

Abstract
An artificial cell-signaling pathway is developed that capitalizes on the T-cell’s innate extravasation ability and transforms it into a vector (T-cell Biofactory) for synthesizing calibrated amounts of engineered proteins in vivo. The modularity of this pathway enables reprogramming of the T-cell Biofactory to target biomarkers on different disease cells, e.g. cancer, viral infections, autoimmune disorders. It can be expected that the T-cell Biofactory leads to a “living drug” that extravasates to the disease sites, assesses the disease burden, synthesizes the calibrated amount of engineered therapeutic proteins upon stimulation by the diseased cells, and reduces targeting of normal cells.
Keywords: adoptive cell therapy, cell engineering, chimeric antigen receptor, drug delivery, nuclear factor of activated T-cell response element
1. Introduction
T cells extravasate to disease sites with cellular resolution[1] and use the T-cell receptor (TCR) to engage the peptide—major histocompatibility complex (pMHC) on the target disease cells with molecular specificity.[2] At the population level, this leads to clonal expansion[3] of pMHC-specific-TCR+ T cells proportional to the disease cell mass.[4] At the single-cell level, this cues the activation pathway in a binary (on/off) event independent of the pMHC density on the surface of the disease cell.[4] This opens an attractive possibility of engineering the T cells to execute a range of effector functions precisely at the disease site and proportionate to the disease burden. We report here on the development of T-cell Biofactories for targeting diseased cells with maximal specificity and autonomously synthesize engineered proteins.
A synthetic immune-regulated human promoter in T cells has been used for site-specific synthesis of interleukin-12 (IL-12),[5] however such T cells are restricted in their use to only target the pMHC complex for which they possess the avidity through its endogenous TCR. A synthetic pathway based on prokaryotic regulatory elements and factors has been recently reported for transforming the T cell into a vector for synthesizing therapeutic proteins in situ.[6] This, however, has the potential to promote immunogenic reactions. We have developed a human immune-regulated artificial cell-signaling pathway whose design transforms the cell into an in vivo living vector (antigen-specific T-cell Biofactory) for synthesizing engineered therapeutic proteins in situ upon interacting with the antigen-presenting target cell (Figure 1A). For ease of manufacture and clinical translation, we designed the signaling pathway as a single DNA sequence (Figure 1B) comprising three constant and two variable domains arranged in cis. The constant domains provide functionality to the T-cell Biofactory and include—a transmembrane molecule (Receptor) that mobilizes the T-cell activation transcriptional machinery (Actuator) to upregulate the effector transgene fused to a signal peptide (Secretor) that assists in transporting it into the extracellular space. The variable domains are responsible for the broad applicability of this platform. They impart specificity to the T-cell Biofactory against particular diseases and include—variable heavy-light (VH−VL) chain (Sensor, part of the transmembrane Receptor) to identify the antigen biomarker on the disease cell independent of pMHC; and the transgene (Effector). They are modular, i.e., the Sensor can be exchanged to reprogram the T-cell Biofactory platform to target biomarkers specific to different cell-based diseases that may alter their pMHC expression to escape the immune attack; and the Effector with a therapeutic transgene to neutralize the pathology that triggered the T-cell Biofactory.
Figure 1.
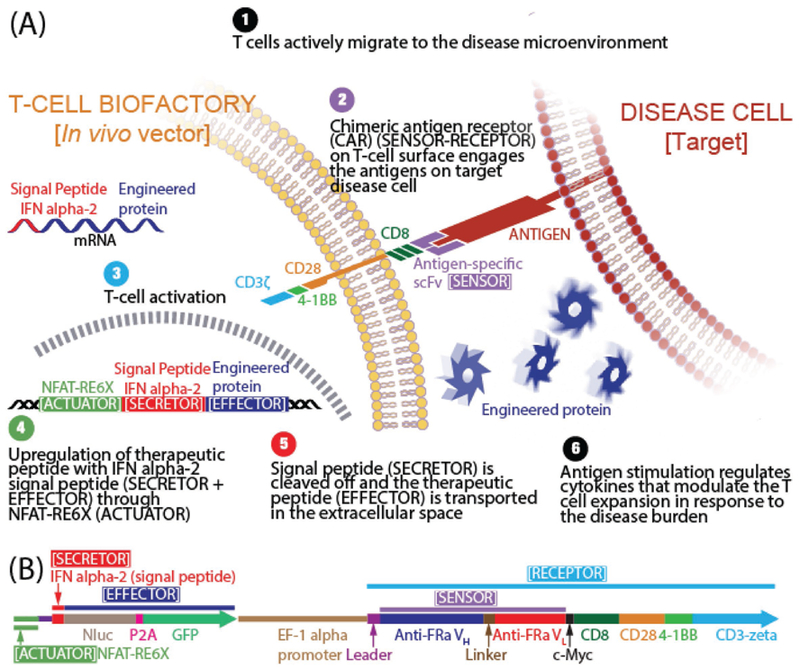
Schematics. A) T-cell Biofactory. B) Artificial cell-signaling pathway comprises of three constant domains (Receptor, Actuator, Secretor) and two variable domains (Sensor, Effector) arranged in cis. [(i) Receptor: Based on the chimeric antigen receptor (CAR) (GenBank: HJ220879.1) from clinical CAR T-cell trials (ref. [11]), containing the CD3-zeta signaling domain of the TCR-CD3 complex that causes intracellular calcium ([Ca2+]i) rise upon engaging its target antigen (ref. [12]). Briefly, the extracellular portion of the CAR is composed of the CD8 segment proximal to the cell membrane. The transmembrane portion that connects the extracellular and intracellular domains is from CD28 and extends as the intracellular costimulatory domain. Additional 4–1BB costimulatory domain found to enhance the therapeutic response of CAR T cells by mitigating early exhaustion of T cells engineered to express antigen-specific CARs was included. (ii) Actuator: Based on six copies of NFAT-RE (ggagga aaaact gtttca tacaga aggcgt) that respond to the [Ca2+]i rise (ref. [10]) and placed upstream of a transcriptional start site for the Effector transgene. (iii) Secretor: Derived from human interferon alpha-2 (IFNa2) (ref. [13]) (GenBank: NM_000605). (iv) Sensor (part of the transmembrane Receptor): Comprises of variable domains from heavy (VH) and light (VL) chain of the antigen-specific antibody. In this work, these were derived from the anti-FRa antibody (ref. [7]) (MORAb-003) or the anti-MSLN antibody (ref. [8]) (MORAb-009). (v) Effector: Comprises of a reporter transgene that can be exchanged with a therapeutic transgene to neutralize the pathology that triggered the T-cell Biofactory. In this work, Nanoluc luciferase reporter was used (Promega, GenBank: JQ437370.1).]
2. Results
Complete details on the development of Actuator, Receptor, and Secretor domains for the T-cell Biofactory are described in the Experimental Section and presented in the Figures S1–S3 (Supporting Information). To demonstrate T-cell Biofactory function (Figure 2A–D), we used VH−VL Sensor sequences from antibodies that recognize human ovarian cancer antigens. We used VH−VL sequences from the anti-folate-receptor alpha (FRa) antibody[7] (MORAb-003) codon-optimized for human expression. To demonstrate flexibility of the T-cell Biofactory platform (Figure 2E), we also redirected its specificity to target mesothelin (MSLN) by exchanging the VH−VL portion with that of the anti-MSLN antibody[8] (MORAb-009). The rationale for the choice of these two antigens was based on the fact that they are overexpressed on multiple cancers and their expression on healthy tissues is rather limited.[9] In vivo activity and specificity of T-cell Biofactories were demonstrated in mice bearing sub-cutaneous OVCAR3 tumors (Figure 3).
Figure 2.
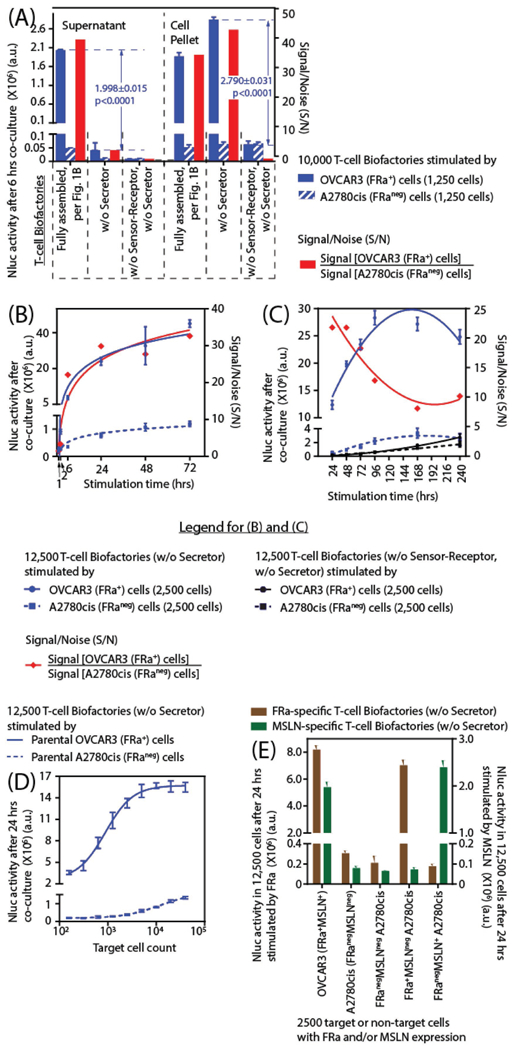
Demonstration and characterization of the T-cell Biofactory function. Bioluminescence due to Nluc activity (n = 4 for all observations, error bar indicates ±1 SD) was a surrogate to quantify the Effector protein expression. Statistical significance for all comparisons was based on two-sample t-tests with common variance. The S/N quantitatively indicated the potential for synthesizing and secreting the engineered protein in the absence of specific antigen-presenting target cells. A) The T-cell Biofactory synthesizes and secretes Effector proteins upon stimulation by antigen-presenting target cells. The activity was statistically significantly different when compared to the control T-cell Biofactories (i) without the Secretor, and (ii) without the Secretor and without the Sensor–Receptor. B) The Effector protein activity was increased within 1 h (p = 0.0032) when stimulated by target versus nontarget cells. C) Sustained activity of the Effector protein was observed for 10 d when stimulated by target versus nontarget cells. D) The Effector protein activity was proportional to the mass of target cells. E) The specificity of the FRa-specific T-cell Biofactory was redirected to target MSLN by exchanging the VH−VL Sensor domain.
Figure 3.
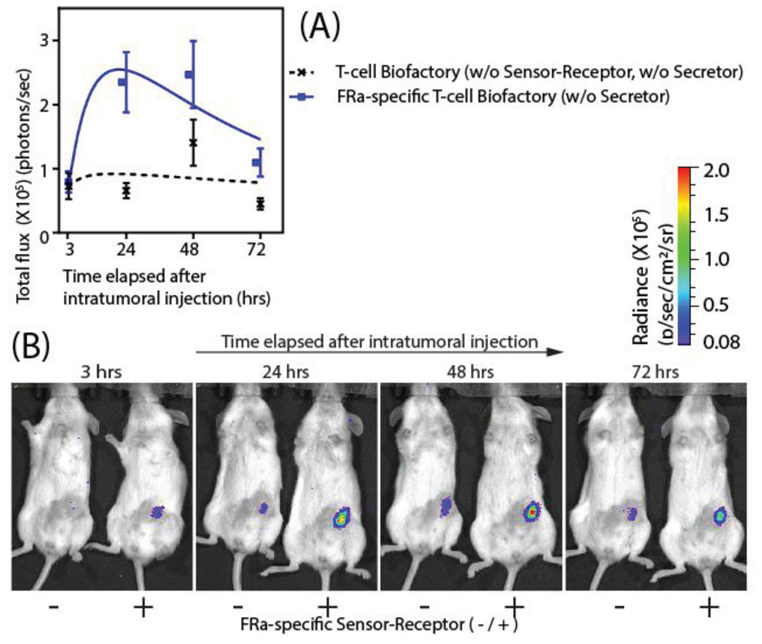
In vivo validation of the T-cell Biofactory: Two differently engineered T-cell Biofactories (20 million each) [(i) with FRa-specific Sensor–Receptor (+); and (ii) without FRa-specific Sensor–Receptor (−)] were injected intratumorally in the two groups of subcutaneous FRa+ OVCAR3 tumors engrafted in NSG mice (n = 10 per group). The mice were imaged for bioluminescent signal at 3, 24, 48, 72, and 240 h after the intratumoral injection. (see Experimental Section “In vivo study/Bioluminescent imaging” for the complete procedure). Bioluminescence due to Nluc activity was a surrogate to quantify the Effector protein expression. A) Effector protein expression in the FRa-specific T-cell Biofactories upon stimulation by OVCAR3 tumors was similar at 3 h (p = 0.809), when compared to the control T-cell Biofactories without FRa specificity, was most different at 24 h (p = 0.003), and continued to be statistically elevated for at least 72 h (p = 0.014). Signal from the two T-cell Biofactories was found to be statistical similar at 48 h (p = 0.112) and 240 h (p = 0.307). The results were expressed as the mean of bioluminescence emission (photons/seconds) (n = 10 per group, error bar indicates ± 1 SEM). Statistical significance for all comparisons were based on two-sample t-tests with common variance. B) Bioluminescent imaging of mice at the indicated time points (see Figure S4, Supporting Information for all images).
2.1. Actuator with Six NFAT-RE Copies Exhibits Highest Signal-to-Noise Ratio
Briefly, we tested three T-cell Biofactories [engineered with either 3, 6, or 9 copies of nuclear factor of activated T-cell response element (NFAT-RE)], all of which were chemically stimulated to respond to the intracellular calcium ([Ca2+]i) rise.[10] We selected the Actuator domain with six copies of NFAT-RE (NFAT-RE6X) placed upstream of a transcriptional start site because it exhibited the highest signal-to-noise ratio (S/N), as defined in the figure (Figure S1, Supporting Information) legend.
2.2. Receptor with Intracellular CD28 and 4–1BB Costimulatory Domains Exhibits Highest Signal-to-Noise Ratio
Next we tested three T-cell Biofactories, each with NFAT-RE6X inducible reporter, but with different Receptor domains. We designed the Receptor domains based on the chimeric antigen receptor (CAR) from clinical CAR T-cell trials,[11] all of which contained the CD3-zeta signaling domain of the TCR-CD3 complex that causes [Ca2+]i rise upon engaging its target antigen.[12] Based on the S/N for the Effector protein expression (Figure S2, Supporting Information) in these T-cell Biofactories, we selected the Receptor domain that also contained intra-cellular CD28 and 4–1BB costimulatory domains (also known as third-generation CAR).
2.3. Secretor with IFNa2 Signal Peptide Exhibits Highest Signal-to-Noise Ratio
Finally, we tested the five T-cell Biofactories, each with NFAT-RE6X inducible reporter and third-generation CAR, but with different Secretor domains. These Secretor domains were derived from the signal peptides of the secretory proteins. Based on the S/N for the secretion of Effector protein secretion (Figure S3, Supporting Information) in the supernatant of these T-cell Biofactories, we selected the Secretor domain derived from human interferon alpha-2 (IFNa2).[13]
2.4. T-cell Biofactory Synthesizes and Secretes Effector Proteins Upon Stimulation by and Proportionate to the Number of Antigen-Presenting Target Cells
OVCAR3 human ovarian cancer cells that express FRa and MSLN were used as target cells and A2780cis human ovarian cancer cells that lack FRa and MSLN were used as nontarget negative controls. The Nluc reporter enzyme was used to represent a quantifiable non-human Effector protein that can be replaced by a human or nonhuman protein or peptide with desired properties. The T-cell Biofactory autonomously synthesizes and releases Effector protein upon engaging its target ligand on the surface of antigen-presenting disease cells (Figure 2A). The function of the Secretor domain for release of the Effector protein was quantified by comparing the Nluc activity in the supernatant to that of a control T-cell Biofac-tory without the Secretor domain and was statistically significantly higher (p < 0.0001) (Figure 2A). Similarly, the effect of the Sensor−Receptor domains on directing the specificity of the T-cell Biofactory was quantified by comparing the Nluc activity in the cell pellet of the T-cell Biofactory with FRa-specific Sensor–Receptor and Actuator domains to the control T-cell Bio-factory with only the Actuator domain. The increase in activity mediated by the Sensor−Receptor domain was also statistically significantly different (p < 0.0001) (Figure 2A). The kinetics of Nluc activity show the protein synthesis from the FRa-specific T-cell Biofactory was increased as early as 1 h (S/N ≈ 1.5 @ 1 h, p = 0.0032; S/N ≈ 3.4 @ 2 h, p < 0.0001) (Figure 2B). It continued to increase until 96 h and was sustained for at least 10 d (Figure 2C). Effector protein activity from the FRaspecific T-cell Biofactories was proportional to the target cell mass (Figure 2D) and statistically significantly different from the FRa-specific T-cell Biofactories stimulated by nontarget cells (p < 0.0001).
2.5. T-cell Biofactory Platform Can be Redirected to Target Different Cell-Surface Antigens with Specificity
Figure 2E demonstrates that the T-cell Biofactory platform can be engineered to recognize different targets on a diseased cell surface. Here, FRa- and MSLN-specific T-cell Biofactories were used to detect FRa or MSLN expression on three A2780cis variant cell lines (engineered with empty vector or with the vector containing FRa or MSLN) as well as parental FRa+MSLN+ OVCAR3 and FRanegMSLNneg A2780cis cells. The results suggest that the two T-cell Biofactories are specific for their targets and statistically significantly different from each another (p < 0.002).
2.6. T-cell Biofactory Platform Responds to the Specific Stimulation by Antigen Presenting Tumors In Vivo
T-cell Biofactories have the potential to synthesize and deliver therapeutics (Effectors) with spatiotemporal resolution since we demonstrate that they can synthesize and secrete an Effector protein upon binding to a specific cell surface target. The dynamic nature of this in vivo living vector is thus expected to prevent systemic toxicity by synthesizing biologic molecules only when and where required and minimizing their delivery to the non-target cells. The in vivo study (n = 10 per group) (Figure 3 and Figure S4, Supporting Information) demonstrated that the Effector protein activity was elevated when FRa-specific T-cell Biofactories were injected intratumorally into OVCAR3 tumors compared to injection of control T-cell Biofactories without FRa-specific Sensor–Receptor. The Effector activity of FRa-specific compared to control biofactories was similar at 3 h (p = 0.809) was elevated and most different from the control at 24 h (p = 0.003), and continued to be statistically significantly elevated for at least 72 h (p = 0.014). The Effector protein activity in FRa-specific T-cell Biofactories was highest at 48 h; however, the elevation was not statistically significant due to a high signal from the control at that time point. Interestingly, the in vivo FRa-mediated T-cell Biofactory response rises rapidly, similar to the in vitro model. The in vivo response however drops after 48 h, which may be due to the loss of T-cell viability subsequent to their intratumoral injection.
3. Discussion
The pathologic state of cell-based diseases is progressive and attributable to dynamic changes that diseased cells undergo in vivo. However, current small-molecule and protein-based therapies are not designed to coevolve with in vivo disease progression.[14] Interpatient variability is also a challenge, and continuous monitoring of the disease state for optimal dosing is not practical. Excess drug in systemic circulation leads to morbidity, and suboptimal levels lead to drug resistance. As such, options for cell-based diseases (e.g., cancer, viral infections, and autoimmune disorders) that evade the immune system or involve its malfunction are limited.
While mammalian cell engineering is poised for clinical impact,[15] challenges still exist, e.g., graft-versus-host disease, cytokine release syndrome, cerebral edema, off-tumor trafficking, and affordable cell manufacturing. Fortunately, the novel solutions in this rapidly evolving area translate across different cell-based strategies. For example, in an analogous T-cell-based approach[6] antigen-specific receptor directly released the intracellular transcriptional activator domain for a therapeutic antibody synthesis upon engaging with the target cell. Similarly, TCR-inducible NFAT promoter in autologous tumor infiltrating lymphocytes was used to secrete IL-12 directly at tumor sites[5] that minimized previously observed clinical toxicities.[16]
The innovation in our work builds on what has been reported earlier.[5,6] The differentiating factor in our T-cell Biofactories is the human artificial cell-signaling pathway that has a modular and variable Sensor–Receptor and signals the synthetic immune-regulated promoters for in situ transgene (Effector) expression. This forms the foundation of this vector technology and greatly expands the potential for targeting many cell-based diseases based on disease-specific biomarkers and independent of the TCR-pMHC signaling pathway. This is of significance because it demonstrates the feasibility of a cell-based vector platform that can be selectively directed against any cell-based disease through its respective biomarker and express any protein.
We expect T-cell Biofactories to be used as living vectors, which by coupling their innate intelligence with engineered capabilities, will lead to a platform for in situ synthesis of therapeutic agents that will circumvent the need to rely on passive diffusion of drugs infused in excess. Our reprogrammable platform will instead autonomously synthesize calibrated amounts of engineered therapeutic proteins from human or nonhuman origin directly at the disease site and limit damage to normal tissues. This may also extend the duration of treatment with the potential to eventually reduce mortality.
4. Experimental Section
Materials and Reagents:
All parental and engineered cell lines [Jurkat Clone E6–1 (ATCC, Cat# TIB-152), OVCAR3 (ATCC, Cat# HTB-161) and A2780cis (Sigma-Aldrich, Cat# 93 112 517)] were maintained in complete RPMI media [RPMI1640 with L−glutamine (Corning, Cat# 10–040CV), and 10% fetal bovine serum (Sigma-Aldrich, Cat# F2442–500ML)]. Puromycin dihydrochloride (ThermoFisher Scientific, Cat# A1113803) or G418 (Corning, Cat# 30 234-CR) was used for selecting stable cells.
Plasmid DNA Production:
Plasmids were designed in SnapGene software (GSL Biotech LLC). Plasmid preparation services (chemical synthesis of DNA insert sequences, subcloning into respective vector backbones, and the amplification) were obtained from Epoch Life Science, Inc. (Missouri City, TX). The gene sequences for transforming the Jurkat Clone E6–1 into the fully assembled (per Figure 1B) or control T-cell Biofactories was subcloned into lentivirus vector plasmid (System Biosciences, CD510B-1) and those for transforming A2780cis into antigen-presenting A2780cis cell line variants were subcloned into PiggyBac transposon vector plasmid (System Biosciences, PB510B-1). Unless otherwise mentioned, puromycin N−acetyltransferase was used as a selection marker in all cases.
Lentivirus Production:
Lentivirus particles carrying synthetic genes were prepared by packaging the corresponding plasmid using the LentiStarter 2.0 kit (System Biosciences, LV051A-1) following the manufacturer’s protocol. Viral supernatant was collected 48 and 72 h after transfection and pooled. Following concentration using PEG-it virus precipitation solution (System Biosciences, Cat# LV810A-1), the viral pellet was resuspended in 1/10th of the original volume in DMEM containing 25 × 10−3 M HEPES buffer (Corning, Cat # 25–060-Cl), and aliquots were stored at −80 °C.
Generation of T-cell Biofactories:
The Jurkat Clone E6–1 suspension cell line was engineered with lentivirus particles carrying the appropriate (fully assembled (Figure 1B) or control) T-cell Biofactory insert in the presence of TransDux (System Biosciences, Cat# LV850A-1) following the manufacturer’s instruction. After 48 h, cells were placed in selection using 0.2 mg mL−1 of puromycin dihydrochloride. The unmodified parental cell line was also placed under selection as a positive control for cell killing by puromycin. Following selection, cells were expanded as required for different assays and frozen using freezing media (complete culture medium and 10% DMSO) for liquid nitrogen stocks.
Generation of A2780cis Cell Line Variants:
Three A2780cis variant cell lines were generated using the PiggyBac Transposon system (System Biosciences, PureFection transfection reagent, Cat # LV750A-1 and PiggyBac Transposase vector Cat # PB210PA-1) following the manufacturer’s protocol. All three plasmids were designed for intracellular expression of Luc2 (GenBank: AY738222.1) and E2Crimson (GenBank: KT878738.1) reporter transgene. One of these plasmids was designed to express FRa (GenBank: NM_01 6725.2) on the A2780cis cell surface, another to express MSLN (GenBank: NM_0 05823.5), and the third was the control plasmid that did not express FRa or MSLN. The unmodified parental cell line was also placed under selection as a positive control for cell killing by the antibiotics. Following selection, Luc2 and E2Crimson expression was verified, and the cells were frozen using freezing media for liquid nitrogen stocks. This resulted into three A2780cis variants (FRa+MSLNnegLuc2–2A-E2Crimson+A2780cis; FRanegMSLN+Luc2–2A-E2Crimson+A2780cis; FRanegMSLNnegLuc2–2AE2Crimson+A2780cis). The A2780cis variant cell lines were expanded as required for different assays.
Stimulation of T-cell Biofactories:
Target or nontarget cells (2500 cells; unless otherwise noted) were cocultured with and used to stimulate the T-cell Biofactories (12500 cells, unless otherwise noted) in 100 mL of complete RPMI in a single well of a 96-well plate. Target and non-target cells comprised of i) engineered A2780cis negative for FRa and MSLN (FRanegMSLNnegLuc2–2A-E2Crimson+A2780cis); ii) engineered A2780cis positive for FRa and negative for MSLN (FRa+MSLNnegLuc2–2A-E2Crimson+A2780cis); and iii) engineered A2780cis negative for FRa and positive for MSLN (FRanegMSLN+Luc2–2A-E2Crimson+A2780cis). Additional cell lines to assess the Effector function of T-cell Biofactories against unmodified parental cell lines were also used. These included iv) parental OVCAR3 cell line with natural expression of FRa and MSLN; and v) parental A2780cis cell line negative for FRa and MSLN. After coculture (24 h, unless otherwise noted), the manufacturer’s protocol was followed to measure NanoLuc (Nluc) activity in the T-cell Biofactories using Nano-Glo assay (Promega, Cat# N1120).
Signal Peptide Experiment and Coculture:
T-cell Biofactories (100 000 cells) were washed three times with serum-free RPMI and resuspended in 200 μL of fresh complete RPMI. Parental OVCAR3 (FRa-presenting cell line) and A2780cis (FRa-negative cell line) were seeded at 12 500 cells per well and similarly treated and resuspended in 200 mL of fresh complete RPMI. Three differently engineered T-cell Biofactories [i) fully assembled per Figure 1B; ii) as in Figure 1B but without the Secretor domain; and iii) as in Figure 1B but without the Secretor domain and without the Sensor–Receptor domain] were mixed with OVCAR3 or A2780cis to create individual cocultures in a 24-well plate with a final volume of 1 mL. After 6 h, a 500-μL aliquot from each coculture was centrifuged in 1.5 mL tube for 5 min at 0.1 RCF. A total of 100 μL of supernatant was drawn without disturbing the cell pellet and transferred to a 96-well plate (four wells were prepared). The Nluc activity was assessed in a 96-well plate with Nano-Glo assay using the manufacturer’s protocol. The cell pellets in each 1.5 mL tube were washed three times with 1 mL of complete RPMI and finally resuspended back to a total of 500 μL of complete RPMI. A total of 100 μL of resuspended cells were drawn from each 1.5 mL tube and transferred to a 96-well plate (4 wells were prepared). The Nluc activity was assessed in the 96-well plate with Nano-Glo assay using the manufacturer’s protocol.
NanoGlo Assay—Nluc Activity:
The manufacturer’s protocol was followed to measure Nluc activity using Nano-Glo assay. Briefly, Nluc substrate was diluted in the cell lysis buffer provided with the Nano-Glo assay. The reconstituted Nluc reagent was added 1:1 to the cells modified to express Nluc enzyme. Following a 3 min incubation, bioluminescence was read on a microplate reader (Perkin Elmer, EnVision Multilabel Plate Reader Model: 2104–0010A).
In Vivo Study:
All mice imaging was handled at SRI International in accordance with guidelines from the Institutional Animal Care and Use Committee.
In Vivo Study: Expansion of T-cell Biofactories and Preparation for Infusion:
Two differently engineered T-cell Biofactories [i) with FRa-specific Sensor–Receptor (as in Figure 1B but without the Secretor domain); and ii) without Sensor–Receptor (as in Figure 1B but without the Secretor domain and without the Sensor–Receptor domain)] were expanded in complete RPMI with 0.5 μg mL−1 of puromycin and seeded at a density of 700 000 cells mL−1. Puromycin was removed from the media two passages before infusion in mice.
In Vivo Study: Tumor Xenografts and Intratumoral Delivery of the T-cell Biofactories:
Female NSG mice (NOD.Cg-Prkdcscid IL2rgtm1wjl/Szj) (age: 6 weeks) were implanted subcutaneously in the left abdominal region just off the mid-line with 3 million parental OVCAR3 (FRa+) cells resuspended in 100 μL serum-free RPMI with 50% Matrigel (Corning, cat # 356 237). The tumors were calipered twice until they reached approximately 60 mm3. The mice were then randomized into two groups of 10 mice per group and each mouse was injected intratumorally with 20 million T-cell Biofactories resuspended in 50 μL, as follows: Group (i)—with FRa-specific Sensor–Receptor; Group (ii)—without Sensor–Receptor. The T-cell Biofactories were tested in vitro for appropriate responses by coculturing them with OVCAR3 cells and A2780cis cells for 24 h, before intratumoral injection. The tumor growth in each group was monitored by caliper measurements for two weeks and found to be statistically similar.
In Vivo Study: Bioluminescence Imaging:
The baseline bioluminescent signal was determined by imaging the mice ventrally 3 h after intratumoral injection of the T-cell Biofactories. Mice were injected intravenously with 10 μg Nano-Glo reagent (1:20 dilution from the stock solution in sterile PBS, equivalent to 0.5 mg kg−1) in the volume of 100 μL and immediately anesthetized with isoflurane inhalation (3–5% for induction and 2% for maintenance) for about 2 min prior to imaging. Imaging was performed in a Lumina XR instrument (Perkin Elmer) with 4 min exposure. The data were quantified by analysis of the region-of-interest (ROI) using Living Image software. The F/Stop was set at 1, the field of view (FOV) was D, and the pixel binning was set as medium. The bioluminescence imaging was repeated at 24, 48, 72, and 240 h post T-cell Biofactories injection.
Experimental Designs and Statistical Analysis:
The experimental design for each panel in the figures is described. GraphPad Prism 6 (GraphPad Software, Inc.) was used to conduct all statistical analysis. Statistical significance for all comparisons was based on two-sample t-tests with common variance and a two-sided p−value of 0.05. There was no adjustment for multiple comparisons.
Experimental Designs and Statistical Analysis: Figure 2A (Functional T-cell Biofactory):
The vertical bars show the mean Nluc activity in three differently engineered T-cell Biofactories when stimulated by two target/nontarget cells [parental OVCAR3 (FRa+) and parental A2780cis (FRaneg) cells]. One T-cell Biofactory was fully assembled (per Figure 1B), a second T-cell Biofactory was a control that lacked the IFNa2 Secretor domain (per Figure 1B but without the Secretor domain), and a third T-cell Biofactory was a control that lacked the IFNa2 Secretor domain and the FRa-specific Sensor–Receptor domain (per Figure 1B but without the Secretor domain and without the Sensor–Receptor domain). The S/N is calculated as the ratio of the mean Nluc activity in the T-cell Biofactories when stimulated by the parental OVCAR3 (FRa+) cells divided by the mean Nluc activity when stimulated by the parental A2780cis (FRaneg) cells. The error bars extend 1 SD above the mean and can also be considered as one half-width of an 86% confidence interval for that mean.
Experimental Designs and Statistical Analysis: Figure 2B (Significant T-cell Biofactory Activation within 1 h):
The Nluc activity in T-cell Biofactories (per Figure 1B but without the Secretor) stimulated by two target/nontarget cells [parental OVCAR3 (FRa+) and parental A2780cis (FRaneg) cells] was fit using the equation Y = a + b * log10(X) where X is the stimulation time in hours. The S/N is calculated as the ratio of the mean Nluc activity in the T-cell Biofactories when stimulated by the parental OVCAR3 (FRa+) cells divided by the mean Nluc activity when stimulated by the parental A2780cis (FRaneg) cells. The error bars are 1 SD above and below the mean and can also be considered as one half-width of an 86% confidence interval for that mean.
Experimental Designs and Statistical Analysis: Figure 2C (Significant T-cell Biofactory Activation for up to 10 d):
The Nluc activity in the two different T-cell Biofactories (per Figure 1B but (i) without the Secretor and without the Sensor–Receptor, or (ii) without the Secretor) stimulated by two target/nontarget cells [parental OVCAR3 (FRa+) and parental A2780cis (FRaneg) cells] was fit using the equation Y = a + b * X + c * X^2, where X is the stimulation time in hours. The S/N is calculated as the ratio of the mean Nluc activity in the T-cell Biofactories when stimulated by the parental OVCAR3 (FRa+) cells divided by the mean Nluc activity when stimulated by the parental A2780cis (FRaneg) cells. The error bars are 1 SD above and below the mean and can also be considered as one half-width of an 86% confidence interval for that mean.
Experimental Designs and Statistical Analysis: Figure 2D (T-cell Biofactory Activation Proportionate to the Target Cell Mass):
The Nluc activity in the T-cell Biofactories (per Figure 1B but without the Secretor) stimulated by two target/nontarget cells [parental OVCAR3 (FRa+) and parental A2780cis (FRaneg) cells] was fit using a four-parameter logistic model Nluc = Nlucmin + (Nlucmax – Nlucmin)/(1 + 10^(b* (X - log10EC50))) where X is the log10 of the target cell count, Nlucmax is an estimated parameter defining a upper asymptote for Nluc, Nlucmin is an estimated parameter defining a lower asymptote for Nluc, b is a “Hill” parameter defining the slope at the inflection point of the fitted curve, and EC50 is an estimated parameter representing the X value corresponding to (Nlucmax – Nlucmin)/2. The error bars are 1 SD above and below the mean and can also be considered as one half-width of an 86% confidence interval for that mean.
Experimental Designs and Statistical Analysis: Figure 2E (Redirected Specificity of the T-cell Biofactory):
The vertical bars show the mean Nluc activity of the two differently engineered T-cell Biofactories (FRaspecific Sensor–Receptor using VH−VL sequence from MORAb-003 or MSLN-specific Sensor−Receptor using VH−VL sequence from MORAb-009) and stimulated by five different target/nontarget cells [FRanegMSLNneg A2780cis; FRa+MSLNneg A2780cis; FRanegMSLN+ A2780cis; parental OVCAR3 (FRa+MSLN+) and parental A2780cis (FRanegMSLNneg) cells]. (All T-cell Biofactories were generated per Figure 1B with FRa- or MSLN-specific Sensor–Receptor but without the Secretor). The error bars extend 1 SD above the mean and can also be considered as one half-width of an 86% confidence interval for that mean.
Experimental Designs and Statistical Analysis: Figure 3 (In Vivo Validation of the T-cell Biofactory):
The Nluc activity in two T-cell Biofactories [(i) with FRa-specific Sensor–Receptor (as in Figure 1B but without the Secretor domain); and (ii) without FRa-specific Sensor–Receptor (as in Figure 1B but without the Secretor domain and without the Sensor–Receptor domain)] stimulated in vivo when injected intratumorally in female NSG mice bearing subcutaneous OVCAR3 (FRa+) ovarian cancer cells was fit (Figure 3A) using the equation Y = amplitude * exp (−0.5 * (ln (X/center)/width)^2) where X is the stimulation time in hours, and the parameters center, width, and amplitude were fit to minimize the squared deviation of the observations from the fitted curve. The parameter center represents the X value corresponding to the peak of the fitted curve, the parameter amplitude represents the height of the fitted curve at the center, and the parameter width represents the width of the fitted distribution. The error bars are one standard error of the mean (SEM) above and below the mean and can also be considered as one half-width of an 86% confidence interval for that mean.
Supplementary Material
Acknowledgements
P.B. was the principal investigator. He conceptualized the project, conducted feasibility studies, obtained funding, analyzed the results, and wrote the manuscript. C.R. and L.B. conducted the experiments, analyzed the results, and wrote the experimental methods section. P.P. conducted the experiments. H.J. conducted statistical examinations and analyzed the results. L.S. analyzed the results. All authors reviewed and commented on the manuscript. Research reported in this publication was supported in part by the National Cancer Institute (R21CA193064) and the National Institute of Biomedical Imaging and Bioengineering (DP2EB024245) of the National Institutes of Health. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. P.B. thanks Drs. Anjana Rao (La Jolla Institute for Allergy and Immunology, La Jolla, CA, USA); Michael Kershaw (Peter MacCallum Cancer Centre, Melbourne, Australia); Dario Campana (National University of Singapore, Singapore); and Sean Yu (Epoch Life Sciences, Missouri City, TX, USA) for helpful discussions.
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Conflict of Interest
The authors declare no conflict of interest.
Contributor Information
Harold Javitz, Education Division, SRI International, Menlo Park, CA 94025, USA.
Lidia Sambucetti, Biosciences Division, SRI International, Menlo Park, CA 94025, USA,.
Parijat Bhatnagar, Biosciences Division, SRI International, Menlo Park, CA 94025, USA, Parijat.Bhatnagar@sri.com; Cancer Imaging and Early Detection Program, Stanford Cancer Institute, Stanford, CA 94305, USA.
References
- [1].Middleton J, Patterson AM, Gardner L, Schmutz C, Ashton BA, Blood 2002, 100, 3853. [DOI] [PubMed] [Google Scholar]
- [2].Liu B, Chen W, Evavold BD, Zhu C, Cell 2014, 157, 357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Denizot F, Wilson A, Battye F, Berke G, Shortman K, Proc. Natl. Acad. Sci. USA 1986, 83, 6089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Kar P, Nelson C, Parekh AB, Curr. Biol 2012, 22, 242. [DOI] [PubMed] [Google Scholar]
- [5].Zhang L, Morgan RA, Beane JD, Zheng ZL, Dudley ME, Kassim SH, Nahvi AV, Ngo LT, Sherry RM, Phan GQ, Hughes MS, Kammula US, Feldman SA, Toomey MA, Kerkar SP, Restifo NP, Yang JC, Rosenberg SA, Clin. Cancer Res 2015, 21, 2278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Roybal KT, Williams JZ, Morsut L, Rupp LJ, Kolinko I, Choe JH, Walker WJ, McNally KA, Lim WA, Cell 2016, 167, 419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].a) Sass PM, Nicolaides N, Grasso L, Routhier E, Gu W, Young J, Yao J, WO/2012/054654, A61K 39/395 (2006.01), C07K 16/30 (2006.01), 2012; [Google Scholar]; b) Ebel W, Routhier EL, Foley B, Jacob S, McDonough JM, Patel RK, Turchin HA, Chao Q, Kline JB, Old LJ, Phillips MD, Nicolaides NC, Sass PM, Grasso L, Cancer Immun 2007, 7, 6. [PMC free article] [PubMed] [Google Scholar]
- [8].a) Ma J, Tang WK, Esser L, Pastan I, Xia D, J. Biol. Chem 2012, 287, 33123; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Hassan R, Ebel W, Routhier EL, Patel R, Kline JB, Zhang J, Chao Q, Jacob S, Turchin H, Gibbs L, Phillips MD, Mudali S, Iacobuzio-Donahue C, Jaffee EM, Moreno M, Pastan I, Sass PM, Nicolaides NC, Grasso L, Cancer Immun 2007, 7, 20. [PMC free article] [PubMed] [Google Scholar]
- [9].a) Cheung A, Bax HJ, Josephs DH, Ilieva KM, Pellizzari G, Opzoomer J, Bloomfield J, Fittall M, Grigoriadis A, Figini M, Canevari S, Spicer JF, Tutt AN, Karagiannis SN, Onco-target 2016, 7, 52553; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Hassan R, Ho M, Eur. J. Cancer 2008, 44, 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].a) Fiering S, Northrop JP, Nolan GP, Mattila PS, Crabtree GR, Herzenberg LA, Genes Dev 1990, 4, 1823; [DOI] [PubMed] [Google Scholar]; b) Müller MR, Rao A, Nat. Rev. Immunol 2010, 10, 645; [DOI] [PubMed] [Google Scholar]; c) Okamura H, Aramburu J, García-Rodríguez C, Viola JPB, Raghavan A, Tahiliani M, Zhang X, Qin J, Hogan PG, Rao A, Mol. Cell 2000, 6, 539; [DOI] [PubMed] [Google Scholar]; d) Hogan PG, Chen L, Nardone J, Rao A, Genes Dev 2003, 17, 2205. [DOI] [PubMed] [Google Scholar]
- [11].a) Kershaw MH, Westwood JA, Darcy PK, Nat. Rev. Cancer 2013, 13, 525; [DOI] [PubMed] [Google Scholar]; b) Campana D, Schwarz H, Imai C, Cancer J 2014, 20, 134; [DOI] [PubMed] [Google Scholar]; c) Imai C, Mihara K, Andreansky M, Nicholson IC, Pui CH, Geiger TL, Campana D, Leukemia 2004, 18, 676. [DOI] [PubMed] [Google Scholar]
- [12].a) Weiss A, Koretzky G, Schatzman RC, Kadlecek T, Proc. Natl. Acad. Sci. USA 1991, 88, 5484; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Weiss A, Littman DR, Cell 1994, 76, 263. [DOI] [PubMed] [Google Scholar]
- [13].Raman R, Miret J, Scalia F, Casablancas A, Lecina M, Cairo JJ, J. Biotechnol 2016, 239, 57. [DOI] [PubMed] [Google Scholar]
- [14].Fischbach MA, Bluestone JA, Lim WA, Sci. Transl. Med 2013, 5, 179ps7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].a) Kemmer C, Gitzinger M, Daoud-El Baba M, Djonov V, Stelling J, Fussenegger M, Nat. Biotechnol 2010, 28, 355; [DOI] [PubMed] [Google Scholar]; b) Saxena P, Heng BC, Bai P, Folcher M, Zulewski H, Fussenegger M, Nat. Commun 2016, 7, 14; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Wroblewska L, Kitada T, Endo K, Siciliano V, Stillo B, Saito H, Weiss R, Nat. Biotechnol 2015, 33, 839; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Culler SJ, Hoff KG, Smolke CD, Science 2010, 330, 1251; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Xie MQ, Haellman V, Fussenegger M, Curr. Opin. Biotechnol 2016, 40, 139. [DOI] [PubMed] [Google Scholar]
- [16].Leonard JP, Sherman ML, Fisher GL, Buchanan LJ, Larsen G, Atkins MB, Sosman JA, Dutcher JP, Vogelzang NJ, Ryan JL, Blood 1997, 90, 2541. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.