

Abstract
This communication describes the design and fabrication of SD-DEP chip with simple components for single-cell manipulation and downstream nucleic acid analysis. The device employed the traditional DEP and insulator DEP to create the local electric field that is tailored to approximately the size of single cells, enabling highly efficient single-cell capture. The multistep procedures of cell manipulation, compartmentalization, lysis, and analysis were performed in the integrated microdevice, consuming minimal reagents, minimizing contamination, decreasing lysate dilution, and increasing assay sensitivity. The platform developed here could be a promising and powerful tool in single-cell research for precise medicine.
Keywords: Single-cell Analysis, Microfluidic Devices, Dielectrophoresis, LAMP, Self-Digitization
Graphical Abstract
Self-digitization dielectrophoretic (SD-DEP) chip was designed by employing the traditional DEP and insulator DEP to create the local electric field for single-cell manipulation and downstream nucleic acid analysis. The multistep procedures of cell manipulation, lysis, and chemical analysis were performed in the integrated microdevice, consuming minimal reagents, minimizing contamination, and increasing assay sensitivity.
Heterogeneity among individual cells has posed significant challenges to traditional bulk assays, which would potentially mask important but rare biological information in heterogeneity at the single-cell level due to the assumption of average outcome, resulting in a misleading interpretation of clinical results.[1] For example, cancer relapse has been correlated to the presence of minimal residual disease (MRD), which is the persistence of disease cells after a patient is considered clinically “cured”.[2] Chemotherapy cannot select for rare resistant sub-clonal populations of cells, thus creating refractory disease, resistant to the initial treatment. Molecular analyses of single cells are required to better understand the function and development of these heterogeneous cell populations in disease research and develop effective therapeutic methods for personalized medicine.[3] An ongoing challenge in single-cell analysis is manipulation (e.g. sorting, trapping, etc.) of the cells of interest and followed by transfer of a single-cell into a designated container with extremely low volume (<1 μL) for downstream molecular analyses, including PCR, genome sequencing, proteomic expression, and secretion profiling.[4] Conventional and commonly used single-cell isolation methods include serial dilution,[5] pipet micromanipulation,[6] fluorescence-activated cell sorting (FACS),[7] and laser-capture microdissection (LCM)[8]. However, most of these strategies are plagued by time cost, operational complexity, unreliable outcome, limited efficiency, deterioration of cell viability, limitations in the dispensing of single cells into nanoliter volume liquids for further downstream analysis, large sample volume requirements, high risk of contamination, and/or the requirement of expensive instruments.
Microfluidic devices, tailored to approximately the size of individual cells with designed unique geometries and precise dimensions, confer inherent advantages over conventional technologies in single cell biology.[9] The capability of integrating the multistep procedures of cell manipulation, lysis, and chemical analysis in a portable and enclosed device could reduce reagent consumption, decrease contamination risk, minimize lysate dilution and increase assay sensitivity, which is critical for assaying low-abundance biomolecules in single cells such as mRNA.[10] Currently, various microfluidic systems have been developed to handle single cells based on a variety of basic micro-manipulation techniques, including hydrodynamic cell traps,[11] valve traps,[12] electrical traps,[13] surface acoustic waves traps,[14] droplets,[15] and micro-wells[5a, 16]. However, the fabrication of an integrated platform for the direct isolation and transfer of single cells into the separate chamber for downstream analysis remains challenging in this field.[17]
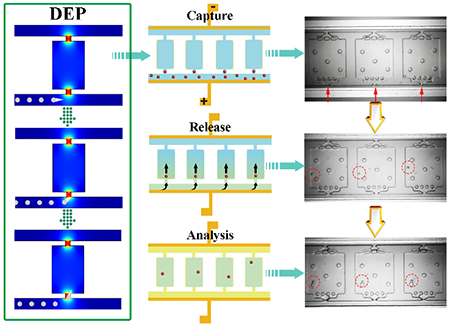
Here, we present the development of a self-digitization dielectrophoretic (SD-DEP) chip that enables rapid, convenient and highly efficient capture and on-demand release of single suspended cells into separated chambers for downstream nucleic-acid analysis using inexpensive and simple components. Compared to the single-cell isolation and analysis devices with multi-layer architecture, the SD-DEP chip described here does not require complex device fabrication and operations such as valves and mixing. The steps of cell analysis using SD-DEP chip are depicted in Scheme 1: A) prime the device with Pluronic F-127 solution to effectively inhibit cell adhesion on PDMS surface, B) fill the trapping channels and chambers with specific buffers and reagents (here, low electrical conductivity (LEC) DEP buffer[18] (8.0% sucrose, 0.3% dextrose, and 0.1% BSA in 1.0 mM Tris (pH 8.0, conductivity σ = 6.2 mS/m)) was applied for cell trapping), C) apply optimized trapping voltage and flow in the cell sample and trap cells at chamber openings, D) flush the excess cells with LEC buffer, turn off the trapping voltage and inject fresh loop mediated isothermal amplification (LAMP) reaction buffer to dispense the trapped cells into the chambers, E) fill the channel with the immiscible phase to isolate cells in the compartments, F) lyse cells and initiate single-cell LAMP reaction. The strength of this experimental design is three-fold. First, the introduction of an immiscible phase takes advantage of an existing robust technology, self-digitization (SD) chip, we developed. [19] This technology employs inherent fluidic phenomena to spontaneously divide an aqueous solution into nanoliter-scale water-in-oil plugs in an array of microfluidic chambers. Each water-in-oil emulsion plug formed on the SD-DEP chip can function as an independent chemical reactor, handling an extremely small volume of liquid containing cells and reagents for following molecular analysis of single cell lysates. Second, the active cell sorting method developed here relies on external DEP force without the use of cell labeling, which provides greater specificity and control of the force employed for cell capture than passive sorting mechanisms or spontaneous digitization alone.[18, 20] This label free cell sorting method has important advantages for conducting high fidelity single cell molecular analyses, such as minimized antibody related costs, applicability to cells lacking biomarkers, and maintaining integrity of endogenous biomarker expression. Finally, features such as the dimensions of the chamber opening and control over DEP force provide means to limit capture in each chamber to one cell. The whole process of cell manipulation was carried out under a microscope, which ensures a high confidence level that a single cell has been compartmentalized.
Scheme 1.

(A-F) Schematic description of each step of the DEP cell trapping and lysis process. (A) Pretreat the device with Pluronic F-127 solution and incubate at 4 °C overnight to effectively inhibit cell adhesion on PDMS surface; (B) Fill the device with low conductivity DEP (LEC) buffer; (C) Trap single cells at the inlet of each chamber with optimized AC voltage and frequency; (D) Turn off the trapping voltage and inject fresh LAMP reaction buffer to flush away the excess cells and dispense the trapped cells into the chambers; (E) Fill the channel with the immiscible phase to compartmentalize cells in chambers; (F) Lyse cells and initiate single-cell LAMP analysis; (G) Schematic of the SD DEP chip showing the dimensions of the key features.
Dielectrophoresis (DEP) is the movement of a polarizable particle caused by the interaction of an induced dipole in the particle with an electric field gradient. [21] The dielectrophoretic force can be either positive (pDEP) towards higher electric field strength or negative (nDEP), in which case the particle is forced down the electric field gradient. The time averaged DEP force, <FDEP>, exerted on a spherical particle by an AC electric field is given by the following equation.
| (1) |
Here, r is the particle radius, εm is the permittivity of the surrounding medium, E is the electric field strength, ω is electric field frequency, and Re[K(ω)] is the real part of the Claussius-Mossotti factor which compares the complex permittivity of the particle (ε*p) and medium (ε*m).
| (2) |
DEP has been employed extensively for the manipulation of biological cells for single-cell analysis [21] and in general, different types of DEP is classified by the strategy employed to generate the field gradient. The current device employs a combination of both traditional DEP, in which the cells are guided by a field gradient at the edge of an electrode, and insulator DEP (iDEP), which employs insulating structures to create local electric field maxima in an externally applied field. More specifically, the device is comprised of two parallel microfluidic channels (50 μm tall × 35 μm wide × 3.2 cm long) interconnected by a series of reaction chambers (50 μm tall × 260 μm long ×250 μm wide) having a narrow opening (15 μm wide) on each end. Filters having 15 μm and 10 μm spacing were located at the inlet and outlet side of the chamber, respectively. A thin film metallic electrode (50 μm wide × 3.2 cm long) extends the entire length of each microchannel and is in contact with the fluid (Scheme 1G). The detailed process for fabrication is shown in Figure S1A. The obtained coin-sized microfluidic device (Figure S1B) was pretreated with Pluronic® F127 overnight to coat the channel. Subsequently, a cell sample in LEC medium was introduced at the inlet of one of the microchannels and an AC voltage was applied at the electrode leads. Under these conditions, local electric field maxima existed both along the electrode edge and at the narrow entrances to the chambers, and thus the chronic myelogenous leukemia K562 cells employed underwent a pDEP response.[21, 22]The cells were attracted to and tracked along the edge of the electrode as they were flowed through the microchannel, and their movement was controlled by a combination of dielectrophoretic and drag forces (FDEP and Fdrag). The electrode edge was positioned sufficiently close to the narrow constrictions that the cells were exposed to the local field gradient there and were attracted to the openings and trapped.
To better understand the influence of the device dimensionality on performance, the design of the SD-DEP chip was guided by the simulation of the resulting electric field using the finite element method (COMSOL Multiphysics 5.2a). As we expected, the simulation results demonstrated that maximum electric field strength is located at the narrow opening of the chamber (Figure S1C). The distribution of the electric field is dominated by the applied voltage pattern (Figure S2), the geometry of the opening (Figure 1A) and the opening size (Figure S3), but is not affected by the dimension of the reaction chamber (Figure S4). Importantly, compared to a tapered entrance, a square-shaped inlet generates an enhanced and defined electric field cage similar in size to a single cell (Figure 1B), discouraging multi-cell capture that may be incurred by a broadly distributed electric field. To maintain pressure balance, the size and shape of the inlet and outlet were identical (15 μm wide, square shaped) for the following experiments. The cell manipulation performance of the SD-DEP chip was further estimated using K562 cells as the model. When no dielectrophoretic force was applied, the K562 cells followed the same path and exited through the outlet. When the dielectrophoretic force was applied, the movement of K562 cells was disturbed by the electric field generated along the channel and finally the cells were attracted to the openings and trapped (Figure 1C-H and Movie S1 in SI). As indicated in eq. (1), the DEP force is sensitive to the size, shape, and dielectric properties of the particles and applied electric field strength. Cells having different sizes experienced a different magnitude of DEP force and larger cells were more strongly deflected (Figure S5 A-F). Furthermore, the trajectories of cells could be regulated by tuning the strength of applied electric field (Figure S5 B and E).
Figure 1.
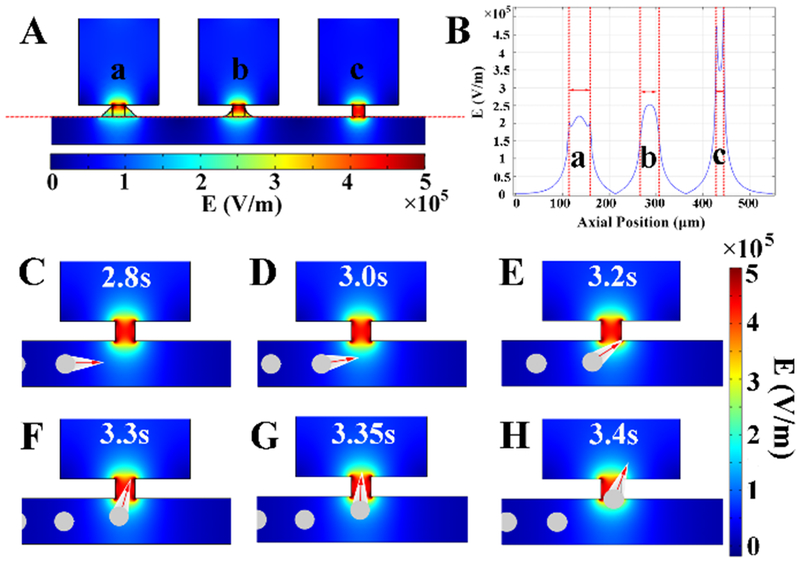
(A) COMSOL simulation of spatial electric-field distribution at the inlet of SD-DEP design with different shape of inlet; (B) The electric-field strength profile along the cut line in (A); (C-H) show the cell trajectories in the channel at different time points. For the sake of visualization, the DEP force applied on cells is demonstrated by the comet label with red arrows on cells. Cells are displayed in grey color. The spatial distribution of electric-field is illustrated in rainbow color. The movie of the simulation results is illustrated as Movie S1 in SI.
Guided by the simulation results, the cell manipulation performance of the as-designed SD-DEP chip was first studied. The Pluronic-pretreated device was rinsed with LEC solution for 15 min. Then, an optimal AC signal with the parameters of 5 Vp-p and 1.5 MHz was applied. A cell sample in LEC solution was pipetted into the inlet reservoir and a height differential was established between the inlet reservoir of the two channels to obtain a desired fluidic flow in the trapping channel. Cell trajectories were observed and recorded by microscopy. Optical images shown in Figure 2 illustrate the response of K562 cells, which could be sequentially trapped at the entrance of each chamber under the optimized flow and DEP force. Single-cell trapping was achieved by controlling the strength of the applied AC electric field, as shown in Figure S6 and Movie S3 in SI. Once the inlet of each chamber was occupied by a cell, the electric field was shielded and could not be perceived by other cells (Figure S7 and Movie S4 in SI). To better confirm the single-cell trapping results, K562 cells were stained with membrane dye FM 1-43, which had a bright fluorescence to better verify the occupation of single cells at the entrance of each chamber (Figure S8). Highly efficient single-cell capture (cell trapping efficiency: 100%; single-cell trapping efficiency: ~92.7%) dominated by DEP force and confined size of the inlet, as shown in Figure S9, allowed the cell populations to be isolated in individual chambers for further molecular characterization.
Figure 2.

(A-D) Multiple series of optical micrographs showing the sequential DEP trapping of K562 cells at the inlet of each chamber. Image slices A-D are taken at different time points for the same chamber. To better illustrate the sequential trapping process, cells are pointed to by colored arrows for easier discrimination. The whole process of the sequential cell trapping with DEP force was shown in Movie S2 in SI.
It is important to note that while a high single-cell capture success rate is key, the ability to reliably dispense the trapped cells into each chamber is also important for single cell analysis. In order to achieve high efficiencies with one single-cell per chamber, fresh low conductivity DEP buffer was first used to flush the excess cells that remained in the channel. Subsequently, complete dispensing of cells into chambers could be achieved by turning off the AC voltage and gradually increasing the flow rate by injection of the LAMP reagents (Movie S5 in SI). The results, as shown in Figure S10 and Figure S11, indicate that the SD-DEP chip enables highly efficient compartmentalization of single cells (single-cell compartmentalization efficiency: ~91.7%) as defined by the entrance geometry. Full dispensing of the captured cells was further supported by introduction of the immiscible-oil into the channel, as shown in Movie S6. These results are significant to confirm that the chip presented here provide secure single cell capture and manipulation for downstream analysis.
LAMP, a promising isothermal nucleic acid amplification method performed under a fixed temperature, simultaneously use 4 to 6 primers, which makes it highly specific for its target in the presence of non-target DNA.[19a] To attain a highly accurate downstream nucleic acids analysis of single cells in each chamber, complete mixing of LAMP reaction reagents with cell lysate is critical. Food dye was added into the LAMP reagent mixture to more clearly demonstrate the solution exchange process, as shown in Figure S12, indicating successful chip filling with LAMP reagents in a short time (about 3 min). The subsequent introduction of multiphase microfluidics provided a route to cell encapsulation by segregating single cells into discrete nanoliter compartments, isolated from other cells and surrounding environments. The filters were necessary to better control the pressure when the immiscible phase was introduced into the channel (Figure S13–15) and to prevent the loss of cells. With these optimized conditions, successful single cell capture, release, and compartmentalization were achieved (Figure 3A-C, Figure S16), offering a robust platform for subsequent on-chip analysis. To verify the performance of the SD-DEP chip for single-cell nucleic acid analysis, the expression of BCR-ABL gene in K562 cells was measured. BCR-ABL oncogene is a fusion gene resulting from a reciprocal translocation between chromosomes 9 and 22 and plays a critical role in the pathogenesis of chronic myeloid leukemia in humans, which is highly expressed in K562 cells.[19b, 23] The cell lysis condition was optimized by performing bulk real-time LAMP amplification of BCR-ABL gene in K562 cells in LAMP reaction buffers without and with different concentrations of cell lysis agent (Triton-X100), as shown in Figure S17. We found LAMP amplification performed in LAMP reaction buffer without the lysis agent, which relied on thermal lysis, showed better performance and a higher output signal. Thus, for the on-chip experiments we chose thermal lysis, which relies solely on heat induced denaturation of cell membrane proteins, thereby opening the cell, avoiding enzymatic or detergent contamination of the intracellular biomolecules.[24] We next evaluated the efficiency and reliability of on-chip cell processing by performing measurements of BCR-ABL oncogene expression from single K562 cells. K562 cells were loaded into the SD-DEP chip followed by capture, dispensing, compartmentalization, and analysis using thermal lysis and one-step LAMP amplification. LAMP amplification significantly increased the fluorescence intensity of the chambers containing a single cell (Figure S18). As shown in Figure 3D-E, successful amplification in 100% of single-cell containing chambers was observed, and further for each chamber that did not contain a cell no amplification was observed, indicating clear separation of individual cells in each chamber (Figure 3D, Figure 3F and Figure S19). Figure S20 illustrates the principal component analysis (PCA) plot of the expression data showing the characteristics of each single cell as a dot. The control group is marked with blue, whereas the K562 cell group is shown in red. In the plot, the two populations are well separated, meaning that distinguishing feature can be found between control group and K562 cells. In this manner, cells with similar expression will be clustered closely together, compared to the control group; dots in K562 cell group are dispersed, suggesting the heterogeneity of gene expression at single-cell level. This result confirms that the strategy described here established the precise measurement of specific RNA in single cells. The reliability of this method was further verified by shortening the LAMP reagent exchange time to generate a concentration gradient from right to left. The chambers with cells but low concentrations of LAMP reagent exhibited lower fluorescence signal (Figure S21), indicating that the output signal was indeed generated by the LAMP reaction. The strategy demonstrated here meets the challenges of high-efficiency single cell capture, on-demand compartmentalization of single cells, and nucleic acid amplification analysis in a simple “all integrated’’ device. In subsequent studies, we aim to exploit the application of SD-DEP chip in determining the distribution of genetic mutations in acute myeloid leukemia (AML) cell populations to better understand drug resistance and minimal residual disease.
Figure 3.

Series of optical micrographs, which demonstrate the DEP trapping of single K562 cell (A), dispensing of single K562 cells into each chamber (B) and compartmentalization of single cells in each chamber with oil (C). To better show the results, enlarged view of figure A-C is provided in Supporting Information as Figure S16. (D) Fluorescence image of the entire device showing 96 reactions of single-cell LAMP amplification. Image is taken after 120 min of LAMP reaction. i) control experiment without K562 cells; ii) and iii) are two parallel tests for the analysis of BCR-ABL gene in single K562 cells in each chamber. (E) The fluorescence intensity profile along the randomly picked chambers from (i) and (ii) in panel D. (F) Optical micrograph of the chambers marked with red square in iii) of panel D before LAMP reaction. There was one empty chamber without a cell in iii), thus further confirming the fluorescence signal was generated by the single-cell LAMP reaction.
In summary, by coupling the specific geometry of the microdevice with label-free dielectrophoretic capture, we developed an inexpensive SD-DEP chip that could rapidly isolate single cells with high efficiency, providing visual confirmation of successful trapping and release. The multistep procedures of cell manipulation, compartmentalization, lysis, and analysis could be performed in a portable and integrated microdevice with simple components, consuming minimal reagents, minimizing contamination, decreasing lysate dilution, and increasing assay sensitivity. The introduction of the self-digitization technology enabled the convenient formation of nanoliter-scale aqueous compartments, serving as the independent chemical reactor for the nucleic acid analysis of single cell lysates. These advancements suggest that this versatile microdevice could be a promising and powerful tool in single-cell research for precision medicine. Further optimization of the as-designed SD-DEP chip and the application of this technique for clinical samples analysis are now in progress in our laboratory.
Supplementary Material
Acknowledgements
We are grateful to the NIH (R21EB018831 to DTC and UG3CA211139 to DTC and JPR) for support of this work.
References
- [1].a) Buettner F, Natarajan KN, Casale FP, Proserpio V, Scial-done A, Theis FJ, Teichmann SA, Marioni JC, Stegle O, Nat. Biotechnol 2015, 33, 155. [DOI] [PubMed] [Google Scholar]; b) Satija R, Shalek AK, Trends Immunol. 2014, 35, 219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Tie J, Wang Y, Tomasetti C, Li L, Springer S, Kinde I, Silli-man N, Tacey M, Wong H-L, Christie M, Kosmider S, Skinner I, Wong R, Steel M, Tran B, Desai J, Jones I, Haydon A, Hayes T, Price TJ, Strausberg RL, Diaz LA, Papadopoulos N, Kinzler KW, Vogelstein B, Gibbs P, Sci. Transl. Med 2016, 8, 346ra92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].a) Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, Cahill DP, Nahed BV, Curry WT, Martuza RL, Louis DN, Rozenblatt-Rosen O, Suvà ML, Regev A, Bernstein BE, Science 2014, 344, 1396. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Khoo BL, Chaudhuri PK, Ramalingam N, Tan DSW, Lim CT, Warkiani ME, Int. J. Cancer 2016, 139, 243. [DOI] [PubMed] [Google Scholar]
- [4].a) Hosic SJ, Murthy SK, Koppes AN, Anal. Chem 2016, 88, 354. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Prakadan SM, Shalek AK, Weitz DA, Nat. Rev. Genet 2017, 18, 345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].a) Gole J, Gore A, Richards A, Chiu Y-J, Fung H-L, Bushman D, Chiang H-I, Chun J, Lo Y-H, Zhang K, Nat. Biotechnol 2013, 31, 1126. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Love JC, Ronan JL, Grotenbreg GM, van der Veen AG, Ploegh HL, Nat. Biotechnol 2006, 24, 703. [DOI] [PubMed] [Google Scholar]
- [6].Zhang K, Han X, Li Y, Li SY, Zu Y, Wang Z, Qin L, J. Am. Chem. Soc 2014, 136, 10858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Rinke C, Lee J, Nath N, Goudeau D, Thompson B, Poulton N, Dmitrieff E, Malmstrom R, Stepanauskas R, Woyke T, Nat. Protoc 2014, 9, 1038. [DOI] [PubMed] [Google Scholar]
- [8].Wang K, Lau TY, Morales M, Mont EK, Straus SE, J. Virol 2005, 79, 14079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].He JC, Brimmo AT, Qasaimeh MA, Chen PY, Chen WQ, Small Methods 2017, 1, DOI: 10.1002/smtd.201700192. [DOI] [Google Scholar]
- [10].a) White AK, VanInsberghe M, Petriv OI, Hamidi M, Si-korski D, Marra MA, Piret J, Aparicio S, Hansen CL, Proc. Natl. Acad. Sci. U S A 2011, 108, 13999. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Toriello NM, Douglas ES, Thaitrong N, Hsiao SC, Francis MB, Bertozzi CR, Mathies RA, Proc. Natl. Acad. Sci. U S A 2008, 105, 20173. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) White AK, Heyries KA, Doolin C, VanInsberghe M, Hansen CL, Anal. Chem 2013, 85, 7182. [DOI] [PubMed] [Google Scholar]; d) Marcy Y, Ouverney C, Bik EM, Lo¨ sekann T, Ivanova N, Martin HG, Szeto E, Platt D, Hugenholtz P, Relman DA, Quake SR, Proc. Natl. Acad. Sci. U S A 2007, 104, 11889. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Fan HC, Wang J, Potanina A, Quake SR, Nat. Biotechnol 2011, 29, 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Kimmerling RJ, Lee Szeto G, Li JW, Genshaft AS, Kazer SW, Payer KR, de Riba Borrajo J, Blainey PC, Irvine DJ, Shalek AK, Manalis SR, Nat. Commun 2016, 7, 10220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].a) Fan HC, Wang J, Potanina A, Quake SR, Nat. Biotechnol 2010, 29, 51. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Ma C, Fan R, Ahmad H, Shi Q, Comin-Anduix B, Chodon T, Koya RC, Liu C-C, Kwong GA, Radu CG, Ribas A, Heath JR, Nat. Med 2011, 17, 738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Toriello NM, Douglas ES, Mathies RA, Anal. Chem 2005, 77, 6935. [DOI] [PubMed] [Google Scholar]
- [14].Collins DJ, Morahan B, Garcia-Bustos J, Doerig C, Plebanski M, Neild A, Nat. Commun 2015, 6, 8686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].a) Mazutis L, Gilbert J, Ung WL, Weitz DA, Griffiths AD, Heyman JA, Nat. Protoc 2013, 8, 870. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Joensson HN, Svahn HA, Angew. Chem. Int. Ed 2012, 51, 12176. [DOI] [PubMed] [Google Scholar]; c) Zhang H, Jenkins G, Zou Y, Zhu Z, Yang CJ, Anal. Chem 2012, 84, 3599. [DOI] [PubMed] [Google Scholar]; d) Fu Y, Li C, Lu S, Zhou W, Tang F, Xie XS, Huang Y, Proc. Natl. Acad. Sci. U S A 2015, 112, 11923. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Novak R, Zeng Y. Shuga J, Venugopalan G Fletcher DA, Smith MT, Mathies RA, Angew. Chem. Int. Ed 2011, 50, 390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Wood DK, Weingeist DM, Bhatia SN, Engelward BP, Proc. Natl. Acad. Sci. U S A 2010, 107, 10008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].a) Kim SH, Yamamoto T, Fourmy D, Fujii T, Small, 2011, 7, 3239. [DOI] [PubMed] [Google Scholar]; b) Kim SH, He X, Kaneda S, Kawada J, Fourmy D, Noji H, Fujii T, Lab Chip, 2014, 14, 730. [DOI] [PubMed] [Google Scholar]; c) Kim SH, Fujii T, Lab Chip, 2016, 16, 2440. [DOI] [PubMed] [Google Scholar]
- [18].Anand RK, Johnson ES, Chiu DT, J. Am. Chem. Soc 2015, 137, 776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].a) Gansen A, Herrick AM, Dimov IK, Lee LP, Chiu DT, Lab Chip 2012, 12, 2247. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Thompson AM, Gansen A, Paguirigan AL, Kreutz JE, Radich JP, Chiu DT, Anal. Chem 2014, 86, 12308. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Schneider T, Yen GS, Thompson AM, Burnham DR, Chiu DT, Anal. Chem 2013, 85, 10417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Li M, Anand RK, J. Am. Chem. Soc 2017, 139, 8950. [DOI] [PubMed] [Google Scholar]
- [21].a) Gagnon ZR, Electrophoresis 2011, 32, 2466. [DOI] [PubMed] [Google Scholar]; b) Li M, Anand RK, Anal. Bioanal. Chem 2018, 410, 2499. [DOI] [PubMed] [Google Scholar]
- [22].a) Suehiroy J, Pethig R, J. Phys. D: Appl. Phys 1998, 31, 3298 [Google Scholar]; b) Altomare L, Borgatti M, Medoro G, Manaresi N, Tartagni M, Guerrieri R, Gambari R, Biotechnol Bioeng. 2003, 82, 474. [DOI] [PubMed] [Google Scholar]
- [23].Ren R, Nat. Rev. Cancer, 2005, 5, 172. [DOI] [PubMed] [Google Scholar]
- [24].Gong Y, Ogunniyi AO, Love JC, Lab Chip 2010, 10, 2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.