

Abstract
Fungal genome projects are revealing thousands of cryptic secondary metabolism (SM) biosynthetic gene clusters that encode pathways that potentially produce valuable compounds. Heterologous expression systems should allow these clusters to be expressed and their products obtained, but approaches are needed to identify the most valuable target clusters. The inp cluster of Aspergillus nidulans contains a gene, inpE, that encodes a proteasome subunit, leading us to hypothesize that the inp cluster produces a proteasome inhibitor and inpE confers resistance to this compound. Previous efforts to express this cluster have failed, but by sequentially replacing the promoters of the genes of the cluster with a regulatable promotor, we have expressed them successfully. Expression reveals that the product of the inp cluster is the proteasome inhibitor fellutamide B, and our data allow us to propose a biosynthetic pathway for the compound. By deleting inpE and activating expression of the inp cluster, we demonstrate that inpE is required for resistance to internally produced fellutamide B. These data provide experimental validation for the hypothesis that some fungal SM clusters contain genes that encode resistant forms of the enzymes targeted by the compound produced by the cluster.
Graphical Absract
Introduction
The sequencing of fungal genomes has revealed that fungi have a vast, and only partially realized, potential to produce secondary metabolite (SM) natural products (NPs). For even well-studied fungi, the SMs that have been isolated and identified after decades of effort are vastly outnumbered by cryptic SM biosynthetic gene clusters (BGCs), encoding biosynthetic pathways the products of which are currently unknown. For example, examination of the Penicillium chrysogenum,1 Aspergillus terreus, Streptomyces avermitilis,2 and Saccharopolyspora erythraea3 genomes suggests that only 10–20% of the SMs potentially generated from these organisms have been identified. The explosion in fungal genome sequencing such as the 1000 Fungal Genomes Project of the Department of Energy Joint Genomes Initiative (http://1000.fungalgenomes.org/home/), moreover, is revealing that many species of fungi have large numbers of heretofore unexplored BGCs. Collectively, these fungi harbor thousands of BGCs that undoubtedly produce many valuable NPs that can, in principle, be accessed. There is a strong need for methods, however, to identify the clusters that are most likely to produce valuable products.
Although emphasis is usually focused on enzymatic genes, many BGCs contain other genes not obviously involved in product synthesis including a category that may be thought of as self-protective and includes transporters, enzymes that detoxify the final product and resistant versions of the protein targeted by the BGC (discussed in 4, 5). The latter category has, in principle, the potential to be particularly useful in identifying the activity of the NP synthesized by the BGC. In fact, resistance gene-guided genome mining of BGCs has recently been demonstrated by Bradley Moore’s group in the marine actinomycete Salinispora.6 In the study, they queried the genomes of 86 Salinispora strains for putative target modifying resistance genes associated with natural product BGCs. They focused on BGCs linked to protein coding genes related to lipid transport and metabolism. Using this approach, they identified a ~22kb PKS-NRPS hybrid gene cluster of 10 open reading frames from S. pacifica CNS-863. They heterologously expressed the gene cluster (renamed tlm) in the S. coelicolor M1152 host and structurally characterized the products as thiolactomycin and analogs which have been previously been shown to be fatty acid synthase II inhibitors.
Links between BGCs and resistance genes have also been observed in filamentous fungi. Regueira et al.7 have noted several instances in which resistance is apparently mediated by a gene within the BGC that encodes a putative resistant allele of the enzyme inhibited by the SM produced by the cluster.8–10 These data led Regueira et al. to predict correctly that a particular SM biosynthetic cluster in Penicillium brevicompactum is responsible for the synthesis of mycophenolic acid, because the cluster contained a gene encoding IMP dehydrogenase, the target of mycophenolic acid. Other putative examples of embedded BGC resistant target genes include the methionine aminopeptidase encoded in the fumagillin gene cluster [11 and our additional database analyses] and the HMG-CoA reductase gene found in the lovastatin/compactin BGC.8, 10
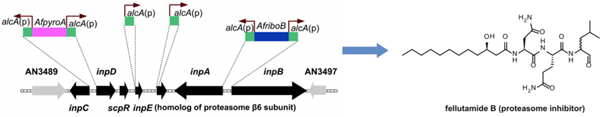
In examining the uncharacterized Aspergillus nidulans BGCs, we noted that one contains a gene (inpE) encoding a putative proteasome subunit, a protein with no obvious role in NP synthesis (Figure 1). This BGC has been designated the inp cluster.12, 13 We hypothesized that inpE might mediate host resistance to the SM product of the inp gene cluster, and the fact that InpE has strong homology to the β6 subunit of the proteasome, which is highly evolutionarily conserved (HomoloGene: 2087, Figure S1) suggested that the product of the inp cluster might be a proteasome inhibitor effective against a wide range of organisms. Proteasome inhibitors have recently come to the fore as cancer chemotherapy agents.14, 15 Bortezomib is a blockbuster drug, recently approved by the FDA, that is effective against multiple myeloma and other cancers. Carfilzomib is another proteasome inhibitor (based on a natural product) that has recently been FDA approved for multiple myeloma and other proteasome inhibitors are in clinical trials.15 We were, thus, particularly interested in determining the product of this BGC.
Figure 1.
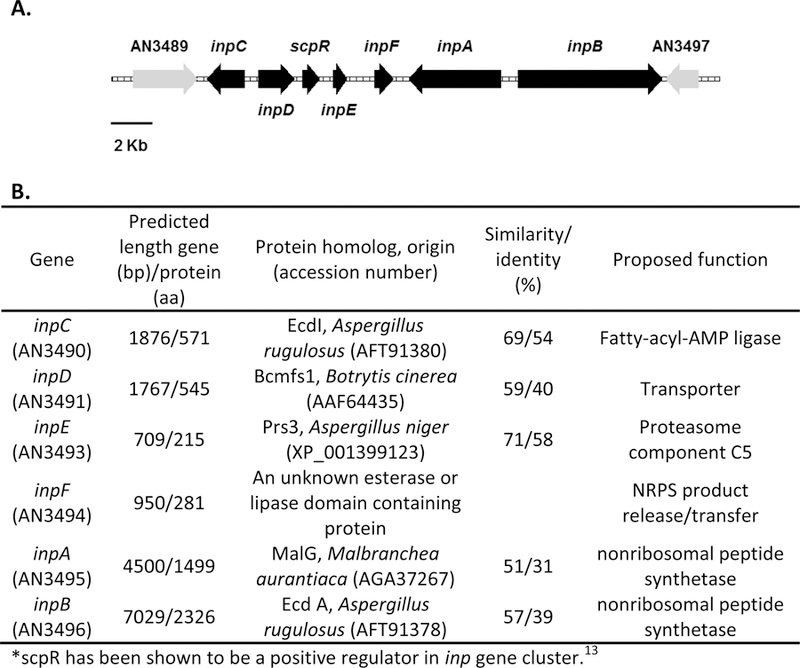
(A) Schematic representation of the inp cluster. Each arrow indicates the direction of transcription and relative sizes of the open reading frames (ORFs). (B) The deduced function of each ORF and the amino acid sequence similarity/identity, as compared with the homologues found in a BLAST search of the NCBI non-redundant protein database.
A pathway-specific regulatory gene, scpR, has been identified in the inp cluster,13 and studies by the Hertweck and Brakhage groups have shown that overexpression of scpR results in the expression of some of the genes in the inp gene cluster.13 However, the activation of the cluster in the scpR overexpression strain was insufficient to enable isolation of the product of the pathway. The final product of the pathway as well as pathway intermediates remained completely unknown.
A variety of approaches have been used to activate expression of fungal BGCs16 but many BGCs have not yet responded to any of these approaches. We have developed a strategy in which we take advantage of high efficiency gene targeting in A. nidulans17, 18 to simply replace the promoters of BGC genes with a regulatable promoter that allows us to activate transcription of each gene to high levels. We were able to use this approach to complete the identification of all the products released by nonreduced PKS genes in A. nidulans.19 These experiments involved replacing the promoters of only 1–3 genes per BGC, however, and many fungal BGCs contain several genes. We wished to determine if it is feasible to use the promoter replacement approach to elucidate larger BGCs. Since a limited number of selectable markers were available to us we need to develop a method of recycling selectable markers for transformation to allow all the promoters of the BGC genes to be replaced.
In this study, we report the use of a serial promoter exchange approach, including a method for recycling a selectable marker, to replace the promoters of six genes of the inp cluster with the regulatable alcA promoter [alcA(p)]. By inducing expression of the genes, we were able to obtain production of the final product of this BCG, which we have determined to be fellutamide B. Fellutamide B is a proteasome inhibitor that is of particular interest because it induces nerve growth factor synthesis.20 By purifying and determining the structures of fellutamide B and the shunt product produced when subsets of the cluster genes are activated by promoter replacement, we are able to propose a biosynthetic pathway for fellutamide B. Through targeted gene deletions, we have found that, as we hypothesized, inpE is not required for fellutamide B production but confers resistance to internally produced fellutamide B. Our results not only provide the first identification and characterization of a fellutamide biosynthetic gene cluster, but they provide additional experimental verification that some fungal SM clusters contain genes encoding resistant alleles of the protein targeted by the SM biosynthesized by the cluster. Therefore, as with bacterial BGCs, we could also apply target-directed genome mining to fungal BGC systems.
Results and Discussion
Determination of the product of the inp gene cluster by serial promoter replacement
The inp gene cluster contains two non-ribosomal peptide synthetase (NRPS) genes, inpA (AN3495) and inpB (AN3496). InpA and InpB have the domain architecture CATR and TCATCAT, respectively.12 C, A, and T refer to a condensation, adenylation, and thiolation domain, respectively.21 The R domain refers to a NAD binding domain that was proposed to release the NRP product via a reductive elimination mechanism. Since there are a total of three A domains, the metabolite associated with this cluster was proposed to be a tripeptide with a reduced C terminus.12
Our starting strain for this work was LO7692 (full genotypes of strains are listed in Table 1). We created this strain by deleting six secondary metabolism gene clusters (sterigmatocystin, emericellamide, asperfuranone, monodictyphenone, terrequinone and one of the portions of the split cluster that encodes the austinol biosynthetic pathway). Deletion of these SM clusters reduces the SM background and facilitates identification of any new metabolite that might be produced as a result of our promoter replacements. We first replaced the promoters of the two NRPS genes, inpA and inpB (Figure 2A). These genes are divergently transcribed, which allowed us to replace both promoters with alcA(p) in a single transformation using a strategy we have previously described19 resulting in strain LO7858 in which inpA and inpB are inducible. Induction of expression of inpA and inpB did not lead to production of compounds detectable by diode array detector (DAD) or mass spectrometer (MS) (Figure 3A). This result suggested that expression of additional gene(s) in the cluster might be necessary for the production of the SM. In the earlier study using scpR overexpression, the inpC and inpD genes were shown to be co-regulated with inpA and inpB, which indicates that they are part of the same SM cluster.13 InpC encodes a putative acyl-AMP ligase that might synthesize an AMP substrate as a starter unit.22 InpD encodes a putative transmembrane transporter that might facilitate the transportation of the substrates or metabolites synthesized by the pathway. Since inpC and D are also divergently transcribed, we replaced the promoters of inpC and D with alcA(p) in an inpA and B promoter replacement strain (Figure 2A) creating strain LO7860 in which inpA, B, C and D are all inducible. Simultaneous induction of expression of inpA, B, C, and D did lead to production of a new DAD and MS detectable peak (compound 1, Figure 3A).
Table 1. A. nidulans strains used in this study.
Note that all strains carry veA1.
| LO1362 | pyrG89, riboB2, pyroA4, nkuA::argB |
| LO7692 | pyroA4, riboB2, pyrG89, nkuA::argB, sterigmatocystin cluster (AN7804-AN7825)∆, emericellamide cluster (AN2545-AN2549)∆, asperfuranone cluster (AN1036-AN1029)∆, monodictyphenone cluster (AN10023-AN10021)∆, terrequinone cluster (AN8513-AN8520)∆, austinol cluster part 1 (AN8379-AN8384)∆ |
| LO7858 | alcA(p)inpB-AfriboB-alcA(p)inpA in LO7692 |
| LO7860 | alcA(p)inpB-AfriboB-alcA(p)inpA, alcA(p)inpC-AfpyroA-alcA(p)inpD in LO7692 |
| LO7870 | alcA(p)inpB-AfriboB-alcA(p)inpA, alcA(p)inpC-AfpyroA-alcA(p)inpD, alcA(p)inpE in LO7692. Note AfpyrG was removed by looping out. |
| LO7975 | pyrG89; pyroA4; riboB2; pyrG89; nkuA::argB; sterigmatocystin cluster (AN7804-AN7825)∆; yA::AtpyrG |
| LO8080 | alcA(p)inpB-AfriboB-alcA(p)inpA, alcA(p)inpC-AfpyroA-alcA(p)inpD, alcA(p)inpF-AfpyrG in LO7692 |
| LO8085 | alcA(p)inpB-AfriboB-alcA(p)inpA, alcA(p)inpC-AfpyroA-alcA(p)inpD, alcA(p)inpE, alcA(p)inpF-AfpyrG in LO7692. |
| LO9169 | pyroA4; riboB2; pyrG89; nkuA::argB; inp cluster (AN3496-AN3490 inclusive)::AtpyrG |
| LO9173 | pyroA4; riboB2; pyrG89; nkuA::argB; inp cluster (AN3496-AN3490 inclusive)::AtpyrG; yA::AN3493-AfriboB |
| LO9222 | alcA(p)inpB-AfriboB-alcA(p)inpA, alcA(p)inpC-Afpyro-alcA(p)inpD, alcA(p)inpF in LO7692 |
| LO9385, 86 | alcA(p)inpB-AfriboB-alcA(p)inpA, alcA(p)inpC-Afpyro-alcA(p)inpD, alcA(p)inpF, inpE::AtpyrG in LO7692 |
| LO9467, 68, 69 | yA::AN3493-ptrA-res in LO9385 |
| LO10079, 80, 81 | alcA(p)inpA::AtpyrG in LO9222 |
| LO10083, 84, 85 | alcA(p)inpB::AtpyrG in LO9222 |
| LO10089, 90, 91 | alcA(p)inpC::AtpyrG in LO9222 |
| LO10093, 96, 97 | alcA(p)inpD::AtpyrG in LO9222 |
| LO10099, 100, 101 | alcA(p)inpF::AtpyrG in LO9222 |
Figure 2.
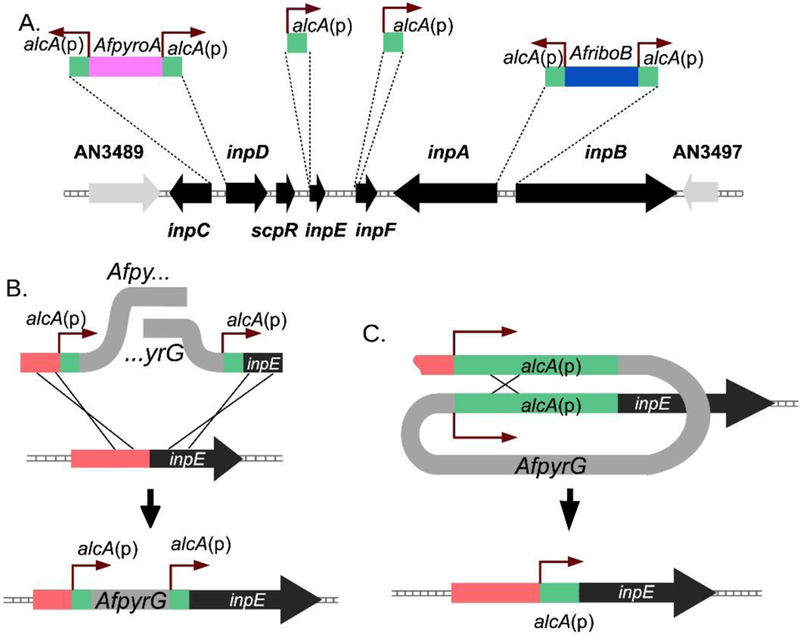
Promoter replacements and strategy. (A) A schematic diagram of the replacements made of the native promoters of the inp cluster. (B) and (C) Promoter replacement with marker recycling strategy used to replace the promoters of inpE and inpF. (B) The endogenous promoter is replaced with a fragment carrying AfpyrG, a marker that can be selected for and against, flanked by direct repeats of the alcA promoter [alcA(p)]. (C) Spontaneous recombination between the alcA(p) repeats results in excision of AfpyrG, which is selected for on 5-fluoroorotic acid resulting in a single copy of alcA(p) driving expression of the target gene, inpE. Since AfpyrG is removed, it can be reused as a selectable marker.
Figure 3.
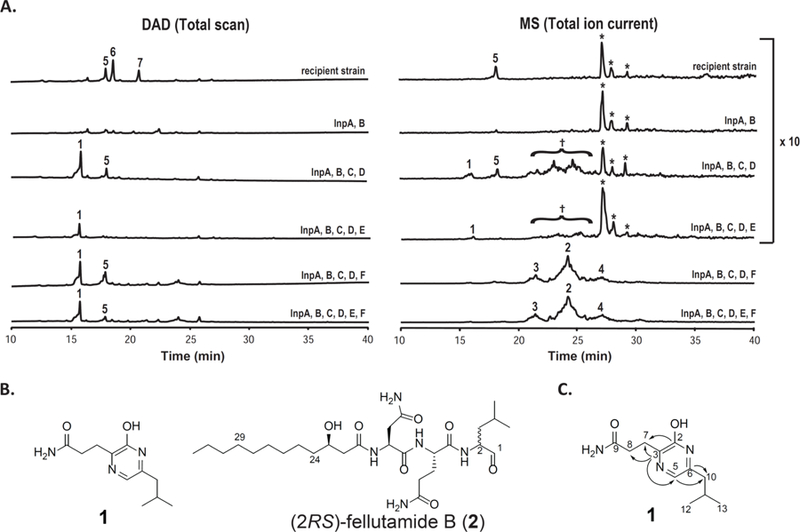
(A) DAD total scan and MS total ion current (TIC) HPLC profiles of culture media extracted from A. nidulans strains expressing inp cluster genes under the control of alcA promoter. The MS TIC profile of recipient; InpA, B; InpA, B, C, D; and InpA, B, C, D, E strains are magnified 10 fold. 1 is 2-hydroxy-5-isobutyl-3-propanamidylpyrazine, 2 is (2RS)-fellutamide B, 3 and 4 are fellutamide B derivatives with different lengths of the aliphatic side chains (see Figure S1), 5 is triacetylfusarinine C, and 6 is lumichrome. 7 has similar UV-Vis spectra with 6 and has 14 amu more than 6, and therefore, 7 could be methyllumichrome. (B) Chemical structures of compounds 1 and 2. (C) Key long-range correlations (gHMBC) of compound 1. *Unknown metabolites detected from MS detector. †Trace amount of felllutamides 2, 3, 4 were detected by MS detector.
Compound 1 was purified from a 1 L culture and its structure was elucidated by spectroscopic methods. The molecular formula of compound 1 was found to be C11H17N3O2 by means of HRESIMS and 13C NMR experiments, representing five indices of hydrogen deficiency (IHD). The 1H, 13C, gHMQC and COSY NMR data of 1 revealed it contained isobutyl [δH 2.34 (2H, d, J = 7.6 Hz, H2-10), 1.93 (1H, m, H-11), and 0.93 (6H, d, J = 6.4 Hz, H3-12 and H3-13); and δC 40.3 (C-10), 29.7 (C-11), and 22.6 (C-12 and C-13)] and propanamidyl [δH 2.99 (2H, t, J = 7.2 Hz, H2-7) and 2.61 (2H, t, J = 7.2 Hz, H2-8); and δC 29.4 (C-7), 32.8 (C-8), and 178.3 (C-9)] moieties (Table S1). Subtracting isobutyl and propanamidyl moieties from the molecular formula of 1, the remaining C4H2N2O represents four IHD, indicating a C4N2 heterocyclic aromatic ring. Compounds containing a pyrazine heterocycle are known in nature and have been thought to derive from condensation of two amino acids.23 In gHMBC spectra, long range 13C-1H correlations between two olefinic carbons [δC 158.9 (C-2) and 156.3 (C-3)] and H2-7 as well as between carbons [δC 140.7 (C-6) and 123.9 (C-5)] and H2-10 established that compound 1 does contain a pyrazine moiety (Figure 3C). Moreover, long-range C-3/H2-8 and C-3/H-5 gHMBC correlations revealed that compound 1 is 2-hydroxy-5-isobutyl-3-propanamidylpyrazine. In addition to compound 1, we identified several MS detectable minor peaks eluted from 20 to 25 min. However, we were unable to characterize their structures due to the low yield of those metabolites (Figure 3A).
Since compound 1 is a dipeptide but not a tripeptide as predicted by the two NRPS domain architectures,12 we suspected that 1 is not the final metabolite of the inp biosynthetic pathway and that, in addition to inpA, B, C, and D, expression of other genes of the inp gene cluster might be necessary for producing the bona fide final metabolite. InpE encodes a putative proteasome component, as mentioned, and the predicted product of inpF is an unknown protein that contains an esterase-lipase domain. Although activation of transcription of inpE and F was not addressed in the previous scpR overexpression work,13 microarray transcription data from A. nidulans grown under various culture conditions has indicated that inpA - F and scpR are part of the same SM gene cluster.24 Because inpE and F are not divergently transcribed, we replaced the promoters of inpE and inpF individually in an inpA, B, C, and D promoter replacement strain. These replacements required the use of a system for recycling the AfpyrG selectable marker (Figure 2B and 2C). Satisfyingly, the inpA, B, C, D, and F promoter replacement strain (LO8080) produced several broad MS detectable peaks upon induction, but the inpA, B, C, D, and E promoter replacement strain (LO7870) did not (Figure 3A). The parent ion of each broad peak has a mass difference of 28 amu and all have related MS/MS fragmentation patterns (Figure S2). These results suggest that the broad peaks are related metabolites synthesized by a single biosynthetic pathway. These metabolites are barely detectable by DAD and therefore they likely contain chromophores with low molar absorptivity or they are produced at a very low level. We were able to purify the major MS detectable metabolite (compound 2) from a large-scale culture. We found that the majority of compound 2 is in the hyphae and we were able to isolate > 20 mg of compound 2 from 1 L of culture. Compound 2 has a molecular formula C27H49N5O7, obtained from its HRESIMS. Searching for compounds with the molecular formula C27H49N5O7 in the SciFinder chemical database revealed a lipotripeptide, fellutamide B, that has a reported MS/MS fragmentation similar to compound 2 (Figure S2).25 Fellutamide B (Figure 3B) was first isolated by Shigemori et al. from the fungus Penicillium fellutanum and the absolute configuration of C-2 was determined to be a mixture of R and S forms.25 Recently, Wu et al. isolated optically pure (2S)-fellutamide B from a marine-derived Penicillium purpurogenum G59.26 Comparison of the 13C spectral data of (2S)-fellutamide B published by Wu et al. with those of compound 2 isolated by us revealed that they had almost identical 13C chemical shifts (Table S2), except that in the 13C NMR spectrum of compound 2, several 13C NMR signals are either broad or split, especially for those carbons near C-2. This suggested that compound 2 we isolated is (2RS)-fellutamide B. Peaks 3 and 4 were determined to be fellutamide B derivatives, with different lengths of aliphatic side chains, from their MS/MS fragmentation data. (Figure S2). Recently, fellutamides were detected from a histone deacetylase, RpdA, knocked down A. nidulans mutant, although the production of fellutamides was not tied to a particular SM gene cluster.27 Fellutamide B has been shown to bind the β-subunits of the 20S proteasome and potently inhibit proteasome catalytic activity.20 We verified that fellutamide B (2) purified from A. nidulans strongly inhibits the activity of mammalian proteasomes in vitro (Figure S3) and fractions containing fellutamide B derivatives also exhibited strong proteasomal inhibition.
A complete synthesis for fellutamide B has been published.28, 29 Our promoter-replaced strain, however, produces > 20 mg/L of fellutamide B with extremely low materials costs. Although there are costs associated with fermentation and purification, it may prove cost effective to produce fellutamides in A. nidulans promoter-replaced strains, and there is likely scope for further strain improvement and improvement in production conditions to increase yields.
To determine if the gene inpE is involved in a biotransformation step in the fellutamide biosynthesis pathway, we constructed strain LO8085 in which all six genes, inpA - F, were placed under the control of alcA(p). The HPLC profile of this strain (Figure 3A) was similar to that of the strain expressing inpA, B, C, D, and F, suggesting that inpE is not involved in a biotransformation step. Our data reveals that the inp cluster is responsible for the biosynthesis of fellutamides.
It is worth pointing out that with our use of alcA(p) to drive expression of several genes, there is potential for recombination between the alcA(p) copies that would loop out some of the genes, resulting in loss of fellutamide production. In fact, we use recombination between repeated alcA(p) sequences in our marker recycling procedure (Figure 2C), although in this instance we use 5-FOA to impose a strong selection for recombinants in which AfpyrG has been looped out. In practice, we have not encountered any lack of stability with our fellutamide producing strains. Most possible loop outs would result in the loss of a selectable marker, which is selected against by growth in minimal medium. The version of alcA(p) we use is only 400 bp in length, and this may minimize recombination. It is also worth pointing out that although our approach worked very efficiently, additional approaches using CRISPR/Cas9 technology have been reported recently.30 These approaches facilitate marker recycling and they may provide a useful alternative approach.
To gain additional understanding of the fellutamide biosynthetic pathway, we augmented the data we obtained from our promoter replacement strains by individually deleting inpA, inpB, inpC, inpD and inpF in strain LO9222. In LO9222, each of the inp genes is under control of the alcA promoter except for inpE, which is under control of its native promoter. By inducing expression and determining the compounds that accumulate we hoped to elucidate the biosynthetic functions of the proteins encoded by each gene. Results are shown in Figure 4. As anticipated, deletion of inpA, inpB, or inpC eliminated the production of fellutamides. Deletion of inpF greatly diminished the production of fellutamides. The fellutamides were produced, however, in the inpD deletion strain so inpD, which putatively encodes a transporter, is not required for fellutamide production. Both of the NRPS genes, inpA or inpB, were required for production of the cyclodipeptide 1 whereas, although not required, deletion of inpC or inpD diminished production of 1. Deletion of inpF does not interfere with production of 1 suggesting that inpF is not required for the production of 1.
Figure 4.
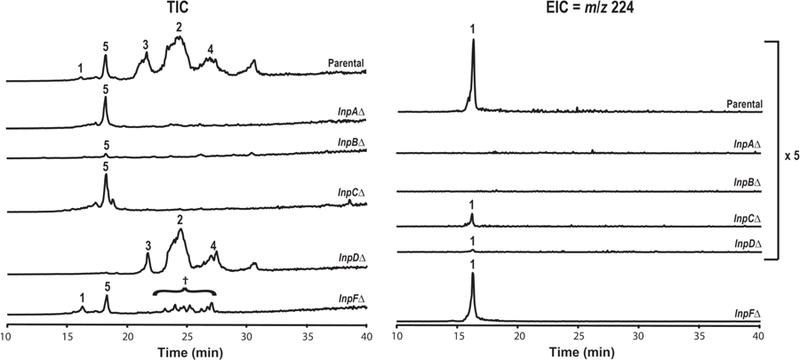
Total ion current (TIC) and extracted ion chromatography (EIC) for compound 1 (EIC = m/z 224) of culture media extracted from A. nidulans strains producing fellutamides (parental, LO9222), and individual inp gene deletion strains cluster genes under the fellutamide producing condition. The EIC profile of parental, InpA, B, C, and D deletion strains are magnified 5 fold. †Trace amount of felllutamides 2, 3, 4 were detected by MS detector.
The overexpression experiments and individual gene knock out experiments in aggregate, enabled us to propose a highly speculative biosynthetic pathway for fellutamide B (2) (Figure 5A). A homolog of InpC, EcdI, has been shown to catalyze lipoinitiation of EcdA, an NRPS involved in echinocandin B biosynthesis.22 Therefore, we propose that in the first step of fellutabmide B biosynthesis InpC activates 3-hydroxydodecanoic acid to generate 3-hydroxydodecanoyl-AMP that is then loaded onto the T0 domain of InpB. The 3-hydroxydodecanoyl-S-phosphopantetheinyl-T0 is sequentially extended with L-Asn and L-Gln by the two CAT modules of InpB. The linear lipodipeptide from InpB is then transferred onto InpA for addition of the third amino acid, L-Leu. Reductive releasing of the lipotripeptide by the R domain of InpA produces (2S)-fellutamide B. Tautomerization of C-2, which might occur in our culture conditions, lead to the formation of (2RS)-fellutamide B (2). It should be noted that there is no fatty acid synthase or highly-reduced PKS (HRPKS) gene in or near the inp gene cluster. Therefore, the 3-hydroxy fatty acid side chain might be derived from primary metabolism. Indeed, feeding commercial available decanoic-d19 acid to the fellutamide producing strain led to the production of 3-d18 (Figure S4). Neither 2-d19 nor 4-d19 was detected in the feeding experiment suggesting that beta-oxidation catabolic processes in mitochondria could be the main source of the 3-hydroxy fatty acid.
Figure 5.
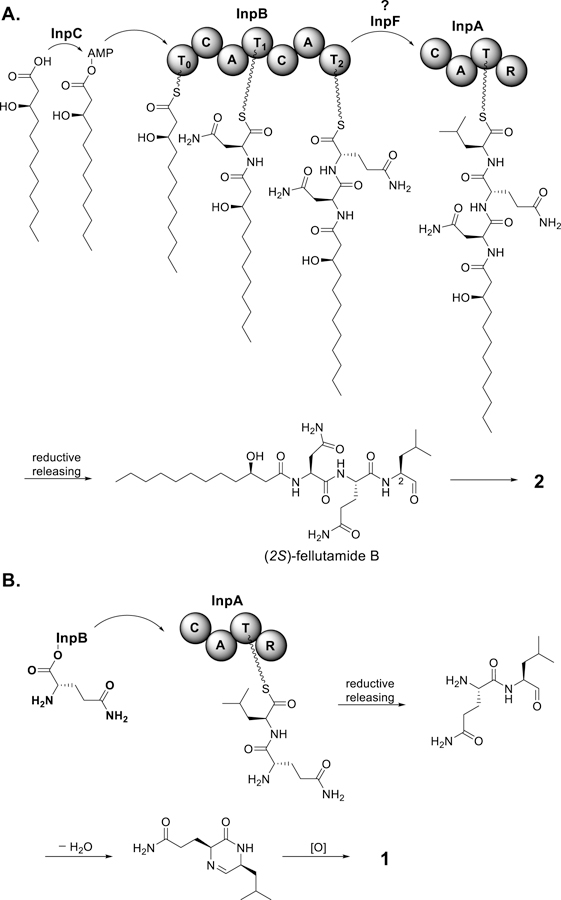
(A) Proposed biosynthesis pathway for (2RS)-fellutamide B (2) and (B) 2-hydroxy-5-isobutyl-3-propanamidylpyrazine (1).
Both heterologous expression and individual gene knock out experiments have demonstrated that InpF, a putative esterase or lipase domain containing protein is also involved in the biosynthesis of fellutamides. We propose that InpF is involved in the release and transfer of the lipodipeptide from InpB to InpA. Fellutamides are detected at a very low level when InpF is not expressed suggesting that the transfer of the lipodipeptide from InpB to InpA is slow and that InpF catalyzes the transfer, leading to the high-level production of fellutamides.
Our individual knock out experiments show that both InpA and InpB are required for the biosynthesis of compound 1. We propose that the C-terminal A domain in InpB recognizes and activates Gln and the A domain in InpA recognizes and activates Leu. The Glu-Leu dipeptide is generated by InpA via condensation of Gln and Leu (Figure 5B). Reductive elimination of the Gln-Leu dipeptide followed by intramolecular Schiff base formation and oxidation leads to 1. Since compound 1 was detected in the InpF deletant strain and InpF expression does not lead to the high production of 1, the transfer of activated Glu to InpA is likely to be InpF independent. Our individual knock out experiments show that both InpC and InpD are not necessary for, but are involved in, the biosynthesis of 1. However, our data does not allow us to deduce the precise roles of InpC and InpD in the biosynthesis of 1.
InpE confers resistance to internally produced fellutamides
The predicted product of inpE (AN3493) is a strong homolog of the conserved β6 proteasome subunit (Figure S1) and, as mentioned, we hypothesized that inpE confers resistance to the product of the inp cluster, fellutamide B. If InpE confers fellutamide resistance, deletion of inpE might confer heightened sensitivity. To test this possibility, we deleted the entire Inp cluster (AN3490-AN3496) by replacing it with the Aspergillus terreus pyrG gene (AtpyrG) in strain LO1362 creating strain LO9169. In this construction inpE/AN3493 is eliminated from the genome. Anticipating that deletion of inpE might increase fellutamide sensitivity, we created a control strain by reinserting inpE at the yA (yellow conidia) locus in LO9169, creating strain LO9173. In broth dilution tests,31 LO9169, LO9173, and LO7975, a control strain with an intact inp cluster, grew at all concentrations of fellutamide B tested (0.5 to 256 μg/mL, at which concentration it precipitated out of solution, data not shown). These data indicate that A. nidulans is highly resistant to externally applied fellutamide B and that the mechanism of resistance does not require inpE. It is possible that the compound simply is not taken up by A. nidulans and does not penetrate the cell wall.
We noted, however, that in our experiments the vast majority of fellutamide B accumulates within hyphae, and we reasoned that A. nidulans must have a mechanism for counteracting the toxicity of internally produced fellutamide B. To test this hypothesis, we deleted inpE in strain LO9222 in which inpA-D and inpF are under control of alcA(p). We designated the resulting deletants LO9385 and LO9386. We tested these inpE deletants under repressing conditions that prevent transcription from alcA(p). Under these conditions no fellutamide B will be produced and any effect of inpEΔ would be independent of fellutamide B production. As shown in Figure 6A, inpEΔ transformants grew normally under alcA(p) repressing conditions, indicating that inpE does not have a function important for growth in the absence of fellutamide B production. We next tested growth of these inpEΔ strains on media that induce transcription from alcA(p), resulting in fellutamide B production. As shown in Figure 6A, growth was strongly inhibited in the inpEΔ strains, revealing that inpE confers resistance to internally produced fellutamide B. As a control, we genetically added back the inpE gene to one of the inpEΔ strains, LO9386, by inserting a copy of inpE, under control of its normal promoter, at the yA locus. The reinserted inpE gene restored growth when fellutamide B production was induced (Figure 6B), confirming that inpE confers resistance to internally produced fellutamide B.
Figure 6.
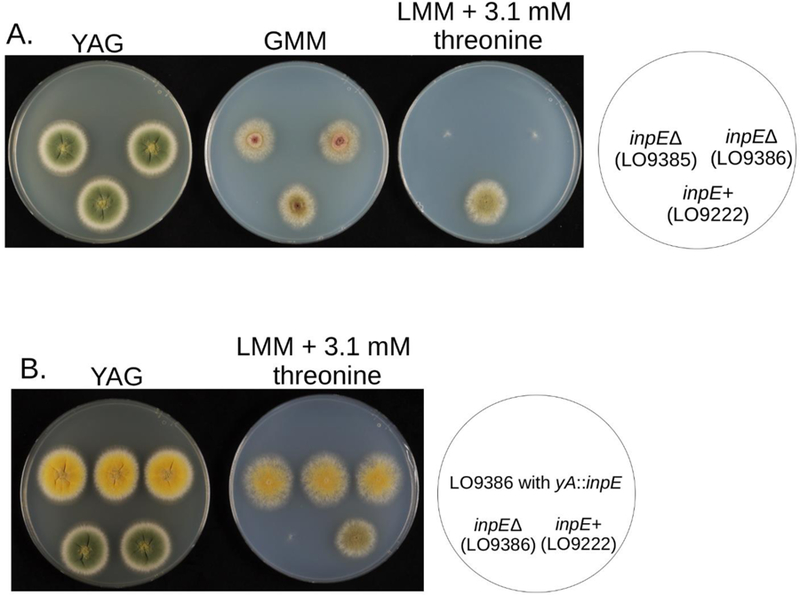
Induction of fellutamide production inhibits growth if inpE is deleted. (A) Two inpE deletant transformants and their parent, LO9222. On complete medium (YAG) the alcA promoter [alcA(p)] is strongly repressed and all strains grow well. All strains grow reasonably well on glucose minimal medium (GMM), which represses alcA(p) slightly less completely. On lactose minimal medium (LMM) with 3.1 mM threonine, fellutamide production is induced and growth of the inpE deletion strains is strongly inhibited. (B) Adding the inpE gene back by inserting it at the yA (yellow spore color) locus restores growth in strains producing fellutamide. The top three colonies are transformants (LO9467, LO9468, LO9469) of the inpEΔ strain, LO9386, in which inpE has been inserted at yA. They grow at least as well as the parental strain LO9222 when fellutamide production is induced on LMM + threonine. Uridine (10 mM) and uracil (1 g/L) were added to all media to fully supplement the pyrG89 mutation that is uncomplemented in some strains.
These data raise a question of why induction of fellutamide production in strains in which inpE is under control of its normal promoter is not harmful. If fellutamide is produced at high levels in cells and inpE were not expressed, the prediction is that the fellutamide would be toxic to the cell. Transcription data on inpE available at the Aspergillus genome database (www.aspergillusgenome.org) and microarray data in the supplemental dataset for reference 24 appear to provide an answer. They reveal that inpE is constitutively expressed. It is not co-regulated with the other genes of the Inp cluster, and the fellutamide-resistant proteasome subunit it encodes is expressed even when the remainder of the genes of the Inp cluster are not expressed at significant levels. InpD, a putative transporter may contribute to fellutamide resistance as well. Deletion of inpD and simultaneous induction of inpA, inpB, inpC and inpF results in a small reduction in growth relative to the parental strain when inpE is intact (Figure S5). The reduction in growth is much less than is the case if inpE is deleted and fellutamide production is induced, however (compare Figure 6 with Figure S5), so inpE appears to be the principal resistance mechanism.
The A. nidulans genome contains another strong β6 homolog, AN5784. We deleted AN5784 in LO9222 (leaving inpE intact) to determine if it had any role in resistance to internally produced fellutamide. Deletion of AN5784 reduced conidiation and slightly reduced growth but did not confer altered sensitivity to internally produced fellutamide (Figure S6). The simplest interpretation of these data is that AN5784 encodes a fellutamide B-sensitive β6 subunit and inpE encodes a fellutamide B-resistant subunit. InpE confers resistance to internally produced fellutamide B and is functional enough to support growth in the absence of AN5784 but growth and conidiation are not normal (i.e. the proteasome is not fully functional if AN5784 is deleted). AN5784 fully supports normal growth, but does not confer fellutamide B resistance.
Conclusions
Our data reveal that the fellutamide B cluster in A. nidulans carries a proteasome subunit that confers resistance to fellutamide B, while another homologous proteasome subunit is present elsewhere in the organism. These data provide experimental confirmation for the hypothesis that in some instances fungal SM clusters carry within them genes that encode resistant alleles of the protein targeted by the compound produced by the cluster. The hallmarks of such clusters are that they contain a gene predicted to encode a protein involved in an essential function unrelated to SM biosynthesis and a homolog of that gene is present elsewhere in the genome. Our result is in agreement with Bradley Moore’s observation in bacterial SM clusters and demonstrates that resistance-gene-guided genome mining approach can be broadly applied to both fungal and bacterial BGCs.6 Our data also reveal that obtaining products from cryptic SM clusters by serial promoter replacement is feasible and practical, and can result in yields that may be commercially useful. Finally, an additional benefit of the serial promoter replacement approach is that we have obtained a shunt product from strains in which the promoters of some of the genes of the inp cluster have been replaced.
Experimental Section
Strains.
Strains used in this study are listed in Table 1. All strains carry the veA1 mutation. Strain FGSC4 used in Figure S5 was obtained from the Fungal Genetics Stock Center in the 1970s. It is wild-type except that it carries the veA1 mutation, as do the vast majority of laboratory strains. The FGSC has changed its designation for Aspergillus nidulans strains by adding an “A” after FGSC and before the number. The FGSC currently carries a wild-type A. nidulans strain designated FGSCA4. This strain is wild-type at the veA locus. Although the strain sequenced in the A. nidulans genome project is usually called FGSCA4, it is actually FGSC4, the strain obtained by the N. R. Morris lab from the FGSC in the 1070s. This is why the veA allele in the sequenced genome is the veA1 allele. The DNA sequenced in the genome project was isolated from FGSC4 by the Oakley lab.
Media.
For induction of alcA on solid medium, we used lactose minimal medium (10 g/L lactose, 6 g/L NaNO3, 0.52 g/L KCl, 0.52 g/L MgSO4·7H2O, 1.52 g/L KH2PO4, 15 g/L agar, and 1 ml/L trace element solution32 (Vishniac and Santer, 1957)) supplemented with threonine at the concentrations indicated in the figure legends. Threonine is an inducer of alcA(p) but is not a substrate for the alcohol dehydrogenase encoded by alcA.33 Repressing solid media were YAG (5 g/L yeast extract, 20 g/L D-glucose, 15 g/L agar and 0.8 ml/L trace element solution)32 or glucose minimal medium (GMM is 10 g/L D-glucose, 6 g/L NaNO3, 0.52 g/L KCl, 0.52 g/L MgSO4·7H2O, 1.52 g/L KH2PO4, 15 g/L agar, and 1 ml/L trace element solution).32
Molecular genetic manipulations.
Transforming fragments were generated by fusion PCR essentially as described.18,34 To reduce the frequency of PCR-induced mutations, we used high-fidelity DNA polymerases KOD DNA polymerase (EMD Biosciences), Phusion DNA polymerase (New England Biolabs or Thermo) or Q5 Hot Start High-Fidelity 2X Master Mix (New England Biolabs). For KOD and Phusion DNA polymerases the annealing temperature was Tm + 2ºC and Tm + 3ºC for Q5 with Tm calculated by the formula Tm = 2(A+T) + 4(G+C). The extension rate was 30 sec/kilobase of expected product.
Production of protoplasts and transformations were carried out as previously described18, 34 except that VinoTaste Pro (Novo) was used at a final concentration of 100 mg/ml for protoplasting.34 Dual promoter exchanges were carried out as previously described.19 Cluster deletions to create strain LO7692 were carried out as previously described using the loop-out approach.35 Selection for excision of fragments carrying AfpyrG on 5-FOA was as previously described.36 Diagnostic PCR was carried out as described using Platinum Taq DNA polymerase (Invitrogen) or OneTaq Hot Start Quickload (New England Biolabs).34 The strategy for deletion of individual inp cluster genes is shown in Figure S7. Inp genes along with the alcA promoter driving their expression were replaced with the Aspergillus terreus pyrG gene (AtpyrG).
Broth microdilution tests for inhibition of growth by fellutamide B were carried out according to the CLSI reference method for filamentous fungi.31 RPMI 1640 medium was supplemented with glutamine, 2% glucose and any supplements required to supplement nutritional markers in the strains.
Fermentation and LC/MS analysis.
For alcA(p) induction, 3 × 107 spores were grown in 30 ml liquid LMM medium [15 g/l lactose, 6g/l NaNO3, 0.52g/l KCl, 0.52g/l MgSO4·7H2O, 1.52g/l KH2PO4, 1 ml/l trace elements solution, supplemented with riboflavin (2.5 mg/l), pyridoxine (0.5 mg/l) or uridine (10 mM) and uracil (1 g/L) when necessary] in 125 ml flasks at 37ºC with shaking at 180 rpm. Cyclopentanone was added to the medium at a final concentration of 10 mM 18 hr after inoculation. Culture medium was collected 48 hr after cyclopentanone induction by filtration and extracted twice with the same volume of EtOAc. EtOAc extract was then evaporated by TurboVap LV (Caliper LifeSciences). The crude extracts were re-dissolved in 0.5 ml of DMSO:MeOH (1:4) and 10 μl was injected for LC-DAD-MS analysis as described previously.37 Details for large-scale purifications of compounds for each strain and compound identification are given in the Supplemental Information as are spectral data. For the isotope feeding experiment, 1 mg of decanoic-d19 acid (Cambridge Isotope Laboratories) in 5 μl of DMSO was added into liquid culture at 0 h (the time of spore inoculation) and 18 h (the time of cyclopentanone induction). Culture medium was collected and analyzed as described above.
Proteasome inhibition assays.
Proteasome activity was assayed with a Millipore 20S Proteasome Activity Assay kit (EMD Millipore catalog # APT280) following the instructions provided with the kit. The assay was performed with proteasomes that were supplied with the kit or, more usually, human 20 proteasomes (BostonBiochem/R & D Systems catalog # E365).
Supplementary Material
Acknowledgement
Supported by grant PO1-GM084077 from the National Institute of General Medical Sciences, by the H. L. Snyder Medical Foundation and by the Irving Johnson Fund of the University of Kansas Endowment.
Footnotes
Supporting information
Detailed experimental procedures and additional tables and figures. This material is available free of charge via the internet at http://pubs.acs.org.
The authors declare no competing financial interest.
References
- 1.van den Berg MA, Albang R, Albermann K, Badger JH, Daran JM, Driessen AJ, Garcia-Estrada C, Fedorova ND, Harris DM, Heijne WH, Joardar V, Kiel JA, Kovalchuk A, Martin JF, Nierman WC, Nijland JG, Pronk JT, Roubos JA, van der Klei IJ, van Peij NN, Veenhuis M, von Dohren H, Wagner C, Wortman J, and Bovenberg RA (2008) Genome sequencing and analysis of the filamentous fungus Penicillium chrysogenum, Nat. Biotechnol 26, 1161–1168. [DOI] [PubMed] [Google Scholar]
- 2.Omura S, Ikeda H, Ishikawa J, Hanamoto A, Takahashi C, Shinose M, Takahashi Y, Horikawa H, Nakazawa H, Osonoe T, Kikuchi H, Shiba T, Sakaki Y, and Hattori M (2001) Genome sequence of an industrial microorganism Streptomyces avermitilis: deducing the ability of producing secondary metabolites, Proc. Natl. Acad. Sci. U. S. A 98, 12215–12220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Oliynyk M, Samborskyy M, Lester JB, Mironenko T, Scott N, Dickens S, Haydock SF, and Leadlay PF (2007) Complete genome sequence of the erythromycin-producing bacterium Saccharopolyspora erythraea NRRL23338, Nat. Biotechnol 25, 447–453. [DOI] [PubMed] [Google Scholar]
- 4.Cimermancic P, Medema MH, Claesen J, Kurita K, Wieland Brown LC, Mavrommatis K, Pati A, Godfrey PA, Koehrsen M, Clardy J, Birren BW, Takano E, Sali A, Linington RG, and Fischbach MA (2014) Insights into secondary metabolism from a global analysis of prokaryotic biosynthetic gene clusters, Cell 158, 412–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Keller NP (2015) Translating biosynthetic gene clusters into fungal armor and weaponry, Nat. Chem. Biol 11, 671–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tang X, Li J, Millan-Aguinaga N, Zhang JJ, O’Neill EC, Ugalde JA, Jensen PR, Mantovani SM, and Moore BS (2015) Identification of Thiotetronic Acid Antibiotic Biosynthetic Pathways by Target-directed Genome Mining, ACS Chem. Biol 10, 2841–2849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Regueira TB, Kildegaard KR, Hansen BG, Mortensen UH, Hertweck C, and Nielsen J (2011) Molecular basis for Mycophenolic Acid Biosynthesis of Penicillium brevicompactum, Appl. Environ. Microbiol 77, 3035–3043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kennedy J, Auclair K, Kendrew SG, Park C, Vederas JC, and Hutchinson CR (1999) Modulation of polyketide synthase activity by accessory proteins during lovastatin biosynthesis, Science 284, 1368–1372. [DOI] [PubMed] [Google Scholar]
- 9.Lowther WT, McMillen DA, Orville AM, and Matthews BW (1998) The anti-angiogenic agent fumagillin covalently modifies a conserved active-site histidine in the Escherichia coli methionine aminopeptidase, Proc. Natl. Acad. Sci. U. S. A 95, 12153–12157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Abe Y, Suzuki T, Mizuno T, Ono C, Iwamoto K, Hosobuchi M, and Yoshikawa H (2002) Effect of increased dosage of the ML-236B (compactin) biosynthetic gene cluster on ML-236B production in Penicillium citrinum, Mol. Genet. Genomics 268, 130–137. [DOI] [PubMed] [Google Scholar]
- 11.Wiemann P, Guo CJ, Palmer JM, Sekonyela R, Wang CC, and Keller NP (2013) Prototype of an intertwined secondary-metabolite supercluster, Proc. Natl. Acad. Sci. U. S. A 110, 17065–17070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.von Dohren H (2009) A survey of nonribosomal peptide synthetase (NRPS) genes in Aspergillus nidulans, Fungal Genet. Biol 46 Suppl 1, S45–52. [DOI] [PubMed] [Google Scholar]
- 13.Bergmann S, Funk AN, Scherlach K, Schroeckh V, Shelest E, Horn U, Hertweck C, and Brakhage AA (2010) Activation of a silent fungal polyketide biosynthesis pathway through regulatory cross talk with a cryptic nonribosomal peptide synthetase gene cluster, Appl. Environ. Microbiol 76, 8143–8149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Goldberg AL (2012) Development of proteasome inhibitors as research tools and cancer drugs, J. Cell Biol 199, 583–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kisselev AF, van der Linden WA, and Overkleeft HS (2012) Proteasome inhibitors: an expanding army attacking a unique target, Chem. Biol 19, 99–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chiang YM, Chang SL, Oakley BR, and Wang CC (2011) Recent advances in awakening silent biosynthetic gene clusters and linking orphan clusters to natural products in microorganisms, Curr. Opin. Chem. Biol 15, 137–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nayak T, Szewczyk E, Oakley CE, Osmani A, Ukil L, Murray SL, Hynes MJ, Osmani SA, and Oakley BR (2006) A versatile and efficient gene-targeting system for Aspergillus nidulans, Genetics 172, 1557–1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Szewczyk E, Nayak T, Oakley CE, Edgerton H, Xiong Y, Taheri-Talesh N, Osmani SA, and Oakley BR (2006) Fusion PCR and gene targeting in Aspergillus nidulans, Nat. Protoc 1, 3111–3120. [DOI] [PubMed] [Google Scholar]
- 19.Ahuja M, Chiang YM, Chang SL, Praseuth MB, Entwistle R, Sanchez JF, Lo HC, Yeh HH, Oakley BR, and Wang CC (2012) Illuminating the diversity of aromatic polyketide synthases in Aspergillus nidulans, J. Am. Chem. Soc 134, 8212–8221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hines J, Groll M, Fahnestock M, and Crews CM (2008) Proteasome inhibition by fellutamide B induces nerve growth factor synthesis, Chem. Biol 15, 501–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fischbach MA, and Walsh CT (2006) Assembly-line enzymology for polyketide and nonribosomal Peptide antibiotics: logic, machinery, and mechanisms, Chem. Rev 106, 3468–3496. [DOI] [PubMed] [Google Scholar]
- 22.Cacho RA, Jiang W, Chooi YH, Walsh CT, and Tang Y (2012) Identification and characterization of the echinocandin B biosynthetic gene cluster from Emericella rugulosa NRRL 11440, J. Am. Chem. Soc 134, 16781–16790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Badrinarayanan S, and Sperry J (2011) Biomimetic synthesis of 2,5-bis(indol-3-ylmethyl)pyrazine via intermolecular amino aldehyde cyclization, Synlett 16, 2339–2342. [Google Scholar]
- 24.Andersen MR, Nielsen JB, Klitgaard A, Petersen LM, Zachariasen M, Hansen TJ, Blicher LH, Gotfredsen CH, Larsen TO, Nielsen KF, and Mortensen UH (2013) Accurate prediction of secondary metabolite gene clusters in filamentous fungi, Proc. Natl. Acad. Sci. U. S. A 110, E99–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shigemori H, Wakuri S, Yazawa K, Nakamura T, Sasaki T, and Kobayashi J (1991) Fellutamide-A and fellutamide-B, cytotoxic peptides from a marine fish-possessing fungus Penicillium-fellutanum, Tetrahedron 47, 8529–8534. [Google Scholar]
- 26.Wu CJ, Li CW, and Cui CB (2014) Seven new and two known lipopeptides as well as five known polyketides: the activated production of silent metabolites in a marine-derived fungus by chemical mutagenesis strategy using diethyl sulphate, Mar. Drugs 12, 1815–1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Albright JC, Henke MT, Soukup AA, McClure RA, Thomson RJ, Keller NP, and Kelleher NL (2015) Large-scale metabolomics reveals a complex response of Aspergillus nidulans to epigenetic perturbation, ACS Chem. Biol 10, 1535–1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schneekloth JS Jr., Sanders JL, Hines J, and Crews CM (2006) Neurotrophic peptide aldehydes: solid phase synthesis of fellutamide B and a simplified analog, Bioorg. Med. Chem. Lett 16, 3855–3858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Giltrap AM, Cergol KM, Pang A, Britton WJ, and Payne RJ (2013) Total synthesis of fellutamide B and deoxy-fellutamides B, C, and D, Mar. Drugs 11, 2382–2397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nødvig CS, Nielsen JB, Kogle ME, and Mortensen UH (2015) A CRISPR-Cas9 system for genetic engineering of filamentous fungi. PLoS One 10, e0133085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.(2008) CLSI. Reference Method for Broth Dilution Antifungal Susceptibility Testing of Filamentous Fungi; Approved Standard—Second edition, Clinical and Laboratory Standards Institute, Wayne, PA. [Google Scholar]
- 32.Vishniac W, Santer M (1957) The thiobacilli. Bacteriol. Rev 21, 195–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Creaser EH, Porter RL, Britt KA, Pateman A,J, and Doy CH (1985) Purification and preliminary characterization of alcohol dehydrogenase from Aspergillus nidulans. Biochem. J 225, 449–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Oakley CE, Edgerton-Morgan H, and Oakley BR (2012) Tools for manipulation of secondary metabolism pathways: rapid promoter replacements and gene deletions in Aspergillus nidulans, Methods Mol. Biol 944, 143–161. [DOI] [PubMed] [Google Scholar]
- 35.Chiang YM, Oakley CE, Ahuja M, Entwistle R, Schultz A, Chang SL, Sung CT, Wang CC, and Oakley BR (2013) An efficient system for heterologous expression of secondary metabolite genes in Aspergillus nidulans, J. Am. Chem. Soc 135, 7720–7731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dunne PW, and Oakley BR (1988) Mitotic gene conversion, reciprocal recombination and gene replacement at the benA, beta-tubulin, locus of Aspergillus nidulans, Mol. Gen. Genet 213, 339–345. [DOI] [PubMed] [Google Scholar]
- 37.Bok JW, Chiang YM, Szewczyk E, Reyes-Domingez Y, Davidson AD, Sanchez JF, Lo HC, Watanabe K, Strauss J, Oakley BR, Wang CC, and Keller NP (2009) Chromatin-level regulation of biosynthetic gene clusters, Nat. Chem. Biol 5, 462–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.