

SUMMARY
FMRP (fragile X mental retardation protein) is a poly-some-associated RNA-binding protein encoded by Fmr1 that is lost in fragile X syndrome. Increasing evidence suggests that FMRP regulates both translation initiation and elongation, but the gene specificity of these effects is unclear. To elucidate the impact of Fmr1 loss on translation, we utilize ribosome profiling for genome-wide measurements of ribosomal occupancy and positioning in the cortex of 24-day-old Fmr1 knockout mice. We find a remarkably coherent reduction in ribosome footprint abundance per mRNA for previously identified, high-affinity mRNA binding partners of FMRP and an increase for terminal oligopyrimidine (TOP) motif-containing genes canonically controlled by mammalian target of rapamycin-eIF4E-binding protein-eIF4E binding protein-eukaryotic initiation factor 4E (mTOR-4EBP-eIF4E) signaling. Amino acid motif- and gene-level analyses both show a widespread reduction of translational pausing in Fmr1 knockout mice. Our findings are consistent with a model of FMRP-mediated regulation of both translation initiation through eIF4E and elongation that is disrupted in fragile X syndrome.
In Brief
Silencing of Fmr1, the gene that encodes FMRP, causes fragile X syndrome. Das Sharma et al. used ribosome profiling in the cortex of 24-day-old Fmr1 knockout mice to dissect FMRP-mediated translational regulation. Fmr1 loss leads to a relief of translational pausing across a large number of genes.
Graphical Abstract
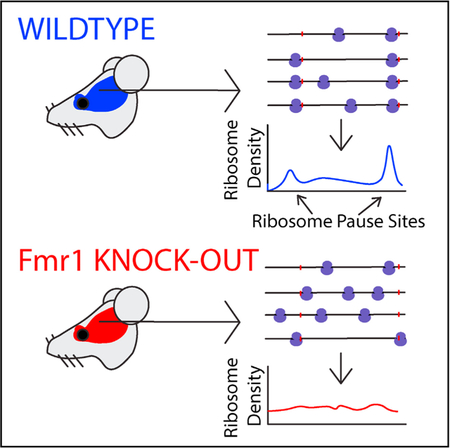
INTRODUCTION
Fragile X syndrome (FXS) is a highly penetrant, heritable form of intellectual disability that is associated with autism. The most common cause of FXS is epigenetic silencing of the FMR1 gene that encodes the fragile X mental retardation protein (FMRP). FMRP is an RNA binding protein that regulates both translation initiation and elongation (Darnell et al., 2011; Khandjian, 1999; Napoli et al., 2008; Stefani et al., 2004). Translation of the majority of cellular mRNAs begins with recognition of the 5′ cap structure m7G(5′)ppp(5′)N by eukaryotic initiation factor 4E (eIF4E). FMRP has been shown to repress translation initiation by interacting with cytoplasmic FMRP-interacting protein 1 (CYFIP1) (Napoli et al., 2008), an eIF4E binding protein that competes with eIF4G for interaction with eIF4E and prevents formation of the initiation complex (Richter and Sonenberg, 2005).
FMRP co-sediments with actively translating ribosomes and polyribosomes in gradient fractionation assays (Feng et al., 1997; Khandjian et al., 1996; Stefani et al., 2004). Recently, a genome-wide analysis of RNA-FMRP interactions was undertaken in the murine brain with high-throughput cross-linking immunoprecipitation (HITS-CLIP) (Darnell et al., 2011). In this study, FMRP was found to bind primarily to protein-coding sequences (CDS) of mRNAs, and no specific binding motif was identified. The highest-affinity mRNA binding partners were enriched in postsynaptic and autism-related genes, including components of the mGluR5 metabotropic glutamate receptor complex and downstream phosphatidylinositol 3-kinase (PI3K) signaling regulator PI3 kinase enhancer (PIKE), both of which are dysregulated in FXS (Bear et al., 2004; Gross et al., 2015). In vitro puromycin runoff experiments on a set of nine high-affinity binding partners showed extensive, FMRP-dependent ribosomal stalling compared with genes with lower HITS-CLIP signal. Furthermore, the in vitro ribosome translocation rate was shown to be significantly higher in brain lysates of Fmr1 knockout (Fmr1-KO) mice than wild-type mice (Udagawa et al., 2013). Studies have also shown elevated rates of protein synthesis in brains of Fmr1-KO mice (Qin et al., 2005) and increased protein expression of many FMRP high-affinity mRNA binding partners (Tang et al., 2015). Taken together, these studies suggest that FMRP represses protein synthesis at the level of translation elongation by acting as a ribosomal brake.
Despite this progress, important questions remain regarding the nature of translational regulation by FMRP in the brain. Although Darnell et al. (2011) have identified high-affinity binding partners, it is unclear whether there is a relationship between FMRP affinity and translational repression. Furthermore, it remains unknown whether FMRP represses translation in the brain through a dominant mechanism or whether both initiation and elongation are significantly affected. Ribosome profiling enables genome-wide measurement of ribosome density on mRNAs with single-nucleotide resolution, allowing simultaneous analysis of the overall ribosome density on each gene and ribosomal stalling. In this study, we conducted ribosome profiling and RNA sequencing (RNA-seq) in wild-type and Fmr1-KO mice to obtain an unbiased, high-resolution assessment of the impact of Fmr1 loss on protein synthesis in the brain.
RESULTS
Translational Landscape of Fmr1 KO Mice
To determine the effect of Fmr1 loss on translation, we conducted ribosome profiling and RNA-seq on the frontal cortex of Fmr1-KO and wild-type male mice at post-natal day 24 (P24). Genome-wide ribosome footprint (RF) and RNA-seq data were highly reproducible across biological replicates with genotype as the principal source of variation (Figures 1A and 1B; Figure S1). As expected, alterations in RF abundance and RNA expression were generally correlated (Figure 1C; Pearson’s r = 0.3), and a handful of genes exhibited particularly large differences in RF abundance between genotypes. For example, we found immediate-early genes, including Arc, Fos, and Egr2, to have significantly elevated RF abundance in Fmr1-KO mice, with much smaller alterations at the RNA level. We also validated our ribosome profiling measurements with polysome profiling and qPCR on genes with high (Arc, Fos) and low (Gabra2) fold changes in RF abundance (Figure S2; Table S1). Consistent with the ribosome profiling data, we observed reduced abundance of Gabra2 transcripts in monosomal and in heavier polysomal fractions (P1 and P2), whereas Arc and Fos show greater abundance in monosome, P1, and P2 fractions of Fmr1-KO samples with slight alterations in global mRNA levels.
Figure 1. Comparison of Transcriptional and Translational States from RNA- and Ribo-Seq in Fmr1-KO versus Wild-Type Mice.

(A and B) Principal component analysis (PCA) of both ribosome profiling (A) and RNA-sequencing (B) libraries from Fmr1-KO and wild-type mice (n = 3 mice/ genotype for ribosome profiling (Ribo-seq) and n = 4 mice/genotype for RNA-seq). Samples are segregated by genotype in principal component 1 (PC1), the axis representing the major source of variation in the data, in both plots.
(C) Comparison of differential ribosome footprint abundance against differential RNA expression levels between genotypes at the level of individual genes. Although ribosome footprint abundance displays a greater range of changes than RNA expression level, these measurements are correlated (r = 0.3). Fmr1, knocked out at the transcript level (by deletion of one exon), shows decreased RNA expression and ribosome density as expected, whereas the immediate-early genes Fos, Arc, and Egr2 show increased ribosome density and RNA expression.
(D and E) Enrichment scores of the top 15 gene ontologies (GOs) enriched in the wild-type or Fmr1-KO brain, determined by GSEA on genes ranked by their fold changes in ribosome footprint abundance (D) or RNA expression (E) as presented in (C). Genes related to protein synthesis are enriched in ribosome density in Fmr1-KO mice compared with wild-type but depleted in RNA expression level (and therefore enriched for in wild-type versus Fmr1-KO mice), whereas ontologies related to neuronal development and morphology show decreased ribosome density in Fmr1-KO mice.
To characterize the effects of Fmr1 loss more broadly, we conducted differential RF abundance and RNA expression analyses. We used gene set enrichment analysis (GSEA) to assess differentially translated and expressed Gene Ontologies (GOs). Interestingly, although GOs associated with protein synthesis had higher RF abundance in Fmr1-KO mice compared with wild-type, translation-associated GOs exhibited lower expression at the RNA level in the Fmr1-KO mice (Figures 1D and 1E). In addition, GOs associated with neuronal projection development, morphology, and extracellular matrix have lower RF abundance in Fmr1-KO mice, whereas neuron-specific genes (Zhang et al., 2014) are not significantly enriched or depleted in RNA-seq (Figure S3B; false discovery rate [FDR] q > 0.05). This suggests that the broad downregulation of specific neuronal ontologies in our ribosome profiling data is unlikely to result from gross changes in cellular composition.
To identify genes with significant alterations in RF abundance per mRNA (RFApm), calculated as the ratio of RF abundance and RNA expression, we used the generalized linear model (GLM) implemented in RiboDiff for joint statistical analysis of the ribosome profiling and RNA-seq data (Table S2). This metric approximates the number of ribosomes bound per mRNA and is commonly referred to as “translation efficiency” (Ingolia et al., 2009). However, RFApm depends on complex relationships between the rates of translation initiation, elongation, and termination that complicate its interpretation (Arava et al., 2005). GSEA revealed that genes involved in protein synthesis have elevated RFApm in Fmr1-KO mice with concomitant reductions in genes associated with extracellular matrix and neuronal function, differentiation, and projection (Figure 2A). Additionally, we found the CDS length to be anticorrelated (Spearman’s correlation = −0.27; p < 0.00001) with the corresponding change in RFApm in response to Fmr1 KO (Figure S3C), consistent with a recent study of Fmr1-dependent translation in Drosophila oocytes (Greenblatt and Spradling, 2018).
Figure 2. Analysis of Differential Translational Regulation from Ribosome Footprint Abundance per mRNA in Fmr1-KO versus Wild-Type Mice.

(A) GSEA performed on genes ranked by their differential ribosome footprint abundance per mRNA (RFApm) between genotypes (n = 3/genotype) reveals increased RFApm of genes related to protein synthesis (ribosome, translation elongation, mitochondrial translation) in Fmr1-KO mice with decreased RFApm of genes involved in neuronal projection development and morphology.
(B and C) Volcano plots comparing the observed effect size of log-fold change in RFApm with adjusted p values for all detected genes.
(B) Shows a trend toward increased RFApm in the 5′-terminal oligopyrimidine (5′ TOP) motif-containing genes (p < 0.00001, GSEA), the canonical targets of mTOR (green).
(C) Demonstrates a uniform, modest reduction in RFApm (p < 0.00001, GSEA) across the top 200 highest-affinity binding partners for FMRP determined by HITS-CLIP in Fmr1-KO mice (orange). Together, (B) and (C) demonstrate the concerted dysregulation of distinct gene sets in opposite directions associated with FMRP loss.
(D) In-depth analysis of differential RFApm results showing the overrepresentation of translational changes dominated by changes in RF abundance.
Translation initiation for effectors of protein synthesis, such as ribosomal proteins and translation factors, is regulated by mammalian target of rapamycin (mTOR) signaling through a cis-regulatory element known as the 5′-terminal oligopyrimidine (5′ TOP) motif found in the corresponding mRNAs (Hsieh et al., 2012; Thoreen et al., 2012). This regulation is mediated by 4E BPs, which, in their dephosphorylated state, sequester the initiation factor eIF4E (Thoreen et al., 2012). FMRP can repress translation via an inhibitory FMRP-CYFIP1-eIF4E complex (Napoli et al., 2008; Santini et al., 2017), and Fmr1-KO mice exhibit increased eIF4E-dependent translation (Sharma et al., 2010). Therefore, we expected that the 5′ TOP motif-containing mRNAs would exhibit increased RFApm in Fmr1-KO mice. Indeed, Figure 2B shows that the set of 5′ TOP transcripts exhibited significantly higher RFApm in Fmr1-KO mice (p < 0.00001, GSEA), consistent with a previously characterized mechanism through which FMRP modulates translation initiation (Napoli et al., 2008). As described above, earlier work showed that FMRP binds to mRNAs that encode proteins associated with synaptic activity and other neuronal functions. The GSEA in Figure 2A suggests a reduction in RFApm for genes with similar ontologies. Indeed, Figure 2C shows that the top 200 highest-affinity FMRP binding partners exhibit significantly reduced RFApm as a group (p < 0.00001, GSEA). We have observed similar characteristics among FMRP-binding partners and 5′ TOP motif-containing transcripts when calculating ratios of RF abundance to RNA for matched replicates (Figure S4). The RFApm changes observed for both 5′ TOP and high-affinity FMRP CLIP targets, and in the transcriptome overall, were largely driven by changes in RF counts rather than changes in transcript levels in our analysis considering all replicates (Figure 2D).
This coherent reduction in ribosome occupancy is surprising, because many of these genes have been shown to be overex-pressed at the protein level in the brains of Fmr1-KO mice (Hou et al., 2006; Schütt et al., 2009; Zalfa et al., 2003; Zhang et al., 2001). One possibility is that protein synthesis from these mRNAs is controlled at the level of translation elongation. For example, a decrease in RFApm could result from a reduction in ribosomal stalling rather than in initiation efficiency (Ingolia et al., 2009).
Alterations in Translation Elongation in Fmr1 KO Mice
Given the previous evidence of FMRP-dependent ribosomal pausing (Darnell et al., 2011) and the results described above, we next quantified ribosomal pausing using the ribosome pause score, proportional to a codon’s RF coverage normalized by the mean per-codon coverage for the transcript in which said codon occurs (Woolstenhulme et al., 2015). As in previous studies (Ishimura et al., 2014), our pause score was modified with additional context specificity, in which the five codons on either side of the codon being scored are considered for normalization to correct for falsely identified pauses due to alternative splicing (see STAR Methods). Specifically, we calculated the ribosome pause score at the level of encoded amino acid sequences, averaging scores across all occurrences of codons corresponding to a given amino acid residue. This metric allows the determination of pause activity due to encoded peptide sequence. Figure 3 compares the distributions of pause scores across mono-, di-, and tri-amino acids between Fmr1-KO and wild-type ribosome occupancy profiles. With few exceptions, sequences exhibited a lower mean pause score in Fmr1-KO than in wild-type profiles, demonstrated by a downward shift away from the main diagonal in Figures 3A–3C. This shift indicates a relief of pausing across a large number of genes associated with Fmr1 loss that is inconsistent with an effect on a limited set of specific binding partners.
Figure 3. Codon-Level Ribosome Pause Score Analysis of Encoded Amino Acid Sequences across Fmr1-KO and Wild-Type Mice.

(A–C) Log-log plots of mean pause scores calculated for (A) single amino acid, (B) di-, and (C) tri-amino acid sequences in Fmr1-KO and wild-type mice (n = 3 for both genotypes), with accompanying p value for the significance of the difference in these two distributions (Mann-Whitney U test). In each plot, the main diagonal is plotted as a blue line representing equal pausing in either genotype, highlighting the downward shift of the mass of individual sequences’ scores and decrease in pause score in Fmr1-KO mice.
(D–F) This shift is visualized differently in (D)–(F), histograms of the log-ratios of mean pause scores for every (D) mono- , (E) di-, and (F) tri-amino acid motif. The downward or rightward shift in (A)–(C) translates to a leftward shift away from the blue vertical line at x = 0, showing decreased pausing for the majority of encoded amino acid motifs in Fmr1-KO versus wild-type mice.
Even though codon-level analysis suggests that alterations in translation elongation are widespread, we further validated these changes directly at the gene level. Gene-level analysis of translational pausing is complicated by the large dynamic range in gene expression, which results in a broad coverage distribution for ribosome profiling across genes. For example, consider two genes with similar translational pausing behavior where one gene is lowly expressed, resulting in a low-coverage ribosome profile. A naive analysis might conclude that this lowly expressed gene has more translational pausing, an artifact of sparse coverage. At low coverage, it is challenging to differentiate noise (which scales inversely with coverage due to counting statistics) from real translational pausing. To address this issue, we developed an analytical method for gene-level analysis of translational pausing that explicitly models the dependence of noise in ribo-some profiles on coverage.
Figures 4A–4C show the dependence of the noise (expressed as coefficient of variation [CV]) in the ribosome profile along the CDS of each gene on coverage (expressed as RF reads per codon). As expected, the CV decreases with increasing coverage regardless of genotype (Figures 4A and 4B; Figures S5A–S5F). We fit the following two-parameter model to the data to accommodate a variety of statistical behaviors for counting noise:
| (Equation 1) |
Figure 4. Gene-Level Modeling of Noise in Ribosome Distribution across Fmr1-KO and Wild-Type Samples.

(A and B) For gene-level analysis of ribosomal pausing, (A) and (B) plot the relationship between noise and coverage for a (A) single wild-type and (B) Fmr1-KO replicate (n = 3/genotype), respectively. In these plots, coverage is the log-mean number of ribosome footprints aligned per codon of a given transcript, noise is represented by log-coefficient of variation, or SD in the number of ribosome footprints per codon divided by mean, and the relationship of these values across each gene is summarized by regression to the two-parameter model in Equation 1, plotted in orange.
(C) Regression curves for each replicate on the same axes (n = 3 for both genotypes), showing both the uniformity of this relationship across biological replicates, as well as the uniform downward shift in coefficient of variation of Fmr1-KO replicates relative to wild-type. This shift, representing a lower degree of coverage variation along the gene body, indicates a widespread reduction in pausing across transcripts.
Figure 5. Gene-Level Ribosome Occupancy Profiles in Fmr1-KO and Wild-Type Mice.

(A–D) At left, nucleotide-resolution plots of P-site occupancy for genes (A) Syn1, (B) Map1b, (C) Rpl4, and (D) Ndel1 for three replicates of both wild-type (blue) and Fmr1-KO (red) mice. These plots are paired on the right with comparisons of the cumulative coefficient of variation (CV), calculated as the CV for the coding sequence up to a given nucleotide position in the CDS. Whereas Syn1 and Map1b both exhibit decreased RFApm in Fmr1-KO mice, Syn1 and Map1b are high-affinity binding partners of FMRP; similarly, Rpl4 and Ndel1 both exhibit increased RFApm in Fmr1-KO mice, but Rpl4 is a 5′-TOP gene and target of mTOR. For all of these genes, the magnitudes of the “spikes” of reproducible, high-frequency P-site alignment, which represent pause sites, are significantly reduced in Fmr1-KO occupancy plots compared with their wild-type counterparts, and this reduction is reflected in a correspondingly diminished increase in cumulative CV at the pause site’s coordinate for Fmr1-KO replicates. The overall decrease in positional noise of aligned P-sites with FMRP loss, represented by the consistent gap in cumulative CV between genotypes at nearly all coordinates, is larger than that which can be explained by large pause reductions alone.
where CV is the coefficient of variation in the ribosome profile of a given gene, μ is mean coverage (RF reads per codon), and α and β are fitting parameters. Importantly, when α = 0 and β = 1, Equation 1 results from a Poisson distribution, whereas α > 0 and β = 1 indicates a negative binomial distribution. Figure 4C shows the fits for all wild-type (n = 3) and Fmr1-KO (n = 3) ribosome profiling datasets. While biological replicates of each genotype are highly reproducible, there is a clear difference between genotypes with the Fmr1-KO mice exhibiting markedly lower CV at higher coverage. Over-dispersion is widely appreciated for RNA counting data derived from high-throughput sequencing, and as expected, α > 0 for all datasets. For highly translated genes, where coverage is drawn from an over-dispersed distribution, CV converges to α 1/2. However, there is a strong genotype effect on α (2.61 ± 0.02 for wild-type and 1.720 ± 0.002 for Fmr1-KO; p = 0.0001). Taken together, these results indicate that the ribo-some profiles of genes in Fmr1-KO brains display less variability in coverage along the CDS than in the wild-type. These findings are consistent with the codon-level analysis described above, reflecting a reduction in translational stalling across a large number of genes in Fmr1-KO mice.
The analysis in Figures 3 and 4 suggests that loss of Fmr1 results in widespread alterations in translation elongation. Figure 5 shows specific examples of this among representative genes from a few different categories. Importantly, there is not a large difference in coverage between wild-type and Fmr1-KO for any of these genes. Figures 5A and 5B show the P-site ribosome profiles for all three wild-type and Fmr1-KO mice for two genes (Syn1, Map1b) with a significant reduction in RFApm and two genes (Rpl4, Ndel1) with a significant enhancement in RFApm. Syn1 and Map1b both are high-affinity FMRP binding partners (Figure 5A), whereas Rpl4 and Ndel1 are not. There are two particularly notable features of these data. First, the Fmr1-KO profiles display a clear reduction in the large, reproducible pauses manifested as “spikes” in the wild-type profiles. Second, as shown in the rightmost panels of Figures 5A and 5B, there is a reproducible, overall reduction in the CV along the gene body that is not explained simply by the reduction in large pauses. Figures 5C and 5D show the same analysis for two genes with a significant increase in RFApm in Fmr1-KO mice. Rpl4 is a TOP-motif gene, which is enriched among genes with an apparent increase in translation efficiency as described above, and the other (Ndel1) is not. For all four genes in Figure 5, we detect stereotyped pauses in the wild-type that are substantially ablated in the KO. We also find a reproducible reduction in cumulative CV along the gene body, suggesting a smoother overall translocation process for the ribosome in the brain of Fmr1-KO mice.
Although many of the high-affinity FMRP binding partners and 5′ TOP motif-containing mRNAs display decreased and increased RFApm, respectively, nearly all of these genes exhibit reduced pausing in Fmr1-KO mice (Figure S5G). Given that FMRP binding partners and non-targets display similar reduction of pausing in Fmr1-KO mice, we also asked whether FMRP binding partners and non-targets exhibit baseline stalling differences in wild-type samples. As shown in Figure S5H, pause scores for FMRP binding partners are only ~5% higher than the remaining transcripts in the wild-type samples on average.
DISCUSSION
Previous studies have shown that FMRP associates with polysomes and the protein-CDS of a large number of transcripts (Brown et al., 2001; Stefani et al., 2004). HITS-CLIP data indicate that FMRP has particularly high affinity for mRNAs involved in synaptic activity and appears to act as a translational brake, stalling ribosomes on these transcripts (Darnell et al., 2011). Prior work has also revealed interactions between FMRP and the translation initiation machinery (Napoli et al., 2008; Santini et al., 2017). Nonetheless, genome-wide measurements of protein synthesis with the resolution to analyze both translation elongation and RFApm have not been undertaken in the brains of Fmr1-KO mice.
We characterized the translational landscape in the cortex of Fmr1-KO mice at a crucial time in post-natal brain development. By P24, the mouse brain has reached its peak synaptic density, and significant pruning of excitatory synapses is taking place, a process known to be dysregulated broadly in autism spectrum disorders (Tang et al., 2014) and specifically in FXS (Comery et al., 1997; He and Portera-Cailliau, 2013). Loss of FMRP-mediated regulation of protein synthesis may be critically linked to the synaptic plasticity and dendritic spine phenotypes observed in FXS (Darnell and Klann, 2013). We discovered a remarkably uniform trend in the RF abundance of FMRP’s high-affinity binding partners with the top 200 FMRP-bound transcripts exhibiting decreased RFApm in Fmr1-KO mice (Figure 2B). This result is surprising because proteins encoded by many of these mRNAs have been shown to be more highly expressed in Fmr1-KO mice (Tang et al., 2015). Reduction in RFApm was not a global effect; for example, the 5′ TOP motif-containing mRNAs, which are comprised mainly of ribosomal protein- and translation factor-encoding transcripts, trended toward increased RFApm with Fmr1 loss (Figure 2C). These genes are known to be controlled at the level of translation initiation by 4E-BP and eIF4E, the latter of which is sequestered by an FMRP-mediated complex and so may be indirectly affected by FMRP loss.
Despite these clear patterns, RFApm is a complicated metric. In many studies, it is interpreted as a measure of translation efficiency that primarily reflects translation initiation. However, this interpretation assumes that initiation is rate limiting and elongation rates are uniform (Arava et al., 2005). Given the potential role of FMRP in regulating translation elongation (Darnell et al., 2011), the apparent reduction in ribosome density for FMRP’s high-affinity binding partners (Figure 2B) may actually result from a relaxation of translational stalling in the absence of FMRP. We took advantage of the nucleotide resolution of ribosome profiling and characterized the noise in wild-type and Fmr1-KO ribosome profiles with both codon motif- and gene-centric analyses. In both cases, we found a significant reduction in translational pausing across a large number of genes in Fmr1-KO mice (Figures 3 and 4). As a genome-wide snapshot of translation in the cortex of Fmr1-KO mice in vivo, our results expand on previous in vitro measurements of ribosome stalling on select mRNAs using puromycin run-off (Darnell et al., 2011) and elongation rate using the ribosome transit time assay (Udagawa et al., 2013). We observed decreases in ribosomal pausing for the FMRP high-affinity binding partners, which exhibited a reduction in RFApm, and for the 5′ TOP motif-containing mRNAs, which showed an increase in RFApm (Figure S5G). We note that our results do not formally rule out the possibility that the FMRP-associated mRNAs are also differentially regulated at the level of translation initiation. However, these results are consistent with a model in which FMRP loss dysregulates ribosomal pausing across a large number of transcripts, and that competition between initiation- (e.g., through FMRP-mediated sequestration of EIF4E) and elongation-level regulation results in disparate alterations in RFApm for certain genes.
Neither our data nor Darnell et al., (2011) study, in which the high-affinity CLIP targets were identified, exclude the possibility that FMRP binds and may influence more transcripts than this limited set. In fact, the increases in the rate of protein synthesis in the Fmr1-KO mouse brain in vivo (Qin et al., 2005) and in fibroblasts sourced from FXS patients and Fmr1-KO mice (Jacquemont et al., 2018), as well as in the ribosome transit rate described in Fmr1-KO mouse brain lysates (Udagawa et al., 2013), are measured globally and are consistent with a broader set of FMRP targets. In addition to fibroblasts lacking expression of the neuronal genes comprising the majority of the CLIP-seq dataset, PAR-CLIP studies in HEK293 cells overexpressing FMRP revealed thousands of FMRP binding partners (Ascano et al., 2012) that only partially overlap the whole set of 842 cortical CLIP-seq targets. Our data support a model in which FMRP interacts simultaneously with its target mRNA (Darnell et al., 2011) and with the ribosome, as described for Drosophila FMRP and ribosomal proteins L5 and L11 (Ishizuka et al., 2002), and more recently validated via cryogenic electron microscopy (cryo-EM) for an N-terminal Drosophila fragile X mental retardation protein (dFMRP) fragment (Chen et al., 2014), as well as in earlier co-sedimentation studies in mammalian cells (Feng et al., 1997; Khandjian et al., 1996). In this context, FMRP may stabilize ribosome pausing events and function as a molecular brake on translation as previously suggested (Darnell and Klann, 2013).
We suggest that therapeutic strategies for FXS should carefully consider the consequences of globally altered protein synthesis. Recent evidence suggests that enhanced translation of certain mRNAs in Fmr1-KO mice may represent compensatory changes, and that enhancing their function may ameliorate disease phenotypes (Thomson et al., 2017). Importantly, our study does not assess whether translational alterations in Fmr1-KO mice are caused by direct loss of FMRP function or by secondary effects arising due to continued absence of FMRP during neural development. A critical aspect is that neuronal activity may be tightly coupled to translational regulation. Several recent studies found translational repression of neuronal mRNAs following fear conditioning in vivo (Cho et al., 2015) and of FMRP binding partners following KCl depolarization in vitro (Dalal et al., 2017). Given extensive evidence of cortical hyperexcitability (Gibson et al., 2008; Hays et al., 2011) and dys-regulation of GABAergic neurotransmission in Fmr1-KO mice (Paluszkiewicz et al., 2011), it is possible that the downregulation of RFApm we observed in FMRP binding partners (Figure 2C) is linked to increased cortical activity. We found enhanced translation of immediate-early genes such as Arc and Fos, as well as decreased translation of Gabra2 (Figure 1C), consistent with previous reports of decreased gamma-Amino-butyric acid A (GABAA) receptor expression and GABA dysfunction in FXS (Braat et al., 2015; D’Hulst et al., 2006). Future studies using knockdown or conditional KO of Fmr1 may be necessary to disentangle the primary effects of acute FMRP loss from secondary alterations in neuronal physiology. Nonetheless, our study shows that Fmr1 loss leads to widespread alterations in mRNA translation, particularly at the level of elongation, during the developmental period of cortical synaptic refinement.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for reagents may be directed to and will be fulfilled by the Lead Contact, Peter A. Sims (pas2182@cumc.columbia.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
All mice were in C57BL/6J background. Camk2a-cre heterozygotes were crossed to homozygous RiboTag mice (Sanz et al., 2009) to obtain Rpl22tm1.1Psam, Camk2a-cre+/− mice, which were further crossed to Fmr1−/y mice (Jax 00325) to generate Fmr1−/y; Rpl22tm1.1Psam,Camk2a-cre+/− females. The Fmr1−/y; Rpl22tm1.1Psam;Camk2a-cre+/− females were then bred to Rpl22tm1.1Psam males to obtain Fmr1−/y; Rpl22tm1.1Psam,Camk2a-cre+/− mice and Rpl22tm1.1Psam,Camk2a-cre+/− control littermates.
Throughout the manuscript, we refer to the Fmr1−/y; Camk2a-Cre+: Rpl22tm1.1Psam (Fmr1 mutants) mice as Fmr1-KO and the Rpl22tm1.1Psam;Camk2a-cre+/− mice as wild-type. All experiments were conducted at postnatal day 24 (P24) on the total frontal cortex of the mouse without immuno-precipitation step. All mouse experimental procedures were reviewed and approved by Columbia University Medical Center Institutional Animal Care and Use Committee (IACUC Protocol AAAR2500).
The mice were genotyped with the following primers for Cre: GCG GTC TGG CAG TAA AAA CTA TC (transgene), GTG AAA CAG CAT TGC TGT CAC TT (transgene), CTA GGC CAC AGA ATT GAA AGA TCT (internal positive control forward), GTA GGT GGA AAT TCT AGC ATC ATC C (internal positive control reverse), and the following primers for RiboTag: GGG AGG CTT GCT GGA TAT G (forward), TTT CCA GAC ACA GGC TAA GTA CAC (reverse). The primers for Fmr1KO mice were: CAC GAG ACT AGT GAG ACG TG (mutant forward); TGT GAT AGA ATA TGC AGC ATG TGA (wild-type forward); CTT CTG GCA CCT CCA GCT T (reverse)
METHOD DETAILS
Tissue Processing for RNA Sequencing and Ribosome Profiling
Mice (n = 10 in total) were sacrificed by cervical dislocation and frozen brain tissue samples were
processed as described previously (Hornstein et al., 2016). Details of the animals used for RNA-Seq and Ribo-Seq experiments and analysis are given in a table format at the end of this section. Brain tissue was processed as described previously (Hornstein et al., 2016). Briefly, snap-frozen frontal cortex (n = 4 mice/genotype for RNA-Seq and n = 3 mice/genotype for ribosome profiling, sample weight ~25mg) was disrupted using a Dounce homogenizer in 1mL of polysome lysis buffer (20 mM Tris-HCl pH 7.5, 250 mM NaCl, 15 mM MgCl2, 1mM DTT, 0.5% Triton X-100, 0.024 U/ml TURBO DNase, 0.48 U/mL RNasin, and 0.1 mg/ml cycloheximide). Homogenates were clarified by centrifugation at 14,000 × g for 10 min at 4°C. Supernatant was collected and used for RNA-Seq and ligation-free ribosome profiling.
Details of replicates used in RNA-Seq and Ribo-Seq experiments
| Sample No. | RNA-Seq? | Ribo-Seq? |
|---|---|---|
| WT1 | Yes | Yes |
| WT2 | Yes | Yes |
| WT3 | Low-complexity library; excluded | Yes |
| WT4 | Yes | No |
| WT5 | Yes | No |
| KO1 | Yes | Yes |
| KO2 | Yes | Yes |
| KO3 | Low-complexity library; excluded | Yes |
| KO4 | Yes | No |
| KO5 | Yes | No |
RNA-Seq Library Construction
Total RNA was isolated from brain lysates using a QIAGEN RNeasy kit and ribosomal RNA was depleted using the Ribo-Zero rRNA removal kit from Illumina according to the manufacturer’s instructions. rRNA depleted total RNA samples were converted to a strand-specific sequencing library using the NEBNext® Ultra Directional RNA Library Prep Kit from Illumina. There were a total of four RNA-Seq libraries generated for each genotype, with each library originating from a different animal. RNA-Seq libraries were quantified using Qubit fluorometer (ThermoFisher) and library size was measured using an Agilent Bioanalyzer.
Sequencing of eight RNA-Seq libraries was performed on an Illumina NextSeq 500 desktop sequencer with a read length of 75 bases. Approximately 20 to 50 million demultiplexed, pass-filtered, single-end reads for each sample were obtained.
Ligation-free Ribosome Profiling
Ribosome-footprints were isolated according to Ingolia et al. (2009) with minor modifications. Briefly, tissue lysates made from frontal cortex were treated with 750U of E. coli RNase I (Thermo Fisher) for ~40 minutes at room-temperature and monosomes were purified by 15%–50% sucrose density gradient centrifugation followed by fractionation using an Isco gradient fractionation system. Monosome samples were size-selected by gel electrophoresis and dephosphorylation reaction was done using T4 Polynucleotide kinase (NEB). Ligation-free ribosome profiling libraries were prepared from dephosphorylated foot-prints (~28–34 nucleotides in length) using a commercially available kit (SMARTer small RNA-Seq Library Preparation Kit, Clontech) following manufacturer’s instructions (Hornstein et al., 2016) . We performed library purification with AMPure XP beads (Beckman Coulter). Libraries were quantified using the Qubit dsDNA High-Sensitivity kit (Life Technologies) and library size was verified with the High-Sensitivity Bioanalyzer DNA chip (Agilent Technologies). Sequencing of six ribosome profiling libraries was done on an Illumina NextSeq 500 desktop sequencer with a read length of 50 bases. We obtained between 20 to 50 million demultiplexed, pass-filtered, single-end reads for each sample.
Polysome Profiling and qPCR Analysis
The clarified brain tissue lysates were loaded onto 15%–50% sucrose gradient and centrifuged at 37,000 RPM in a SW41 rotor for 3.5 hr at 4°C. Polysome gradients were fractionated and the optical density at 254 nm was continuously recorded using Isco-UA5 fluorescence/absorbance monitoring system. Fractions containing disomes to pentasomes were pooled together and designated as P1. Fractions heavier than pentasomes were pooled as P2 in our experiment. RNA was purified from fraction −80S, P1 and P2 using phenol-chloroform extraction followed by ethanol precipitation. Reverse transcription of RNAs and real time PCR experiments were performed according to Hornstein et al. (2016) with three probes representing genes with either high or low footprint density as found by ribosome profiling: Gabra2 (Mm00433435_m1, Cat no:4448892), Arc (Mm00487425_m1, Cat no:4453320), Fos (Mm01205647_g1, Cat no: 4453320). The qPCR data were analyzed by first computing a quantitative cycle number Cq for each gene for each of three technical replicates per biological replicate. We then averaged these Cq values across technical replicates. We then computed ΔCq values by subtracting the average Cq value for the gene Actb (Mm01205647_g1, Cat no: 4453320) from the average Cq value across technical replicates. For each gene, this gave us three biological replicate ΔCq values for each gene for wild-type and three biological replicate ΔCq values for each gene in the KO. In addition, this gave us a standard error in the mean (SEM) for each of these ΔCq values. Finally, we computed a ΔΔCq by averaging the three ΔCq for the wild-type biological replicates and subtracting the average ΔCq for the three KO biological replicates such that: ΔΔCq = ((WT ± SEMWT) - (KO ± SEMKO)) We used a two-sample Student’s t test on the ΔCq values across biological replicates to compute p values for the comparison of wild-type to KO. To obtain the error bars shown in Figure S2, we propagated the error in our wild-type and KO measurements for ΔΔCq such that: SEMΔΔCq = (SEMWT2 + SEMKO2)1/2 Samples were not paired for statistical testing. The standard methods described above for differential expression in qPCR were carried out using the Prism statistical software package.
QUANTIFICATION AND STATISTICAL ANALYSIS
High-Throughput Sequencing Data Processing
Bioinformatics analysis was performed following a protocol from Hornstein et al. (2016) (Ingolia et al., 2012) with minor modifications. Ribosome profiling libraries were processed by removing the first 4 and last 10 positions of each sequenced read with the following command to fastx-trimmer:
fastx_trimmer -f 4 -l 40 -Q33 -i INFILE -o OUTLFILE
following which we trimmed remaining poly(A) sequence from the 3′ end, discarding trimmed reads shorter than 25 nucleotides. Libraries were then depleted of ribosomal RNA by alignment to an rRNA reference library comprised of rRNA sequences from mm9 with bowtie2, allowing for one alignment error. Unaligned reads were retained and aligned to the mm10 assembly of the mouse genome and Gencode-annotated transcriptome with STAR (Dobin et al., 2013). Alignments to the exons and CDSs of genes were counted with the featureCounts (Liao et al., 2014) program from the subread suite (see Table S3), yielding between 4 and 10 million reads uniquely mapped to the CDS per ribosome profiling library.
Statistical Analysis of RNA Expression, RF Abundance, and RF Abundance per mRNA (RFApm)
We used DESeq2 (Love et al., 2014) to analyze differential expression from uniquely aligned RNA-Seq reads (see Table S4) and differential RF abundance from ribosome profiling reads that aligned uniquely to the CDS of each gene (see Table S5). We used the generalized linear model in RiboDiff (Zhong et al., 2017) to analyze differential ribosome RF per mRNA (RFApm). For this analysis, only reads that aligned uniquely to the CDS were used for both RNA-Seq and ribosome profiling. We used the Java implementation of gene set enrichment analysis (GSEA) (Subramanian et al., 2005) to assess the statistical enrichment of gene ontologies. Specifically, we pre-ranked each gene by fold change and used “classic” mode to compute normalized enrichment scores and corrected p values for gene sets in the MSigDB C5 gene ontology collection.
We additionally performed GSEA of the proteins differentially synthesized in cortical synaptosomes at P17 due to Fmr1 loss (Tang et al., 2015) in our RF and footprints per mRNA (RFApm) data, and also of the top 200 neuron-specific genes (Zhang et al., 2014) in our RNA-Seq data after Hornstein et al. (2016). Neither the up- nor downregulated proteins from the Tang et al. dataset exhibit significant enrichment or depletion in our data at either the level of RF or RFApm (FDR q > 0.05; Figure S3A, enrichment plots not shown). Differences in the ages of the mice involved (P17 versus P24 in our study) and the cellular subcompartment analyzed (synaptosome versus total cortical homogenate) may explain this lack of concordance between studies. Neuron-specific GSEA revealed no significant change as a function of Fmr1 expression (FDR q > 0.05; Figure S3B).
Codon Motif-Level Analysis of Pausing
Ribosome profiling libraries were first aligned to the transcriptome using the -quantmode TranscriptomeSAM option in STAR v2.5 as follows:
STAR–readFilesCommand zcat–genomeDir STAR_INDEX–runThreadN 12–outSAMtype BAM SortedByCoordinate–readFilesIn INFILE–outSAMprimaryFlag AllBestScore–outSAMattrIHstart 0–quantMode TranscriptomeSAM–outFileNamePrefix OUTFILE
Transcriptome-aligned libraries were then filtered by removing reverse-complemented (SAM flag 272 or 16), suboptimal, and non-CDS-aligned reads.
We chose one representative transcript and CDS for each gene by summing counts for all transcripts independently, then choosing the transcript with the highest sum of counts for each gene. To reduce reads from ~28–30nt footprints to A-site locations, we used the psite script from the plastid library for ribosome profiling analysis (Dunn and Weissman, 2016). This script calculates the location of a ribosomal P-site relative to the 5′ end of a footprint based on its length; increasing the calculated P-site offset by 3 nucleotides yields the A-site offset. We obtained codon occupancy profiles by summing over A-sites overlapping the 0, +1, and −1 nucleotide positions relative to the codon start, then merged them by summation across samples within either condition (wild-type or Fmr1-KO), collapsing six samples to two overall profiles with greatly increased coverage. We then limited the set of transcripts under consideration to those with mean coverage of at least 0.1 A-sites per codon for the first 150 codons in both profiles, yielding 8,967 total transcripts, and calculated pause scores for all but the first and last 10 codons within each.
Ribosome pause scores were calculated following the approach described by Woolstenhulme et al. (2015), modified to correct for potential differences in splicing across profiles in line with Ishimura et al. (2014). The pause score S of any codon x is calculated by the following equation:
Where c(x) is the number of A-sites aligned within codon x, BGt is equivalent to the mean number of A-sites per codon in the transcript to which codon x belongs (< c > ), and BGL/R are the median number of A-sites per codon for the five codons to the left and right of codon x, respectively. We calculated context-specific pause scores for every codon of every CDS by dividing the codon’s ribosome occupancy by the maximum of three values: the mean occupancy of the first 150 codons of the transcript and the median occupancies of the five codons 5′ and 3′ to the codon in question. To obtain a mean pause score for each amino acid, we averaged scores across all occurrences of codons encoding that amino acid residue; di- and tri-amino acids with a minimum of 100 occurrences across the transcripts considered were similarly summarized. For mono-, di-, and tri-amino acid datasets, we performed a Mann-Whitney U-test to determine statistical significance of the difference in the distributions of pause scores between genotypes.
Gene-Level Analysis of Translational Pausing
We used Ribo-TISH (Zhang et al., 2017) to determine the ribosome P-site offsets for each fragment length and P-site ribosome profiles for each transcript in our ribosome profiling data. For the initial quality control step, we used the following command:
ribotish quality -b BAMFILE -g GTF -p 16
followed by a prediction step with:
ribotish predict -b BAMFILE -g GTF -f GENOME_FASTA -o OUTPUT_FILE -p 16 -transprofile PROFILE_OUTPUT_FILE -framebest -seq -aseq
We then restricted our analysis to annotated ORFs, and for each isoform of each gene, we computed the mean coverage (number of RFs per codon) and the CV in coverage (standard deviation in the number of RFs per codon divided by mean). For each gene, we selected the isoform with the lowest CV. Isoforms with extremely non-uniform coverage, which can result from low usage or exclusion of a subset of exons, are typically not the dominantly expressed isoform. Finally, as described under Results, we fit Equation 1 to a plot of log2(CV) versus log2(mean coverage) to assess the genome-wide dependence of noise along the CDS on coverage using the curve_fit function in SciPy.
DATA AND SOFTWARE AVAILABILITY
All software are freely or commercially available. The accession number for the data reported in this paper is GEO: GSE114064.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, Peptides, and Recombinant Proteins | ||
| TURBO DNase | Thermo Fisher Scientific | Cat no: AM2238 |
| RNasin Ribonuclease Inhibitor | Promega | Cat no: N2515 |
| Cyoloheximide | Sigma | Cat no: C1988-1G |
| Triton X -100 | Sigma | CAS No: 9002-93-1, Pack Size: X100-100ML |
| RNase I | Thermo Fisher Scientific | Cat no: AM2294 |
| SUPERaseIN | Thermo Fisher Scientific | Cat no: AM2696 |
| T4 Polynucleotide kinase | NEB | Cat no: M0201S |
| Protease Inhibitor Cocktail | Sigma | Cat no: P8340-1ML |
| Acid-Phenol:Chloroform | Thermo Fisher Scientific | Cat no: AM9722 |
| GlycoBlue Co-precipitant | Thermo Fisher Scientific | Cat no: AM9516 |
| AMPure XP beads | Beckman Coulter | Cat no: A63881 |
| Critical Commercial Assays | ||
| RNeasy Mini Kit | QIAGEN | Cat no: 74104 |
| Ribo-Zero rRNA removal kit | Illumina | Cat no: MRZH11124 |
| NEBNext® Ultra Directional RNA Library Prep Kit | NEB | Cat no: E7420S |
| SMARTer small RNA-Seq Library Preparation Kit | Clontech | Cat no. 635029 |
| TaqMan Universal Maser Mix II, with UNG | Applied Biosystems | Cat no. 4440038 |
| High-Capacity RNA-to-cDNA kit | Applied Biosystems | Cat no. 4387406 |
| Qubit RNA HS Assay Kit | Thermo Fisher Scientific | Cat no: Q32852 |
| Qubit dsDNA HS Assay Kit | Thermo Fisher Scientific | Cat no: Q32854 |
| Agilent High Sensitivity DNA Kit | Agilent | Cat no: 5067–4627 |
| Agilent RNA 6000 Pico Kit | Agilent | Cat no: 5067–1513 |
| Agilent Small RNA kit | Agilent | Cat no: 5067–1548 |
| Deposited Data | ||
| Raw and analyzed data | This Paper | GEO:GSE114064 |
| Experimental Models: Organisms/Strains | ||
| Mouse: Camk2a-cre | The Jackson Laboratory | 005359 |
| Mouse: Rpl22tm1.1Psam | The Jackson Laboratory | 011029 |
| Mouse: Fmr1−/y | The Jackson Laboratory | 00325 |
| Mouse: Rpl22tm1.1Psam,Camk2a-cre+/− | This Paper | N/A |
| Mouse:Fmr1Vy; Rpl22tm1.1Psam,Camk2a-cre+/− | This Paper | N/A |
| Oligonucleotides | ||
| Gabra2,Taqman Gene Expression Assay | Applied Biosystems | Mm00433435_m1, Cat no:4448892 |
| Arc, Taqman Gene Expression Assay | Applied Biosystems | Mm00487425_m1, Cat no:4453320 |
| Fos, Taqman Gene Expression Assay | Applied Biosystems | Mm01205647_g1, Cat no: 4453320) |
| Software and Algorithms | ||
| bowtie2 | N/A | https://github.com/BenLangmead/bowtie2 |
| STAR | N/A | https://github.com/alexdobin/STAR |
| featureCounts | N/A | http://subread.sourceforge.net/ |
| plastid | N/A | https://plastid.readthedocs.io/en/latest/installation.html |
| Samtools | N/A | http://samtools.sourceforge.net/ |
| Ribo-TISH | N/A | https://github.com/zhpn1024/ribotish |
| Ribodiff | N/A | https://github.com/ratschlab/RiboDiff |
| DeSeq2 | N/A | https://github.com/mikelove/DESeq2 |
| GSEA | Broad Institute | http://software.broadinstitute.org/gsea |
Highlights.
Genome-wide snapshot of cortical translation in Fmr1-KO mice by ribosome profiling
Translational alterations of FMRP CLIP-seq targets, 5′ TOP motif-containing genes
Reduction in translational pausing in Fmr1-KO mice across many genes
ACKNOWLEDGMENTS
We acknowledge the JP Sulzberger Columbia Genome Center and the Institute for Comparative Medicine for technical support. P.A.S., G.T., and D.S. were supported by 345915 from the Simons Foundation. P.A.S. was supported by K01EB016071 from NIH/NIBIB. D.S. was supported by R01DA07418 from NIH/NIDA, R01MH108186 from NIH/NIMH, and the JPB Foundation. G.T. was supported by W81XWH-16–1-0263 from DOD and K01MH096956 from NIH/NIMH. N.H. was supported by F31NS089106 from NIH/NINDS.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found with this article online at https://doiorg/10.1016/j.celrep.2019.02.086.
REFERENCES
- Arava Y, Boas FE, Brown PO, and Herschlag D (2005). Dissecting eukaryotic translation and its control by ribosome density mapping. Nucleic Acids Res. 33, 2421–2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ascano M Jr., Mukherjee N, Bandaru P, Miller JB, Nusbaum JD, Corcoran DL, Langlois C, Munschauer M, Dewell S, Hafner M, et al. (2012). FMRP targets distinct mRNA sequence elements to regulate protein expression. Nature 492, 382–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bear MF, Huber KM, and Warren ST (2004). The mGluR theory of fragile X mental retardation. Trends Neurosci. 27, 370–377. [DOI] [PubMed] [Google Scholar]
- Braat S, D’Hulst C, Heulens I, De Rubeis S, Mientjes E, Nelson DL, Willemsen R, Bagni C, Van Dam D, De Deyn PP, and Kooy RF (2015). The GABAA receptor is an FMRP target with therapeutic potential in fragile X syndrome. Cell Cycle 14, 2985–2995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown V, Jin P, Ceman S, Darnell JC, O’Donnell WT, Tenenbaum SA, Jin X, Feng Y, Wilkinson KD, Keene JD, et al. (2001). Microarray identification of FMRP-associated brain mRNAs and altered mRNA translational profiles in fragile X syndrome. Cell 107, 477–487. [DOI] [PubMed] [Google Scholar]
- Chen E, Sharma MR, Shi X, Agrawal RK, and Joseph S (2014). Fragile X mental retardation protein regulates translation by binding directly to the ribo-some. Mol. Cell 54, 407–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho J, Yu NK, Choi JH, Sim SE, Kang SJ, Kwak C, Lee SW, Kim JI, Choi DI, Kim VN, and Kaang BK (2015). Multiple repressive mechanisms in the hippocampus during memory formation. Science 350, 82–87. [DOI] [PubMed] [Google Scholar]
- Comery TA, Harris JB, Willems PJ, Oostra BA, Irwin SA, Weiler IJ, and Greenough WT (1997). Abnormal dendritic spines in fragile X knockout mice: maturation and pruning deficits. Proc. Natl. Acad. Sci. USA 94, 5401– 5404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalal JS, Yang C, Sapkota D, Lake AM, O’Brien DR, and Dougherty JD (2017). Quantitative nucleotide level analysis of regulation of translation in response to depolarization of cultured neural cells. Front. Mol. Neurosci 10, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darnell JC, and Klann E (2013). The translation of translational control by FMRP: therapeutic targets for FXS. Nat. Neurosci 16, 1530–1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darnell JC, Van Driesche SJ, Zhang C, Hung KY, Mele A, Fraser CE, Stone EF, Chen C, Fak JJ, Chi SW, et al. (2011). FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell 146, 247–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Hulst C, De Geest N, Reeve SP, Van Dam D, De Deyn PP, Hassan BA, and Kooy RF (2006). Decreased expression of the GABAA receptor in fragile X syndrome. Brain Res. 1121, 238–245. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn JG, and Weissman JS (2016). Plastid: nucleotide-resolution analysis of next-generation sequencing and genomics data. BMC Genomics 17, 958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y, Absher D, Eberhart DE, Brown V, Malter HE, and Warren ST (1997). FMRP associates with polyribosomes as an mRNP, and the I304N mutation of severe fragile X syndrome abolishes this association. Mol. Cell 1, 109–118. [DOI] [PubMed] [Google Scholar]
- Gibson JR, Bartley AF, Hays SA, and Huber KM (2008). Imbalance of neocortical excitation and inhibition and altered UP states reflect network hyperexcitability in the mouse model of fragile X syndrome. J. Neurophysiol 100, 2615–2626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenblatt EJ, and Spradling AC (2018). Fragile X mental retardation 1 gene enhances the translation of large autism-related proteins. Science 361, 709–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross C, Chang CW, Kelly SM, Bhattacharya A, McBride SM, Danielson SW, Jiang MQ, Chan CB, Ye K, Gibson JR, et al. (2015). Increased expression of the PI3K enhancer PIKE mediates deficits in synaptic plasticity and behavior in fragile X syndrome. Cell Rep. 11, 727–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hays SA, Huber KM, and Gibson JR (2011). Altered neocortical rhythmic activity states in Fmr1 KO mice are due to enhanced mGluR5 signaling and involve changes in excitatory circuitry. J. Neurosci 31, 14223–14234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He CX, and Portera-Cailliau C (2013). The trouble with spines in fragile X syndrome: density, maturity and plasticity. Neuroscience 251, 120–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornstein N, Torres D, Das Sharma S, Tang G, Canoll P, and Sims PA (2016). Ligation-free ribosome profiling of cell type-specific translation in the brain. Genome Biol. 17, 149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou L, Antion MD, Hu D, Spencer CM, Paylor R, and Klann E (2006). Dynamic translational and proteasomal regulation of fragile X mental retardation protein controls mGluR-dependent long-term depression. Neuron 51, 441–454. [DOI] [PubMed] [Google Scholar]
- Hsieh AC, Liu Y, Edlind MP, Ingolia NT, Janes MR, Sher A, Shi EY, Stumpf CR, Christensen C, Bonham MJ, et al. (2012). The translational landscape of mTOR signalling steers cancer initiation and metastasis. Nature 485, 55–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia NT, Ghaemmaghami S, Newman JR, and Weissman JS (2009). Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science 324, 218–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia NT, Brar GA, Rouskin S, McGeachy AM, and Weissman JS (2012). The ribosome profiling strategy for monitoring translation in vivo by deep sequencing of ribosome-protected mRNA fragments. Nat. Protoc 7, 1534–1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishimura R, Nagy G, Dotu I, Zhou H, Yang XL, Schimmel P, Senju S, Nishimura Y, Chuang JH, and Ackerman SL (2014). RNA function. Ribo-some stalling induced by mutation of a CNS-specific tRNA causes neurode-generation. Science 345, 455–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishizuka A, Siomi MC, and Siomi H (2002). A Drosophila fragile X protein interacts with components of RNAi and ribosomal proteins. Genes Dev. 16, 2497–2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacquemont S, Pacini L, Jonch AE, Cencelli G, Rozenberg I, He Y, D’Andrea L, Pedini G, Eldeeb M, Willemsen R, et al. (2018). Protein synthesis levels are increased in a subset of individuals with fragile X syndrome. Hum. Mol. Genet 27, 2039–2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khandjian EW (1999). Biology of the fragile X mental retardation protein, an RNA-binding protein. Biochem. Cell Biol 77, 331–342. [PubMed] [Google Scholar]
- Khandjian EW, Corbin F, Woerly S, and Rousseau F (1996). The fragile X mental retardation protein is associated with ribosomes. Nat. Genet 12, 91–93. [DOI] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, and Shi W (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Napoli I, Mercaldo V, Boyl PP, Eleuteri B, Zalfa F, De Rubeis S, Di Marino D, Mohr E, Massimi M, Falconi M, et al. (2008). The fragile X syndrome protein represses activity-dependent translation through CYFIP1, a new 4E-BP. Cell 134, 1042–1054. [DOI] [PubMed] [Google Scholar]
- Paluszkiewicz SM, Martin BS, and Huntsman MM (2011). Fragile X syndrome: the GABAergic system and circuit dysfunction. Dev. Neurosci 33, 349–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin M, Kang J, Burlin TV, Jiang C, and Smith CB (2005). Postadoles-cent changes in regional cerebral protein synthesis: an in vivo study in the FMR1 null mouse. J. Neurosci 25, 5087–5095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter JD, and Sonenberg N (2005). Regulation of cap-dependent translation by eIF4E inhibitory proteins. Nature 433, 477–480. [DOI] [PubMed] [Google Scholar]
- Santini E, Huynh TN, Longo F, Koo SY, Mojica E, D’Andrea L, Bagni C, and Klann E (2017). Reducing eIF4E-eIF4G interactions restores the balance between protein synthesis and actin dynamics in fragile X syndrome model mice. Sci. Signal 10, eaan0665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanz E, Yang L, Su T, Morris DR, McKnight GS, and Amieux PS (2009). Cell-type-specific isolation of ribosome-associated mRNA from complex tissues. Proc. Natl. Acad. Sci. USA 106, 13939–13944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schütt J, Falley K, Richter D, Kreienkamp HJ, and Kindler S (2009). Fragile X mental retardation protein regulates the levels of scaffold proteins and glutamate receptors in postsynaptic densities. J. Biol. Chem 284, 25479–25487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma A, Hoeffer CA, Takayasu Y, Miyawaki T, McBride SM, Klann E, and Zukin RS (2010). Dysregulation of mTOR signaling in fragile X syndrome. J. Neurosci 30, 694–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefani G, Fraser CE, Darnell JC, and Darnell RB (2004). Fragile X mental retardation protein is associated with translating polyribosomes in neuronal cells. J. Neurosci 24, 7272–7276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang B, Wang T, Wan H, Han L, Qin X, Zhang Y, Wang J, Yu C, Berton F, Francesconi W, et al. (2015). Fmr1 deficiency promotes age-dependent alterations in the cortical synaptic proteome. Proc. Natl. Acad. Sci. USA 112, E4697–E4706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang G, Gudsnuk K, Kuo SH, Cotrina ML, Rosoklija G, Sosunov A, Sonders MS, Kanter E, Castagna C, Yamamoto A, et al. (2014). Loss of mTOR-dependent macroautophagy causes autistic-like synaptic pruning deficits. Neuron 83, 1131–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson SR, Seo SS, Barnes SA, Louros SR, Muscas M, Dando O, Kirby C, Wyllie DJA, Hardingham GE, Kind PC, et al. (2017). Cell-type- specific translation profiling reveals a novel strategy for treating fragile X syndrome. Neuron 95, 550–563.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thoreen CC, Chantranupong L, Keys HR, Wang T, Gray NS, and Sabatini DM (2012). A unifying model for mTORC1-mediated regulation of mRNA translation. Nature 485, 109–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udagawa T, Farny NG, Jakovcevski M, Kaphzan H, Alarcon JM, Anil-kumar S, Ivshina M, Hurt JA, Nagaoka K, Nalavadi VC, et al. (2013). Genetic and acute CPEB1 depletion ameliorate fragile X pathophysiology. Nat. Med 19, 1473–1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolstenhulme CJ, Guydosh NR, Green R, and Buskirk AR (2015). High-precision analysis of translational pausing by ribosome profiling in bacteria lacking EFP. Cell Rep. 11, 13–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zalfa F, Giorgi M, Primerano B, Moro A, Di Penta A, Reis S, Oostra B, and Bagni C (2003). The fragile X syndrome protein FMRP associates with BC1 RNA and regulates the translation of specific mRNAs at synapses. Cell 112, 317–327. [DOI] [PubMed] [Google Scholar]
- Zhang P, He D, Xu Y, Hou J, Pan BF, Wang Y, Liu T, Davis CM, Ehli EA, Tan L, et al. (2017). Genome-wide identification and differential analysis of translational initiation. Nat. Commun 8, 1749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O’Keeffe S, Phatnani HP, Guarnieri P, Caneda C, Ruderisch N, et al. (2014). An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci 34, 11929–11947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YQ, Bailey AM, Matthies HJ, Renden RB, Smith MA, Speese SD, Rubin GM, and Broadie K (2001). Drosophila fragile X-related gene regulates the MAP1B homolog Futsch to control synaptic structure and function. Cell 107, 591–603. [DOI] [PubMed] [Google Scholar]
- Zhong Y, Karaletsos T, Drewe P, Sreedharan VT, Kuo D, Singh K, Wendel HG, and Ra¨tsch G (2017). RiboDiff: detecting changes of mRNA translation efficiency from ribosome footprints. Bioinformatics 33, 139–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All software are freely or commercially available. The accession number for the data reported in this paper is GEO: GSE114064.