treatment (Table 2).
Table 2.
Impact of I-R injury after either vehicle- or Ac2-26 treatment, on body and organ weights at endpoint, either 48 h or 7-days following I-R.
| Sham | I-R + vehicle | I-R + Ac2-26 | ||
|---|---|---|---|---|
| 48 h myocardial I-R | ||||
| n | 6 | 7 | 8 | |
| Body Weight (BW, g) | 26.9 ± 1.3 | 29.0 ± 0.4 | 28.2 ± 1.0 | |
| Organ weight/BW (mg/g) | Heart | 4.2 ± 0.1 | 4.8 ± 0.2# | 5.0 ± 0.2# |
| Atria | 0.3 ± 0.0 | 0.4 ± 0.0 | 0.3 ± 0.0 | |
| LV | 3.1 ± 0.1 | 3.6 ± 0.1 | 3.8 ± 0.2 | |
| RV | 0.8 ± 0.1 | 0.8 ± 0.1 | 0.8 ± 0.0 | |
| lung | 5.2 ± 0.2 | 5.3 ± 0.1 | 5.0 ± 0.3 | |
| 7-days myocardial I-R | ||||
| n | 4 | 6 | 6 | |
| Body Weight (BW, g) | 26.3 ± 0.7 | 25.5 ± 0.6 | 25.6 ± 1.0 | |
| Organ weight/BW (mg/g) | Heart | 4.4 ± 0.2 | 5.2 ± 0.2# | 5.0 ± 0.2# |
| Atria | 0.2 ± 0.0 | 0.4 ± 0.00# | 0.3 ± 0.0 | |
| LV | 3.4 ± 0.1 | 3.9 ± 0.2# | 4.0 ± 0.2# | |
| RV | 0.8 ± 0.1 | 0.9 ± 0.1 | 0.7 ± 0.1 | |
| Lung | 5.8 ± 0.3 | 5.6 ± 0.16 | 5.7 ± 0.2 | |
#P < 0.05 vs. sham. One-way ANOVA with Dunnett’s post-hoc test. Data were presented as mean ± SEM, n indicates number of mice at endpoint.
Impact of Ac2-26 on MI-Induced Cardiac Dysfunction in vivo
The effects of Ac2-26 were assessed on severe cardiac injury was induced by 4 weeks permanent LAD occlusion (cohort 4). All mice in this cohort were included in survival analysis (see Supplementary Figures S1, S2). There was no statistical difference in survival between vehicle and Ac2-26 treated mice after MI (Supplementary Figure S2). End-point body and organ weights after sham surgery or 4 weeks post-MI, are shown in Table 3. Surgical MI or treatment with Ac2-26 did not affect BW or normalized RV or lung weight. HW, LV and atria weights were significantly increased to similar levels in both vehicle and Ac2-26-treated hearts (Figure 9 and Table 3).
Table 3.
Impact of MI in vivo in the absence and presence of Ac2-26 administration, on endpoint body and organ weights 4 weeks following permanent LAD occlusion, as well as echocardiographic parameters of LV function in anesthetized mice at 1 and 4 weeks post MI.
| Sham | MI | MI+ Ac2-26 | ||
|---|---|---|---|---|
| n | 6 | 10 | 12 | |
| Body Weight (BW, g) | 28.5 ± 0.7 | 28.3 ± 0.5 | 29.0 ± 0.5 | |
| Organ weight/BW (mg/g) | Atria | 0.3 ± 0.0 | 0.5 ± 0.0# | 0.5 ± 0.0# |
| RV | 0.8 ± 0.1 | 0.9 ± 0.1 | 0.9 ± 0.0 | |
| Lung | 5.9 ± 0.2 | 6.2 ± 0.2# | 6.4 ± 0.2# | |
| Echocardiographic analysis (1 week post MI) | ||||
| Sham | MI | MI+ Ac2-26 | ||
| HR (beats per min) | 518 ± 32 | 572 ± 24 | 551 ± 12 | |
| LVEDD (mm) | 4.27 ± 0.23 | 4.65 ± 0.16 | 4.55 ± 0.07 | |
| LVPW (mm) | 0.64 ± 0.02 | 0.67 ± 0.02 | 0.71 ± 0.02 | |
| LVESD (mm) | 2.97 ± 0.24 | 3.62 ± 0.17# | 3.31 ± 0.07 | |
| Echocardiographic analysis (4 weeks post MI) | ||||
| HR (beats per min) | 608 ± 7 | 582 ± 20 | 600 ± 18 | |
| LVEDD (mm) | 4.03 ± 0.08 | 4.92 ± 0.19## | 4.73 ± 0.14# | |
| LVPW (mm) | 0.68 ± 0.02 | 0.79 ± 0.02 | 0.77 ± 0.02 | |
| LVESD (mm) | 2.59 ± 0.10 | 3.86 ± 0.22### | 3.65 ± 0.16## | |
LVEDD, LV end-diastolic dimension; LVESD, LV end-systolic dimension; LVPW, LV posterior wall thickness;#P < 0.05, ##P < 0.01, ###P < 0.001 vs. sham. One-way ANOVA with Dunnett’s post hoc test. Data were presented as mean ± SEM, n indicates number of mice at endpoint.
Figure 9.
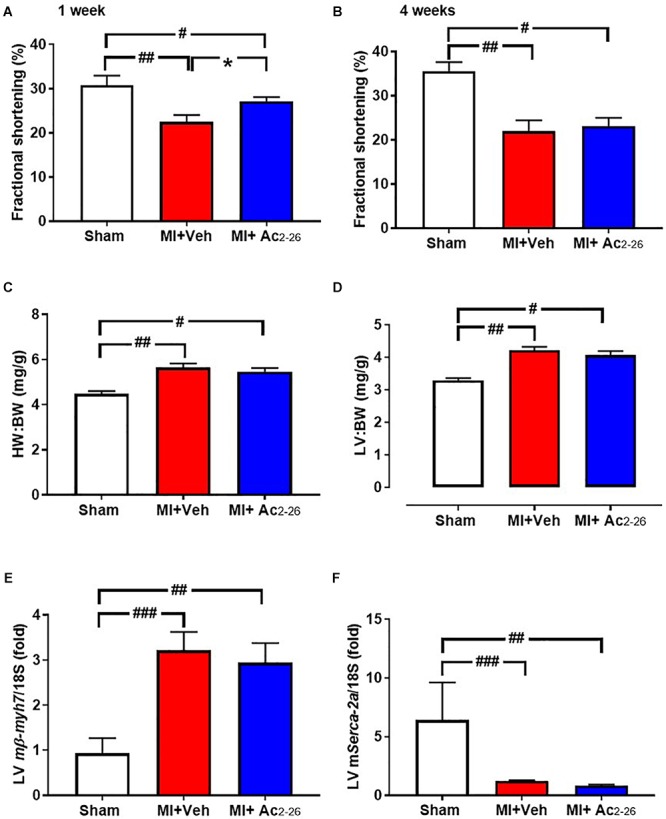
Impact of Ac2-26 on cardiac response to MI over the long-term. Cardiac dysfunction was assessed after 4 weeks of permanent LAD occlusion. MI significantly impaired fractional shortening (A) 1 week and (B) 4 weeks post LAD ligation. (C) HW:BW, (D) LV:BW and (E) LV mmyh7 were increased and (F) LV mSerca-2a was decreased in response to MI. Administration of Ac2-26 significantly improved cardiac function 1 week post LAD ligation (A), but not other changes in parameters 4 weeks post MI (B–F). #P < 0.05, ##P < 0.01, ###P < 0.001 vs. sham and ∗P < 0.05 vs. vehicle-treated MI mice. One-way ANOVA with Tukey’s post-hoc test. Data were presented as mean ± SEM, with number of mice per group. n = 6 (sham), n = 10 (I-R + vehicle), and n = 12 (I-R + Ac2-26).
Echocardiographic analysis of cardiac dimensions and LV function was performed in anesthetized mice both at 1 week post MI and at study end point (Table 3 and Figure 9 LV chamber dimensions measured as LVESD and LVEDD thickness were increased by MI 4 wks post MI (Table 3). Although there was no change in heart rate, %FS was reduced at both 1 and 4 weeks post MI (Figure 9A,B). This was partially protected with 1 week, but not 4 weeks, of Ac2-26 treatment. The significant MI-induced increase in hypertrophic mmyh7-gene expression, and reduction in LV sarco/endoplasmic reticulum Ca2+-ATPase (mSerca-2a gene expression), both of which play an important role in cardiac contractile function, were not prevented by Ac2-26 treatment (Figure 9E p = 0.01 overall on one-way ANOVA; Figure 9F, p = 0.005 overall on one-way ANOVA). In addition, the MI-induced increase in pro-fibrotic mCtgf gene expression in the non-infarct area at the 28-day timepoint was no longer evident in mice administered Ac2-26 (Supplementary Table S2). Ac2-26 also significantly reduced transforming growth factor-β (mTgf-β expression; Supplementary Table S2). Further, the significantly increased gene expression of the pro-inflammatory macrophage marker mS100a9 at this timepoint (p < 0.05) was no longer evident in mice treated with Ac2-26 for 4 weeks post MI (Supplementary Table S2).
Discussion
The major finding of the present study revealed that the N-terminal peptide of ANX-A1, Ac2-26, not only reduces cardiomyocyte death in vitro, but also reduces cardiac necrosis, inflammation, cardiac fibrosis and apoptosis, in a preclinical mouse model of myocardial I-R injury as summarized in Figure 10. Further, Ac2-26 delays the onset of cardiac dysfunction after MI in mice in vivo. These early cardioprotective properties of Ac2-26 in the first few days after myocardial ischemic insults in particular may reveal clinical insights into protective add-on approaches for management of MI.
Figure 10.

Proposed mechanism of Ac2-26-induced cardioprotection. Ac2-26 reduces cardiomyocyte death, myocardial inflammation, and limits early cardiac remodeling against myocardial ischemic insult. Schema created using a modification to an image provided by Servier Medical Art by Servier (https://smart.servier.com/image-set-download/), licensed under a Creative Commons Attribution 3.0 Unported Licence (https://creativecommons.org/licences/by/3.0/).
We have previously demonstrated that FPRs are functionally expressed in the whole heart and in isolated cardiomyocyte preparations (Qin et al., 2013). Given that our prior in vitro experiments lacked the concomitant presence of immune cells, it is thus likely that the cardioprotective action of Ac2-26 in vivo could be mediated via both FPR1- and FPR2-dependent mechanisms in vivo (Qin et al., 2015). We have previously shown that administration of Ac2-26 at the time of reperfusion in vitro preserves LV contractile function in isolated buffer-perfused rodents hearts subjected to I-R, via an FPR1-dependent mechanism (Qin et al., 2013). In contrast, the anti-inflammatory effects of Ac2-26 are considered predominantly mediated via FPR2 (Gavins et al., 2003a). Here, we investigated the impact of Ac2-26 on gene expression of both FPR1 and FPR2 subtypes in cardiomyocytes, and observed that Ac2-26 significantly increased FPR2 gene expression over 48 h. These data are consistent with an acute Ac2-26-mediated activation of cardiac FPRs to preserve LV function (within minutes, especially FPR1), whilst stabilizing cardiac FPR mRNA expression within days following its administration, especially given the reported short (90 min) half-life of FPR mRNA in unstimulated cells (Mandal et al., 2005; Waechter et al., 2012).
A number of studies have previously investigated the cardioprotective actions of Ac2-26 and full-length ANX-A1 protein in vivo (La et al., 2001a; Gavins et al., 2005; D’Amico et al., 2000; Dalli et al., 2012), at least over the short-term. The duration of reperfusion following ischemia employed in these earlier I-R studies in vivo has ranged from 45 min to 2 h (La et al., 2001a; Gavins et al., 2003a, 2005). A small number of studies have shown that Ac2-26 reduces infarct size and leukocyte recruitment when administrated at reperfusion (D’Amico et al., 2000; La et al., 2001b; Gavins et al., 2005). However, the cardioprotective effect of Ac2-26 against myocardial I-R injury at later timepoints (beyond the first few hours) has not been previously determined. The current study demonstrated for the first time that Ac2-26 reduces myocardial infarct size, and levels of the circulating cardiomyocyte injury marker, cTnl, 24 h post I-R, indicating that Ac2-26 likely preserves cardiomyocyte viability in vivo. This suggests that Ac2-26 might offer potential protection of cardiomyocyte viability in clinical settings.
Inflammatory cell infiltration into the myocardium is observed early, in the first few hours of reperfusion following ischemia (Dreyer et al., 1991; Hawkins et al., 1996; La et al., 2001b; Gavins et al., 2005). Infiltrating neutrophils play a central role in damage of MI. They are the first cells to be summoned to the insult region, resulting in exaggerated levels of both reactive oxygen species (ROS) and pro-inflammatory responses, leading to microvascular injury, cardiomyocyte death, extracellular matrix degradation and adverse cardiac remodeling (Gao et al., 2012). Neutrophil activation in MI promotes expression of adhesion molecules, leading to adherence of neutrophils to the endothelium, followed by transmigration, and direct interaction with cardiomyocytes (Ansari et al., 2018). Several studies have demonstrated the anti-neutrophil potential effects of ANX-A1 and Ac2-26. Administration of Ac2-26 was previously shown to reduce neutrophil infiltration into cardiac tissue after 1 h reperfusion in a mouse model of I-R (Gavins et al., 2006). The resultant increased cardiac accumulation of neutrophils and macrophages likely contributes to myocardial damage following the insult, consistent with our observations in the present study, 48 h after I-R. Our study now demonstrates that Ac2-26-induced significant reduction of local myocardial inflammatory cell infiltration, especially neutrophils, is still evident at a much later timepoint than previously observed, consistent with the early inhibition of neutrophil infiltration into mouse myocardial injury after the insult (Gavins et al., 2003a).
In the present study, a significant increase in circulating total and differential WBC following 48 h reperfusion was observed in response to I-R in mice, predominately attributed to increased circulating lymphocytes and neutrophils. Ac2-26 administration significantly reduced circulating WBC and neutrophils, suggesting this peptide may reduce systemic inflammation in response to an ischemic insult. An ischemic insult stimulates the expansion and mobilization of haematopoietic stem progenitor cells (HSPCs), to increase supply of myeloid cells to the injured myocardium (Dutta et al., 2012); endogenous ANX-A1 plays an important role in this progress (Qin et al., 2017a). It is thus possible that Ac2-26 may limit HSPCs expansion and mobilization, however, this was beyond the scope of the current study.
In this study, we have measured the levels of leukocytes (Ly6B+), macrophage (CD68+) neutrophil (Ly6G+) and inflammatory monocytes (Ly6C+) 48 h and 7 days post ischemic insult (Lee et al., 2013). In our hands, levels of infiltrating inflammatory cells peaks at 48 h, whilst levels of inflammatory monocytes/macrophages remain elevated 7 days after the insult. Ac2-26 reduced the total number of leucocytes, especially macrophages, but not the total number of neutrophils infiltrating the heart. However, the function of neutrophils could be affected by Annexin A1 and Ac2-26, as has been demonstrated in human neutrophils previously (Dalli et al., 2012); this was not, however, sought here.
Cardiac fibrosis is a key hallmark of the progression to HF following ischemic insults (Gao et al., 2012). This is attributed to necrotic and apoptotic cardiomyocytes being replaced by extracellular matrix proteins following the injury. In the present study, a significant increase in LV collagen deposition was clearly evident 7-days post I-R. Excitingly, our analysis revealed that Ac2-26 blunted this I-R-induced LV collagen deposition, suggesting that Ac2-26 has the potential to elicit relatively long-term cardioprotective effects, particularly at the level of adverse remodeling. Cardiomyocyte apoptosis was also significantly elevated in mice 7-days post I-R, consistent with previous reports (Qin et al., 2017b). This was significantly reduced by Ac2-26, suggesting that Ac2-26 may be able to enhance the survival of cardiomyocytes.
Left ventricular contractile impairment is a causal contribution of progression to HF following ischemic injury (Gao et al., 2012). In the present study, Ac2-26 delayed cardiac dysfunction in response to permanent LAD occlusion, as observed by protection of FS 1 week after MI, whilst this was no longer apparent after 4 weeks. This is consistent with our previous report that the permanent ligation-induced impairment in cardiac function was only significantly exaggerated in ANX-A1 deficient mice at 1 week, but not 4 weeks, post MI (Qin et al., 2017a). It is also possible that the full length ANX-A1 protein (or other, more stable, FPR agonists) is required to rescue the cardiac function over a longer-term, as suggested by previous reports of cardioprotection with the small-molecule FPR agonist Cmpd17b (Qin et al., 2017b) and the cleavage resistant peptide, CR-Ac2-50 (Dalli et al., 2013).
Limitations of the Study
In this study, we demonstrated that Ac2-26 attenuates cardiac necrosis, inflammation, and early remodeling against myocardial insults in vitro and in vivo. Whilst Ac2-26 delayed cardiac dysfunction by 1 week, we did not obtain additional echocardiographs between the 1 and 4 week timepoints, thus the maximum amount of time that cardiac dysfunction was delayed by the intervention could not be determined. In addition, higher doses, or more metabolically stable forms, of Ac2-26,, which might provide additional protection of LV function over the longer-term (Dalli et al., 2013) were not investigated here. It is also important to note that determination of gene expression in ischemic myocardium at the end of 4 weeks post MI, such as in this study, may be limited by the tissue being too necrotic and/or fibrotic for treatment modalities to penetrate. However, examination of gene expression at earlier timepoints, or within the at-risk but non-infarcted myocardium, have provided additional insights into the cardioprotection mechanisms of Ac2-26. Further, changes observed at the level of gene expression are not always accompanied by parallel changes in protein levels. The level of FPR expression in the heart is widely accepted to be relatively low compared to inflammatory cells and other tissues, e.g., kidney (Qin et al., 2015). Whilst only levels of FPR gene expression (and not protein) were reported in the present study, this was precluded by the less sensitive nature of conventional protein detection methods such as immunohistochemistry or Western blot, compared to qPCR measurement of gene expression, regardless of the target assessed. Lastly, we suggest that future studies explore more detailed mechanistic interrogation, to further elucidate the mechanism which ANX-A1 and its mimetics might promote active resolution in the injured myocardium. In addition, whether delayed intervention of Ac2-26 (e.g., 1–4 h post MI), in keeping with the timing of percutaneous intervention in the clinic, could enhance its translational therapeutic potential.
Conclusion
This study suggests that the ANX-A1 system may represent a novel therapeutic target for ischemic heart disease, given its ability to abrogate cardiomyocyte necrosis, inflammation and remodeling in a relevant preclinical model in vivo (illustrated in Figure 10). We have also provided new insights into the opportunities offered by ANX-A1-based approaches to limit early cardiac contractile dysfunction after an ischemic insult in vivo. This delay in progression of LV systolic dysfunction is a likely consequence of the favorable pro-resolving effects of Ac2-26. Development of ANX-A1 mimetics that are resistant to degradation are particularly attractive for future translational studies, particularly for treating MI in the early hours after an ischemic event (while the injury is still evolving), alone or concurrent with standard care, to reduce progression to HF and death in affected patients.
Author Contributions
CQ, XG, and RR conceived and designed the study. CQ, SR, NC, MD, JW, AA, DH, RL, HK, X-JD, and XG performed the experiments. CQ, NC, MD, MT, RL, JW, DD, and RR analyzed the data. CQ, XG, X-JD, XG, and RR interpreted the results. CQ and JW prepared the figures. CQ and RR drafted the manuscript. CQ, JB, YY, DD, HK, ML, AM, X-JD, XG, and RR edited and revised the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We acknowledge the Monash Micro-Imaging Facility (provision of instrumentation and training). No other persons have made substantial contributions to this manuscript.
Footnotes
Funding. This work was supported in part by both the National Health and Medical Research Council (NHMRC) of Australia, including APP1045140 (to RR, XG, and YY), and the Victorian Government’s Operational Infrastructure Support Program. RR and X-JD are NHMRC Senior Research Fellows (APP1059960 and APP1043026 respectively), CQ is a Baker Fellow. AM was supported Centenary Award from CSL. ML was supported by a post-doctoral fellowship from the National Heart Foundation.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2019.00269/full#supplementary-material
References
- Ansari J., Kaur G., Gavins F. N. E. (2018). Therapeutic potential of annexin A1 in ischemia reperfusion injury. Int. J. Mol. Sci. 19:E1211. 10.3390/ijms19041211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker E. L., Forouhar F. A., Grunnet M. L., Boulay F., Tardif M., Bormann B. J., et al. (1998). Broad immunocytochemical localization of the formylpeptide receptor in human organs, tissues, and cells. Cell Tissue Res. 292 129–135. 10.1007/s004410051042 [DOI] [PubMed] [Google Scholar]
- Chatterjee B. E., Yona S., Rosignoli G., Young R. E., Nourshargh S., Flower R. J., et al. (2005). Annexin 1-deficient neutrophils exhibit enhanced transmigration in vivo and increased responsiveness in vitro. J. Leukoc. Biol. 78 639–646. 10.1189/jlb.0405206 [DOI] [PubMed] [Google Scholar]
- Cooray S. N., Gobbetti T., Montero-Melendez T., McArthur S., Thompson D., Clark A. J., et al. (2013). Ligand-specific conformational change of the G-protein-coupled receptor, A. L. X.,/FPR2 determines proresolving functional responses. Proc. Natl. Acad. Sci. U.S.A. 110 18232–18237. 10.1073/pnas.1308253110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corminboeuf O., Leroy X. (2015). FPR2/ALXR agonists and the resolution of inflammation. J. Med. Chem. 58 537–559. 10.1021/jm501051x [DOI] [PubMed] [Google Scholar]
- Dalli J., Consalvo A. P., Ray V., Di Filippo C., D’Amico M., Mehta N., et al. (2013). Proresolving and tissue-protective actions of annexin A1-based cleavage-resistant peptides are mediated by formyl peptide receptor 2/lipoxin A(4) receptor. J. Immunol. 190 6478–6487. 10.4049/jimmunol.1203000 [DOI] [PubMed] [Google Scholar]
- Dalli J., Montero-Melendez T., McArthur S., Perretti M. (2012). Annexin A1 N-terminal derived peptide ac2-26 exerts chemokinetic effects on human neutrophils. Front. Pharmacol. 3:28. 10.3389/fphar.2012.00028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalli J., Norling L. V., Renshaw D., Cooper D., Leung K. Y., Perretti M. (2008). Annexin 1 mediates the rapid anti-inflammatory effects of neutrophil-derived microparticles. Blood 112 2512–2519. 10.1182/blood-2008-02-140533 [DOI] [PubMed] [Google Scholar]
- D’Amico M., Di Filippo C., La M., Solito E., McLean P. G., Flower R. J., et al. (2000). Lipocortin 1 reduces myocardial ischemia-reperfusion injury by affecting local leukocyte recruitment. FASEB J. 14 1867–1869. 10.1096/fj.99-0602fje [DOI] [PubMed] [Google Scholar]
- Donner D. G., Kiriazis H., Du X. J., Marwick T. H., McMullen J. R. (2018). Improving the quality of preclinical research echocardiography: observations, training, and guidelines for measurement. Am. J. Physiol. Heart Circ. Physiol. 315 H58–H70. 10.1152/ajpheart.00157.2018 [DOI] [PubMed] [Google Scholar]
- Dreyer W. J., Michael L. H., West M. S., Smith C. W., Rothlein R., Rossen R. D., et al. (1991). Neutrophil accumulation in ischemic canine myocardium. Insights into time course, distribution, and mechanism of localization during early reperfusion. Circulation 84 400–411. 10.1161/01.CIR.84.1.400 [DOI] [PubMed] [Google Scholar]
- Drucker D. J. (2016). Never waste a good crisis: confronting reproducibility in translational research. Cell Metab. 24 348–360. 10.1016/j.cmet.2016.08.006 [DOI] [PubMed] [Google Scholar]
- Dufton N., Hannon R., Brancaleone V., Dalli J., Patel H. B., Gray M., et al. (2010). Anti-inflammatory role of the murine formyl-peptide receptor 2: ligand-specific effects on leukocyte responses and experimental inflammation. J. Immunol. 184 2611–2619. 10.4049/jimmunol.0903526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dufton N., Perretti M. (2010). Therapeutic anti-inflammatory potential of formyl-peptide receptor agonists. Pharmacol. Ther. 127 175–188. 10.1016/j.pharmthera.2010.04.010 [DOI] [PubMed] [Google Scholar]
- Dutta P., Courties G., Wei Y., Leuschner F., Gorbatov R., Robbins C. S., et al. (2012). Myocardial infarction accelerates atherosclerosis. Nature 487 325–329. 10.1038/nature11260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flower R. J., Blackwell G. J. (1979). Anti-inflammatory steroids induce biosynthesis of a phospholipase A2 inhibitor which prevents prostaglandin generation. Nature 278 456–459. 10.1038/278456a0 [DOI] [PubMed] [Google Scholar]
- Gao X. M., Liu Y., White D., Su Y., Drew B. G., Bruce C. R., et al. (2011). Deletion of macrophage migration inhibitory factor protects the heart from severe ischemia-reperfusion injury: a predominant role of anti-inflammation. J. Mol. Cell Cardiol. 50 991–999. 10.1016/j.yjmcc.2010.12.022 [DOI] [PubMed] [Google Scholar]
- Gao X. M., Ming Z., Su Y., Fang L., Kiriazis H., Xu Q., et al. (2010). Infarct size and post-infarct inflammation determine the risk of cardiac rupture in mice. Int. J. Cardiol. 143 20–28. 10.1016/j.ijcard.2009.01.019 [DOI] [PubMed] [Google Scholar]
- Gao X. M., White D. A., Dart A. M., Du X. J. (2012). Post-infarct cardiac rupture: recent insights on pathogenesis and therapeutic interventions. Pharmacol. Ther. 134 156–179. 10.1016/j.pharmthera.2011.12.010 [DOI] [PubMed] [Google Scholar]
- Gao X. M., Xu Q., Kiriazis H., Dart A. M., Du X. J. (2005). Mouse model of post-infarct ventricular rupture: time course, strain- and gender-dependency, tensile strength, and histopathology. Cardiovasc. Res. 65 469–477. 10.1016/j.cardiores.2004.10.014 [DOI] [PubMed] [Google Scholar]
- Gavins F. N., Yona S., Kamal A. M., Flower R. J., Perretti M. (2003a). Leukocyte antiadhesive actions of annexin 1: ALXR- and FPR-related anti-inflammatory mechanisms. Blood 101 4140–4147. [DOI] [PubMed] [Google Scholar]
- Gavins F. N. E., Yona S., Kamal A. M., Flower R. J., Perretti M. (2003b). Leukocyte antiadhesive actions of annexin 1: ALYR- and FPR-related anti-inflammatory mechanisms. Blood 101 4140–4147. [DOI] [PubMed] [Google Scholar]
- Gavins F. N. E., Kamal A. M., D’Amico M., Oliani S. M., Perretti M. (2005). Formyl-peptide receptor is not involved in the protection afforded by annexin 1 in murine acute myocardial infarct. FASEB J. 19 100–102. 10.1096/fj.04-2178fje [DOI] [PubMed] [Google Scholar]
- Gavins F. N. E., Leoni G., Getting S. J. (2006). Annexin 1 and melanocortin peptide therapy for protection against ischaemic-reperfusion damage in the heart. ScientificWorldJournal 6 1008–1023. 10.1100/tsw.2006.196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris J. G., Flower R. J., Perretti M. (1995). Alteration of neutrophil trafficking by a lipocortin 1 N-terminus peptide. Eur. J. Pharmacol. 279 149–157. 10.1016/0014-2999(95)00145-B [DOI] [PubMed] [Google Scholar]
- Hausenloy D. J., Botker H. E., Condorelli G., Ferdinandy P., Garcia-Dorado D., Heusch G., et al. (2013). Translating cardioprotection for patient benefit: position paper from the working group of cellular biology of the heart of the european society of cardiology. Cardiovasc. Res. 98 7–27. 10.1093/cvr/cvt004 [DOI] [PubMed] [Google Scholar]
- Hawkins H. K., Entman M. L., Zhu J. Y., Youker K. A., Berens K., Dore M., et al. (1996). Acute inflammatory reaction after myocardial ischemic injury and reperfusion. Development and use of a neutrophil-specific antibody. Am. J. Pathol. 148 1957–1969. [PMC free article] [PubMed] [Google Scholar]
- Heusch G. (2013). Cardioprotection: chances and challenges of its translation to the clinic. Lancet 381 166–175. 10.1016/S0140-6736(12)60916-7 [DOI] [PubMed] [Google Scholar]
- Huynh K., Kiriazis H., Du X. J., Love J. E., Jandeleit-Dahm K. A., Forbes J. M., et al. (2012). Coenzyme Q10 attenuates diastolic dysfunction, cardiomyocyte hypertrophy and cardiac fibrosis in the db/db mouse model of type 2 diabetes. Diabetologia 55 1544–1553. 10.1007/s00125-012-2495-3 [DOI] [PubMed] [Google Scholar]
- Irvine J. C., Cao N., Gossain S., Alexander A. E., Love J. E., Qin C. X., et al. (2013). HNO/cGMP-dependent antihypertrophic actions of isopropylamine-NONOate in neonatal rat cardiomyocytes: potential therapeutic advantages of HNO overNO. Am. J. Physiol. Heart Circ. Physiol. 305 H365–H377. 10.1152/ajpheart.00495.2012 [DOI] [PubMed] [Google Scholar]
- Irvine J. C., Ganthavee V., Love J. E., Alexander A. E., Horowitz J. D., Stasch J. P., et al. (2012). The soluble guanylyl cyclase activator Bay 58-2667 selectively limits cardiomyocyte hypertrophy. PLoS One 7:e44481. 10.1371/journal.pone.0044481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- La M., D’Amico M., Bandiera S., Di Filippo C., Oliani S. M., Gavins F. N., et al. (2001a). Annexin 1 peptides protect against experimental myocardial ischemia-reperfusion: analysis of their mechanism of action. FASEB J. 15 2247–2256. [DOI] [PubMed] [Google Scholar]
- La M., Tailor A., D’Amico M., Flower R. J., Perretti M. (2001b). Analysis of the protection afforded by annexin 1 in ischaemia-reperfusion injury: focus on neutrophil recruitment. Eur. J. Pharmacol. 429 263–278. [DOI] [PubMed] [Google Scholar]
- Lee P. Y., Wang J. X., Parisini E., Dascher C. C., Nigrovic P. A. (2013). Ly6 family proteins in neutrophil biology. J. Leukoc. Biol. 94 585–594. 10.1189/jlb.0113014 [DOI] [PubMed] [Google Scholar]
- Lima K. M., Vago J. P., Caux T. R., Negreiros-Lima G. L., Sugimoto M. A., Tavares L. P., et al. (2017). The resolution of acute inflammation induced by cyclic AMP is dependent on annexin A1. J. Biol. Chem. 292 13758–13773. 10.1074/jbc.M117.800391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maderna P., Cottell D. C., Toivonen T., Dufton N., Dalli J., Perretti M., et al. (2010). FPR2/ALX receptor expression and internalization are critical for lipoxin A4 and annexin-derived peptide-stimulated phagocytosis. FASEB J. 24 4240–4249. 10.1096/fj.10-159913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal P., Novotny M., Hamilton T. A. (2005). Lipopolysaccharide induces formyl peptide receptor 1 gene expression in macrophages and neutrophils via transcriptional and posttranscriptional mechanisms. J. Immunol. 175 6085–6091. 10.4049/jimmunol.175.9.6085 [DOI] [PubMed] [Google Scholar]
- Perretti M., Flower R. J. (1995). Anti-inflammatory lipocortin-derived peptides. Agents Actions Suppl. 46 131–138. 10.1007/978-3-0348-7276-8_13 [DOI] [PubMed] [Google Scholar]
- Perretti M., Leroy X., Bland E. J., Montero-Melendez T. (2015). Resolution pharmacology: opportunities for therapeutic innovation in inflammation. Trends Pharmacol. Sci. 36 737–755. 10.1016/j.tips.2015.07.007 [DOI] [PubMed] [Google Scholar]
- Perretti M., Wheller S. K., Choudhury Q., Croxtall J. D., Flower R. J. (1995). Selective inhibition of neutrophil function by a peptide derived from lipocortin 1 N-terminus. Biochem. Pharmacol. 50 1037–1042. 10.1016/0006-2952(95)00238-U [DOI] [PubMed] [Google Scholar]
- Qin C. X., Buxton K. D., Pepe S., Cao A. H., Venardos K., Love J. E., et al. (2013). Reperfusion-induced myocardial dysfunction is prevented by endogenous annexin-A1 and its N-terminal-derived peptide Ac-ANX-A1(2-26). Br. J. Pharmacol. 168 238–252. 10.1111/j.1476-5381.2012.02176.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin C. X., Finlayson S. B., Ai-Sharea A., Tate M., De Blasio M. J., Deo M., et al. (2017a). Endogenous annexin-A1 regulates haematopoietic stem cell mobilisation and inflammatory response post myocardial infarction in mice in vivo. Sci. Rep. 7:16615. 10.1038/s41598-017-16317-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin C. X., May L. T., Li R., Cao N., Rosli S., Deo M., et al. (2017b). Small-molecule-biased formyl peptide receptor agonist compound 17b protects against myocardial ischaemia-reperfusion injury in mice. Nat. Commun. 8:14232. 10.1038/ncomms14232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin C. X., Yang Y. H., May L., Gao X., Stewart A. G., Tu Y., et al. (2015). Cardioprotective potential of annexin-A1 mimetics in myocardial infarction. Pharmacol. Ther. 148 47–65. 10.1016/j.pharmthera.2014.11.012 [DOI] [PubMed] [Google Scholar]
- Ritchie R. H., Gordon J. M., Woodman O. L., Cao A. H., Dusting G. J. (2005). Annexin-1 peptide Anx-1(2-26) protects adult rat cardiac myocytes from cellular injury induced by simulated ischaemia. Br. J. Pharmacol. 145 495–502. 10.1038/sj.bjp.0706211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie R. H., Sun X. L., Bilszta J. L., Gulluyan L. M., Dusting G. J. (2003). Cardioprotective actions of an N-terminal fragment of annexin-1 in rat myocardium in vitro. Eur. J. Pharmacol. 461 171–179. 10.1016/S0014-2999(03)01314-1 [DOI] [PubMed] [Google Scholar]
- Ritchie R. H., Sun X. L., Dusting G. J. (1999). Lipocortin-1 preserves myocardial responsiveness to beta-adrenergic stimulation in rat papillary muscle. Clin. Exp. Pharmacol. Physiol. 26 522–524. 10.1046/j.1440-1681.1999.03067.x [DOI] [PubMed] [Google Scholar]
- Sugimoto M. A., Vago J. P., Teixeira M. M., Sousa L. P. (2016). Annexin A1 and the resolution of inflammation: modulation of neutrophil recruitment, apoptosis, and clearance. J. Immunol. Res. 2016:8239258. 10.1155/2016/8239258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waechter V., Schmid M., Herova M., Weber A., Gunther V., Marti-Jaun J., et al. (2012). Characterization of the promoter and the transcriptional regulation of the lipoxin A4 receptor (FPR2/ALX) gene in human monocytes and macrophages. J. Immunol. 188 1856–1867. 10.4049/jimmunol.1101788 [DOI] [PubMed] [Google Scholar]
- Ye R. D., Boulay F., Wang J. M., Dahlgren C., Gerard C., Parmentier M., et al. (2009). International union of basic and clinical pharmacology. LXXIII. Nomenclature for the formyl peptide receptor (FPR) Family. Pharmacol. Rev. 61 119–161. 10.1124/pr.109.001578 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.