

Abstract
The nematode Caenorhabditis elegans has been central to the understanding of metazoan biology. However, C. elegans is but one species among millions and the significance of this important model organism will only be fully revealed if it is placed in a rich evolutionary context. Global sampling efforts have led to the discovery of over 50 putative species from the genus Caenorhabditis, many of which await formal species description. Here, we present species descriptions for 10 new Caenorhabditis species. We also present draft genome sequences for nine of these new species, along with a transcriptome assembly for one. We exploit these whole‐genome data to reconstruct the Caenorhabditis phylogeny and use this phylogenetic tree to dissect the evolution of morphology in the genus. We reveal extensive variation in genome size and investigate the molecular processes that underlie this variation. We show unexpected complexity in the evolutionary history of key developmental pathway genes. These new species and the associated genomic resources will be essential in our attempts to understand the evolutionary origins of the C. elegans model.
Keywords: C. elegans, genomics, phylogenomics, morphology, species description
Impact summary.
Caenorhabditis elegans is a tiny, free‐living nematode, or roundworm, which is been used extensively in biological research. Recent years have seen a focused effort to discover new species that are closely related to C. elegans in the hope that they will help us understand how this important model organism evolved. We present formal species descriptions and names for 10 new species of Caenorhabditis isolated from across the world. We sequenced the genomes and transcriptomes of each new species and use the data to reconstruct the evolutionary history of these and other Caenorhabditis species. We demonstrate the utility of these new species and their associated data by analyzing the evolution of morphology, the evolution of important developmental genes, and the evolution of genome size. These new resources will be essential to our attempts to understand the evolutionary origins of this important nematode.
Caenorhabditis elegans has become one of the preeminent model organisms in modern biology, but only recently have we started to understand its natural ecology and evolutionary history (Félix & Braendle 2010). Studying C. elegans within the context of its wild ecology and alongside its closest relatives will provide an important evolutionary context for the particular systems analyzed within the laboratory. A large collection of wild‐caught strains of C. elegans is now available for comparative exploration of natural variation (Cook et al. 2016, 2017), and ecological interactions are being investigated through co‐analysis of nematodes and microbial associates from natural systems (Schulenburg & Félix 2017). In parallel, the last decade has seen a focused search for new species in the genus Caenorhabditis.
The discovery of new Caenorhabditis species was, for many years, hindered by a poor understanding of the natural ecology of these nematodes (Félix & Braendle 2010). Surveys of natural populations of C. elegans and Caenorhabditis briggsae revealed that, rather than being “soil nematodes,” Caenorhabditis species thrive in microbe‐rich environments, such as rotting fruits, flowers, and stems (Kiontke et al. 2011; Félix & Duveau 2012; Félix et al. 2014; Ferrari et al. 2017). This new understanding, combined with extensive worldwide sampling efforts, has led to the discovery of more than 50 species (Kiontke et al. 2011; Félix et al. 2014; Ferrari et al. 2017; MAF, LF, MVR, CB, unpublished; John Wang, Michael Ailion, Erik Andersen, Asher Cutter, pers. comm.) of Caenorhabditis, many of which await formal species description.
Although morphology remains fundamental to species diagnosis, closely related Caenorhabditis species are often morphologically very similar (Sudhaus & Kiontke 2007) and many morphological characters are homoplasious within the genus (Kiontke et al. 2011). This has motivated the use of mating tests for species definition and comparisons of molecular sequences such as ribosomal DNA (rDNA) or internal transcribed spacer (ITS) sequences, in addition to morphology, for species diagnosis (Kiontke et al. 2011; Félix et al. 2014). These molecular sequences have also enabled a reconstruction of the Caenorhabditis phylogeny (Cho et al. 2004; Kiontke et al. 2004, 2011). Recently, whole‐genome data have been exploited for phylogenomic analysis (Slos et al. 2017).
Genome sequences of species closely related to C. elegans have furthered our understanding of C. elegans biology and revealed insights into the evolutionary forces that have shaped its genome. The publication of the C. briggsae genome in 2003 enabled the first comparative genomics studies of Caenorhabditis, which revealed an unusually high rate of intrachromosomal rearrangement but comparatively rare interchromosomal rearrangement (Stein et al. 2003). Additional genome sequences have been published from species across the genus (Mortazavi et al. 2010; Fierst et al. 2015; Slos et al. 2017; Kanzaki et al. 2018; Ren et al. 2018; Yin et al. 2018). Comparisons between genomes of hermaphroditic species such as C. elegans and their outcrossing relatives have exposed the genomic consequences of a switch in reproductive mode from gonochorism to autogamous hermaphroditism, including changes in overall genome structure, gene structure, and protein‐coding gene content (Thomas et al. 2012; Fierst et al. 2015; Kanzaki et al. 2018; Yin et al. 2018).
Here, we use mating tests, accompanied by molecular and morphological analyses, to characterize and describe 10 new Caenorhabditis species isolated from across the world. We present draft genome sequences for nine of the 10 new species, and a transcriptome assembly for one, and use the data to reconstruct the Caenorhabditis phylogeny. By studying morphology in the context of this phylogenetic tree, we find further examples of homoplasious morphological characters in the genus. We use the genome sequences to study variation in genome size and the evolution of genes involved in a key developmental pathway. These new species and their draft genome sequences will become an important resource for the growing number of evolutionary biologists who use Caenorhabditis in their research.
Results
TEN NEW SPECIES DECLARATIONS
By sampling a variety of substrates in diverse geographic locations, we found 10 new species of Caenorhabditis, most of them from rotting fruit. While initial selection of isolates for further analysis was based on morphological or molecular assignment to Caenorhabditis, as argued and implemented in Félix et al. (2014), the justification for raising species is based on a biological species concept that can be easily implemented for these culturable nematodes. Thus, for each new isolate, we attempted crosses with previously described species in culture that have the most similar ribosomal RNA cistron internal transcribed spacer 2 (rDNA ITS2) sequence (Table 1 or Table S1). The results of the crosses are shown in Table S2. For two putative species, Caenorhabditis parvicauda sp. n. and Caenorhabditis uteleia sp. n., that also had striking morphological novelty (see below), no alignment of the rDNA ITS2 region could be obtained with default parameters in NCBI nucleotide BLAST (word size 28, gap penalty 1,2, gap costs 0,2.5). We considered these highly divergent from any known species and did not perform mating tests. Based on the phylogenetic relationships detailed below, the 10 species all belong to the clade of species whose most basally branching member is Caenorhabditis monodelphis (Slos et al. 2017) and are therefore in the genus Caenorhabditis (Osche 1952; Dougherty 1953).
Table 1.
Caenorhabditis species names, reference isolates and sequenced strains
| Caenorhabditis species name | Temp. species number | Abbreviation | Reference isolate | Sequenced strain (inbreeding rounds) | Genome accession number | rDNA accession number(s) |
|---|---|---|---|---|---|---|
| Caenorhabditis parvicauda sp. n. | C. sp. 21 | Cpv | NIC134 | NIC534 (25x) | PRJEB12595 | MH800325 |
| Caenorhabditis zanzibari sp. n. | C. sp. 26 | Cza | JU2161 | JU2190 (26x) | PRJEB12596 | MH809973, MH809941, MH809969 |
| Caenorhabditis panamensis sp. n. | C. sp. 28 | Cpa | QG702 | QG2080 (25x) | PRJEB28259 | MH809974, MH809942, MH809970 |
| Caenorhabditis becei sp. n. | C. sp. 29 | Cbe | QG704 | QG2083 (25x) | PRJEB28243 | MH809975, MH809943, MH809971 |
| Caenorhabditis uteleia sp. n. | C. sp. 31 | Cut | JU2469 | JU2585 (25x) | PRJEB12600 | MH800326 |
| Caenorhabditis sulstoni sp. n. | C. sp. 32 | Csu | SB454 | JU2788 (25x) | PRJEB12601 | MH800333 |
| Caenorhabditis quiockensis sp. n. | C. sp. 38 | Cqu | JU2745 | JU2809 (25x) | PRJEB11354 | MH800334 |
| Caenorhabditis waitukubuli sp. n. | C. sp. 39 | Cwt | NIC564 | NIC564 (isofemale) | PRJEB12602 | MH800335 |
| Caenorhabditis tribulationis sp. n. | C. sp. 40 | Ctb | JU2774 | JU2818 (25x) | PRJEB12608 | MH809976, MH809944, MH809972, |
| Caenorhabditis vivipara sp. n. | C. sp. 43 | Cvv | NIC1070 | NIC1070 | PRJEB12605 | MH800336 |
Specific name referents were derived as follows: 21: Reduced tail (in the male). 26: First isolated in Zanzibar. 28: First isolated in Panama. 29: First isolated on Barro Colorado Island, Panama. 31: U‐shape male tail. 32: In honor of John Sulston. 38: First isolated on Quiock river trail, Guadeloupe. 39: First isolated on the island of Dominique, in the local native American language. 40: First isolated in the region of Cape Tribulation, Australia. 43: Viviparous.
The formal descriptions of each species can be found in Document S1. We describe the following new species (see Table 1 for the correspondence with the informal numbering system used to refer to the species in previous publications):
Caenorhabditis becei sp. n.
Caenorhabditis panamensis sp. n.
Caenorhabditis parvicauda sp. n.
Caenorhabditis quiockensis sp. n.
Caenorhabditis sulstoni sp. n.
Caenorhabditis tribulationis sp. n.
Caenorhabditis uteleia sp. n.
Caenorhabditis vivipara sp. n.
Caenorhabditis waitukubuli sp. n.
Caenorhabditis zanzibari sp. n.
GENOME SEQUENCES OF NINE NEW CAENORHABDITIS SPECIES
We sequenced the genomes of all newly described species to high coverage (100–350×) using short‐read Illumina technology. After identifying and removing reads from nontarget organisms, we generated draft assemblies for each species, employing heterozygosity‐aware assembly approaches where necessary. Draft assemblies were scaffolded using assembled transcripts or long‐insert (or “mate‐pair”) data, where available. The sequence data generated for Caenorhabditis vivipara were not of sufficient quality to generate a reference assembly and are not discussed further. All assemblies have been submitted to DDBJ/ENA/GenBank (Table 1).
Assembly span indicated substantial variation in genome size among species (Table 2). At 65.1 Mb, the genome of Caenorhabditis sulstoni is the smallest Caenorhabditis genome published thus far, and nearly 35 Mbp smaller than the C. elegans genome. For all species other than C. waitukubuli (see below), assembly spans were consistent with semi‐independent estimates based on kmer spectra analysis (Fig. S1). The contiguity of the resulting assemblies was highly variable. The assemblies of Caenorhabditis becei and C. panamensis, which were scaffolded with long‐insert data, are the most contiguous, with N50 lengths of 487 and 768 kbp, respectively. The assembly of Caenorhabditis waitukubuli is the least contiguous, with an N50 length of 15.1 kbp. Kmer spectrum analysis (Fig. S2) indicated extensive heterozygosity present in the genome of this strain, which has not been fully collapsed during assembly. The proportion of undetermined bases (i.e., gaps denoted as Ns) was low in all cases. Despite considerable differences in assembly contiguity, Benchmarking Universal Single‐Copy Orthologs (BUSCO) and Core Eukaryotic Genes Mapping Approach (CEGMA) indicated that all assemblies were of high gene‐level completeness.
Table 2.
Genome assembly and annotation metrics for nine species of Caenorhabditis
| Species | Species number | Sequenced strain | Assembly span (Mbp) | Scaffold count (n) | N50 (kbp) | Ns% | BUSCO genome complete/fragmented % | Protein‐coding gene count (n) | BUSCO proteome complete/fragmented % |
|---|---|---|---|---|---|---|---|---|---|
| C. parvicauda | 21 | NIC534 | 93.7 | 5,719 | 44.4 | 1.11 | 89.5/5 | 16,412 | 89.9/6.4 |
| C. zanzibari | 26 | JU2190 | 101.1 | 3,128 | 91.3 | 0.22 | 98.2/1.2 | 22,198 | 98.6/1 |
| C. panamensis | 28 | QG2080 | 79.0 | 986 | 487.2† | 1.16 | 97.3/1.8 | 17,134* | 91.9/3.6 |
| C. becei | 29 | QG2083 | 87.9 | 1,567 | 767.5† | 1.74 | 97.5/1.4 | 18,669* | 91.9/3.9 |
| C. uteleia | 31 | JU2585 | 104.0 | 3,222 | 177.0 | 0.99 | 96/3.1 | 27,614 | 96.1/3.1 |
| C. sulstoni | 32 | JU2788 | 65.1 | 2,044 | 136.7 | 0.59 | 97.9/1 | 18,192 | 95.3/3.3 |
| C. quiockensis | 38 | JU2809 | 100.4 | 4,890 | 139.4 | 0.09 | 96.1/3.2 | 22,278 | 95.7/3.4 |
| C. waitukubuli | 39 | NIC564 | 91.4 | 21,203 | 15.1 | 1.27 | 92.5/4.8 | 30,089 | 92.7/5.5 |
| C. tribulationis | 40 | JU2818 | 101.2 | 3,276 | 224.5 | 0.15 | 97.7/1.1 | 24,787 | 97.9/1.4 |
| C. elegans | ‐ | N2 | 100.2 | 7 | 17,493.8 | 0.00 | 98.4/1.0‡ | 20,094 | 99.7/0.3‡ |
*RNA‐seq data were not generated and therefore not used to guide gene prediction.
†long‐insert (or “mate‐pair”) data were used during assembly. BUSCO version 3.0.2 was used with the ‘Nematoda_odb9’ dataset for both genome and proteome completeness assessment. Scaffolds shorter than 500 bp were discarded prior to annotation. Wormbase version WS264 of the C. elegans genome was used (Lee et al. 2018).
‡BUSCO does not find core genes in the complete C. elegans N2 genome. More detailed metrics are presented in Table S3.
RNA‐seq data were used to guide gene prediction for all species except C. becei and C. panamensis. The number of protein‐coding genes predicted in each assembly varied considerably. The genome of C. parvicauda has the fewest predicted genes, at 16,412. This is likely an underestimate of the true gene number, as ∼5% of BUSCO genes were absent from the assembly. C. waitukubuli has 30,089 predicted protein‐coding genes. Overall 19.8% of BUSCO genes found in this assembly were present in multiple copies, suggesting that this high gene number is an artifact arising from regions of uncollapsed heterozygosity present in the assembly. The lack of RNA‐seq data for C. becei and C. panamensis resulted in less complete gene sets, with a larger percentage of BUSCO genes missing from the gene sets (4.5% and 4.2%, respectively) than from the draft assemblies (0.9% and 1.1%, respectively).
PHYLOGENETIC RELATIONSHIPS WITHIN THE GENUS CAENORHABDITIS
Previous analyses of Caenorhabditis phylogeny have used morphology or small numbers of loci and have defined subgeneric groups of taxa (Kiontke et al. 2011): the Elegans supergroup, which contains the Japonica and Elegans groups, and the Drosophilae supergroup, which contains the Drosophilae and Angaria groups. We exploited our new and existing genomic data to re‐examine the phylogenetic structure of Caenorhabditis. We performed orthology clustering of 781,865 protein sequences predicted from the genomes and transcriptomes of all 10 newly described species, 22 other Caenorhabditis species (C. elegans Sequencing Consortium 1998; Stein et al. 2003; Mortazavi et al. 2010; Kanzaki et al. 2018; Yin et al. 2018), and from the outgroup taxon Diploscapter coronatus (Hiraki et al. 2017). We identified 1988 single‐copy orthologues, each of which was present in at least 27 of the 33 taxa, and aligned their amino‐acid sequences. We performed maximum likelihood (ML) and Bayesian inference (BI) analyses on a concatenated alignment of these loci. We also employed a supertree approach by estimating gene trees for all single‐copy loci using ML analysis and providing the resulting topologies to ASTRAL‐III (Mirarab & Warnow 2015) to estimate the species tree.
The three analyses yielded highly congruent, well‐supported topologies that displayed very few inconsistencies, discussed below (Fig. 1; Fig. S3–5). The majority of relationships, including the monophyly of both the Elegans and Japonica groups, were recovered with maximal support (bootstrap support of 100 and Bayesian posterior probability values of 1) regardless of method. All approaches recovered a clade of C. guadeloupensis + C. uteleia as sister to the Elegans supergroup. We found C. parvicauda to be the second‐most basally arising species in the genus. We recovered C. sulstoni, C. becei, C. waitukubuli, and C. panamensis as members of the Japonica group. The sister taxa C. zanzibari and C. tribulationis were placed as members of the Elegans group, being most closely related to C. sinica. We recovered C. quiockensis as sister to C. angaria + C. castelli.
Figure 1.
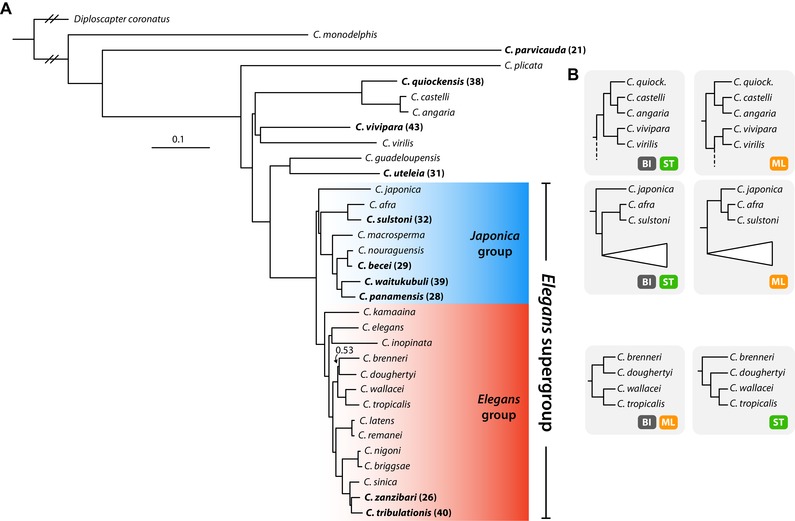
Phylogenetic relationships of 32 Caenorhabditis species and D. coronatus. (A) Phylogeny inferred using Bayesian inference with the CAT‐GTR+Γ substitution model. Species described here are highlighted in bold, with previous species numbers in parentheses. Bayesian posterior probabilities are 1.0 unless noted as branch annotations. Scale is in substitutions per site. (B) Alternative hypotheses and support from each analysis approach. ML, Maximum likelihood inference using GTR+Γ substitution model; BI, Bayesian inference using the CAT‐GTR+Γ substitution model; ST, Supertree approach, using gene trees as input (substitution model selected automatically for each alignment).
Three relationships, which tended to have lower‐than‐maximal support, were inconsistent across methods (Fig. 1B). Both the supertree approach and BI recovered the C. vivipara + C. virilis group as sister to the clade containing C. quiockensis, C. castelli, and C. angaria, while ML analysis recovered this group as sister to the group consisting of the sister taxa C. guadeloupensis + C. uteleia and the Elegans supergroup. Second, the placement of C. japonica as sister to other members of the Japonica group was recovered by both the supertree approach and by BI, whereas ML analysis recovered C. japonica as sister to C. afra + C. sulstoni. Finally, BI and ML analysis recovered C. brenneri and C. doughertyi as sister taxa, whereas the supertree approach recovered C. brenneri as sister to the clade containing C. doughertyi, C. wallacei, and C. tropicalis.
MORPHOLOGICAL NOVELTY IN CAENORHABDITIS PARVICAUDA SP. N. AND CAENORHABDITIS UTELEIA SP. N
Morphological features of C. parvicauda are displayed in Fig. 2. Most striking is the absence of a fan in the tail of C. parvicauda adult males (Fig. 2F, I, Fig. S6; compare with Figs. 3C,D and 4A) and a left–right asymmetry in the locations of caudal papillae. The caudal papillae include two pairs anterior to the cloaca (v1, v2) followed by groups of two (v3, v4), two (v5 and ad), and three pairs (v6, v7, and pd [posterior dorsal]) as seen in Fig. 2F, I and Fig. S6. Papillae ad (anterior dorsal) and v5 are both open to the outside. The positions of the dorsal papillae ad (anterior dorsal) and pd generally differ between the right and left sides at two levels. First, whereas papilla v5 has a similar anterior–posterior position on the right and left sides, papilla ad is generally anterior to v5 on the right side and at the same position or posterior to v5 on the left side (Fig. 2F, I and Fig. S6). Second, while papilla pd has a similar anterior–posterior position on the right and left sides, ventral papillae v6 and v7 are generally located posterior to it on the right side and anterior on the left side (Fig. 2I and Fig. S6). The spicules are thick, with a complex tip (Fig. 2G). Simple pre‐ and post‐cloacal sensilla can be seen (Fig. 2H). The males mate in a spiral position. This left–right asymmetry is highly unusual. The outer side of the mouth of C. parvicauda is endowed with the usual set of sensory organs disposed in a concentric manner, namely six labial sensillae, two amphids, and four male‐specific cephalic sensillae (Fig. 2A). The buccal cavity is short compared to most other Caenorhabditis (Sudhaus & Kiontke 1996), with two teeth at the base. The pharyngeal sleeve extends anteriorly to half of the buccal cavity (Fig. 2B). Three cuticular ridges can be seen in the lateral field of adults of both sexes (Fig. 2C, D). The adult female tail end is long and thin (Fig. 2E).
Figure 2.
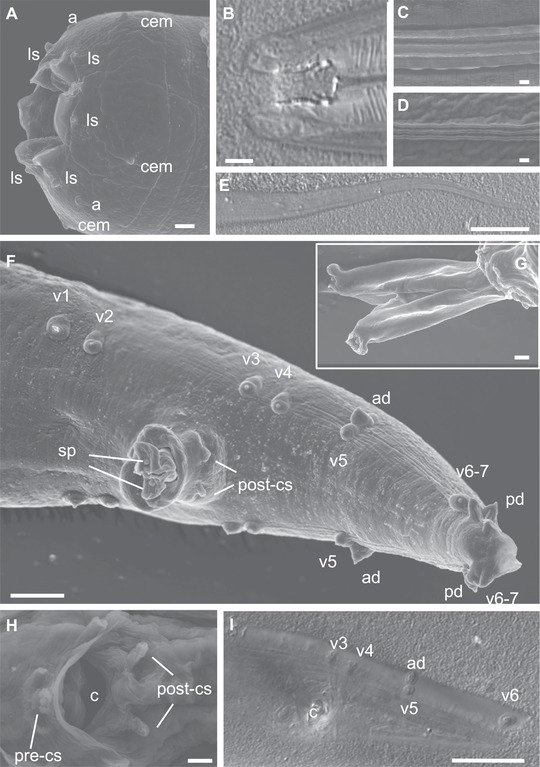
Morphology of Caenorhabditis parvicauda sp. n. by scanning electron microscopy (SEM) and Nomarski optics (DIC). (A and B) Mouth of an adult male (A, SEM; B, DIC). (C) Cuticular lateral ridges of a dauer juvenile (SEM). (D) Cuticular lateral ridges of an adult male (SEM). (E) Female tail (DIC). (F) Male tail, ventro‐lateral view (SEM). (G) Genital opening with extruded spicules. (H) Male genital opening. (I) Male tail in ventral view (DIC). Anterior is to the left in (A, B, E–I). The animals are from strain JU2070. a, amphid; ad, anterior dorsal papilla; c, cloaca; cem, male cephalic sensillum (absent in females); ls, labial sensillum; pd, posterior dorsal papilla; pre/post‐cs, pre/post cloacal sensillum; v1, etc.: ventral papilla 1, etc.; sp, spicule. Scale bars: 1 μm, except in (E, F, and I): 5 μm. See also Figure S6.
Figure 3.
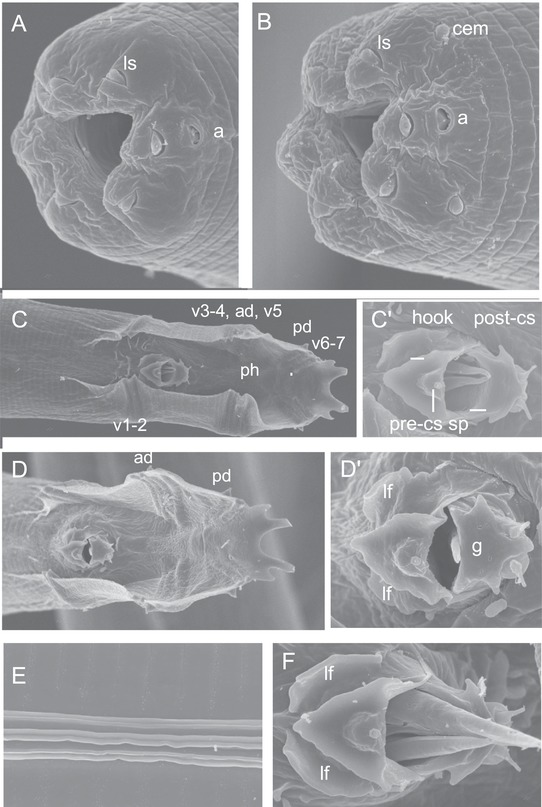
Scanning electron microscopy of Caenorhabditis uteleia sp. n. (A) Mouth of an adult female. (B) Mouth of an adult male. (C and D) Male tail in ventral view. (C’ and D’) Higher magnification of the corresponding male genital openings. (E) Cuticular lateral ridges, adult male. (F) Male genital opening. The animals are from strain JU2469. Anterior is to the left. a, amphid; cem, male cephalic sensillum (absent in females); lf, lateral fold on either side of the hook; ls, labial sensillum; r1, etc., ray 1, etc.; ph, phasmid; sp, spicule; pre/post‐cs, pre/post cloacal sensillum; g, posterior end of the gubernaculum. Bars: 1 μm, except in (C and D): 5 μm.
Figure 4.
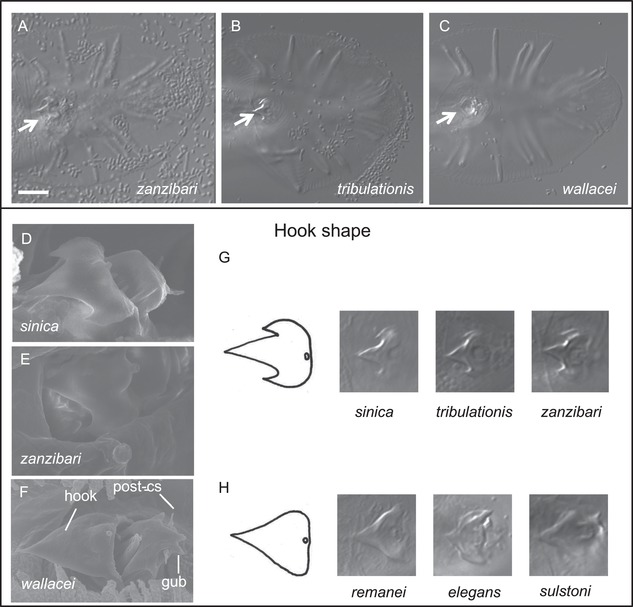
Male hook shape in Elegans group species. (A–C) Ventral views of the male tail of Caenorhabditis zanzibari strain JU2161 (A), C. tribulationis strain JU2774 (B), and C. wallacei JU1873 (C), in Nomarski optics. The arrow points to the precloacal hook. Bar: 10 μm. (D–F) Scanning electron micrographs of the hooks and post‐cloacal sensilla (post‐cs) in C. sinica JU727 (D), C. zanzibari JU2161 (E), and C. wallacei JU1873 (F). “gub”: forked posterior end of gubernaculum. G,H: Drawings of hook shape. (G) Refers to the trilobed shape in C. sinica, C. tribulationis, and C. zanzibari sp. n, while (H) refers to the simpler shape in most other Elegans supergroup species such as C. wallacei, C. elegans, C. remanei, and C. sulstoni.
The morphology of C. uteleia is remarkable for the contour of its male tail fan (Fig. 3). The posterior margin shows a distinctive complex shape with one large central valley and two smaller ones on the left and right sides (Fig. 3C, D). The fan is well developed, opened anteriorly, and shows a smooth, unserrated margin. One group of two pairs of rays is found anterior to the cloaca, followed by two groups of four and three pairs. The dorsal rays are in antero‐posterior positions 5 and 7. The pre‐cloacal sensillum is on a hook, itself in between two characteristic lateral folds (Fig. 3C’, D’, F show the hook, the gubernaculum ventral end with two lateral ears, and the pair of post‐cloacal sensilla). The spicule tip is pointed (Fig. 3C’, F).
MORPHOLOGICAL CHARACTER EVOLUTION
What are the implications of the new species and the new phylogenetic tree for character evolution in the genus? Character evolution in Caenorhabditis was first studied in Sudhaus & Kiontke (1996) based on a morphology‐based phylogenetic tree. Kiontke et al. (2011) and Félix et al. (2014) mapped morphology onto a previous six‐gene phylogenetic tree and recently, Slos et al. (2017) discussed the stem species pattern using a morphological comparison with the sister group Protoscapter.
Starting from the base of the genus, defined by the placement of the sister taxon Protoscapter (Slos et al. 2017), containing Diploscapter, we now have as successive branches C. monodelphis (likely to be associated with the presently unavailable Caenorhabditis sonorae; Slos et al. 2017), then C. parvicauda and then C. plicata. Species in these three branches share characters that are ancestral in the genus (plesiomorphous), such as the absence of a hook. All four species differ greatly in morphology and show private characters and combinations thereof. C. parvicauda is unusual for Caenorhabditis because the male tail does not include an extended fan and papilla v5 does not appear broader than the others. However, in contrast to C. monodelphis, C. parvicauda displays some characters that are shared with other Caenorhabditis species, such as three lateral cuticular ridges. C. parvicauda also shares some characters with some other species that are likely homoplastic. For example, C. parvicauda mate in a spiral fashion on an agar plate, as do species of the Angaria group (Fig. 5A).
Figure 5.
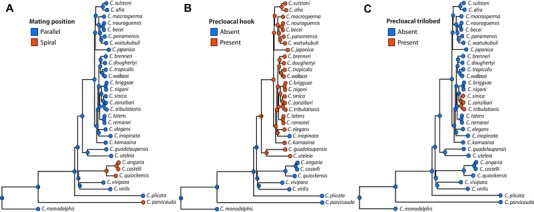
Ancestral state reconstruction of precloacal hook morphology and mating position. (A) Mating position. (B) Precloacal lip in the shape of a hook (with a pointed ventral tip). (C) Precloacal lip in the shape of a trilobed hook. Ancestral state reconstruction was performed by generating 1000 stochastic character maps of each morphological character on the phylogenetic tree in Figure 1A using the equal rates model of evolution. Pie charts on internal nodes represent posterior probabilities of ancestral states.
C. quiockensis is typical for the Angaria group (C. angaria, C. castelli; Kiontke et al. 2011; Félix et al. 2014) in displaying spiral mating and having a distinctive male tail morphology (Fig. S7A–C). The fan has an oval shape in ventral view and is open anteriorly. Rays 4 (= ad) and 7 (= pd) open dorsally to the velum.
C. vivipara is distinct in its viviparity. In laboratory conditions, the females do not lay embryonated eggs (however, we cannot rule out that they lay embryos in some environment). Instead, L1 larvae exit their mother's body through the vulva, as, for example, in the genus Panagrellus in family Panagrolaimidae (Andrássy 1983; Félix et al. 2018). In the closely related species C. virilis, females lay late‐stage embryonated eggs compared to most Caenorhabditis species (which accumulate few embryos in their uteri) but are not viviparous in standard laboratory conditions. Like C. virilis, however, C. vivipara has a wide and heart‐shaped male fan, similar to the male fans of species in the Elegans supergroup yet with no terminal notch (compare Figs. S7E and S8).
C. uteleia has a previously unknown combination of characters that do not all match its most closely related known species C. guadeloupensis (Kiontke et al. 2011; Félix et al. 2014). For example, the male fan of C. uteleia is open and unserrated on the anterior side as observed in many species in the Drosophilae supergroup and in basally branching species. Conversely, the ad ray is in the fifth position in Elegans supergroup species and basally branching species such as C. monodelphis (Slos et al. 2017) but not in C. guadeloupensis. The latter, such as C. plicata, C. drosophilae, the Angaria group, C. virilis, and C. vivipara, shows a dorsal ray in anteroposterior position 4 (Kiontke et al. 2011; Félix et al. 2014; present data). Thus, these characters remain homoplastic in the new phylogenetic tree (Fig. S9) and will most likely remain so. Finally, the new clade that includes C. guadeloupensis, C. uteleia, and the Japonica + Elegans groups is distinguished by the presence of a hook bearing the pre‐cloacal sensillum on the anterior margin of the cloaca (Fig. 5B). This character also appears in C. portoensis but not in C. virilis (Kiontke et al. 2011; Félix et al. 2014) or C. vivipara (Fig. S7F, G), and thus it will be important to affirm the position of C. portoensis (currently resolved in a clade including C. virilis; Kiontke et al. 2011; Félix et al. 2014) to define how many times this character has evolved.
In contrast, the clade including the Elegans and Japonica groups displays little variation except for the tropical fig species C. inopinata (Kanzaki et al. 2018). One character variation that is unique to the Elegans group is the independent evolution of hermaphroditism in three species (Kiontke et al. 2011; Félix et al. 2014). The present work does not add new hermaphroditic species, as all 10 new species reproduce through females and males. However, using the large set of species studied here including six new species in the Elegans and Japonica groups, we distinguished characters in the male tail that vary in either the Elegans or Japonica group. Interestingly, none of these characters correlate with reproductive mode. Within the Elegans group, the small clade containing C. sinica, C. zanzibari, and C. tribulationis displays a characteristic shape of the male hook with three marked lobes (Fig. 4). The hook of other species of the Elegans supergroup such as C. elegans displays a single lobe. The complex shape of the hook as first described for C. sinica as a specific character (Huang et al. 2014) now constitutes a clear apomorphy (derived character), with no pattern of convergent evolution (Fig. 5C).
In the Japonica group, ray v4 is shorter in the clade including C. becei, C. waitukubuli, C. panamensis, and C. nouraguensis, compared to C. sulstoni, C. afra, C. elegans, and many other species in which ray v4 displays a similar length to the ad (anterior dorsal) ray (see also drawings in Kiontke et al. (2011). Ray v4 is also short in C. japonica (Kiontke et al. 2002), but longer in C. macrosperma JU1857, which implies events of homoplasy in any of the configurations of the phylogenetic tree for this latter species (Fig. 1).
Kiontke et al. (2011) and Félix et al. (2014) noted that the ventral tip of the spicules was broad and complex outside the Elegans supergroup as well as in C. japonica and C. afra. The ventral tip of the spicules is wide in C. afra as well as its sister species C. sulstoni (Fig. S10). It also seems broader and bent at an angle in C. becei, more so than in C. panamensis or C. waitukubuli (Fig. S10). This character thus varies in the Japonica group, while the tip remains pointed in the Elegans group, and given the present phylogeny, some homoplasy must also be present in this character.
GENOME SIZE VARIATION IN CAENORHABDITIS
The molecular mechanisms and evolutionary forces that underlie interspecific variation in genome size remain poorly understood. Hermaphroditism has evolved three times independently in Caenorhabditis (Kiontke et al. 2011), and this switch in reproductive mode is hypothesized to impact genome size and content (Lynch & Conery 2003). Previous studies have revealed that genomes and transcriptomes of hermaphroditic species are smaller than those of their outcrossing relatives (Thomas et al. 2012; Fierst et al. 2015). A comparison of the genomes of the closely related sister taxa C. nigoni and C. briggsae revealed that the genome of C. briggsae has undergone extensive contraction after the evolution of hermaphroditism, which has largely been driven by loss of genes with male‐biased expression (Yin et al. 2018).
To study genome size variation in the genus, we first identified genome assemblies that had an excess of single‐copy loci present in two or more copies. The presence of such duplicates suggests that an assembly includes significant uncollapsed haploid allelic segments, and thus represents an inflated genome size estimate. We thus excluded five genomes from subsequent analyses. By comparing genome size of the remaining species within the context of the Caenorhabditis phylogeny, we explored the extensive variation in genome size, ranging from 65 Mb (the genomes of the sister taxa C. afra and C. sulstoni) to 140 Mb (the genome of C. doughertyi; Fig. 6A). The genomes of all three hermaphroditic species (C. briggsae, C. elegans, and C. tropicalis) are smaller than their most closely related outcrossing relatives (C. nigoni, C. inopinata, and C. doughertyi, respectively). We note that the genome of C. wallacei, the closest known relative of C. tropicalis, is not yet publicly available and was not included in this analysis.
Figure 6.
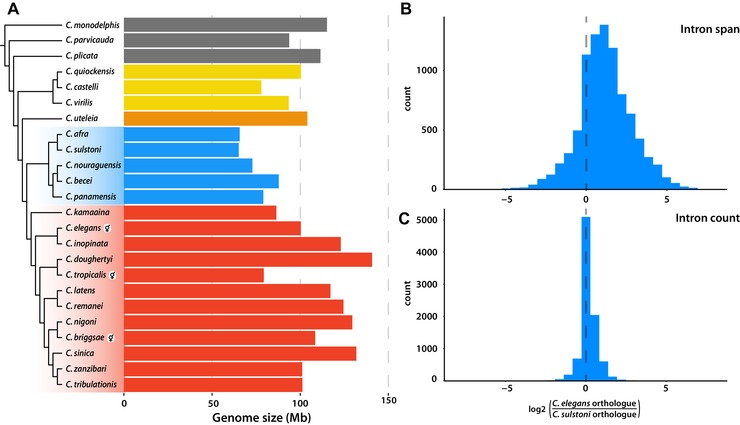
Genome size and gene structure variation in Caenorhabditis. (A) Genome size variation in the context of the Caenorhabditis phylogeny. Hermaphroditic species are highlighted. Phylogenetic tree is based on Figure 1A, with major clades highlighted. (B) Histogram of the log2‐transformed ratio of intron span in 8954 genes in C. elegans compared to their orthologues in C. sulstoni. (C) Histogram of the log2‐transformed ratio of intron count in 8954 genes in C. elegans compared to their orthologues in C. sulstoni.
To investigate the unusually small genome of C. sulstoni, we compared the content of its genome with that of C. elegans (Table S5). Relative to C. elegans, C. sulstoni has 2170 fewer genes, 1694 (78%) of which are due to a reduction in gene family size. By comparing gene structure in 8,954 single‐copy orthologues between C. sulstoni and C. elegans, we found that genes in C. sulstoni are on average shorter, largely because introns have undergone contraction (Fig. 6B). In contrast, intron–exon structure appears to be largely conserved, with limited evidence of decreased intron count in C. sulstoni (Fig. 6C). C. sulstoni also had a reduction in mean intergenic distance (578 bp vs. 1047 bp in C. elegans) and in estimated repeat content (12% vs. 18% in C. elegans).
To explain genome size variation across the genus while taking species’ relationships into account, we used phylogenetic generalized least squares (PGLS) analysis of genome size against protein‐coding gene number, intron count, intron size, intergenic distance, and repeat content (Fig. S11; Table S6). Only protein‐coding gene number and proportion of repetitive DNA were significantly positively correlated with genome size (Figs. S11A and S11B).
THE EVOLUTION OF NOTCH/LIN‐12 SIGNALLING PROTEINS IN CAENORHABDITIS
The new genome data and the resolved phylogeny permit detailed examination of the origins and diversification of genes and gene families. Developmental genetics in C. elegans has identified many genetic systems critical across Metazoa, but has also identified idiosyncrasies that are not shared. As an example of the power of these new data, we examine Notch signaling.
Notch signaling is a highly conserved intercellular signaling pathway involved in an array of cell fate decisions during animal development (Artavanis‐Tsakonas et al. 1999). Basic components of this pathway were characterized in C. elegans at the same time as in Drosophila melanogaster (Greenwald 1985, 2005). Central to this pathway are the Notch receptors (Greenwald 1985; Yochem et al. 1988; Yochem & Greenwald 1989). These transmembrane proteins bind extracellular ligands of the Delta/Serrate/LAG‐2 (DSL) family. Ligand binding results in cleavage and nuclear translocation of an intracellular domain that associates with transcription factors to influence the expression of genes involved in cell‐fate decisions (Greenwald 2005). C. elegans possesses two Notch‐like receptor loci, lin‐12 and glp‐1, which have overlapping but not identical biological roles (Lambie & Kimble 1991). The two loci are the product of a gene duplication event, which has been followed by some degree of subfunctionalization (Rudel & Kimble 2002). We used the new genomic data to study the evolution of these two loci.
From an orthology clustering analysis including 27 Caenorhabditis species and the outgroup taxon Diploscapter coronatus, we identified the orthogroup containing the C. elegans proteins LIN‐12 and GLP‐1. We aligned the protein sequences of each member of the orthogroup and reconstructed a phylogenetic tree using maximum likelihood inference. The resulting topology (Fig. 7A) reaffirms that lin‐12 and glp‐1 are the product of a gene duplication event, and indicates that this occurred at the base of the Elegans supergroup. The genomes of several species outside the Elegans supergroup, including the basal taxa C. monodelphis and C. parvicauda, encode only a single Notch‐like receptor. We also find evidence for a second duplication of glp‐1 at the base of the Elegans supergroup, followed by loss in species belonging to the Elegans group, including C. elegans (Fig. 7B). Genomes of species belonging to the Japonica group have retained both copies of the glp‐1‐like gene, and therefore encode three Notch‐like receptor genes in total. Both duplication branches had maximal bootstrap support. Further, within‐species duplications of the Notch‐like receptor genes appear to have occurred in several species, including in C. uteleia. In those cases in which the subtending branches are extremely short, such as in D. coronatus, these putative within‐species duplications are likely to be an artifact arising from regions of uncollapsed heterozygosity present in the genome assembly.
Figure 7.
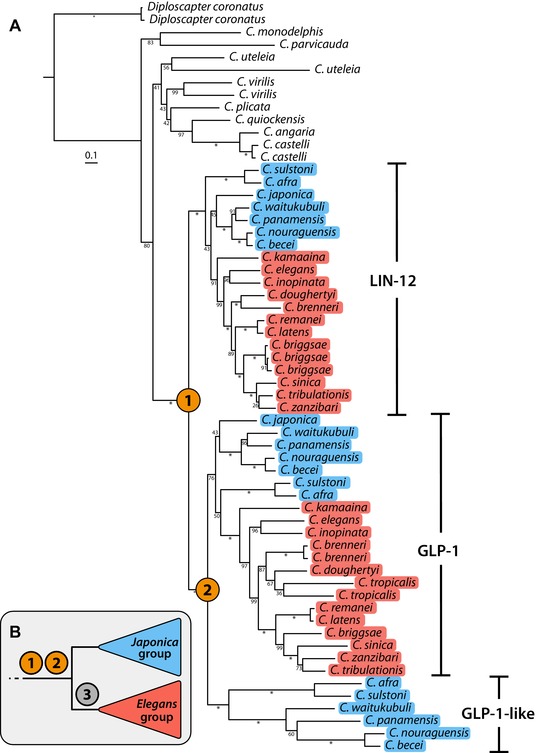
Maximum likelihood gene tree of Notch‐like receptors in Caenorhabditis. (A) Gene tree of the orthogroup containing C. elegans proteins LIN‐12 (CELEG.R107.8) and GLP‐1 (CELEG.F02A9.6) inferred using maximum likelihood (JCCMut+Γ substitution model). Elegans and Japonica groups are highlighted in red and blue, respectively. Duplication events are denoted by orange circles. Branch lengths represent the number of substitutions per sites; scale is shown. Bootstrap values are displayed as branch annotations, “*” = 100. (B) Inferred events: (1) Duplication of ancestral Notch‐like receptor gene; (2) Duplication of glp‐1 gene; (3) Loss of one of the two duplicated glp‐1 genes in the Elegans group.
Both Elegans supergroup duplication events were followed by divergence in the rate of substitution of the paralogues, as indicated by branch lengths (Fig. 7A). GLP‐1 and its orthologues underwent increased rates of substitution after the first duplication event relative to LIN‐12 and orthologues, with means of 0.93 and 0.58 substitutions per amino acid site, respectively (Table S7). The paralogues of GLP‐1 found in the Japonica group have also undergone increased rates of substitution relative to GLP‐1 after the second duplication event, with means of 1.22 and 0.78 substitutions per site, respectively.
The binding of DSL ligands by Notch receptor proteins is mediated by extracellular epidermal growth factor (EGF)‐like repeats (Rebay et al. 1991). In C. elegans, LIN‐12 and GLP‐1 differ in the number of EGF‐like repeats, with 13 and 10, respectively. We counted the number of EGF‐like repeats in all orthologues of LIN‐12 and GLP‐1 using InterProScan and used a stochastic character mapping approach to infer the structure of the ancestral Caenorhabditis Notch‐like receptor. We infer that the ancestral receptor possessed 13 EGF‐like repeats and was thus similar in structure to LIN‐12 and its orthologues and to the single Notch‐like receptors of those species that do not belong to the Elegans supergroup (Fig. S12; Table S8). We also infer that the loss of three EGF‐like repeats in GLP‐1 and its orthologues occurred prior to the divergence of the Elegans supergroup and to the second duplication event (Fig. S12).
Discussion
PHYLOGENETIC RELATIONSHIPS IN THE GENUS CAENORHABDITIS
Using genomic and transcriptomic data from 32 species of Caenorhabditis and the outgroup taxon D. coronatus, we have generated the most comprehensive Caenorhabditis phylogenetic tree published to date, in terms of number of taxa and number of orthologues sampled. The majority of relationships were recovered with maximal support by all analyses and are consistent with previous studies (Kiontke et al. 2011; Slos et al. 2017). The 10 species described here are sampled from all previously defined major clades in the genus, and include the second‐most early diverging Caenorhabditis, C. parvicauda.
Our results corroborate the monophyly of the Japonica and Elegans groups (and therefore the Elegans supergroup) as defined in Kiontke et al. (2011). The majority of relationships within these groups are consistent with previous studies (Kiontke et al. 2011; Slos et al. 2017). However, C. kamaaina (sp. 15), which in our analyses was recovered as the most early diverging species in the Elegans group, has previously been recovered as sister to the Japonica group (Kiontke et al. 2011). Our finding that the C. guadeloupensis + C. uteleia group is sister to the Elegans supergroup is consistent with the tree topology in Slos et al. (2017) and provides evidence that the Drosophilae supergroup, as recovered by the analysis of Kiontke et al. (2011), is paraphyletic.
Interestingly, relationships at three nodes were inconsistent across analyses. The placement of C. japonica as sister to C. afra + C. sulstoni and the placement of the clade containing C. vivipara + C. virilis as sister to the Elegans supergroup were recovered by ML analysis only. Both of these relationships disagree with all previously published Caenorhabditis phylogenetic trees (Kiontke et al. 2011; Slos et al. 2017). The sister relationship of C. brenneri and C. doughertyi, as recovered by BI and ML, was recovered in the analyses of Kiontke et al. (2011), but not by Slos et al. (2017).
It is not clear why the different approaches generate inconsistent topologies, and whether these inconsistencies are due to conflict in the data or due to low resolution, which result in ambiguous regions of the topology. Concatenated alignments of large numbers of loci, such as the one used in our BI and ML analyses, may contain genes that have different histories due to processes such as incomplete lineage sorting or introgression through hybridization in the early stages of speciation (Maddison & Wiens 1997). Given sufficient conflicting signals, concatenation approaches are known to lead to inaccurate topologies (Kubatko & Degnan 2007). Coalescent‐based summary approaches, such as ASTRAL‐III, are able to accommodate conflicting signals in gene trees arising from incomplete lineage sorting (Mirarab et al. 2014), but may be sensitive to gene tree estimation error (Gatesy & Springer 2014; Roch & Warnow 2015). The different models used in BI and ML analysis (CAT‐GTR and GTR, respectively) are presumably one source of conflict between these two analyses. The CAT‐general‐time reversible model (GTR), unlike the GTR model, is able to accommodate heterogeneity in the amino acid substitution process at different sites in the alignment (Lartillot & Philippe 2004). Further work is required to determine whether accounting for such heterogeneity is necessary for this dataset.
We feel it premature to redefine the groups and supergroups in Caenorhabditis until we have a more complete representation of the known species, and particularly of species currently placed in the Drosophilae group. Our data could be used to support a broadening of the Elegans supergroup to include C. uteleia and C. guadeloupensis. Alternately, the Elegans supergroup could be retained as the clade including the last common ancestor of C. elegans and C. japonica and their respective groups, but not C. uteleia and C. guadeloupensis. Many more species outside the Elegans and Japonica groups are now available in culture for genome sequencing and subsequent placement on the phylogenetic tree (Kiontke et al. 2011; Félix et al. 2014). As sequencing technologies improve, and chromosomal assembly becomes more achievable (Kanzaki et al. 2018), we would also hope to use a chromosomally resolved genomic tree to uncover additional patterns and reveal novel processes in Caenorhabditis evolution.
GENOME SIZE VARIATION IN CAENORHABDITIS
By comparing 24 Caenorhabditis species, we found evidence for substantial variation in genome size within the genus, with genome sizes ranging from 65 to 140 Mb.
The smallest genome, that of C. sulstoni, differed by 35 Mb from that of the reference C. elegans because of reduction in protein‐coding gene number, intron size, intergenic distances, and proportion of repetitive DNA. Surprisingly, while we found that introns in C. sulstoni appear to have undergone contraction, we did not find evidence for decreased intron count in its small genome. Using PGLS analysis, we found evidence that genome size is significantly positively correlated with protein‐coding gene number and with proportion of repetitive DNA.
Our analysis of genome size variation assumes that we have accurately estimated the span of each genome in our draft assemblies. In Caenorhabditis, in which levels of nucleotide diversity are particularly high in outcrossing species (Cutter et al. 2006; Dey et al. 2013), several published draft genomes are known to contain high levels of uncollapsed heterozygosity, and thus have larger‐than‐expected assembly spans (Barrière et al. 2009). Consistent with this, we found that the genomes of C. brenneri and C. japonica (with assembly spans of 190 and 166 Mb, respectively) contained high numbers of duplicated loci, as did the genomes of C. angaria, C. waitukubuli. The genome of the outgroup taxon, Diploscapter coronatus, also has elevated duplication rates. D. coronatus is a parthenogen, and its assembly represents two copies of the original haploid genome (Hiraki et al. 2017). These species were excluded from our analyses.
Several studies have established a link between reproductive mode and genome size in Caenorhabditis, suggesting that hermaphroditic species have smaller genomes due to gene loss (Thomas et al. 2012; Fierst et al. 2015; Yin et al. 2018). C. briggsae appears to have undergone extensive genome contraction after the evolution of hermaphroditism compared to its gonochoristic sister C. nigoni. This genome contraction in C. briggsae is largely the result of loss of genes with male‐biased expression (Yin et al. 2018). In contrast, C. elegans does not show a significant reduction in protein‐coding gene number when compared with its outcrossing sister taxon C. inopinata. Instead, it appears that a substantial fraction of the 22.1 Mb difference in genome size between these two species is the result of the expansion of transposable elements in the C. inopinata genome (Kanzaki et al. 2018).
Given our findings and those of previous studies, genome size variation within Caenorhabditis is driven by multiple, interacting, underlying mechanisms. The relative contributions of contrasting molecular processes, such as gene loss versus gene family expansion, or intergenic contraction versus repeat expansion, and their evolutionary drivers, such as a switch in reproductive mode, must differ between different lineages in the genus. A further, more detailed analysis, involving genomes of other species not analyzed here, including that of C. wallacei, the outcrossing sister taxon of C. tropicalis, will be required before we are able to identify general trends, and specific mechanisms, responsible for genome size variation within the genus.
THE EVOLUTION OF NOTCH/LIN‐12 SIGNALLING PROTEINS IN CAENORHABDITIS
C. elegans is a major developmental biology model, and understanding how its particular instance of nematode (and animal) development was established may help reveal the logic of the evolution of development and the importance of processes such as developmental systems drift, wherein similar phenotypes are formed by divergent developmental processes (True & Haag 2001). We studied the evolution of Notch‐like receptor genes in the genomes of 27 species of Caenorhabditis and in D. coronatus, and found that the C. elegans genes lin‐12 and glp‐1 are the product of a gene duplication event that occurred at the base of the Elegans supergroup. We also found evidence of a subsequent duplication of glp‐1 on the same branch, followed by loss at the base of the Elegans group. The genomes of Japonica group species have three Notch‐like receptor genes. We found that lin‐12 and its orthologues were more conserved, with a lower rate of substitution compared to glp‐1 and its orthologues and an EGF‐like repeat structure that is more similar to that of the inferred ancestral Notch‐like receptor.
Previous genetic analyses of lin‐12 and glp‐1 identified several shared and receptor‐specific developmental functions, suggesting subdivision of ancestral roles after duplication (Lambie & Kimble 1991). While LIN‐12 and GLP‐1 proteins have diverged in sequence, it is likely that a substantial amount of this divergence in function is due to differences in expression patterns of these two proteins as GLP‐1 is capable of performing LIN‐12‐specific roles when placed under the control of lin‐12 regulatory sequences (Fitzgerald et al. 1993). The extent to which ancestral roles have been subdivided (subfunctionalization), or whether novel roles have been acquired (neofunctionalization), and to what extent these changes are due to changes in expression pattern, could be addressed by studying the function of the ancestral Notch‐like receptor gene in species outside the Elegans supergroup. Particularly exciting are the third Notch‐like receptor genes present in the genome of Japonica group species that appear to have undergone a substantial increase in substitution relative to glp‐1 and its orthologues after duplication. Genetic analyses of Notch‐like receptors, which have yet to be carried out in these species, could reveal the implications of this increased rate of substitution, including whether further subdivision of ancestral roles has occurred or novel roles have been acquired.
GENOMICS, SPECIES DESCRIPTION, AND CAENORHABDITIS BIOLOGY
By describing new species of Caenorhabditis with high‐quality genomic data we hope to promote not only understanding of the evolution of this exciting genus, but also exploitation of these new species in deepening understanding of the pattern and process of organismal evolution. In particular, the ability to interfere with gene function through RNAi and accurately modify genomes using CRISPR‐Cas editing is routine in C. elegans and becoming established in other species. Using the genomic data presented here, specific loci can be deleted or altered to test hypotheses of gene function in a rich phylogenetic context.
Currently, strains corresponding to about 50 species of Caenorhabditis are available in culture. Our goal is to provide genome sequences and formal species descriptions for all these species. The assemblies presented here are not chromosomal, unlike the genome of C. elegans, because of the inability of short‐read data to bridge and resolve large repeats. Recent developments in genomic technologies make phased, chromosomal assemblies of all Caenorhabditis species’ genomes an achievable goal, and we and others are using long read and other approaches to advance this goal.
Approximately 20,000 new species are described each year. We also suggest that, wherever technically possible, the generation of genomic data as part of the introduction of new taxa should become standard. Complete sequencing of all taxa on earth has been proposed (Lewin et al. 2018), and active support should be offered to taxonomists to add their new species to this global effort.
Methods
SAMPLING AND ISOLATION
See Kiontke et al. (2011) and (Barrière & Félix 2014) for details of sampling strategies. Briefly, rotting vegetable matter samples were collected and stored in plastic bags. The samples were then analyzed in the laboratory by placing them onto 90 mm NGM agar plates (Stiernagle 2006), seeded in the center with Escherichia coli OP50. Caenorhabditis species are generally attracted by the E. coli lawn. Isofemale lines were established by isolation of a single female that was already mated or by co‐culturing one female and one male. A list of isolates is available in Table S1.
CULTURE AND FREEZING
Nematodes were cultured on NGM agar plates and frozen with standard C. elegans protocols (Stiernagle 2006). Some species, such as C. parvicauda do not grow well on E. coli OP50 and are better kept with some of their original microbial environment. Like other species in the Angaria group, C. quiockensis does not survive well freezing and thawing with the standard C. elegans protocol and was heat‐shocked for 1–2 hrs at 37°C before freezing.
CROSSES AND ASSIGNMENT OF ISOLATES TO SPECIES
For each of the interspecific crosses with results presented in Table S2, five L4 females and five L4 or adult males were placed together on a 55 mm agar plate seeded with E. coli OP50. The plate was checked regularly for presence and cross‐fertility of the progeny. To assign additional isolates (listed in Table S1), either crosses were performed, or barcode sequence alone was considered sufficient if it was identical to the barcode of a reference strain.
INBREEDING AND NUCLEIC ACID PREPARATION
For inbreeding, one L4 female and one male were used to seed each generation. Inbreeding was performed for 25–26 generations.
After thawing, each inbred strain was bleached and grown on 90 mm NGM plates enriched with agarose (for 1 L: 3 g NaCl, 5 g bacto‐peptone, 10 g agar, 7 g agarose, 1 mL cholesterol 5 mg/mL in ethanol, 1 mL CaCl2 1 M, 1 mL MgSO4 1 M, 25 mL KPO4 1 M). Worms were harvested just after starvation and washed in M9 several times to remove E. coli.
For genomic DNA extraction, the nematode pellets were resuspended in 600 μL of Cell Lysis Solution (Qiagen) complemented with 5 μL of proteinase K (20 μg/μL) and incubated overnight at 56°C with shaking. The day after, the lysates were incubated for one hour at 37°C with 10 μL of RNAse A (20 μg/μL) and the proteins were precipitated with 200 μL of protein precipitation solution (Qiagen). After centrifugation, the supernatants were collected in new tubes and genomic DNA were precipitated with 600 μL of isopropanol. The pellets were washed in ethanol 70% and dried one hour before being resuspended in 50 μL of DNAse free‐water.
For RNA extraction, 100 μL of nematode pellet was resuspended in 500 μL of Trizol (5 volumes of Trizol per volume of pelleted nematodes). The Trizol suspension was frozen in liquid nitrogen and then transferred to a 37°C water bath to be thawed completely. This freezing/thawing process was repeated four to five times and the suspension was vortexed for 30 sec and let rest for 30 sec (five cycles). A total of 100 μL chloroform was added and the tubes were shaken vigorously by hand for 15 sec and incubated for 2–3 min at room temperature. After centrifugation (15 min at 13,000 rpm and 4°C), the aqueous (upper) phase containing the RNA was transferred to a new tube and precipitated with 250 μL of isopropanol. The pellets were washed in 70% ethanol and dried 15–20 min before being resuspended with 50–100 μL of RNAse‐free water. An aliquot of each DNA and RNA preparation was run on agarose gel to check their quality and quantitated with Qubit (Thermo Scientific)
GENOME SEQUENCING
For C. parvicauda, C. quiockensis, C. sulstoni, C. tribulationis, C. uteleia, C. waitukubuli, and C. zanzibari, two short‐insert (insert sizes of 300 and 600 bp, respectively) genomic libraries and a single short‐insert (150 bp) RNA library were prepared using Illumina Nextera reagents and sequenced (125 bases, paired‐end) on an Illumina HiSeq 2500 at Edinburgh Genomics (Edinburgh, UK). For C. becei and C. panamensis, a short‐insert (insert size of 600 bp) genomic library and a long‐insert (insert size of 4 kb) genomic library were prepared using Illumina Nextera reagents and sequenced (100 bases, paired‐end) on an Illumina HiSeq 2500 at the NYU Center for Genomics and Systems Biology GenCore facility (New York, USA). All raw data have been deposited in the relevant International Nucleotide Sequence Database Collaboration databases (Table 1).
DE NOVO GENOME ASSEMBLY AND GENE PREDICTION
Detailed methods for each species, along with all software tools used (including versioning and command line options), are available in Supporting Information 2. We performed quality control of all raw sequence data using FastQC (Andrews 2010) and used Skewer (Jiang et al. 2014) and FASTQX Toolkit (Gordon & Hannon 2010) to remove low‐quality bases and adapter sequence. Adapters were removed from long‐insert data using NextClip (Leggett et al. 2014). For each species, we identified contaminants using taxon‐annotated, GC‐coverage plots as implemented in blobtools (Laetsch & Blaxter 2017), preliminary assemblies were generated using CLC Bio (CLCBio, Copenhagen, Denmark), and likely taxon origin determined using NCBI‐BLAST+ (Camacho et al. 2009) or using Kraken (Wood & Salzberg 2014). Reads originating from contaminant genomes were discarded. We estimated the optimal k‐mer length for assembly independently for each genome using KmerGenie (Chikhi & Medvedev 2014). Preliminary assemblies were generated using several de Bruijn graph assemblers, including Velvet (Zerbino & Birney 2008) and Platanus (Kajitani et al. 2014), across several parameter values. The resulting assemblies were assessed using numerical metrics, and two biological completeness metrics, CEGMA (Parra et al. 2007), and BUSCOs (Simão et al. 2015; using the ‘Nematoda_ob9’ dataset). For each species, the highest quality assembly was selected and, where possible, scaffolded with assembled transcripts using SCUBAT2 (https://github.com/GDKO/SCUBAT2) or with long‐insert “mate‐pair” data using Platanus.
We identified repeats independently in each genome using RepeatModeler (Smit & Hubley 2010). After filtering sequences that likely originated from protein‐coding genes, we combined each repeat library with known Rhabditida repeats obtained from RepBase (Jurka et al. 2005). This concatenated repeat library was then provided to RepeatMasker (Smit et al. 1996) for masking. If RNA‐seq data were available, reads were aligned to the assembly using STAR (Dobin et al. 2013) and the resulting BAM file provided to BRAKER (Hoff et al. 2016), which performed final gene prediction. If RNA‐seq data were not available, genes were predicted initially using MAKER2 (Hoff et al. 2016) with a training set composed of ab initio predictions from GeneMark (Lukashin & Borodovsky 1998), gene models identified by CEGMA (Parra et al. 2007), and the C. elegans protein sequence set. The resulting gene models were used to train AUGUSTUS (Keller et al. 2011), which generated the final gene set.
PHYLOGENETIC ANALYSIS
Accessions to all data used in phylogenomics analysis are available in Table S9. For those species with available genome sequences, we identified and collected the protein sequence of the longest isoform of each protein‐coding gene. For those species for which only transcriptome data was available (C. guadeloupensis and C. vivipara), open reading frames and putative protein sequences were predicted using TransDecoder (Haas & Papanicolaou 2012). OrthoFinder (Emms & Kelly 2015; using the default inflation value of 1.5) was used to cluster all protein sequences into putatively orthologous groups (OGs). OGs containing loci present as single copy in at least 27 of the 33 species (except in those species with genome assemblies known to contain regions of uncollapsed heterozygosity [i.e., C. angaria, C. brenneri, C. japonica, C. waitukubuli, and D. coronatus] and in those species for which only transcriptome data were available, where counts of two were allowed) were selected.
To identify paralogous sequences, we aligned the protein sequences of each OG using FSA (Bradley et al. 2009) and generated a maximum likelihood tree along with 100 rapid bootstraps using RAxML (Stamatakis 2014)(using best‐fitting amino acid substitution, as selected by the model testing component of RAxML, and gamma‐distributed rate variation among sites). Each tree was screened by PhyloTreePruner (Kocot et al. 2013; collapsing nodes with bootstrap support <50), and any OGs containing paralogues were discarded. If two representative sequences were present for any species (i.e., “in‐paralogues”) after this paralogue screening step, the longest of the two sequences was retained and the other discarded.
The protein sequences of each one‐to‐one OG were then aligned using FSA and gene trees estimated as previously described. ASTRAL‐III (Mirarab & Warnow 2015), a coalescent‐based method, was then used to reconstruct the species phylogeny using individual genes trees as an input. We also reconstructed the species tree using a concatenation approach. TrimAl (Capella‐Gutiérrez et al. 2009) was used to remove spuriously aligned regions from each alignment, which were subsequently concatenated using catfasta2phyml (available at https://github.com/nylander/catfasta2phyml). ML analysis was performed with RAxML (general‐time reversible model (GTR) (Tavaré 1986) with gamma‐distributed rate variation among sites) along with 100 bootstrap replicates. Bayesian inference was carried out using the site‐heterogeneous CAT‐GTR substitution model (Lartillot & Philippe 2004) (with gamma‐distributed rate‐variation among sites) implemented in PhyloBayes MPI (Lartillot et al. 2013), with two independent chains. Convergence was assessed using Tracer (Rambaut et al. 2007). A posterior consensus tree was estimated using samples from both chains, with the initial 10% of all trees discarded as burn‐in. Newick trees were visualized using the iTOL web server (Letunic & Bork 2016).
MORPHOLOGICAL CHARACTER MAPPING
Morphological characters and phenotypes were encoded as “1” or “0” depending on their presence or absence (Table S4). Ancestral morphological character states were inferred using the stochastic mapping implemented in the phytools package (Revell 2012). Using the equal rates model of evolution, we simulated 1000 character histories on the species tree in Fig. 1A and summarized the character histories as posterior probabilities on internal nodes.
GENOME SIZE ANALYSIS
For those species for which gene structure information was available, we collected the longest isoform of each protein‐coding gene. As previously described, we clustered protein sequences into putatively orthologous groups using OrthoFinder. To identify genome assemblies that might contain uncollapsed haploid segments and thus expanded genome spans, we selected orthogroups containing loci that were present at least 22 of the 28 species and that were on average single copy. For each species, we counted the number of loci present in the selected orthogroups, and divided this count by the total number of orthogroups that contained a representative sequence for that species. We excluded genome assemblies that had a duplication ratio of >1. 2 (C. angaria, C. brenneri, C. japonica, C. waitukubuli, and Diploscapter coronatus) from downstream analyses of genome size (Table S10). We collected summary statistics for each genome assembly using custom scripts and performed PGLS analyses using the ape R package in (Paradis & Schliep 2018), using the Brownian model of evolution and the phylogenetic tree in Figure 1A.
NOTCH‐LIKE RECEPTOR ANALYSIS
In an existing orthology clustering set, we identified the OG that contained the C. elegans proteins LIN‐12 (R107.8) and GLP‐1 (F02A9.6) and collected the protein sequences of each member. After removing sequences that were shorter than 700 amino acids, we generated an amino acid alignment using FSA. We performed a maximum likelihood analysis using RAxML, allowing the substitution model to be automatically selected, and conducted 100 rapid bootstrap replicates. Branch lengths were extracted using a custom Python script (available at https://github.com/lstevens17/caeno-ten-descriptions), making use of the ete3 module (Huerta‐Cepas et al. 2016). We identified conserved domains in each protein sequence using InterProScan (Jones et al. 2014). Counts of EGF‐like repeats were obtained from the ProSiteProfiles database (release 2017_09; Sigrist et al. 2013). To infer the ancestral EGF‐like repeat count, we used the stochastic mapping approach implemented in the phytools package (Revell 2012). Using the equal rates model of evolution, we simulated 1000 character histories on the gene tree inferred by RAxML, and summarized the character histories as posterior probabilities on internal nodes.
SCANNING ELECTRON MICROSCOPY
Nematode cultures were resuspended and washed twice in M9 solution, then fixed overnight at 4°C in M9 or 50 mM phosphate pH 7.0 + glutaraldehyde 2.5 to 4%, depending on the batch. The fixed animals were rinsed twice in M9 and dehydrated through an ethanol series, pelleting them at each step at 1 g in a tube. The samples were processed through critical point drying and coating with 20 nm of Au/Pd, and then observed with a JEOL 6700F microscope at the Ultrastructural Microscopy Platform of the Pasteur Institute.
NOMARSKI MICROGRAPHS
The Nomarski micrographs were taken using an AxioImager 2 (Zeiss) after mounting the animals on a Noble agar pad as described in (Shaham 2006). The pictures showing extruded spicules were taken after exposing the animals for 2 sec in the microwave before adding the coverslip.
Associate Editor: S. Wright
Supporting information
Table S1. List of isolates and their origin.
Table S2. Mating tests. This table contains several sheets, showing the results of crosses between isolates of different species. Successful crosses are labeled in green.“100s of embryos” refer to unhatched dead embryos remaining on the plate.
Table S3. Detailed genome assembly and gene prediction statistics.
Table S4. Morphological characters used for ancestral state reconstruction. ‘1’ denotes presence or existence; ‘0’ denotes absence.
Table S5. Genome contents of C. sulstoni and C. elegans. Gene structure statistics were calculated using the longest isoform of each protein‐coding gene. UTR regions were not annotated in C. sulstoni and so were not considered in either species.
Table S6. Genome statistics used in PGLS analysis. Gene structure statistics were calculated using the longest isoform of each protein‐coding gene. UTR regions were not considered as they were not annotated in several species. Repeat content was estimated de novo using RepeatModeler and RepeatMasker.
Table S7. Mean branch lengths from Maximum likelihood gene tree of all Notch‐like proteins. Branch lengths were extracted using a custom Python script (available at https://github.com/lstevens17/caeno-ten-descriptions).
Table S8. EGF‐like repeat counts for LIN‐12/GLP‐1 homologues. Counts of EGF‐like repeats were obtained from were obtained from the ProSiteProfiles database (release 2017_09).
Table S9. Accessions and links to data used in phylogenomic analysis.
Table S10. Completeness and duplication statistics for 28 Caenorhabditis species based on 8,286 orthologues. We selected groups of orthologues which were present in at least 22 species and had a mean count of 1. The duplication ratio was calculated by dividing the total number of sequences present for each species by the total number of orthogroups which contained a representative sequence for that species.
Figure S1. Assembly spans and genome size estimates. Kmers of length 19 were counted using KMC (v2.3). Genome size was estimated using GenomeScope (Vurture et al. 2017). The model used by GenomeScope to estimate genome size did not converge for C. waitukubuli (presumably due to high heterozygosity) and is not shown.
Figure S2. Kmer spectra for C. waitukubuli (sp. 39). Kmers were counted using KMC (v2.3). Plotted in R using the ggplot2 package.
Figure S3. PhyloBayes phylogenetic tree. Phylogenetic tree inferred using Bayesian inference with the CAT‐GTR+Γ substitution model. Bayesian posterior probabilities are 1.0 unless noted as branch annotations. Scale is in substitutions per site.
Figure S4. RAxML phylogenetic tree. Maximum likelihood phylogenetic inferred using RAxML with the GTR+Γ substitution model. Bootstrap support values (100 replicates) are 100 unless noted as branch annotations. Scale in substitutions per site.
Figure S5. ASTRAL‐III phylogenetic tree. Phylogenetic tree inferred using ASTRAL‐III, by providing maximum likelihood gene trees (inferred using RAxML with the substitution model selected automatically) as input. As ASTRAL‐III outputs trees with branch lengths in coalescent units, branch lengths in substitutions per site were estimated using RAxML with the GTR+Γ substitution model and the concatenated alignment. Bayesian posterior probabilities are 1.0 unless noted as branch annotations. Scale is in substitutions per site.
Figure S6. Morphology of C. parvicauda male tail. A, A’: Two focal planes in Nomarski optics of the same adult male tail of strain NIC134. B: Scanning electron microscopy, strain JU2070. ad: anterior dorsal papilla; pd: posterior dorsal papilla; vr1, etc.: ventral papilla 1, etc. These photographs exemplify the left‐right asymmetry of the posterior papillae, observed in strains NIC134 and JU2070, as well as JU1766 (not shown).
Figure S7. Morphology of C. quiockensis and C. vivipara. A‐C: Nomarski micrographs of male tails of C. quiockensis strain JU2745. D: C. castelli JU1426 is shown for comparison. Same scale in A‐D. E: C. vivipara NIC1070 male tail F‐H: Details of pericloacal regions of other animals. Note the much larger and wider fan in C. vivipara. Ad: anterior dorsal papilla; gub: gubernaculum; pd: posterior dorsal papilla; spic: spicule; v1, etc.: ventral papillae.(A,B,D, H) are lateral views. (C) and (E‐G) are ventral views. I: Gravid female adult with late‐stage embryos in the uterus and a recently laid L1 stage larva. Bars: 10 μm, except 5 μm for (F,G) and 50 μm for (I). Anterior is to the left except in H where the anterior is to the top.
Figure S8. Male tails of species of the Elegans supergroup. Ventral views by Nomarski optics of C. becei strain QG704, C. waitukubuli strain NIC564, C. panamensis strain QG702, C. sulstoni strain SB454 and C. afra JU1199 male tails. Note the variation in the respective lengths of rays 4 and 5. All pictures at are at the same scale. Bar: 10 μm. The correspondence between the v1‐7, ad, pd papilla nomenclature and that of rays used in C. elegans is indicated on the C. becei picture. v1‐7 denote ventral papillae, while ad and pd denote the anterior and posterior dorsal papillae, respectively. In the C. elegans nomenclature, rays are instead numbered r1‐r9 without distinction between ventral and dorsal rays.
Figure S9. Ancestral state reconstruction of male tail characters. A: Antero‐posterior position of the dorsal ray. B: Closed fan. Ancestral state reconstruction was performed by generating 1000 stochastic character maps of each morphological character on the phylogenetic tree in Fig. 1A using the equal rates model of evolution. Pie charts on internal nodes represent poster probabilities of ancestral states. The fan of C. vivipara was ambiguous and thus excluded from the open fan character mapping analysis.
Figure S10. Spicule tip shape in Elegans supergroup species. Nomarski micrographs. Left column: ventral view. Right column: ventro‐lateral view. The tip of spicules in C. afra and C. sulstoni (as in several species of the Japonica group; Kiontke et al. 2011) is broad and bent with a discontinuity on the curvature, compared to a thin and continuously bent tip in species of the Elegans group such as C. zanzibari or C. elegans. That of C. panamensis strain QG702 is quite thin, while that of C. becei strain QG704 is broad. Bar for all panels: 5 μm.
Figure S11. Phylogenetic generalized least squares (PGLS) analysis of genome size and contents.
A: Protein‐coding gene number. B: Estimated repeat content. C: Mean intron count per gene. D: Mean intron size. E: Mean intergenic distance. Phylogenetic tree presented in Fig. 1A was used in all analyses; points are coloured based on major clades. Gene structure statistics were calculated using the longest isoform of each protein‐coding gene. UTR regions were not considered as they were not annotated in several species. Repeat content was estimated de novo using RepeatModeler and RepeatMasker.
Figure S12. Ancestral state reconstruction of EGF‐like repeat structure. Ancestral state reconstruction was performed by generating 1000 stochastic character maps of EGF‐like repeat count on the gene tree presented in Fig. 7A using the equal rates model of evolution. Pie charts on internal nodes represent poster probabilities of ancestral states.
Document S1. Species declarations.
Document S2. Detailed bioinformatics methods. Methods, versions and relevant parameters used in genome assembly, gene prediction, phylogenomic analysis, genome size analysis, and Notch‐receptor analysis.
ACKNOWLEDGEMENTS
We dedicate this article, and Caenorhabditis sulstoni, to John Sulston, a mentor and colleague. We thank Adeline Mallet for assistance at the Ultrastructural Microscopy Platform of the Pasteur Institute. We are very grateful to Fabrice Besnard, Ludmilla Lokmane, Jean‐Baptiste Pénigault, Clotilde Gimond, Howard Baylis, Kevin Howan, Sarah Mühlberger, and Amir Yassin for samples. We thank Taisei Kikuchi, Janna Fierst, and Erich Schwarz for pre‐publication access to genomic data. We gratefully acknowledge the Republic of Panama and the Smithsonian Tropical Research Institute for supporting collections on Barro Colorado Island. Samples were exported from Panama under Scientific Permit SEX/A‐25‐12 from the Autoridad Nacional del Ambiente. We thank the staff of Edinburgh genomics for expert support. Edinburgh Genomics has core funding from the UK Natural Environment Research Council (UKSBS PR18037). We also thank the three anonymous reviewers for their helpful comments on an earlier version of this manuscript. L.S. was supported by a Baillie Gifford studentship. L.F., A.R., and M.A.F. were supported by the Centre National de la Recherche Scientifique (CNRS), the Ecole Normale Supérieure, and by Agence Nationale pour la Recherche grant ANR‐11‐BSV3‐013. C.B. and S.F. were supported by the CNRS. K.C.K. and D.H.A.F. were supported by NSF grant NSF IOS‐1656736. M.V.R. was supported by NIH grant R01GM121828.
AUTHOR CONTRIBUTIONS
C.B., M.A.F., L.F., C.G., T.K., M.V.R., and W.S. collected and isolated nematodes. C.B., L.F., S.F., M.A.F., M.V.R., and T.K. generated inbred lines. C.B., M.A.F., M.V.R., and L.F. performed mating tests. C.B., S.F., L.N., and A.R. isolated DNA and RNA. T.B., C.C., M.D.N., L.N., and L.S. performed genome assembly and annotation. M.A.F., D.H.A.F., K.K., and W.S. studied morphology. L.S. performed phylogenomic and comparative genomic analysis. L.S., M.A.F., and M.B. wrote the manuscript. All authors read, provided comments on, and approved the final version of the manuscript.
DATA ARCHIVING
All raw sequence data, reference genome assemblies and rDNA sequences have been deposited in the relevant INSDC databases (see Table 1 for accession numbers). Accessions and links to all data used in the phylogenomics analysis are available in Table S9. Data files associated with this study have been deposited in Zenodo under the accession 10.5281/zenodo.1402254. All assemblies and annotations described in this article are available to browse, query, and download at http://www.caenorhabditis.org. Information about morphology, geographic distribution, selected gene sequence data, and taxonomy of these and other Caenorhabditis species has been deposited in RhabditinaDB (http://rhabditina.org).
LITERATURE CITED
- Andrássy, I. 1983. A taxonomic review of the suborder Rhabditina (Nematoda: Secernentia). Office de la Recherche Scientifique et Technique Outre‐Mer (ORSTOM), Paris. [Google Scholar]
- Andrews, S. 2010. FastQC: a quality control tool for high throughput sequence data.
- Artavanis‐Tsakonas, S. , Rand M. D., and Lake R. J.. 1999. Notch signaling: cell fate control and signal integration in development. Science, 284, 770–776. [DOI] [PubMed] [Google Scholar]
- Barrière, A. , and Félix M.‐A.. 2014. Isolation of C. elegans and related nematodes. WormBook, 2, 1–19. [DOI] [PubMed] [Google Scholar]
- Barrière, A. , Yang S.‐P., Pekarek E., Thomas C. G., Haag E. S., and Ruvinsky I.. 2009. Detecting heterozygosity in shotgun genome assemblies: lessons from obligately outcrossing nematodes. Genome Res., 19, 470–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley, R. K. , Roberts A., Smoot M., Juvekar S., Do J., Dewey C., et al. 2009. Fast statistical alignment. PLoS Comput. Biol., 5, e1000392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camacho, C. , Coulouris G., Avagyan V., Ma N., Papadopoulos J., Bealer K., et al. 2009. BLAST+: architecture and applications. BMC Bioinformatics, 10, 421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capella‐Gutiérrez, S. , Silla‐Martínez J. M., and Gabaldón T.. 2009. trimAl: a tool for automated alignment trimming in large‐scale phylogenetic analyses. Bioinformatics, 25, 1972–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- C. elegans Sequencing Consortium . 1998. Genome sequence of the nematode C. elegans: a platform for investigating biology. Science, 282, 2012–2018. [DOI] [PubMed] [Google Scholar]
- Chikhi, R. , and Medvedev P.. 2014. Informed and automated k‐mer size selection for genome assembly. Bioinformatics, 30, 31–37. [DOI] [PubMed] [Google Scholar]
- Cho, S. , Jin S.‐W., Cohen A., and Ellis R. E.. 2004. A phylogeny of Caenorhabditis reveals frequent loss of introns during nematode evolution. Genome Res., 14, 1207–1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook, D. E. , Zdraljevic S., Roberts J. P., and Andersen E. C.. 2017. CeNDR, the Caenorhabditis elegans natural diversity resource. Nucleic Acids Res., 45, D650–D657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook, D. E. , Zdraljevic S., Tanny R. E., Seo B., Riccardi D. D., Noble L. M., et al. 2016. The genetic basis of natural variation in Caenorhabditis elegans telomere length. Genetics, 204, 371–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cutter, A. D. , Baird S. E., and Charlesworth D.. 2006. High nucleotide polymorphism and rapid decay of linkage disequilibrium in wild populations of Caenorhabditis remanei . Genetics, 174, 901–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dey, A. , Chan C. K. W., Thomas C. G., and Cutter A. D.. 2013. Molecular hyperdiversity defines populations of the nematode Caenorhabditis brenneri . Proc. Natl. Acad. Sci. USA, 110, 11056–11060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin, A. , Davis C. A., Schlesinger F., Drenkow J., Zaleski C., Jha S., et al. 2013. STAR: ultrafast universal RNA‐seq aligner. Bioinformatics, 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dougherty, E. C. 1953. The genera of the subfamily Rhabditinae Micoletzky, 1922 (Nematoda) In: Thapar commemoration volume. Univ. of Lucknow, Lucknow, India: Pp. 69–76. [Google Scholar]
- Emms, D. M. , and Kelly S.. 2015. OrthoFinder: solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol., 16, 157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Félix, M.‐A. , Ailion M., Hsu J.‐C., Richaud A., and Wang J.. 2018. Pristionchus nematodes occur frequently in diverse rotting vegetal substrates and are not exclusively necromenic, while Panagrellus redivivoides is found specifically in rotting fruits. PLoS One, 13, e0200851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Félix, M.‐A. , and Braendle C.. 2010. The natural history of Caenorhabditis elegans . Curr. Biol., 20, R965–9. [DOI] [PubMed] [Google Scholar]
- Félix, M.‐A. , Braendle C., and Cutter A. D.. 2014. A Streamlined system for species diagnosis in Caenorhabditis (Nematoda: Rhabditidae) with name designations for 15 distinct biological species. PLoS One, 9, e94723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Félix, M.‐A. , and Duveau F.. 2012. Population dynamics and habitat sharing of natural populations of Caenorhabditis elegans and C. briggsae . BMC Biol., 10, 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari, C. , Salle R., Callemeyn‐Torre N., Jovelin R., Cutter A. D., and Braendle C.. 2017. Ephemeral‐habitat colonization and neotropical species richness of Caenorhabditis nematodes . BMC Ecol., 17, 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fierst, J. L. , Willis J. H., Thomas C. G., Wang W., Reynolds R. M., Ahearne T. E., et al. 2015. Reproductive mode and the evolution of genome size and structure in Caenorhabditis nematodes . PLoS Genet., 11, e1005323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgerald, K. , Wilkinson H. A., and Greenwald I.. 1993. glp‐1 can substitute for lin‐12 in specifying cell fate decisions in Caenorhabditis elegans . Development, 119, 1019–1027. [DOI] [PubMed] [Google Scholar]
- Gatesy, J. , and Springer M. S.. 2014. Phylogenetic analysis at deep timescales: unreliable gene trees, bypassed hidden support, and the coalescence/concatalescence conundrum. Mol. Phylogenet. Evol., 80, 231–266. [DOI] [PubMed] [Google Scholar]
- Gordon, A. , and Hannon G. J.. 2010. Fastx‐toolkit. FASTQ/A short‐reads pre‐processing tools. Available at: http://hannonlab.cshl.edu/fastx_toolkit, 5.
- Greenwald, I. 1985. lin‐12, a nematode homeotic gene, is homologous to a set of mammalian proteins that includes epidermal growth factor. Cell, 43, 583–590. [DOI] [PubMed] [Google Scholar]
- Greenwald, I. 2005. LIN‐12/Notch signaling in C. elegans . WormBook, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas, B. , and Papanicolaou A.. 2012. Transdecoder.
- Hiraki, H. , Kagoshima H., Kraus C., Schiffer P. H., Ueta Y., Kroiher M., et al. 2017. Genome analysis of Diploscapter coronatus: insights into molecular peculiarities of a nematode with parthenogenetic reproduction. BMC Genomics, 18, 478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoff, K. J. , Lange S., Lomsadze A., Borodovsky M., and Stanke M.. 2016. BRAKER1: unsupervised RNA‐Seq‐Based Genome Annotation with GeneMark‐ET and AUGUSTUS. Bioinformatics, 32, 767–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, R.‐E. , Ren X., Qiu Y., and Zhao Z.. 2014. Description of Caenorhabditis sinica sp. n. (Nematoda: Rhabditidae), a nematode species used in comparative biology for C. elegans. PLoS One, 9, e110957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huerta‐Cepas, J. , Serra F., and Bork P.. 2016. ETE 3: reconstruction, analysis, and visualization of phylogenomic data. Mol. Biol. Evol., 33, 1635–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang, H. , Lei R., Ding S.‐W., and Zhu S.. 2014. Skewer: a fast and accurate adapter trimmer for next‐generation sequencing paired‐end reads. BMC Bioinformatics, 15, 182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, P. , Binns D., Chang H.‐Y., Fraser M., Li W., McAnulla C., et al. 2014. InterProScan 5: genome‐scale protein function classification. Bioinformatics, 30, 1236–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurka, J. , Kapitonov V. V., Pavlicek A., Klonowski P., Kohany O., and Walichiewicz J.. 2005. Repbase update, a database of eukaryotic repetitive elements. Cytogenet. Genome Res., 110, 462–467. [DOI] [PubMed] [Google Scholar]
- Kajitani, R. , Toshimoto K., Noguchi H., Toyoda A., Ogura Y., Okuno M., et al. 2014. Efficient de novo assembly of highly heterozygous genomes from whole‐genome shotgun short reads. Genome Res., 24, 1384–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanzaki, N. , Tsai I. J., Tanaka R., Hunt V. L., Liu D., Tsuyama K., et al. 2018. Biology and genome of a newly discovered sibling species of Caenorhabditis elegans . Nat. Commun., 9, 3216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller, O. , Kollmar M., Stanke M., and Waack S.. 2011. A novel hybrid gene prediction method employing protein multiple sequence alignments. Bioinformatics, 27, 757–763. [DOI] [PubMed] [Google Scholar]
- Kiontke, K. C. , Félix M.‐A., Ailion M., Rockman M. V., Braendle C., Pénigault J.‐B., et al. 2011. A phylogeny and molecular barcodes for Caenorhabditis, with numerous new species from rotting fruits. BMC Evol. Biol., 11, 339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiontke, K. , Gavin N. P., Raynes Y., Roehrig C., Piano F., and Fitch D. H. A.. 2004. Caenorhabditis phylogeny predicts convergence of hermaphroditism and extensive intron loss. Proc. Natl. Acad. Sci. USA, 101, 9003–9008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiontke, K. , Hironaka M., and Sudhaus W.. 2002. Description of Caenorhabditis japonica n. sp.(Nematoda: Rhabditida) associated with the burrower bug Parastrachia japonensis (Heteroptera: Cydnidae) in Japan. Nematology, 4, 933–941. [Google Scholar]
- Kocot, K. M. , Citarella M. R., Moroz L. L., and Halanych K. M.. 2013. PhyloTreePruner: a phylogenetic tree‐based approach for selection of orthologous sequences for phylogenomics. Evol. Bioinform. Online, 9, 429–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubatko, L. S. , and Degnan J. H.. 2007. Inconsistency of phylogenetic estimates from concatenated data under coalescence. Syst. Biol., 56, 17–24. [DOI] [PubMed] [Google Scholar]
- Laetsch, D. R. , and Blaxter M. L.. 2017. BlobTools: Interrogation of genome assemblies. F1000Res., 6, 1287. [Google Scholar]
- Lambie, E. J. , and Kimble J.. 1991. Two homologous regulatory genes, lin‐12 and glp‐1, have overlapping functions. Development, 112, 231–240. [DOI] [PubMed] [Google Scholar]
- Lartillot, N. , and Philippe H.. 2004. A Bayesian Mixture Model for Across‐Site Heterogeneities in the Amino‐Acid Replacement Process. Mol. Biol. Evol., 21, 1095–1109. [DOI] [PubMed] [Google Scholar]
- Lartillot, N. , Rodrigue N., Stubbs D., and Richer J.. 2013. PhyloBayes MPI: phylogenetic reconstruction with infinite mixtures of profiles in a parallel environment. Syst. Biol., 62, 611–615. [DOI] [PubMed] [Google Scholar]
- Lee, R. Y. N. , Howe K. L., Harris T. W., Arnaboldi V., Cain S., Chan J., et al. 2018. WormBase 2017: molting into a new stage. Nucleic Acids Res., 46, D869–D874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leggett, R. M. , Clavijo B. J., Clissold L., Clark M. D., and Caccamo M.. 2014. NextClip: an analysis and read preparation tool for Nextera Long Mate Pair libraries. Bioinformatics, 30, 566–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letunic, I. , and Bork P.. 2016. Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res., 44, W242–W245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewin, H. A. , Robinson G. E., John Kress W., Baker W. J., Coddington J., Crandall K. A., et al. 2018. Earth BioGenome project: sequencing life for the future of life. Proc. Natl. Acad. Sci. USA, 201720115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukashin, A. V. , and Borodovsky M.. 1998. GeneMark.hmm: new solutions for gene finding. Nucleic Acids Res., 26, 1107–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch, M. , and Conery J. S.. 2003. The origins of genome complexity. Science, 302, 1401–1404. [DOI] [PubMed] [Google Scholar]
- Maddison, W. P. , and Wiens J. J.. 1997. Gene Trees in Species Trees. Syst. Biol., 46, 523–536. [Google Scholar]
- Mirarab, S. , Reaz R., Bayzid M. S., Zimmermann T., Swenson M. S., and Warnow T.. 2014. ASTRAL: genome‐scale coalescent‐based species tree estimation. Bioinformatics, 30, i541–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirarab, S. , and Warnow T.. 2015. ASTRAL‐II: coalescent‐based species tree estimation with many hundreds of taxa and thousands of genes. Bioinformatics, 31, i44–i52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortazavi, A. , Schwarz E. M., Williams B., Schaeffer L., Antoshechkin I., Wold B. J., et al. 2010. Scaffolding a Caenorhabditis nematode genome with RNA‐seq. Genome Res., 20, 1740–1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osche, G. 1952. Systematik und phylogenie der gattung Rhabditis (Nematoda). Zoologische Jahrbucher ‐ Abt. Zool. Phys. der Tiere, 81, 190–280. [Google Scholar]
- Paradis, E. , and Schliep K.. 2018. ape 5.0: an environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics. [DOI] [PubMed] [Google Scholar]
- Parra, G. , Bradnam K., and Korf I.. 2007. CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics, 23, 1061–1067. [DOI] [PubMed] [Google Scholar]
- Rambaut, A. , Drummond A. J., and Suchard M.. 2007. Tracer v1. 6. http://tree.bio.ed.ac.uk/software/tracer/
- Rebay, I. , Fleming R. J., Fehon R. G., Cherbas L., Cherbas P., and Artavanis‐Tsakonas S.. 1991. Specific EGF repeats of Notch mediate interactions with Delta and Serrate: implications for Notch as a multifunctional receptor. Cell, 67, 687–699. [DOI] [PubMed] [Google Scholar]
- Ren, X. , Li R., Wei X., Bi Y., Ho V. W. S., Ding Q., et al. 2018. Genomic basis of recombination suppression in the hybrid between Caenorhabditis briggsae and C. nigoni . Nucleic Acids Res., 46, 1295–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Revell, L. J. 2012. phytools: an R package for phylogenetic comparative biology (and other things). Methods Ecol. Evol., 3, 217–223. [Google Scholar]
- Roch, S. , and Warnow T.. 2015. On the robustness to gene tree estimation error (or lack thereof) of coalescent‐based species tree methods. Syst. Biol., 64, 663–676. [DOI] [PubMed] [Google Scholar]
- Rudel, D. , and Kimble J.. 2002. Evolution of discrete Notch‐like receptors from a distant gene duplication in Caenorhabditis . Evol. Dev., 4, 319–333. [DOI] [PubMed] [Google Scholar]
- Schulenburg, H. , and Félix M.‐A.. 2017. The Natural Biotic Environment of Caenorhabditis elegans . Genetics, 206, 55–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaham, S. 2006. WormBook: Methods in cell biology, WormBook, ed. The C. elegans Research Community. WormBook, doi/10. 1895/wormbook, 1.
- Sigrist, C. J. A. , de Castro E., Cerutti L., Cuche B. A., Hulo N., Bridge A., et al. 2013. New and continuing developments at PROSITE. Nucleic Acids Res., 41, D344–D347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simão, F. A. , Waterhouse R. M., Ioannidis P., Kriventseva E. V., and Zdobnov E. M.. 2015. BUSCO: assessing genome assembly and annotation completeness with single‐copy orthologs. Bioinformatics, 31, 3210–3212. [DOI] [PubMed] [Google Scholar]
- Slos, D. , Sudhaus W., Stevens L., Bert W., and Blaxter M.. 2017. Caenorhabditis monodelphis sp. n.: defining the stem morphology and genomics of the genus Caenorhabditis . BMC Zool., 2, 4. [Google Scholar]
- Smit, A. F. A. , Hubley R., and Green P.. 1996. RepeatMasker . Available at: http://www.repeatmasker.org/.
- Smit, A. , and Hubley R.. 2010. RepeatModeler Open‐1.0 . Available at: http://www.repeatmasker.org/RepeatModeler/.
- Stamatakis, A. 2014. RAxML version 8: a tool for phylogenetic analysis and post‐analysis of large phylogenies. Bioinformatics, 30, 1312–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein, L. D. , Bao Z., Blasiar D., Blumenthal T., Brent M. R., Chen N., et al. 2003. The genome sequence of Caenorhabditis briggsae: a platform for comparative genomics. PLoS Biol., 1, e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stiernagle, T. 2006. Maintenance of C. elegans . WormBook, 11, 1–11. [DOI] [PMC free article] [PubMed]
- Sudhaus, W. , and Kiontke K.. 1996. Phylogeny of Rhabditis subgenus Caenorhabditis (Rhabditidae, Nematoda)* . J. Zoolog. Syst. Evol. Res., 34, 217–233. [Google Scholar]
- Sudhaus, W. , and Kiontke K.. 2007. Comparison of the cryptic nematode species Caenorhabditis brenneri sp. n. and C. remanei (Nematoda: Rhabditidae) with the stem species pattern of the Caenorhabditis elegans group. Zootaxa, 1456, 45–62. [Google Scholar]
- Tavaré, S. 1986. Some probabilistic and statistical problems in the analysis of DNA sequences. Lectures on mathematics in the life sciences. [Google Scholar]
- Thomas, C. G. , Li R., Smith H. E., Woodruff G. C., Oliver B., and Haag E. S.. 2012. Simplification and desexualization of gene expression in self‐fertile nematodes. Curr. Biol., 22, 2167–2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- True, J. R. , and Haag E. S.. 2001. Developmental system drift and flexibility in evolutionary trajectories. Evol. Dev., 3, 109–119. [DOI] [PubMed] [Google Scholar]
- Vurture, G. W. , Sedlazeck F. J., Nattestad M., Underwood C. J., Fang H., Gurtowski J., et al. 2017. GenomeScope: fast reference‐free genome profiling from short reads. Bioinformatics, 33, 2202–2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood, D. E. , and Salzberg S. L.. 2014. Kraken: ultrafast metagenomic sequence classification using exact alignments. Genome Biol., 15, R46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin, D. , Schwarz E. M., Thomas C. G., Felde R. L., Korf I. F., Cutter A. D., et al. 2018. Rapid genome shrinkage in a self‐fertile nematode reveals sperm competition proteins. Science, 359, 55–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yochem, J. , and Greenwald I.. 1989. glp‐1 and lin‐12, genes implicated in distinct cell‐cell interactions in C. elegans, encode similar transmembrane proteins. Cell, 58, 553–563. [DOI] [PubMed] [Google Scholar]
- Yochem, J. , Weston K., and Greenwald I.. 1988. The Caenorhabditis elegans lin‐12 gene encodes a transmembrane protein with overall similarity to Drosophila Notch. Nature, 335, 547–550. [DOI] [PubMed] [Google Scholar]
- Zerbino, D. R. , and Birney E.. 2008. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res., 18, 821–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. List of isolates and their origin.
Table S2. Mating tests. This table contains several sheets, showing the results of crosses between isolates of different species. Successful crosses are labeled in green.“100s of embryos” refer to unhatched dead embryos remaining on the plate.
Table S3. Detailed genome assembly and gene prediction statistics.
Table S4. Morphological characters used for ancestral state reconstruction. ‘1’ denotes presence or existence; ‘0’ denotes absence.
Table S5. Genome contents of C. sulstoni and C. elegans. Gene structure statistics were calculated using the longest isoform of each protein‐coding gene. UTR regions were not annotated in C. sulstoni and so were not considered in either species.
Table S6. Genome statistics used in PGLS analysis. Gene structure statistics were calculated using the longest isoform of each protein‐coding gene. UTR regions were not considered as they were not annotated in several species. Repeat content was estimated de novo using RepeatModeler and RepeatMasker.
Table S7. Mean branch lengths from Maximum likelihood gene tree of all Notch‐like proteins. Branch lengths were extracted using a custom Python script (available at https://github.com/lstevens17/caeno-ten-descriptions).
Table S8. EGF‐like repeat counts for LIN‐12/GLP‐1 homologues. Counts of EGF‐like repeats were obtained from were obtained from the ProSiteProfiles database (release 2017_09).
Table S9. Accessions and links to data used in phylogenomic analysis.
Table S10. Completeness and duplication statistics for 28 Caenorhabditis species based on 8,286 orthologues. We selected groups of orthologues which were present in at least 22 species and had a mean count of 1. The duplication ratio was calculated by dividing the total number of sequences present for each species by the total number of orthogroups which contained a representative sequence for that species.
Figure S1. Assembly spans and genome size estimates. Kmers of length 19 were counted using KMC (v2.3). Genome size was estimated using GenomeScope (Vurture et al. 2017). The model used by GenomeScope to estimate genome size did not converge for C. waitukubuli (presumably due to high heterozygosity) and is not shown.
Figure S2. Kmer spectra for C. waitukubuli (sp. 39). Kmers were counted using KMC (v2.3). Plotted in R using the ggplot2 package.
Figure S3. PhyloBayes phylogenetic tree. Phylogenetic tree inferred using Bayesian inference with the CAT‐GTR+Γ substitution model. Bayesian posterior probabilities are 1.0 unless noted as branch annotations. Scale is in substitutions per site.
Figure S4. RAxML phylogenetic tree. Maximum likelihood phylogenetic inferred using RAxML with the GTR+Γ substitution model. Bootstrap support values (100 replicates) are 100 unless noted as branch annotations. Scale in substitutions per site.
Figure S5. ASTRAL‐III phylogenetic tree. Phylogenetic tree inferred using ASTRAL‐III, by providing maximum likelihood gene trees (inferred using RAxML with the substitution model selected automatically) as input. As ASTRAL‐III outputs trees with branch lengths in coalescent units, branch lengths in substitutions per site were estimated using RAxML with the GTR+Γ substitution model and the concatenated alignment. Bayesian posterior probabilities are 1.0 unless noted as branch annotations. Scale is in substitutions per site.
Figure S6. Morphology of C. parvicauda male tail. A, A’: Two focal planes in Nomarski optics of the same adult male tail of strain NIC134. B: Scanning electron microscopy, strain JU2070. ad: anterior dorsal papilla; pd: posterior dorsal papilla; vr1, etc.: ventral papilla 1, etc. These photographs exemplify the left‐right asymmetry of the posterior papillae, observed in strains NIC134 and JU2070, as well as JU1766 (not shown).
Figure S7. Morphology of C. quiockensis and C. vivipara. A‐C: Nomarski micrographs of male tails of C. quiockensis strain JU2745. D: C. castelli JU1426 is shown for comparison. Same scale in A‐D. E: C. vivipara NIC1070 male tail F‐H: Details of pericloacal regions of other animals. Note the much larger and wider fan in C. vivipara. Ad: anterior dorsal papilla; gub: gubernaculum; pd: posterior dorsal papilla; spic: spicule; v1, etc.: ventral papillae.(A,B,D, H) are lateral views. (C) and (E‐G) are ventral views. I: Gravid female adult with late‐stage embryos in the uterus and a recently laid L1 stage larva. Bars: 10 μm, except 5 μm for (F,G) and 50 μm for (I). Anterior is to the left except in H where the anterior is to the top.
Figure S8. Male tails of species of the Elegans supergroup. Ventral views by Nomarski optics of C. becei strain QG704, C. waitukubuli strain NIC564, C. panamensis strain QG702, C. sulstoni strain SB454 and C. afra JU1199 male tails. Note the variation in the respective lengths of rays 4 and 5. All pictures at are at the same scale. Bar: 10 μm. The correspondence between the v1‐7, ad, pd papilla nomenclature and that of rays used in C. elegans is indicated on the C. becei picture. v1‐7 denote ventral papillae, while ad and pd denote the anterior and posterior dorsal papillae, respectively. In the C. elegans nomenclature, rays are instead numbered r1‐r9 without distinction between ventral and dorsal rays.
Figure S9. Ancestral state reconstruction of male tail characters. A: Antero‐posterior position of the dorsal ray. B: Closed fan. Ancestral state reconstruction was performed by generating 1000 stochastic character maps of each morphological character on the phylogenetic tree in Fig. 1A using the equal rates model of evolution. Pie charts on internal nodes represent poster probabilities of ancestral states. The fan of C. vivipara was ambiguous and thus excluded from the open fan character mapping analysis.
Figure S10. Spicule tip shape in Elegans supergroup species. Nomarski micrographs. Left column: ventral view. Right column: ventro‐lateral view. The tip of spicules in C. afra and C. sulstoni (as in several species of the Japonica group; Kiontke et al. 2011) is broad and bent with a discontinuity on the curvature, compared to a thin and continuously bent tip in species of the Elegans group such as C. zanzibari or C. elegans. That of C. panamensis strain QG702 is quite thin, while that of C. becei strain QG704 is broad. Bar for all panels: 5 μm.
Figure S11. Phylogenetic generalized least squares (PGLS) analysis of genome size and contents.
A: Protein‐coding gene number. B: Estimated repeat content. C: Mean intron count per gene. D: Mean intron size. E: Mean intergenic distance. Phylogenetic tree presented in Fig. 1A was used in all analyses; points are coloured based on major clades. Gene structure statistics were calculated using the longest isoform of each protein‐coding gene. UTR regions were not considered as they were not annotated in several species. Repeat content was estimated de novo using RepeatModeler and RepeatMasker.
Figure S12. Ancestral state reconstruction of EGF‐like repeat structure. Ancestral state reconstruction was performed by generating 1000 stochastic character maps of EGF‐like repeat count on the gene tree presented in Fig. 7A using the equal rates model of evolution. Pie charts on internal nodes represent poster probabilities of ancestral states.
Document S1. Species declarations.
Document S2. Detailed bioinformatics methods. Methods, versions and relevant parameters used in genome assembly, gene prediction, phylogenomic analysis, genome size analysis, and Notch‐receptor analysis.