

 Thiosemicarbazones reveal strong anti-tyrosinase activity.
Thiosemicarbazones reveal strong anti-tyrosinase activity.
Abstract
Tyrosinase plays an essential role in melanogenesis. Excess production of melanin can be a reason for hyperpigmentation skin disorders in mammals and enzymatic browning in plant-derived foods. Catalyzing the rate-limiting step of melanin synthesis, tyrosinase has become the most studied target for melanogenesis inhibition. Over the past ten years, a number of synthetic thiosemicarbazone derivatives have been reported to possess strong tyrosinase inhibitory properties with IC50 values below 1 μM, placing them among the most potent tyrosinase inhibitors. This review gives an overview of tyrosinase activity and describes tyrosinase-inhibiting thiosemicarbazones in terms of their structure–activity relationships, kinetics of enzyme inhibition and mechanism of action. Results of the studies of thiosemicarbazones as tyrosinase inhibitors from over 20 research articles have been analyzed, compared and summarized in the present paper. Using thiosemicarbazones as tyrosinase inhibitors is a promising approach in developing anti-melanogenetic agents for skin-whitening cosmetics and anti-browning agents for food.
1. Introduction
Interest in skin whitening agents among the world population is growing. Around 15% of both men and women use skin whitening products. Global Industry Analysts, Inc. predicts that the global market for skin lighteners will reach US$23 billion by 2020.1
Melanins are the pigments responsible for skin color. They are some of the most widely encountered pigments in nature. Melanins can be found in bacteria, fungi, plants and animals. Their colors vary from yellow to black. Synthesis process of this biopolymer is called melanogenesis and it takes place in melanocytes, special cells within the basal layer of the dermis. Melanins are then secreted to surrounding keratinocytes. The amount and the type of pigment synthesized in melanocytes and its distribution in keratinocytes determines the color of the human skin and hair. Melanins also protect the mammalian skin from UV light by absorbing radiation.2 However, the accumulation of overproduced pigment in the skin can be the reason for hyperpigmentation problems and skin diseases, including freckles, age spots, melasma and melanoma.3
Melanin is formed via a series of enzymatic and non-enzymatic reactions and those, catalyzed by tyrosinase, are the most important among them. Tyrosinase hydroxylates monophenols to o-diphenols and oxidizes the o-diphenols to the corresponding o-quinones.4
Tyrosinase is also responsible for enzymatic browning in fruits, vegetables, mushrooms and beverages. Formation of dark pigment shortens the shelf life of fresh products, spoiling the smell and taste. Moreover, o-quinones can bind to protein residue groups, changing the nutrition characteristics of the protein.5
Tyrosinase inhibition is the most common approach for skin hyperpigmentation treatment, as the enzyme catalyzes the rate-limiting step of the pigmentation process.6
Over the past 30 years, tyrosinase has received considerable attention as a target for compounds preventing hyperpigmentation. Some valuable reviews have appeared on tyrosinase and melanogenesis inhibitors from natural and synthetic sources,1,6–18 but none of them focused on thiosemicarbazones in details. Thiosemicarbazones represent a very interesting class of ligands. They are well-known metal-chelating compounds. They have a great variety of pharmacological and biological properties as metal complexes and free ligands.19,20 This versatile group of compounds has been reported to be used in antimicrobial,21 antimalarial22 and anticancer23,24 treatment.
Over the past ten years, thiosemicarbazones have been also intensively studied as potential tyrosinase inhibitors.25–45 A number of synthetic thiosemicarbazone derivatives have been reported to possess strong tyrosinase inhibitory properties. Results of the studies on thiosemicarbazones as tyrosinase inhibitors from over 20 research articles have been analyzed, compared and summarized in the present paper. This review provides a brief overview of tyrosinase characteristics and describes its inhibition by thiosemicarbazones along with structure–activity relationships, kinetics of enzyme inhibition and mechanism of action.
2. Tyrosinase overview
Tyrosinase is a copper-containing enzyme belonging to the oxygen oxidoreductase family (EC 1.14.18.1).46 Proteins containing copper can be assigned to one of three classes based on their spectroscopic and structural properties. Tyrosinase is type-3 metalloprotein, which means that its active site contains two copper atoms and an activated oxygen atom.47
Tyrosinase is a bifunctional polyphenol oxidase (PPO), able to catalyze two different reactions: oxidation of monophenols (monophenolase activity) to o-quinones and oxidation of o-diphenols to o-quinones (diphenolase activity),48 as shown in Scheme 1. Both activities arise from the binding of dioxygen to the two copper atoms (identified as Cu(A) and Cu(B), Fig. 1), located in the active site.49
Scheme 1. Reactions catalyzed by tyrosinase: (a) monooxygenation of monophenols to o-quinones (monophenolase activity) and (b) oxidation of o-diphenols to o-quinones (diphenolase activity).

Fig. 1. Crystal structure of Agaricus bisporus mushroom tyrosinase (PDB:; 2Y9X): A) heterotetramer structure, B) catalytic subunit, and C) active site structure. Color representation: orange – copper, grey – carbon, blue – nitrogen, red – oxygen, and yellow – sulphur.

2.1. Tyrosinase structure
Tyrosinase from A. bisporus is a heterotetramer, consisting of two heavy (H) and two light (L) subunits (Fig. 1A), with total molecular mass of 120 kDa. Heavy subunit is the catalytic one and the function of l-subunit is still not known.50,51
Tyrosinases have three domains: N-terminal, C-terminal and central. The central domain is conserved in all tyrosinases from different sources, and this is the only conserved part of the enzyme. The central domain contains two copper binding sites called Cu(A) and Cu(B) (Fig. 1B).10 Each of two copper atoms in the tyrosinase molecule is ligated to three conserved histidine residues (Fig. 1C).51,52
As the protein structure is stabilized mostly by sulfide linkages, the location of cysteine residues is very important. The number of cysteines and their location in the central, C- or N-terminal domains varies between organisms.51,53
2.2. Melanins
Melanins have been investigated for a very long time as they are some of the most widely distributed pigments in nature (from bacteria to mammals) and play diverse roles in the lives different organisms.54–57
Melanins are a family of pigments with diverse structures and origin, produced by oxidation and polymerization of tyrosine in animals or phenolic compounds in lower organisms.
In nature, melanin exists in few different forms out of which eumelanin and pheomelanin are discussed in more detail. Eumelanin is a black-to-brown pigment. It is polymerized by oxidation of l-dopa or l-tyrosine via 5,6-dihydroxyindole or 5,6-dihydroxyindole-2-carboxylic acid (DHICA, Fig. 2). Eumelanin has some unique properties, such as electrical conductivity, paramagnetism, radical scavenging, metal ion binding and photoelectronic capability. Because of such a number of great properties, eumelanin is considered as a material for making functional devices.
Fig. 2. Pathway of melanin biosynthesis (Tyr–tyrosinase, Tyrp1–DHICA oxidaze, Tyrp2–dopachrome tautomerase).

Pheomelanin is a yellow-to-reddish pigment, found in red hair or freckles. It is polymerized via oxidation of 5-S-cysteinyldopa (l-tyrosine is polymerized with l-cysteine). The main difference between this pigment and eumelanin is the presence of sulfur in aromatic ring structures of pheomelanin.
Melanins (in particular pheomelanin) may have toxic properties upon exposure to UV radiation. Pheomelanin tends to undergo photodegradation and can contribute to damage of DNA in melanocytes, as it can generate hydrogen peroxide and superoxide anions.
Melanin composition (chemical phenotype) correlates with hair color (visual phenotype). Eumelanin predominates in dark hair (black, dark brown, brown, light brown, blond, and the smallest amount in red), whereas pheomelanin level stays low but constant. As a lighter pigment, pheomelanin, can be found in red hair and the redder areas of the skin, e.g. lips. The ratio of these pigments determines different hair, eye and skin color. The mixing of pigments varies not only between people but also for one person.
The skin of people whose melanocytes produce smaller amount of dark pigment is easily burnt after exposure to sun. White hair contains no melanin at all and grey contains only a few melanin granules.
Melanocytes are responsible for the synthesis of melanin within specialized membrane-bound organelles termed melanosomes and the subsequent transfer of the melanosomes to surrounding epidermal cells, the keratinocytes.
The function of melanin in mammals is protection of the skin from damage caused by ultraviolet radiation. Melanin is also a free radical scavenger, reducing the production of reactive oxygen species (ROS).
Mammalian eyes also contain melanin-producing cells. Another type of melanin is found in humans, and the highest amount is found in substantia nigra and locus coeruleus, regions of the brain that are the main targets of Parkinson's disease.
Melanins were also found in some birds, reptiles, amphibians, and fish. Black insoluble eumelanin is the component of cephalopod ink. Melanogenesis in insects is mainly related to cuticle sclerotization and innate immune response.
Plants need melanin to strengthen their cell wall. In contrast to animal pigments, plant melanin does not contain nitrogen and its color may range from dark brown to totally black. In the sclerotization reactions of plants, tyrosinase is released from vesicles in response to injury and catalyses oxidation of tannins to produce quinones, which bind to protein groups (e.g. thiol or amine) and produce metalloprotein. The result is blackening and hardening at a site of injury. This reaction is called an enzymatic browning.
Melanins are also found in fungi and bacteria.
2.3. Melanin biosynthesis pathway
Pathway of melanin biosynthesis is also called melanogenesis. The scheme of reactions leading to eumelanin and pheomelanin is shown in Fig. 2.55,58–62 Eumelanin and pheomelanin are both derived from dopaquinone, the common precursor which is formed from l-tyrosine in reaction catalysed by tyrosinase.
It has been believed that the first stage of melanogenesis leads to dopaquinone production by, first, hydroxylation of l-tyrosine to 3,4-dihydroxyphenylalanine (l-dopa) and subsequent oxidation of l-dopa to dopaquinone. However, it was reported by Cooksey et al.63 that dopaquinone and other o-quinones are formed directly form the phenolic substrate during the initial stage of melanin biosynthesis.
Dopaquinone is a very reactive compound, and in the absence of sulfhydryl compounds it undergoes cyclization (the intramolecular addition of the amino group) to produce leucodopachrome (cyclodopa).
A redox exchange between cyclodopa and another dopaquinone leads to dopachrome, the first color (orange) intermediate of melanogenesis. The second product of this reaction is l-dopa. l-dopa, produced in abovementioned reaction, is considered to be the source of dopa formed in this synthesis pathway.
Dopachrome then gradually rearranges to generate 5,6-dihydroxyindole (DHI) that is subsequently oxidized to indole-5,6-quinone. Alternative reaction is catalysed by dopachrome tautomerase and produces dihydroxyindole-2-carboxylic acid (DHICA) from dopachrome. DHICA is then oxidized in a redox reaction to form indole-5,6-quinine-carboxylic acid. DHI is produced in higher quantity than DHICA. DHI and DHICA are further polymerized to form eumelanin. Eumelanin derivatives of DHI and DHICA differ from each other. DHI-derived eumelanin is flocculent and black, whereas DHICA-derived eumelanin is finely dispersed and light brown.64
Regarding the production of pheomelanin, the presence of sulfhydryl compounds (cysteine) leads only to thiol adducts of dopa (cysteinyldopas). Further oxidation of the thiol adducts leads to the formation of pheomelanin via benzothiazine intermediates.
2.4. Tyrosinase inhibitors and their applications
Literature presents many compounds with tyrosinase inhibitory activity from both natural and synthetic sources.1,6–18 Natural tyrosinase inhibitors like flavonols (e.g. kaempferol and quercetin),65 chalcones (e.g. glabridin),66 coumarins (e.g. aloesin),67 stilbenes (e.g. oxyresveratrol),68 and aldehydes (e.g. cinnamaldehyde, cuminaldehyde and anisaldehyde)69,70 mostly have their origin in plants. Kojic acid71 and azelaic acid72 are fungus metabolites. Kojic acid is often used as a positive control in the literature for comparing the strength of the newly discovered inhibitors.
Potency of kojic acid as a tyrosinase inhibitor varies a little between the enzymes from different sources and the substrates used. For inhibition of tyrosinase diphenolase activity from A. bisporus, IC50 of kojic acid is around 28 μM.37,38 Kinetic studies revealed that kojic acid inhibits mushroom tyrosinase activity in a competitive manner with Ki of 0.03 mM and l-tyrosine as a substrate, and in a mixed manner with Ki of 0.02 mM and l-dopa as a substrate.73
Inhibitors of synthetic origin (usually drugs or simple chemicals) were also reported as tyrosinase inhibitors. Captopril (known drug for hypertension),74 tropolone (Cu2+ chelator, one of the most potent tyrosinase inhibitors),75 4-substituted resorcinols76,77 and methimazole (drug for thyroid problems)78 are examples of chemicals with antityrosinase activity.
Structures of some natural and synthetic tyrosinase inhibitors are presented in Fig. 3.
Fig. 3. Structures of some tyrosinase inhibitors.

Structural investigations of tyrosinase inhibitors with enzymes reveal that tyrosinase can be effectively inhibited without direct interaction between copper ions and inhibitors.
Crystal structures of complexes of tyrosinase with kojic acid79 (A) and tropolone51 (B) are presented in Fig. 4. In both cases, inhibitors interact with amino acid residues in the active site but don't chelate with any of the catalytic coopers.
Fig. 4. Crystal structures of tyrosinase active site with A) kojic acid (tyrosinase from Bacillus megaterium, PDB:; 3NQ1) and B) tropolone (tyrosinase from Agaricus bisporus, PDB:; 2Y9X). Color representation: orange – copper ions, grey – carbon, blue – nitrogen, red – oxygen, yellow – sulphur, and green – inhibitors.

Investigations of tyrosinase inhibitors as well as discovery and characterization of the new ones are very useful for their potential applications. Moreover, understanding the regulation of this enzyme gives a possibility of control of the whole biosynthesis pathway of melanin production.
Tyrosinase is responsible for formation of dark pigment in fungi- and plant-derived foods. This process is called enzymatic browning and affects the shelf life of fresh products spoiling their smell and taste. Products of tyrosinase reaction can bind to sulfhydryl and amino groups in proteins leading to destruction of essential amino acids residues. Tyrosinase inhibitors may be considered as preservatives for foods.5
Use of tyrosinase inhibitors is becoming increasingly important in the cosmetic industry due to their skin-whitening effect. Unfortunately, because of toxicity most of described in literature tyrosinase inhibitors can't be used orally or applied to skin. Kojic acid, a known ingredient of skin-lightening cosmetics, has been reported to be safe for human skin at a concentration of up to 1%, but depigmentation effect was seen at a concentration of 4%.80
Hydroquinone, another strong depigmenting agent, has been reported to be dangerous above concentration of 1% and it should not be used in leave-on cosmetic products.81
In medicine, tyrosinase inhibitors are used to treat some dermatological disorders, such as age spots, melasma and sites of actinic damage. They can also be found among the agents used to treat depigmentation after sunburn.8 Moreover, many defects in melanin biosynthesis pathway are related to some neurodegenerative diseases, such as Alzheimer's, Parkinson's and Huntington's.82
3. Thiosemicarbazones as tyrosinase inhibitors
In most cases, mushroom tyrosinase from Agaricus bisporus has been used in enzymatic assays. Mushroom tyrosinase is most often applied as a model of human tyrosinase as it has the highest homology with the mammalian tyrosinase.83 Other arguments in favour of using the enzyme from Agaricus bisporus as a model system for studying tyrosinase activity and inhibition are availability of convenient enzymatic assay, high activity and commercial availability in purified form.8,84
For the purpose of screening, known tyrosinase inhibitors (kojic acid, tropolone, and ascorbic acid) were used as references and positive controls. l-Tyrosine or l-dopa has been applied as substrates to identify the monophenolase or diphenolase activity of the tyrosinase, respectively. It should also be pointed out that in most cases, only diphenolase activity was investigated.
IC50 was used as the standard parameter characterizing the inhibitory potency of inhibitors. In some cases, more detailed investigations were performed, suppling kinetic information about enzyme–inhibitor interactions, such as reversibility, inhibition type (competitive, uncompetitive, noncompetitive or mixed) and inhibition constant Ki.
3.1. Thiosemicarbazones
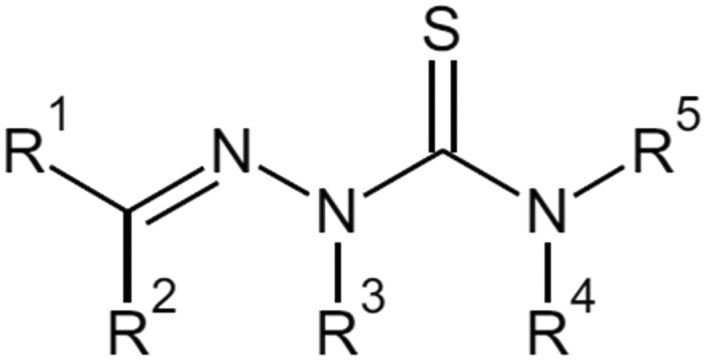
Thiosemicarbazones are derivatives of thiosemicarbazone with at least one substituted proton (Fig. 5). There are 5 possibilities for proton substitution and many compounds with at least one substitution were reported as potent tyrosinase inhibitors. The presence of electronegative oxygen, sulfur or nitrogen atoms on the ligand enhances the coordination ability of ligands. Thiosemicarbazones usually act as chelating agents with transition metal ions bonding via the sulfur or hydrazine nitrogen atoms located on the thiosemicarbazide moiety.85 As tyrosinase possesses two copper ions in its active site, it is possible for thiosemicarbazones to chelate with the ions and decrease catalytic activity of the enzyme.25
Fig. 5. General structure of thiosemicarbazone derivatives with all possible substitution positions.
3.2. Benzaldehyde thiosemicarbazone and its derivatives
Benzaldehyde thiosemicarbazone (1) (Fig. 6) was reported a few times as a potent tyrosinase inhibitor.26–30 Substitution of proton with aromatic ring may enhance the affinity of thiosemicarbazones for the enzyme because of higher similarity to natural tyrosinase substrates, l-tyrosine and l-dopa also bearing a phenyl group. High affinity of such compounds for the enzyme is the result of the van der Waals interactions of the aromatic ring with the hydrophobic residues present in the tyrosinase cavity.25
Fig. 6. Structures of hydroxy- and methoxy-substituted benzaldehyde thiosemicarbazone derivatives as tyrosinase inhibitors with their IC50 values.

IC50 values for benzaldehyde thiosemicarbazone vary in the literature from 0.84 to 9 μM.26,28–30 This compound exhibits noncompetitive inhibition with Ki of 3.7 μM27 or mixed-type inhibition with Ki of 0.93 and Kis of 93 μM.26
Based on the manual docking and the following molecular dynamic simulation, Buitrago et al. suggested that benzaldehyde inhibits tyrosinase via interaction of the thiosemicarbazide moiety with copper ions in the tyrosinase active site,26 whereas based on the results of molecular docking,27 it was reported that benzaldehyde interacts with the active site via the phenyl ring.
Substitution of a phenyl ring with a variety of substituents has been a very popular strategy for developing novel tyrosinase inhibitors.
3.2.1. Hydroxy- and methoxy-substitution of benzaldehyde thiosemicarbazone
Yi et al.30 investigated a group of mono-, di- and tri-substituted benzaldehyde thiosemicarbazone derivatives with hydroxy- and methoxy-groups on the benzene ring. Structures with the best IC50 values are presented in Fig. 6.
In general, monosubstitution is more favorable than di- and trisubstitution. Para position substitution seems to yield the best results for substitution of the hydroxyl group (IC50 of 0.41 μM). Bromine atom in para position enhances inhibitory properties, decreasing the value of IC50 to 0.28 μM.
In most cases, disubstitution of the phenyl ring decreases inhibitory properties of investigated compounds, but hydroxyl groups in ortho and para positions yield very good results with IC50 of 0.18 μM.
Trisubstitution of the phenyl ring drastically increases IC50 values, which is in accordance with research paper by Xue et al.,29 also describing the influence of substitution of the phenyl ring of benzaldehyde thiosemicarbazone on tyrosinase activity, but the source of the enzyme is P. rapae, not A. bisporus.
As studies show that the 4-substitution of the phenyl ring is the most favorable for tyrosinase inhibitors, Chen et al.31 investigated the kinetics of interaction of 4-hydroxy and 4-methoxybenzaldehyde thiosemicarbazones with tyrosinase. IC50 of 4-hydroxybenzaldehyde thiosemicarbazone for monophenolase activity of the enzyme was 0.76 μM, and inhibition was achieved only by limiting the rate of the reaction as the lag time prolongation was not observed. IC50 for diphenolase activity of this compound was 3.80 μM. This compound is a reversible, mixed-type inhibitor of tyrosinase with Ki and Kis values of 2.82 and 6.79 μM, respectively. 4-Methoxybenzaldehyde thiosemicarbazone has IC50 value of 7.0 μM, but in this case, prolongation of the lag period was observed. IC50 for diphenolase activity was 2.62 μM, and for this activity the kinetics parameters were measured also. 4-Methoxybenzaldehyde thiosemicarbazone turned out to be a reversible, mixed-type inhibitor of diphenolase activity of tyrosinase with Ki and Kis values of 1.47 and 15.10 μM, respectively. Both compounds also exhibited tyrosinase in B16 mouse melanoma cells. However, methoxybenzaldehyde thiosemicarbazone turned out to be a more potent inhibitor with an IC50 value of 139 μM.
3.2.2. 2-Chloro- and 4-chlorobenzaldehyde thiosemicarbazone derivatives
Li et al.32 reported inhibition kinetics for 2-chlorobenzaldehyde thiosemicarbazone (15) and 4-chlorobenzaldehyde thiosemicarbazone (16). Their structures are shown in Fig. 7. Both compounds revealed reversible mechanism of inhibition. IC50 was determined for both mono- and diphenolase activities of tyrosinase. For (15), IC50 values were 15.4 and 1.22 μM for mono- and diphenolase activity of the enzyme, respectively, and for (16), They were 6.7 and 1.82 μM. Compound (15) is a noncompetitive inhibitor with Ki value of 1.20 μM, and (16) shows mixed-type inhibition with Ki and Kis of 1.25 and 2.49 μM, respectively. For monophenolase activity, (15) and (16) decreased the reaction rate but did not influence the lag time.
Fig. 7. Structures of 2-chlorobenzaldehyde thiosemicarbazone (15), 4-chlorobenzaldehyde thiosemicarbazone (16), trans-cinnamaldehyde thiosemicarbazone (17), 4-dimethylaminobenzaldehyde thiosemicarbazone (18) and 4-dimethylaminobenzaldehyde-N-phenyl-thiosemicarbazone (19).

3.2.3. trans-Cinnamaldehyde thiosemicarbazone
Zhu et al.33 reported that the derivative of cinnamaldehyde and thiosemicarbazide (Fig. 7), compound (17), is a reversible mixed-typed inhibitor of diphenolase tyrosinase activity. Its Ki and Kis values are 4.45 and 8.85 μM, respectively. For monophenolase activity, inhibitor not only decreases the steady-state rate but also lengthens the lag time.
3.2.4. 4-Dimethylaminobenzaldehyde thiosemicarbazone and 4-dimethylaminobenzaldehyde-N-phenyl-thiosemicarbazone
Yang et al.34 examined the inhibitory activity of two 4-dimethylaminobenzaldehyde thiosemicarbazone derivatives (Fig. 7) and the influence of substitution of proton of thiosemicarbazide terminal group with a phenyl moiety. In this case, IC50 values were determined for both mono- and diphenolase tyrosinase activities, and they were 1.54 and 2.02 μM for (18) and 1.78 and 0.80 μM for (19). Both compounds revealed a reversible mode of inhibition. (18) decreased tyrosinase monophenolase activity in a mixed-type mode with Ki and Kis values of 1.77 and 6.49 μM, respectively. Substitution of proton in position R5 with phenyl group changed the way (19) inhibits tyrosinase to a noncompetitive type inhibition, enhancing inhibitory potency (Ki 0.77 μM). Both compounds inhibited tyrosinase activity but slightly prolonged the lag time.
3.2.5. 4-O-Substituted benzaldehyde thiosemicarbazones
Yi et al.35 investigated a group of 4-O-substituted derivatives of benzaldehyde thiosemicarbazone. Structures of the most potent tyrosinase inhibitors are shown in Fig. 8. The most active inhibitor has IC50 of 0.34 μM. All presented compounds can be considered very potent inhibitors, but introducing either oxygen or hydroxyl group into the structure definitely decreases the IC50 value. Branching of alkyl chain has a positive impact when the chain is short. Elongation of alkyl chain to 8, 10 and 12 carbon atoms drastically decreased inhibitory properties of the compounds. When the thiosemicarbazide moiety of the most active compound (22) was replaced by semicarbazide moiety, the compound lost all its activity. This observation suggests that the sulphur atom in thiosemicarbazide moiety is crucial for biological activity of thiosemicarbazones.
Fig. 8. Structures of 4-O-substituted derivatives of benzaldehyde thiosemicarbazone as tyrosinase inhibitors with their IC50 values.

3.3. Acetophenone thiosemicarbazone and its derivatives
3.3.1. Paeonol thiosemicarbazone derivatives
Zhu et al.25 investigated a series of methoxy- and hydroxy-substituted paeonol thiosemicarbazone analogues for their ability to inhibit mushroom tyrosinase. Structures of the group of such compounds with methyl group in R5 position are presented in Fig. 9. Disubstitution of the phenyl ring with hydroxyl groups on C-2 and C-4 was the most beneficial for inhibition. The IC50 value for (39) was 6 μM. The advantage of such type of substitution was confirmed by the results described for compound (8). However, when methyl group in R5 was replaced by a phenyl ring, inhibition activity drastically decreased and IC50 for all analogues was higher than 300 μM. Nonetheless, in both cases, inhibition activity was much higher than that of parent paoenol compounds where IC50 for most structures was above 1000 μM. Kinetic studies have been performed for (39). The inhibitor exhibited reversible and competitive inhibition with Ki value of 5 μM. Competitive inhibition indicates that (39) competes with l-dopa for the access to the active site. In addition, fluorescence studies confirmed that the inhibitor chelates with copper ion in the active site.
Fig. 9. Structures of paeonol derivatives as tyrosinase inhibitors with their IC50 values.

3.3.2. Aminoacetophenone thiosemicarbazone derivatives
You et al.36 investigated 3- and 4-aminoacetophenone thiosemicarbazones and their modifications, leading to 3- and 4-amidoacetophenone thiosemicarbazone derivatives. Fig. 10 portrays chemical structures of 4-substituted phenyl ring of aminoacetophenone thiosemicarbazones as this group showed better inhibitory properties than compounds with 3-substitution.
Fig. 10. Structures of 4-substituted phenyl ring of aminoacetophenone thiosemicarbazones as tyrosinase inhibitors with their IC50 values.

Kinetic studies were performed for compound (52), which had the lowest IC50 value of 0.291 μM. Compound (52) turned out to be a reversible noncompetitive inhibitor. Unfortunately, the inhibition constant has not been reported by the authors. Modification of amino group and formation of amide group had a very positive impact. There was no obvious tendency of the acyl group to influence the inhibitory strength, so it can be assumed that the amide group was crucial for enhancing the inhibitory potency of investigated compounds.
3.3.3. 4-Alkoxy and 4-acyloxyacetophenone thiosemicarbazones
You et al.37 reported a series of 4-alkoxy and 4-acyloxyacetophenone thiosemicarbazones. Plenty of substituents were tested to find a potent tyrosinase inhibitor. The influence of the length of methylene linker between the phenyl ring and the thiosemicarbazone moiety was also examined. As can be seen in Fig. 11, there is no obvious relationship between the distance of phenyl group from the scaffold of the compound and inhibitory activity. Introduction of a proper 4-alkoxy or 4-acyloxy substituent can significantly enhance inhibition potency of investigated compounds as their presence results in IC50 values below 1 μM. In turn, substitution of phenyl ring with 4-hydroxy group in the case of (76) decreased inhibitory ability. Authors also tested the effect of replacing a proton from the terminal NH2 group on thiosemicarbazide moiety by an amino group on (56), (57) and (60) activity. In all cases this substitution resulted in drastic decrease of inhibition potency. Compound (71) showed the best inhibitory properties with IC50 of 0.072 μM, which in suggests that 4-OCOCH3 substitution on the phenyl ring (distant from the thiosemicarbazone scaffold of two methylene groups) was the most favorable. Kinetics studies have been also performed for (66) and (79), which bear the same 4-substituent and exhibit similar IC50 values, to check if the number of methylene groups in the linker influences the mechanism of inhibition. Both compounds turned out to be reversible competitive inhibitors of tyrosinase.
Fig. 11. Structures of 4-alkoxy and 4-acyloxyacetophenone thiosemicarbazones as tyrosinase inhibitors and their IC50 values.

3.3.4. 4-Substituted acetophenones thiosemicarbazones
When methyl group is in position R2, a variety of 4-substitutions on the phenyl ring yields low IC50 values (below 1 μM). Some interesting examples are presented in Fig. 12.38,39 Even the addition of a huge biphenyl moiety with various substituents (90–94) resulted in low IC50 values39 (Fig. 12) when methyl was in R2 position.
Fig. 12. Structures of acetophenone thiosemicarbazone derivatives as tyrosinase inhibitors with their IC50 values.

3.4. Substitution of thiosemicarbazone scaffold with alkyl chain in R2 position
Song et al.38 and Hałdys et al.27 investigated the influence of elongation and branching of alkyl chain in R2 position on tyrosinase inhibition activity. Both papers present consistent results. No matter if R1 is a phenyl ring27 or a substituted phenyl ring (4-methyl, 4-butyl or 4-cyclohexyl),39 the elongation of the alkyl chain decreases the ability of the compound to inhibit tyrosinase, leading to a total loss of biological activity. However, methyl substituent is the most favorable, resulting in greater potency than a proton in R2 position.27,38,39 Starting with ethyl group, substitution inhibition activity decreases with chain elongation.
It was reported27 that in spite of the negative impact of elongation of alkyl chain on inhibition (in vitro), the tendency of melanogenesis inhibition is opposite in B16 mouse melanoma cells (Fig. 13). Thus, it can be predicted that those compounds inhibit melanin biosynthesis also at stages other than the step catalyzed by tyrosinase, for example, affecting activity of other key enzymes in melanin biosynthesis, such as dopachrome tautomerase, microphthalmia associated transcription factor or tyrosinase related protein 1.86
Fig. 13. Structures of thiosemicarbazones with substitution in R2 position with IC50 values of melanogenesis inhibition in B16 mouse melanoma cells.

Kinetics parameters for (95) were measured. Compound (95) exhibited reversible competitive inhibition with Ki of 0.38 μM. When phenyl ring was substituted with 3-naphthyl ring, inhibition potency was maintained, and it was reported that this inhibitor was a mixed-type with Ki and Kis of 0.83 and 1.4 μM, respectively.27
3.5. Aromatic heterocyclic thiosemicarbazones
3.5.1. Five-membered aromatic heterocyclic thiosemicarbazones
Thiosemicarbazones with aromatic heterocyclic substituents have gained interest of researchers, presenting an alternative for aromatic benzene moiety. Xie et al.40 investigated influence of thiophene, furan and pyrrole rings (Fig. 14) on the inhibition potency of investigated compounds towards tyrosinase. IC50 values for (104), (105) and (106) were 3.99, 2.35, and 0.43 μM, respectively. In all those cases, replacing one of the protons on a terminal NH2 group of thiosemicarbazide moiety with a methyl or a phenyl group drastically decreased inhibitory properties of the compounds. More detailed investigations have been performed for (104), (105) and (106). All the compounds turned out to be reversible mixed inhibitors of tyrosinase, having higher affinity for the free enzyme than for the enzyme–substrate complex. Inhibition constants were Ki 0.08 μM and Kis 0.29 μM, Ki 0.60 μM and Kis 1.18 μM, and Ki 167 μM and Kis 2.59 μM for (104), (105) and (106), respectively. Fluorescence quenching studies showed that the investigated compounds bound to tyrosinase active site via static forces and formed complexes with copper ions. Molecular docking and 1H NMR titration revealed that the sulfur atom of thiourea moiety of (106) and dicopper ions of the active site formed a complex, and the NH group of the pyrrole ring forms hydrogen bonds with amino acid residue His 224 in the active site of tyrosinase.
Fig. 14. Structures of thiosemicarbazone derivatives with 5-membered aromatic heterocyclic substituents as tyrosinase inhibitors and their IC50 values.

Based on the abovementioned results, Dong et al.41,42 performed further investigation of aromatic heterocycle thiosemicarbazones (Fig. 14). Replacement of a proton in position R2 with a methyl group slightly decreased the inhibitory potency of (104) and (106) and produced IC50 values of 4.148 and 1.406 μM for (107) and (109), respectively. For the compound with a furan ring, replacing a proton with a methyl group in R2 position resulted in decrease of IC50 to 0.824 μM (108).
It was reported28,41,42 that 6-membered aromatic heterocycles or fused-ring aromatic heterocycles in position R1 on a thiosemicarbazone skeleton decrease inhibitory potency of these compounds.
3.5.2. Thiophene-2-carbaldehyde thiosemicarbazone derivatives
As thiophene ring turned out to be most promising substituent in position R1, a group of thiosemicarbazones with a modified thiophene ring was investigated41–44 to find a moiety that enhances inhibitory properties. Some electronegative groups in position C-4 on a thiophene ring (Fig. 15) greatly contributed to enhanced biological activity. Addition of sterically bulky substituents decreased IC50 to 0.068 μM, indicating that (121) is the most potent inhibitor. Such a low IC50 value also suggests that (121) is one of the most potent tyrosinase inhibitors of all. Changing the position of substituent of (110), (111), (113) and (115) on a thiophene ring from C-4 to C-5 decreased inhibitory activity. Carbonyl group seems to be crucial for enhancing inhibition properties. Kinetic studies performed for compounds (110–113) revealed that they are mixed-type reversible inhibitors of tyrosinase diphenolase activity.
Fig. 15. Structures of 4- and 5-functionalized thiophene-2-carbaldehyde thiosemicarbazone derivatives as tyrosinase inhibitors with their IC50 values.

4. Conclusions and future prospects
Tyrosinase is the most studied target for melanogenesis inhibition. Catalyzing the rate-limiting step of melanin synthesis, tyrosinase has become one of the most important targets for the development of anti-hyperpigmentation agents.
Literature presents a number of studies devoted to the thiosemicarbazone derivatives possessing great capability of inhibiting the tyrosinase enzyme. Considering the general structure of thiosemicarbazone derivatives (see Fig. 16), some general points regarding structure–activity relationship of thiosemicarbazones can be mentioned based on the above minireview. Thiosemicarbazide moiety is a crucial group in the inhibition process. Specifically, a sulfur atom has the ability to chelate copper ions in the active site of tyrosinase. The most favorable substituent in position R5 is a proton or a phenyl ring. Longer chains decrease inhibitory properties of the compound. The biggest groups of thiosemicarbazones as tyrosinase inhibitors are reported for benzaldehyde or acetophenone derivatives what indicates that proton or methyl group is favorable in position R2. Phenyl ring in this position also seems to have a positive impact in some cases. Elongation or branching of the chain in R2 leads to a loss of biological activity.
Fig. 16. General points regarding structure–activity relationship of thiosemicarbazones with mushroom tyrosinase.

A number of substitutions have been made in position R1, leading to great inhibitory activity of modified analogues. The presence of aromatic ring seems to be crucial as it can interact with pocket of tyrosinase. Substitutions of the phenyl ring improve inhibitor parameters. Substituents in para position yield the best results. Among many investigated modifications, some of them deserve to be mentioned. Substitution with hydroxyl groups makes a strong impact on thiosemicarbazone inhibition activity, but it seems to be obvious as such moiety is similar to natural tyrosinase substrates, l-tyrosine and l-dopa. 4-O- and 4-N-substitutions also have been intensively investigated and should be pointed out as beneficial for inhibitory activity. Halogen substitution of phenyl ring definitely deserves further investigation by researchers. Another big group of substituents is aromatic heterocycles. Five-membered aromatic heterocyclic compounds, especially functionalized thiophene rings, should be considered as substituents for thiosemicarbazones when developing new tyrosinase inhibitors.
There is no information available in the literature regarding substitutions of protons in R3 and R4 positions in the context of tyrosinase inhibition.
As it was presented, many thiosemicarbazones have IC50 values lower than 1 μM, which places them among the best described tyrosinase inhibitors.
Kinetic investigations have been made for plenty of potent analogues. All of them reveal reversible mechanism of inhibition. Most often, inhibition was mixed or noncompetitive, indicating that thiosemicarbazones have the ability to interact with both the free enzyme and the enzyme–substrate complex. For mixed inhibition, Ki value was always lower than Kis, suggesting higher affinity of thiosemicarbazones for the free enzyme than the enzyme–substrate complex.
Inhibition of tyrosinase enzyme has been the most common approach in depigmenting research. Unfortunately, only a few tyrosinase inhibitors, described in the literature, found a practical application. Efficiency in enzyme inhibition in vitro is not the only factor that should be considered. Parameters, related to stability, solubility, cytotoxicity, absorption and penetration also have a great impact on the real possibility of using the inhibitors in the industry.6
In vitro effectiveness of inhibitors is not always reflected in in vivo research and clinical trials. Differences in results of in vitro and in vivo investigations may suggest that a new strategy combining two or more inhibitors with different mechanisms is needed.6 Active thiosemicarbazones could inhibit the enzyme much better in synergistic effect with already known inhibitors.
One of the main issues in effective depigmentation is finding a proper drug formulation and delivery system, which ensures that proper compound concentrations will reach the melanocyte. Possible examples include liposomes as carriers for lipophilic thiosemicarbazones or encapsulation of thiosemicarbazone.6
The enzyme from Agaricus bisporus is highly homologous with the mammalian one, so it is commonly used as a model for studies on melanogenesis. Actually, almost all studies on tyrosinase inhibition have been performed using mushroom tyrosinase because of the enzyme's commercial availability in purified form, existence of a convenient enzymatic assay and high activity.8,83,84
However, there are also limitations of such approach. First, it is known that the affinity of inhibitors for mammalian tyrosinase is commonly lower than the affinity for mushroom tyrosinase.6 Mushroom tyrosinase is, therefore, a good model system for selecting potential compounds as good inhibitors, but testing them with human enzyme will be eventually necessary.
Moreover, in spite of high homology and conserved active site of mushroom and human tyrosinases, slight structural differences might influence the activity and toxicity of thiosemicarbazones of interest. The human tyrosinase is membrane bound while the mushroom enzyme is located in cytosol. In addition, in contrast to the tetramer form of mushroom tyrosinase, human enzyme is a monomer undergoing glycosylation during its maturation process.10
There is no information on thiosemicarbazones as human or mammalian tyrosinase inhibitors. As it was concluded, the success of inhibiting of melanogenesis by thiosemicarbazones may be related to inhibition of other stages of melanin biosynthesis in addition to the reaction, catalyzed by tyrosinase.
Results of some in vivo research indicate that thiosemicarbazones can inhibit melanogenesis in mammalian cell lines. However, it is not clear if tyrosinase is the main target in the melanin biosynthesis process. Further investigations should be made as the compounds are usually tested on mushroom tyrosinase and some differences in structures between mushroom and human tyrosinases may influence the inhibition process and safety of the cells.
We hope that this review will be useful to medicinal chemists, developing novel melanogenesis inhibitors with drug-like properties, and for researchers looking for compounds with applications in food preservatives and cosmetics industries.
Conflicts of interest
There are no conflicts to declare.
Acknowledgments
The authors acknowledge the support from the Polish Ministry of Science and Higher Education (Ministerstwo Nauki i Szkolnictwa Wyższego) for the Faculty of Chemistry of Wrocław University of Science and Technology.
References
- Pillaiyar T., Manickam M., Namasivayam V. J. Enzyme Inhib. Med. Chem. 2017;32:403–425. doi: 10.1080/14756366.2016.1256882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pongkai P., Saisavoey T., Sangtanoo P., Sangvanich P., Karnchanatat A. Food Sci. Biotechnol. 2018;26:1199–1208. doi: 10.1007/s10068-017-0186-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hridya H., Amrita A., Mohan S., Gopalakrishnan M., Dakshinamurthy T. K., Doss G. P., Siva R. Int. J. Biol. Macromol. 2016;86:383–389. doi: 10.1016/j.ijbiomac.2016.01.098. [DOI] [PubMed] [Google Scholar]
- Lai X., Wichers H. J., Soler-Lopez M., Dijkstra B. W. Chem. – Eur. J. 2018;24:47–55. doi: 10.1002/chem.201704410. [DOI] [PubMed] [Google Scholar]
- Xu H., Zhang X., Karangwa E., Xia S. J. Sci. Food Agric. 2017;97:4210–4218. doi: 10.1002/jsfa.8295. [DOI] [PubMed] [Google Scholar]
- Solano F., Briganti S., Picardo M., Ghanem G. Pigm. Cell Res. 2006;19:550–571. doi: 10.1111/j.1600-0749.2006.00334.x. [DOI] [PubMed] [Google Scholar]
- Kim Y. J., Uyama H. Cell. Mol. Life Sci. 2005;62:1707–1723. doi: 10.1007/s00018-005-5054-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parvez S., Kang M., Chung H. S., Bae H. Phytother. Res. 2007;21:805–816. doi: 10.1002/ptr.2184. [DOI] [PubMed] [Google Scholar]
- Nesterov A., Zhao J., Jia Q. Drugs Future. 2008;33:945–954. [Google Scholar]
- Chang T. S. Int. J. Mol. Sci. 2009;10:2440–2475. doi: 10.3390/ijms10062440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loizzo M. R., Tundis R., Menichini F. Compr. Rev. Food Sci. Food Saf. 2012;11:378–398. [Google Scholar]
- Chang T. S. Materials. 2012;5:1661–1685. [Google Scholar]
- Mendes E., Perry M. de J., Francisco A. P. Expert Opin. Drug Discovery. 2014;9:533–554. doi: 10.1517/17460441.2014.907789. [DOI] [PubMed] [Google Scholar]
- Orhan I. E., Khan M. T. H. Curr. Top. Med. Chem. 2014;14:1486–1493. doi: 10.2174/1568026614666140523120741. [DOI] [PubMed] [Google Scholar]
- Pillaiyar T., Manickam M., Jung S. H. Expert Opin. Ther. Pat. 2015;25:775–788. doi: 10.1517/13543776.2015.1039985. [DOI] [PubMed] [Google Scholar]
- Ullah S., Son S., Yun H. Y., Kim D. H., Chun P., Moon H. R. Expert Opin. Ther. Pat. 2016;26:347–362. doi: 10.1517/13543776.2016.1146253. [DOI] [PubMed] [Google Scholar]
- Lee S. Y., Baek N., Nam T. G. J. Enzyme Inhib. Med. Chem. 2016;31:1–13. doi: 10.3109/14756366.2015.1004058. [DOI] [PubMed] [Google Scholar]
- Pillaiyar T., Namasivayam V., Manickam M., Jung S. H. J. Med. Chem. 2018;61:7395–7418. doi: 10.1021/acs.jmedchem.7b00967. [DOI] [PubMed] [Google Scholar]
- Lobana T. S. RSC Adv. 2015;5:37231–37274. [Google Scholar]
- Pelosi G. Open Crystallogr. J. 2010;3:16–28. [Google Scholar]
- Yusuf M., Jain P. Arabian J. Chem. 2014;7:525–552. [Google Scholar]
- Chellan P., Nasser S., Vivas L., Chibale K., Smith G. S. J. Organomet. Chem. 2010;695:2225–2232. [Google Scholar]
- Zhao H. C., Shi Y. P., Liu Y. M., Li C. W., Xuan L. N., Wang P., Zhang K., Chen B. Q. Bioorg. Med. Chem. Lett. 2013;23:6577–6579. doi: 10.1016/j.bmcl.2013.10.062. [DOI] [PubMed] [Google Scholar]
- Yu Y., Suryo Rahmanto Y., Richardson D. R. Br. J. Pharmacol. 2012;165:148–166. doi: 10.1111/j.1476-5381.2011.01526.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu T. H., Cao S. W., Yu Y. Y. Int. J. Biol. Macromol. 2013;62:589–595. doi: 10.1016/j.ijbiomac.2013.09.056. [DOI] [PubMed] [Google Scholar]
- Buitrago E., Vuillamy A., Boumendjel A., Yi W., Hardré G., Philouze C., Serratrice G., Jamet H., Regliér M., Belle C. Inorg. Chem. 2014;53:12848–12858. doi: 10.1021/ic501829s. [DOI] [PubMed] [Google Scholar]
- Hałdys K., Goldeman W., Jewgiński M., Wolińska E., Anger N., Rossowska J., Latajka R. Bioorg. Chem. 2018;81:577–586. doi: 10.1016/j.bioorg.2018.09.003. [DOI] [PubMed] [Google Scholar]
- Yi W., Dubois C., Yahiaoui S., Haudecoeur R., Belle C., Song H., Hardré R., Réglier M., Boumendjel A. Eur. J. Med. Chem. 2011;46:4330–4335. doi: 10.1016/j.ejmech.2011.07.003. [DOI] [PubMed] [Google Scholar]
- Xue C. B., Zhang L., Luo W. C., Xie X. Y., Jiang L., Xiao T. Bioorg. Med. Chem. 2007;15:2006–2015. doi: 10.1016/j.bmc.2006.12.038. [DOI] [PubMed] [Google Scholar]
- Yi W., Cao R. H., Chen Z. Y., Yu L., Ma L., Song H. C. Chem. Pharm. Bull. 2009;57:1273–1277. doi: 10.1248/cpb.57.1273. [DOI] [PubMed] [Google Scholar]
- Chen L. H., Hu Y. H., Song W., Song K. K., Liu X., Jia Y. L., Zhuang J. X., Chen Q. X. J. Agric. Food Chem. 2012;60:1542–1547. doi: 10.1021/jf204420x. [DOI] [PubMed] [Google Scholar]
- Li Z. C., Chen L. H., Yu X. J., Hu Y. H., Song K. K., Zhou X. W., Chen Q. X. J. Agric. Food Chem. 2010;58:12537–12540. doi: 10.1021/jf1033625. [DOI] [PubMed] [Google Scholar]
- Zhu Y. J., Song K. K., Li Z. C., Pan Z. Z., Guo Y. J., Zhou J. J., Wang Q., Liu B., Chen Q. X. J. Agric. Food Chem. 2009;57:5518–5523. doi: 10.1021/jf9007554. [DOI] [PubMed] [Google Scholar]
- Yang M. H., Chen C. M., Hu Y. H., Zheng C. Y., Li Z. C., Ni L. L., Sun L., Chen Q. X. J. Biosci. Bioeng. 2013;115:514–517. doi: 10.1016/j.jbiosc.2012.11.019. [DOI] [PubMed] [Google Scholar]
- Yi W., Cao R.-H., Chen Z. Y., Yu L., Wen H., Yan Q., Ma L., Song H. Chem. Pharm. Bull. 2010;58:752–754. doi: 10.1248/cpb.58.752. [DOI] [PubMed] [Google Scholar]
- You A., Song S., Zhu G., Song H., Yi W. Eur. J. Med. Chem. 2015;93:255–262. doi: 10.1016/j.ejmech.2015.02.013. [DOI] [PubMed] [Google Scholar]
- You A., Zhou J., Song S., Zhu G., Song H., Yi W. Bioorg. Med. Chem. 2015;23:924–931. doi: 10.1016/j.bmc.2015.01.024. [DOI] [PubMed] [Google Scholar]
- Song S., You A., Chen Z., Zhu G., Wen H., Song H., Yi W. Eur. J. Med. Chem. 2017;139:815–825. doi: 10.1016/j.ejmech.2017.08.033. [DOI] [PubMed] [Google Scholar]
- Liu J., Yi W., Wan Y., Ma L., Song H. Bioorg. Med. Chem. 2008;16:1096–1102. doi: 10.1016/j.bmc.2007.10.102. [DOI] [PubMed] [Google Scholar]
- Xie J., Dong H., Yu Y., Cao S. Food Chem. 2016;190:709–716. doi: 10.1016/j.foodchem.2015.05.124. [DOI] [PubMed] [Google Scholar]
- Dong H., Liu J., Liu X., Yu Y., Cao S. J. Mol. Struct. 2018;1151:353–365. [Google Scholar]
- Dong H., Liu J., Liu X., Yu Y., Cao S. Bioorg. Chem. 2017;75:106–117. doi: 10.1016/j.bioorg.2017.07.002. [DOI] [PubMed] [Google Scholar]
- Liu J., Li M., Yu Y., Cao S. Int. J. Biol. Macromol. 2017;103:1096–1106. doi: 10.1016/j.ijbiomac.2017.05.036. [DOI] [PubMed] [Google Scholar]
- Xu J., Liu J., Zhu X., Yu Y., Cao S. Food Chem. 2017;221:1530–1538. doi: 10.1016/j.foodchem.2016.10.140. [DOI] [PubMed] [Google Scholar]
- El-Sadek M. M., Hassan S. Y., Abdelwahab H. E., Yacout G. A. Molecules. 2012;17:8378–8396. doi: 10.3390/molecules17078378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomon E. I., Baldwin M. J., Lowery M. D. Chem. Rev. 1992;92:521–542. [Google Scholar]
- Siegbahn P. E. R. J. Biol. Inorg. Chem. 2003;8:567–576. doi: 10.1007/s00775-003-0449-4. [DOI] [PubMed] [Google Scholar]
- Claus H., Decker H. Syst. Appl. Microbiol. 2006;29:3–14. doi: 10.1016/j.syapm.2005.07.012. [DOI] [PubMed] [Google Scholar]
- Ismaya W. T., Rozeboom H. J., Schurink M., Boeriu C. G., Wichers H., Dijkstra B. W. Acta Crystallogr., Sect. F: Struct. Biol. Cryst. Commun. 2011;67:575–578. doi: 10.1107/S174430911100738X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strothkemp K. G., Jolley R. L., Mason H. S. Biochem. Biophys. Res. Commun. 1976;70:519–524. doi: 10.1016/0006-291x(76)91077-9. [DOI] [PubMed] [Google Scholar]
- Ismaya W. T., Rozeboom H. J., Weijn A., Mes J. J., Fusetti F., Wichers H. J., Dijkstra B. W. Biochemistry. 2011;50:5477–5486. doi: 10.1021/bi200395t. [DOI] [PubMed] [Google Scholar]
- Jackman M. P., Hajnal A., Lerch K. Biochem. J. 1991;274:707–713. doi: 10.1042/bj2740707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Gelder C. W. G., Flurkey W. H., Wichers H. J. Phytochemistry. 1997;45:1309–1323. doi: 10.1016/s0031-9422(97)00186-6. [DOI] [PubMed] [Google Scholar]
- Eom T., Woo K., Shim B. S. Kongop Hwahak. 2016;27:115–122. [Google Scholar]
- d'Ischia M., Wakamatsu K., Cicoira F., Di Mauro E., Garcia-Borron J. C., Commo S., Galvan I., Ghanem G., Kenzo K., Meredith P., Pezzella A., Santato C., Sarna T., Simon J. D., Zecca L., Zucca F. A. Pigm. Cell Melanoma Res. 2015;28:520–544. doi: 10.1111/pcmr.12393. [DOI] [PubMed] [Google Scholar]
- Ito S., Wakamatsu K., Ozeki H. Pigm. Cell Res. 2000;13(Suppl. 8):103–109. doi: 10.1034/j.1600-0749.13.s8.19.x. [DOI] [PubMed] [Google Scholar]
- Ito S., Wakamatsu K. Photochem. Photobiol. 2008;84:582–592. doi: 10.1111/j.1751-1097.2007.00238.x. [DOI] [PubMed] [Google Scholar]
- Prota G. Med. Res. Rev. 1988;8:525–556. doi: 10.1002/med.2610080405. [DOI] [PubMed] [Google Scholar]
- Prota G. Prog. Chem. Org. Nat. Prod. 1995;64:93–148. doi: 10.1007/978-3-7091-9337-2_2. [DOI] [PubMed] [Google Scholar]
- Raper H. S. Biochem. J. 1927;21:89–96. doi: 10.1042/bj0210089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason H. S. J. Biol. Chem. 1948;172:83–99. [PubMed] [Google Scholar]
- Ito S., Prota G. Experientia. 1977;33:1118–1119. doi: 10.1007/BF01946005. [DOI] [PubMed] [Google Scholar]
- Cooksey C. J., Garrratt P. J., Land E. J., Pavel S., Ramsden C. A., Riley P. A., Smit N. P. M. J. Biol. Chem. 1997;272:26226–26235. doi: 10.1074/jbc.272.42.26226. [DOI] [PubMed] [Google Scholar]
- Aroca P., García-Borrón J. C., Solano F., Lozano J. A. Biochim. Biophys. Acta. 1990;1035:266–275. doi: 10.1016/0304-4165(90)90088-e. [DOI] [PubMed] [Google Scholar]
- Kubo I., Kinsy-Hori I., Chaudhuri S. K., Kubo Y., Sánchez Y., Ogura T. Bioorg. Med. Chem. 2000;8:1749–1755. doi: 10.1016/s0968-0896(00)00102-4. [DOI] [PubMed] [Google Scholar]
- Chen J. M., Yu X. J., Huang Y. F. Spectrochim. Acta, Part A. 2016;168:111–117. doi: 10.1016/j.saa.2016.06.008. [DOI] [PubMed] [Google Scholar]
- Jones K., Hughes J., Hong M., Jia Q., Orndorff S. Pigm. Cell Res. 2002;15:335–340. doi: 10.1034/j.1600-0749.2002.02014.x. [DOI] [PubMed] [Google Scholar]
- Kim Y. M., Yun J., Lee C. K., Lee H., Min K. R., Kim Y. J. Biol. Chem. 2002;277:16340–16344. doi: 10.1074/jbc.M200678200. [DOI] [PubMed] [Google Scholar]
- Kubo I., Kinst-Hori I. J. Agric. Food Chem. 1998;46:5338–5341. [Google Scholar]
- Kubo I., Kinst-Hori I. J. Agric. Food Chem. 1999;47:4574–4578. doi: 10.1021/jf990165v. [DOI] [PubMed] [Google Scholar]
- Chen J. S., Wei C. I., Marshall M. R. J. Agric. Food Chem. 1991;39:1897–1901. [Google Scholar]
- Schallreuter K. U., Wood J. W. Arch. Dermatol. Res. 1990;282:168–171. doi: 10.1007/BF00372617. [DOI] [PubMed] [Google Scholar]
- Eom T., Woo K., Shim B. S. Kongop Hwahak. 2016;27:115–122. [Google Scholar]
- Espín J. C., Wichers H. J. Biochim. Biophys. Acta. 2001;1544:289–300. doi: 10.1016/s0167-4838(00)00230-2. [DOI] [PubMed] [Google Scholar]
- Kahn V., Andrawis A. Phytochemistry. 1985;24:905–908. [Google Scholar]
- McEvily J. A., Iyengar R., Otwell W. S. Crit. Rev. Food Sci. Nutr. 1992;32:253–273. doi: 10.1080/10408399209527599. [DOI] [PubMed] [Google Scholar]
- Frankos V. H., Schmitt D. F., Haws L. C., McEvily A. J., Iyengar R., Miller S. A. Regul. Toxicol. Pharmacol. 1991;14:202–212. doi: 10.1016/0273-2300(91)90007-i. [DOI] [PubMed] [Google Scholar]
- Andrawis A., Kanh V. Biochem. J. 1986;235:91–96. doi: 10.1042/bj2350091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sendovski M., Kanteev M., Ben-Yosef V. S., Adir N., Fishman A. J. Mol. Biol. 2011;405:227–237. doi: 10.1016/j.jmb.2010.10.048. [DOI] [PubMed] [Google Scholar]
- Burnett C. L., Bergfeld W. F., Belsito D. V., Hill R. A., Klaassen C. D., Liebler D. C., Marks Jr. J. G., Shank R. C., Slaga T. J., Snyder P. W., Andersen F. A. Int. J. Toxicol. 2010;29:244S–273S. doi: 10.1177/1091581810385956. [DOI] [PubMed] [Google Scholar]
- Andersen F. A., Bergfeld W. F., Belsito D. V., Hill R. A., Klaassen C. C. D., Liebler D. C., Marks Jr. J. G., Shank R. C., Slaga T. J., Snyder P. W. Int. J. Toxicol. 2010;29(6 Suppl):274S–287S. doi: 10.1177/1091581810385957. [DOI] [PubMed] [Google Scholar]
- Halaban R., Patton R. S., Cheng E., Svedine S., Trombetta E. S., Wahl M. L., Ariyan S., Hebert D. N. J. Biol. Chem. 2002;277:14821–14828. doi: 10.1074/jbc.M111497200. [DOI] [PubMed] [Google Scholar]
- Bagherzadeh K., Talari F. S., Sharifi A., Ganjali M. R., Saboury A. A., Amanlou M. J. Biomol. Struct. Dyn. 2015;33:487–501. doi: 10.1080/07391102.2014.893203. [DOI] [PubMed] [Google Scholar]
- Briganti S., Camera E., Picardo M. Pigm. Cell Res. 2003;16:101–110. doi: 10.1034/j.1600-0749.2003.00029.x. [DOI] [PubMed] [Google Scholar]
- Arslan H., Duran N., Borekci G., Ozer C. K., Akbay C. Molecules. 2009;14:519–527. doi: 10.3390/molecules14010519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehraiki A., Abbe P., Cerezo M., Rouaud F., Regazzetti C., Chignon-Sicard B., Passeron T., Bertolotto C., Ballotti R., Rocchi S. J. Invest. Dermatol. 2014;134:2589–2597. doi: 10.1038/jid.2014.202. [DOI] [PubMed] [Google Scholar]