

Abstract
Oxidized low-density lipoprotein (oxLDL) and associated oxidized phosphocholine-headgroup phospholipids (oxPCs) activate blood platelets through ligation of the scavenger receptor CD36. Previously, we found that oxLDL stimulated phosphorylation of phospholipase Cγ2 (PLCγ2). However, the functional relevance of PLCγ2 phosphorylation in oxLDL-mediated platelet hyperactivity remained elusive. Here, we set out to explore the functional importance of PLCγ2 in oxLDL-mediated platelet activation using human and genetically modified murine platelets. The CD36-specific oxidized phospholipid (oxPCCD36) triggered the generation of reactive oxygen species (ROS) in platelets under static and arterial flow conditions. The ROS generation in response to oxPCCD36 was sustained for up to 3 h but ablated in CD36- and PLCγ2-deficient platelets. The functional importance of ROS generation in response to atherogenic lipid stress was examined through measurement of P-selectin expression. OxPCCD36 induced P-selectin expression, but required up to 60 min incubation, consistent with the timeline for ROS generation. P-selectin expression was not observed in CD36- and PLCγ2-deficient mice. The ability of oxPCCD36 and oxLDL to stimulate P-selectin expression was prevented by incubation of platelets with the ROS scavenger N-acetyl-cysteine (NAC) and the NOX-2 inhibitor gp91ds-tat, but not with the NOX-1 inhibitor ML171. In summary, we provide evidence that prolonged exposure to oxLDL-associated oxidized phospholipids induces platelet activation via NOX-2-mediated ROS production in a CD36- and PLCγ2-dependent manner.
Keywords: OxLDL, oxPCCD36, platelet hyperactivity, PLCγ2, reactive oxygen species
Introduction
Studies in mice and humans suggest strongly that dyslipidemia induces a prothrombotic phenotype in which platelet hyperactivity plays a major role (1–3). Dyslipidemia is characterized by increased levels of atherogenic oxidized lipid-rich particles, which are proposed as potential mediators of altered platelet function (2–4). Oxidized low-density lipoprotein (oxLDL) and oxidized phosphocholine-headgroup phospholipids (oxPCs) represent sterile danger-associated molecular patterns that promote platelet hyperactivity in dyslipidemia and coronary artery disease (2,3,5). Our work, and that of others, has shown that the scavenger receptor CD36 acts to functionally link oxLDL and oxPCs to platelet activation (2,6–9). However, the detailed mechanisms driving platelet activation in response to the oxLDL–CD36 signaling node are yet to emerge. We recently demonstrated that NOX-2-dependent reactive oxygen species (ROS) production by oxLDL and oxPCs leads to CD36-mediated platelet hyperactivity by reducing sensitivity to cGMP signaling (6). Importantly, intracellular ROS serve as essential cell-signaling molecules that drive platelet aggregation, P-selectin expression, platelet–neutrophil interactions, and TxB2 production (10–13). Previously, we found that the oxLDL–CD36 signaling node is linked to the phosphorylation and activation of PLCγ2 and, therefore, hypothesized that PLCγ2 activation is required for oxLDL-mediated ROS induction (6,7,13). The aim of this study was to determine the role of PLCγ2 in oxLDL- and oxPCCD36-induced ROS production and to investigate its functional relevance.
Methods
Materials
Native LDL (nLDL) was isolated from drug-free, healthy donors and oxidized as previously described (14). P-Selectin was from BD Bioscience (Oxford, UK), and oxPCCD36 (KoDiA-PC), 1-palmitoyl-2-arachidonoyl-sn-phosphatidylcholine (PAPC), and anti-pPLCγ2 1217 antibody were from Santa Cruz (Wembley, UK). E06 anti-oxPC antibody was from Avanti Polar Lipids (Alabama, USA). Anti-β-tubulin was from Merck Millipore (Watford, UK). ROS detection kit and N-acetyl-cysteine (NAC) was from Enzo Life Sciences (Exeter, UK). ML171 was from Tocris (Bristol, UK). GP91ds-tat was from AnaSpec (Fremont, USA). Collagen was from Takeda (Wien, Austria). Thrombin and indomethacin were from Sigma (Munich, Germany).
Immunoblotting
All human and animal work was approved by the Hull York Medical School Ethics Committee. Human blood was taken from drug-free volunteers. Human and murine platelets (5 × 108 platelets/ml) from wild-type (WT) and PLCγ2-deficient mice were isolated and lysed as described previously (13). Lysates were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to polyvinylidene difluoride (PVDF) membranes. Membranes were blocked with 5% milk and then incubated with anti-beta tubulin (1:1000) or anti-PLCγ2 pTyr 1217 (1:1000) (6). Membranes were developed and analyzed using a LICOR CLx Imaging System (Cambridge, UK). For dot blots, 3 µg of nLDL and oxLDL were loaded onto a PVDF membrane and blotted for anti-oxPC by E06 (1:1000).
Experimental animals
PLCγ2−/- and CD36−/- mice were provided by Professor S. Watson (University of Birmingham, UK) and Professor M. Febbriao, (University of Alberta, Canada), respectively, with WT (C57Bl/6) animals from Charles River (Kent, UK). All mice were used at 8 weeks of age.
Flow cytometry
Isolation of human and murine platelets was performed as described previously (6). For flow cytometry experiments, platelet-rich plasma (PRP) was incubated for 1 h in the presence or absence of oxLDL (50 µg/ml), nLDL (50 µg/ml), oxPCCD36 (25 µM), or PAPC (25 µM). In some cases, PRP was preincubated with NAC (5 mM), gp91ds-tat (2 µM), and ML171 (5 µM) for 30 min and P-selectin expression was analyzed using a BD LSRFortessa Cytometer.
Analysis of ROS generation
Washed murine platelets (2 × 108 platelets/ml) were incubated with a previously validated ROS Detection Probe (5 µM) at 37°C for 30 min (Enzo Life Sciences, Exeter, UK) and pretreated with oxPCCD36 or PAPC (5–25 µM) for 15 min before reconstitution with autologous red blood cells (50/50; v/v) (6). Blood was perfused through fibrinogen (1 mg/ml)-coated biochips (Cellix Ltd.) at arterial shear 1000 s−1 for 2 min and analyzed for surface coverage and mean fluorescence intensity using ImageJ v2.0. Alternatively, washed murine platelets (1 × 108 platelets/ml) were incubated with an ROS Detection Probe and oxPCCD36 for up to 3 h in suspension and fluorescence was measured using a microplate reader (Tecan Group Ltd.).
Statistics
Data are presented as means ± standard error of the mean of at least three independent experiments. Differences between groups were assessed using the Student’s t-test, Mann–Whitney U-test, or ANOVA and statistical significance taken at p ≤ 0.05.
Results
Oxidized LDL and associated oxidized phospholipids induce PLCγ2 phosphorylation
Incubation of human platelets with oxLDL (50 µg/ml), but not nLDL (50 µg/ml), led to a significant phosphorylation of PLCγ2 (Figure 1(ai) and (aii)). To confirm these findings in murine platelets, we used oxPCCD36, an oxidized phospholipid present in oxLDL (15), to account for potential differences in sensitivity between human and murine platelets to human oxLDL. Concordantly, oxPCCD36 (25-50μM), but not the non-oxidized lipid PAPC (25-50μM) mimicked oxLDL induced PLCγ2 phosphorylation on human and murine platelets. The presence of oxPC within our oxLDL preparation was confirmed using the oxPC-specific antibody (E06) (Figure 1(d)) (16).
Figure 1.
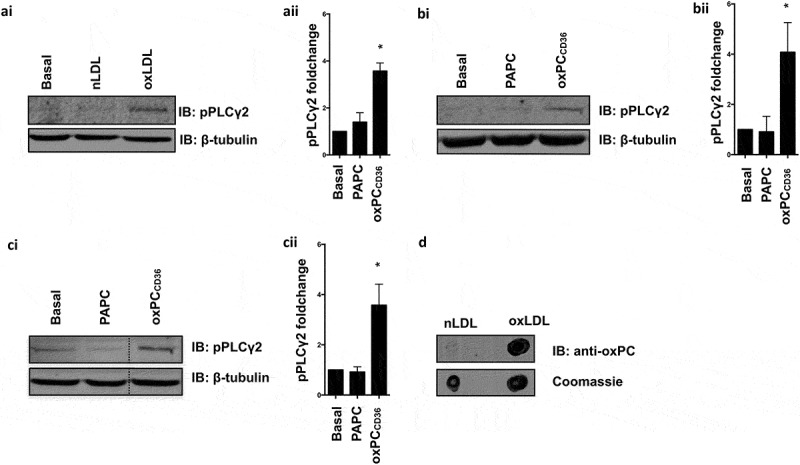
Oxidized LDL and oxidized phospholipids induce phosphorylation of PLCγ2 in human and murine platelets.
(ai) Washed human platelets (5 × 108 platelets/ml) incubated with apyrase, indomethacin, EGTA, and tirofiban were treated with nLDL (50 µg/ml) or oxLDL (50 µg/ml) for 2 min. Lysates were separated by SDS-PAGE and immunoblotted for pPLCγ2-tyr1217 (1:1000) and β-tubulin (1:1000). (aii) Quantification of three independent experiments, *p < 0.05. (bi) Human platelets treated with oxPCCD36 (50 µM) or PAPC (50 µM) for 15 min analyzed as previously described. (bii) Quantification of three independent experiments, *p < 0.05. (ci) Murine platelets treated with oxPCCD36 (25 µM) or PAPC (25 µM) for 15 min and analyzed as previously described. (cii) Quantification of three independent experiments, *p < 0.05. All immunoblots are representative of three independent experiments. (d) nLDL (3 µg) and oxLDL (3 µg) blotted for oxidized phospholipids by anti-oxPC antibody E06 (1:1000).
oxPCCD36 induces sustained ROS production in a CD36- and PLCγ2-specific manner
We next explored the functional importance of this phosphorylation event using platelets from genetically modified mice lacking PLCγ2 (Figure 2(a)). Focusing on CD36-dependent signaling, we evaluated the role of PLCγ2 in ROS production under flow and static conditions, which we have shown to be important for platelet hyperactivity (6,7). The perfusion of whole blood from WT or PLCγ2-deficient mice over immobilized fibrinogen led to similar levels of platelet adhesion, which was unaltered by 15 min incubation with oxPCCD36 (Figure 2(b)). However, treatment of blood with oxPCCD36, but not PAPC, led to early accumulation of ROS in WT animals. In contrast, ROS generation in response to oxPCCD36 was blunted significantly in PLCγ2-deficient mice (Figure 2(c)), suggesting that oxPCCD36 induces ROS generation in a PLCγ2-dependent manner. We next quantified ROS production in suspended platelets. Unlike the rapid and extensive ROS generation by collagen and thrombin (Supplementary Figure S1), OxPCCD36 (25 µM) induced slow and sustained ROS production for up to 3 h in WT platelets (longest time tested; p < 0.001) (Figure 2(di) and (dii)). In contrast with OxPCCD36, the unoxidized phospholipid PAPC did not induce ROS production in WT platelets (Supplementary Figure S2). Treatment of platelets deficient in either CD36 or PLCγ2 with oxPCCD36 did not lead to ROS production above basal levels. To confirm that genetic deletion of CD36 and PLCγ2 did not interfere with the ability or capacity of platelets to produce ROS, platelets were treated with pyocyanin (250 µM) that stimulates ROS production via an acute cyclic non-enzymatic reduction by NAD(P)H (17). ROS generation in response to pyocyanin was not affected by the absence of CD36 or PLCγ2 (Figure 2(e)), suggesting that oxPCCD36-induced ROS production specifically requires both CD36 and downstream activation of PLCγ2.
Figure 2.
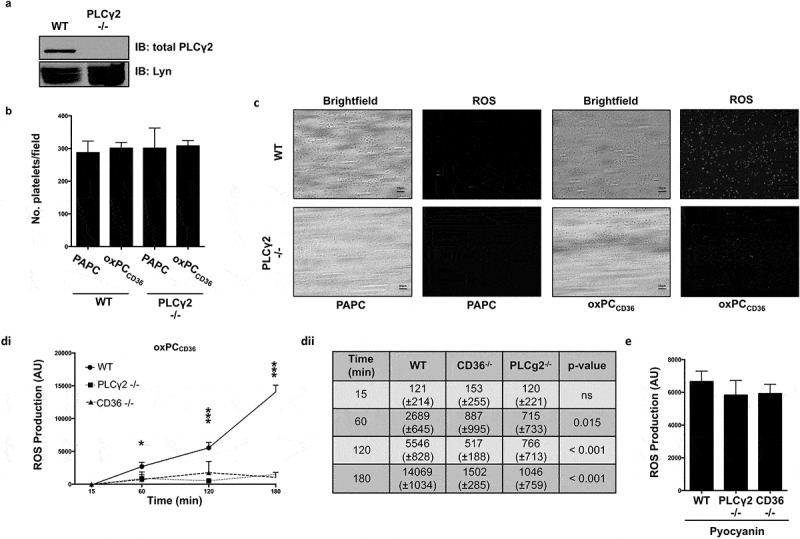
The oxidized phospholipid induces ROS generation in murine platelets in a CD36- and PLCγ2-dependent manner.
(a) Washed murine platelets from WT and PLCγ2−/- animals were lysed and blotted for total PLCγ2 (1:1000) and Lyn (1:1000). (b) Wild-type and PLCγ2−/- murine platelets were stained with ROS-detection probe and then treated with oxPCCD36 or PAPC (5 µM). Reconstituted blood was perfused through fibrinogen-coated capillary tubes, and images of adherent platelets were then taken under bright-field microscopy and the number of adherent platelets enumerated (n = 3). (c) As in (b), except platelets were visualized by fluorescence microscopy at 60x magnification. (di) Washed murine platelets (1 × 108 platelets/ml) from WT, CD36−/-, and PLCγ2−/- animals were incubated with the ROS-detection probe for 30 min at 37°C, then treated with oxPCCD36 (25 µM) for up to 180 min, and fluorescence measured at 520 nm (n = 3). Data expressed as ROS production above N-acetyl-cysteine-treated platelets. *p < 0.05 and ***p < 0.001 compared to N-acetyl-cysteine-treated platelets. (dii) Quantification of ROS production expressed as AU over time. ROS expressed as AU. (e) Washed murine platelets (1 × 108 platelets/ml) from WT, CD36−/-, and PLCγ−/- animals were treated for 1 h with pyocyanin (250 µM) and subsequent fluorescence measured and presented as in (d) (n = 3).
oxLDL and associated oxPCCD36 induce P-selectin expression by intracellular production of ROS
Having established that PLCγ2 was essential for intracellular ROS production in response to oxidized lipid stress, we next examined how this affected platelet activation. Incubation of WT murine platelets with oxPCCD36 (25 µM), in the absence of other agonists, caused a significant increase in P-selectin expression (p = 0.015), while PAPC (25 µM) had no effect (Figure 3(a)). Again, the ability of the oxidized phospholipid to stimulate P-selectin was lost in CD36- and PLCγ2 deficient platelets. In contrast, the stimulatory effects of ADP were similar in all genotypes. Interestingly, oxPCCD36-induced P-selectin expression required extended incubation times and was detected after 60 min incubation. We next explored the mechanism of oxPCCD36-induced P-selectin expression. Since the expression of P-selectin in response to oxPCCD36 occurred in the same time frame as that of ROS generation (Figure 2(dii)), we hypothesized that the two observations may be linked. To explore this hypothesis, we used the general ROS scavenger NAC (5 mM). Pretreatment of WT platelets with NAC caused a reduction of approximately 80% in oxPCCD36-stimulated P-selectin expression (Figure 3(b), p = 0.019). This is in contrast to the classical hemostatic agonists, collagen (5 µg/ml) and thrombin (0.05 U/ml), where inhibition by NAC induced approximately a 5–10% reduction in P-selectin expression and the secondary agonist ADP (10 µM) that could be reduced by approximately 25–30% (Supplementary Figure S3). These data suggest that within platelets, the sustained generation of ROS response to oxPC is required for subsequent expression of P-selectin but are less important for hemostatic agonists. To test whether cyclooxygenases (COX) are a potential source of oxPCCD36-induced ROS production, we analyzed ROS production in the presence of the pan-COX inhibitor indomethacin (10 µM) and subsequent stimulation by oxPCCD36. ROS generation was not affected by COX inhibition confirming that oxPCCD36-induced ROS generation is COX independent (Supplementary Figure S4). Having established that oxidized phospholipids induced ROS-dependent P-selectin expression in murine platelets, we examined its relevance in human platelets. Treatment of human platelets with oxLDL (50 µg/ml) led to a significant increase in P-selectin expression above basal levels (p < 0.001), but again required incubation times of up to 1 h. Consistent with the observation in murine platelets, P-selectin expression was blocked by NAC. Next, we examined the potential source of ROS using pharmacological inhibitors of NOX1 (ML171; 5 µM) and NOX2 (gp91ds-tat; 2 µM), employed under conditions that blunt platelet ROS production (6). OxLDL-induced P-selectin expression was ablated in the presence of NAC (p = 0.034) and gp91ds-tat (p = 0.027), but not ML171 (Figure 3(ci) and (cii)). These data confirm the previous observation and distinguish NOX2 as the likely the source of oxLDL-induced ROS production in human platelets.
Figure 3.
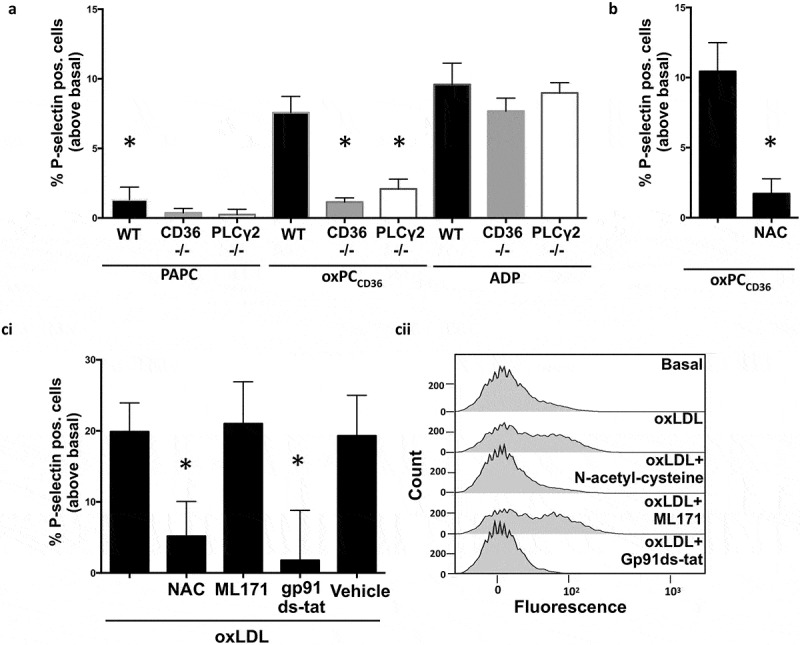
OxLDL- and oxPCCD36-induced P-selectin expression is linked to ROS production.
(a) PRP from WT, CD36−/-, and PLCγ2−/- animals was treated with PAPC/oxPCCD36 (25 µM) or ADP (10 µM) for 60 min before fixation and flow cytometric analysis for P-selectin surface expression. *p < 0.05 compared to oxPCCD36-treated WT (n = 3). (b) As in (a), except PRP from WT mice was pretreated with N-acetyl-cysteine (5 mM) for 30 min prior to incubation with oxPCCD36 (25 µM) (n = 3), *p < 0.05. (ci) Human PRP pretreated with N-acetyl-cysteine (5 mM), ML171 (5 µM), and gp91ds-tat (2 µM) for 30 min and stimulated with oxLDL (50 µg/ml) for 60 min before fixation and flow cytometric analysis for P-selectin surface expression (n = 3). *p < 0.05 compared to oxLDL-treated sample. (cii) Representative histograms of human platelet flow cytometric experiments.
Discussion
The aim of this study was to confirm the key functional role of PLCγ2 in the transduction of atherogenic lipid stress into platelet hyperactivity. Several groups, including ours, have indicated that PLCγ2 is downstream of oxidized lipid signaling; however, the functional consequence of this signaling event remained obscure (6,7,13). Here, we demonstrate that the PLCγ2 lies downstream of CD36 ligation and show that the lipase is required for oxPC-induced ROS production and subsequent platelet activation. To the best of our knowledge, this is the first observation linking lipid stress to PLCγ2- and ROS-induced platelet activation and suggests a new role of ROS in aberrant platelet activation. Nevertheless, in the current study, we focused on PLCγ2 exclusively and cannot preclude that other isoforms may play a role in oxPC-induced ROS generation. However, given the current lack of specific PLC inhibitors, further characterization of PLC isoforms in platelets remains difficult.
Interestingly, our experiments imply that hemostatic agonists such as collagen and thrombin rely on ROS production in only a limited capacity (Supplementary Figure S3). In contrast, in oxPCCD36- and oxLDL-treated platelets, ROS appears to be an essential driver that is obligatory for platelet activation. Therefore, when ROS production is either inhibited by gp91ds-tat or scavenged by NAC, oxidized lipid-induced platelet activation is reduced by approximately 80% in both human and murine platelets. This suggests a causal relationship between ROS and platelet activity in the context of oxidized lipid stress. Many studies have focused on the ability of oxLDL to synergize with hemostatic agonists to promote platelet hyperactivity (18–20). However, we show that prolonged incubation of platelets with oxLDL and oxPCCD36 alone leads to activation and contrasts with the rapid activation induced by hemostatic agonists. This observation demonstrates a clear distinction from ROS induction by physiological agonists, where ROS are induced rapidly by both thrombin and collagen within minutes (12). Recently, the presence of oxidatively modified LDL has been demonstrated in the circulation of patients with acute coronary syndromes and stroke (21–23). In this context, our data could suggest that the prolonged exposure of platelets to oxidized lipoproteins could contribute to the sustained low-level platelet hyperactivity observed in patients (24–27). Indeed, the ROS-dependent increase in P-selectin expression may underpin an increased capacity of platelets for PSGL-1-dependent interactions with leukocytes and subsequent endothelial cell activation (28). Therefore, the current findings extend our understanding of the oxLDL–CD36 node to present a potential mechanism for a platelet-mediated prothrombotic shift, facilitated by oxidized phospholipid ligation and subsequent intracellular ROS production.
Funding Statement
This project has been funded by the Rotations-Program of the Medical Faculty of RWTH Aachen University and the British Heart Foundation (PG/13/90/20578 and PG/12/49/29441).
Declaration of interest
The authors report no conflicts of interest.
Supplementary material
Supplemental data for this article can be accessed here.
References
- 1.Carvalho AC, Colman RW, Lees RS.. Platelet function in hyperlipoproteinemia. N Engl J Med. 1974;290:434–438. doi: 10.1056/NEJM197402212900805. [DOI] [PubMed] [Google Scholar]
- 2.Podrez EA, Byzova TV, Febbraio M, Salomon RG, Ma Y, Valiyaveettil M, Poliakov E, Sun M, Finton PJ, Curtis BR, Chen J, Zhang R, Silverstein RL, Hazen SL.. Platelet CD36 links hyperlipidemia, oxidant stress and a prothrombotic phenotype. Nat Med. 2007;13:1086–1095. doi: 10.1038/nm1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Colas R, Sassolas A, Guichardant M, Cugnet-Anceau C, Moret M, Moulin P, Lagarde M, Calzada C. LDL from obese patients with the metabolic syndrome show increased lipid peroxidation and activate platelets. Diabetologia. 2011;54:2931–2940. doi: 10.1007/s00125-011-2272-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Podrez EA, Poliakov E, Shen Z, Zhang R, Deng Y, Sun M, Finton PJ, Shan L, Gugiu B, Fox PL, Hoff HF, Salomon RG, Hazen SL. Identification of a novel family of oxidized phospholipids that serve as ligands for the macrophage scavenger receptor CD36. J Biol Chem. 2002;277:38503–38516. doi: 10.1074/jbc.M203318200. [DOI] [PubMed] [Google Scholar]
- 5.Tsimikas S, Brilakis ES, Miller ER, McConnell JP, Lennon RJ, Kornman KS, Witztum JL, Berger PB. Oxidized phospholipids, Lp(a) lipoprotein, and coronary artery disease. N Engl J Med. 2005;353:46–57. doi: 10.1056/NEJMoa043175. [DOI] [PubMed] [Google Scholar]
- 6.Magwenzi S, Woodward C, Wraith KS, Aburima A, Raslan Z, Jones H, McNeil C, Wheatcroft S, Yuldasheva N, Febbriao M, Kearney M, Naseem KM. Oxidized LDL activates blood platelets through CD36/NOX2-mediated inhibition of the cGMP/protein kinase G signaling cascade. Blood. 2015;125:2693–2703. doi: 10.1182/blood-2014-05-574491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wraith KS, Magwenzi S, Aburima A, Wen Y, Leake D, Naseem KM. Oxidized low-density lipoproteins induce rapid platelet activation and shape change through tyrosine kinase and Rho kinase–signaling pathways. Blood. 2013. doi: 10.1182/blood-2013-04-491688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen K, Febbraio M, Li W, Silverstein RL. A specific CD36-dependent signaling pathway is required for platelet activation by oxidized low-density lipoprotein. Circ Res. 2008;102:1512–1519. doi: 10.1161/CIRCRESAHA.108.172064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Korporaal SJ, Gorter G, Van Rijn HJ, Akkerman J-WN-W. Effect of oxidation on the platelet-activating properties of low-density lipoprotein. Arterioscler Thromb Vasc Biol. 2005;25:867–872. doi: 10.1161/01.ATV.0000158381.02640.4b. [DOI] [PubMed] [Google Scholar]
- 10.Seno T, Inoue N, Gao D, Okuda M, Sumi Y, Matsui K, Yamada S, Hirata K-I, Kawashima S, Tawa R, Imajoh-Ohmi S, Sakurai H, Yokoyama M. Involvement of NADH/NADPH oxidase in human platelet ROS production. Thromb Res. 2001;103:399–409. doi: 10.1016/s0049-3848(01)00341-3. [DOI] [PubMed] [Google Scholar]
- 11.Walsh TG, Berndt MC, Carrim N, Cowman J, Kenny D, Metharom P. The role of Nox1 and Nox2 in GPVI-dependent platelet activation and thrombus formation. Redox Biol. 2014;2:178–186. doi: 10.1016/j.redox.2013.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Delaney MK, Kim K, Estevez B, Xu Z, Stojanovic-Terpo A, Shen B, Ushio-Fukai M, Cho J, Du X. Differential roles of the NADPH-oxidase 1 and 2 in platelet activation and thrombosis. Arterioscler Thromb Vasc Biol. 2016. doi: 10.1161/ATVBAHA.116.307308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zimman A, Titz B, Komisopoulou E, Biswas S, Graeber TG, Podrez EA. Phosphoproteomic analysis of platelets activated by pro-thrombotic oxidized phospholipids and thrombin. PloS One. 2014;9. doi: 10.1371/journal.pone.0084488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wilkins GM, Leake DS. The oxidation of low density lipoprotein by cells or iron is inhibited by zinc. FEBS Lett. 1994;341:259–262. doi: 10.1016/0014-5793(94)80468-0. [DOI] [PubMed] [Google Scholar]
- 15.Podrez EA, Poliakov E, Shen Z, Zhang R, Deng Y, Sun M, Finton PJ, Shan L, Febbraio M, Hajjar DP, Silverstein RL, Hoff HF, Salomon RG, Hazen SL. A novel family of atherogenic oxidized phospholipids promotes macrophage foam cell formation via the scavenger receptor CD36 and is enriched in atherosclerotic lesions. J Biol Chem. 2002;277:38517–38523. doi: 10.1074/jbc.M205924200. [DOI] [PubMed] [Google Scholar]
- 16.Hörkkö S, Bird DA, Miller E, Itabe H. Monoclonal autoantibodies specific for oxidized phospholipids or oxidized phospholipid–protein adducts inhibit macrophage uptake of oxidized low-density lipoproteins. J Clin Invest. 1999. doi: 10.1172/JCI4533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hall S, McDermott C, Anoopkumar-Dukie S, McFarland AJ, Forbes A, Perkins AV, Davey AK, Chess-Williams R, Kiefel MJ, Arora D, Grant GD. Cellular effects of pyocyanin – a secreted virulence factor of Pseudomonas aeruginosa. Toxins. 2016;8:236. doi: 10.3390/toxins8080236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tornvall P, Chirkova L, Toverud KD, Horowitz JD, Chirkov Y. Native and oxidized low density lipoproteins enhance platelet aggregation in whole blood. Thromb Res. 1999;95:177–183. doi: 10.1016/S0049-3848(99)00036-5. [DOI] [PubMed] [Google Scholar]
- 19.Volf I, Roth A, Moeslinger T, Cooper J, Schmid W, Zehetgruber M, Koller E. Stimulating effect of biologically modified low density lipoproteins on ADP-induced aggregation of washed platelets persists in absence of specific binding. Thromb Res. 2000;97:441–449. doi: 10.1016/S0049-3848(99)00197-8. [DOI] [PubMed] [Google Scholar]
- 20.Ardlie NG, Selley ML, Simons LA. Platelet activation by oxidatively modified low density lipoproteins. Atherosclerosis. 1989;76:117–124. [DOI] [PubMed] [Google Scholar]
- 21.Imazu M, Ono K, Tadehara F, Kajiwara K, Yamamoto H, Sumii K, Tasaki N, Oiwa J, Shimohara Y, Gomyo Y, Itabe H. Plasma levels of oxidized low density lipoprotein are associated with stable angina pectoris and modalities of acute coronary syndrome. Int Heart J. 2008;49:515–524. [DOI] [PubMed] [Google Scholar]
- 22.Chan H-C-C, Ke L-Y-Y, Chu C-S-S, Lee A-S-S, Shen M-Y-Y, Cruz MA, Hsu J-F-F, Cheng K-H-H, Chan H-CB-C, Lu J, Lai W-T-T, Sawamura T, Sheu S-H-H, Yen J-H-H, Chen C-H-H. Highly electronegative LDL from patients with ST-elevation myocardial infarction triggers platelet activation and aggregation. Blood. 2013;122:3632–3641. doi: 10.1182/blood-2013-05-504639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shen M-Y-Y, Chen F-Y-Y, Hsu J-F-F, Fu R-H-H, Chang C-M-M, Chang C-T-T, Liu C-H-H, Wu J-R-R, Lee A-S-S, Chan H-C-C, Sheu J-R-R, Lin S-Z-Z, Shyu W-C-C, Sawamura T, Chang K-C-C, Hsu CY, Chen C-H-H. Plasma L5 levels are elevated in ischemic stroke patients and enhance platelet aggregation. Blood. 2016;127:1336–1345. doi: 10.1182/blood-2015-05-646117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pawelczyk M, Baj Z, Chmielewski H, Kaczorowska B, Klimek A. The influence of hyperlipidemia on platelet activity markers in patients after ischemic stroke. Cerebrovasc Dis. 2009;27:131–137. doi: 10.1159/000177920. [DOI] [PubMed] [Google Scholar]
- 25.dPawelczyk M, Kaczorowska B, Baj Z. The impact of hyperglycemia and hyperlipidemia on plasma P-selectin and platelet markers after ischemic stroke. Arch Med Sci. 2017. doi: 10.5114/aoms.2017.65816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bröijersén A, Hamsten A, Eriksson M, Angelin B, Hjemdahl P. Platelet activity in vivo in hyperlipoproteinemia – importance of combined hyperlipidemia. Thromb Haemost. 1998;79:268–275. [PubMed] [Google Scholar]
- 27.Sener A, Ozsavci D, Oba R, Demirel GY, Uras F, Yardimci KT. Do platelet apoptosis, activation, aggregation, lipid peroxidation and platelet-leukocyte aggregate formation occur simultaneously in hyperlipidemia? Clin Biochem. 2005;38:1081–1087. doi: 10.1016/j.clinbiochem.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 28.Dole VS, Bergmeier W, Patten IS, Hirahashi J, Mayadas TN, Wagner DD. PSGL-1 regulates platelet P-selectin-mediated endothelial activation and shedding of P-selectin from activated platelets. Thromb Haemost. 2007;98:806–812. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.