

Abstract
From early unicellular organisms that formed in salty water environments to complex organisms that live on land away from water, cells have had to protect a homeostatic internal environment favorable to the biochemical reactions necessary for life. In this chapter, we will outline what steps were necessary to conserve the water within our cells and how mechanisms have evolved to maintain and regulate our cellular and organismal volume. We will first examine whole body water homeostasis and the relationship between kidney function, regulation of blood pressure, and blood filtration in the process of producing urine. We will then discuss how the composition of the lipid-rich bilayer affects its permeability to water and salts, and how the cell uses this differential to drive physiological and biochemical cellular functions. The capacity to maintain cell volume is vital to epithelial transport, neurotransmission, cell cycle, apoptosis, and cell migration. Finally, we will wrap up the chapter by discussing in some detail specific channels, cotransporters, and exchangers that have evolved to facilitate the movement of cations and anions otherwise unable to cross the lipid-rich bilayer and that are involved in maintaining or regulating cell volume.
1. Introduction
The conventional theory of how life began some 3.8 billion years ago involves a “primordial soup” of inanimate matter, present in the oceans of early Earth, that upon addition of external energy in the form of lightning or ultraviolet light caused simple molecules to combine into more complex compounds (Haldane, 1929). Haldane further postulated that these organic compounds accumulated to concentrations sufficient to produce macromolecules and ultimately fully fledged cells. However, the ionizing UV radiation required to stimulate these early chemical reactions would inherently destroy as much as it creates, indicating that a “surface” primordial soup would not be the best environment for the emergence of life.
Volcanic deep submarine hydrothermal vents discovered in the late 1970s provided another potential source of primordial life-promoting energy (Corliss, et al., 1979). However, their extreme temperatures, low pH, and short lifespans made them problematic as sites for the origin of life (Bada, 2004; Orgel, 2008). Russell and colleagues proposed in 1993 that emergence of life in the depths of the oceans would require warm alkaline hydrothermal fluids rich in dissolved hydrogen and light hydrocarbons such as methane and acetate (Russell, Daniel & Hall, 1993; Russell, Daniel, Hall & Sherringham, 1994). The discovery of a second type of hydrothermal vent in 2000 (Kelley, et al., 2001) produced by the geochemical process of olivine hydroxylated to serpentine provided just such an alkaline hydrothermal environment (Bach, et al., 2006; Sleep, Meibom, Fridriksson, Coleman & Bird, 2004). These “first cells” were most likely autotrophs that derived their energy needs and carbon from the chemical reduction of CO2 with H2 electrons (Lane & Martin, 2012; Martin & Russell, 2007; Russell & Martin, 2004). Indeed, the hydrogenation of carbon dioxide to form methane or acetate via the reductive acetyl CoA pathway is present in the most ancient prokaryotes (Shock & Schulte, 1998).
Seawater contains in mequiv/L: 540 Cl−, 460 Na+, 50 Mg2+, 30 SO42−, 10 Ca2+, and 10 K+. Thus, the first biochemical reactions occurred in a salt-rich environment. Primordial cells isolated themselves from this “salty” outside environment by creating a lamellar phospholipid bilayer matrix to protect their biological processes. Depending on the molecular structure, water content, pH, ionic strength, temperature, and pressure, this lipid-rich barrier allowed these cells to keep an inside environment that is conducive to protein synthesis and enzymatically driven biochemical reactions (Daniel, Oger & Winter, 2006). This basic cellular architecture has remained intact all throughout evolution, from simple unicellular to complex multicellular organisms. As unicellular cells combined and evolved into specific organ systems within multicellular organisms, this lipid-rich membrane acquired additional components and properties, such as a membrane potential that cells have espoused to communicate between themselves, and/or an apical-basolateral polarity that cells utilize for trans-membrane solute transport.
By mass, water is the most abundant molecule inside a cell and consequently, the volume of water constitutes the major parameter in the volume of a cell. The water content of animal tissues obtained by drying ranges from 70% to 80% (Cook, Cramer & Kenyon, 1952), which corresponds to some 3 kg of water per kg of dry weight (3/(3+1) = 75%). The water content of the human body is a bit lower (60%), due to the lower water content of bones (30%). As animals have transitioned from aquatic ponds, rivers, lakes, and oceans to terrestrial landmasses of earth, they had to find ways to conserve this water (Figure 1A). In the next section, we will examine whole body water homeostasis, a critical function of all creatures, especially terrestrial species subjected to continuous water loss. In section 3, we will discuss cell volume homeostasis. Despite a relatively stable internal environment in the bodies of terrestrial species, individual cells have retained throughout evolution mechanisms that allow them to maintain and regulate their volume.
Figure 1. Terrestrial species evolved from creatures that originated in the ocean.

A. As life invaded land, it carried with it some of the ocean water. The drawing was taken from the Delpire laboratory website at Vanderbilt University Medical center: https://www.vumc.org/delpire-lab/. B. In humans, and most terrestrial species, water is constantly lost through perspiration/evaporation from the skin, exhalation from the lungs, urination from the bladder, and feces from the colon. To maintain water homeostasis (in = out), an equivalent amount of water needs to be consumed through drinking and food.
2. Whole body water homeostasis
In 1885, the Belgian physiologist Léon Fredericq noted that as you climb the evolutionary ladder, the number of regulatory mechanisms became greater and more complex and tended to free organisms from harmful influences and changes of their environment (Fredericq, 1885). He noted that for invertebrates, this independence to the external milieu was only relative. Experiments showing a lower salt content in the blood of fresh water versus seawater crustaceans supported this observation, whereas “the blood from sea water fish was not saltier to the taste than the blood of fresh water fish”. Our reason to mention salt in the first paragraph of this section is that salt and water are tightly interconnected.
The vast majority (70–75%) of water in the human body is intracellular or located within cells. The remaining 25–30% is extracellular water, with 1/5 found in plasma and 4/5 found in the interstitial space. As a terrestrial creature, humans lose water through urine (1.5 liters), feces (0.1 liters), respiration and perspiration (0.5 – 0.8 liters). To maintain water balance, this loss is countered by water intake through eating and drinking (Figure 1B). How exactly does the body sense the loss of water? Specialized “sensors” in the vasculature and other organs detect a decrease in extracellular volume as well as an increase in the plasma osmolarity.
2.1. Blood Pressure
Blood pressure refers to the pressure of blood in the large arteries of the circulatory system. Cardiac output, arterial stiffness, and peripheral resistance all influence the body’s blood pressure. One can simplify the circulatory system by considering it as a “closed” circuit consisting of a pump (heart) and pipes (arteries and veins). The pressure that exists in the system depends upon the cardiac output, vascular resistance, and the amount of liquid in the system. The reason that “closed” is written with quotations is because unlike the non-permeable plumbing in our house, blood vessels are able to exchange fluid, gases, and solutes with the interstitial milieu. Moreover, the circulatory system is in contact with the outside through lung, intestinal tract, and kidney endothelia and epithelia, and as such, has the capacity for environmental exchange of fluids, gases, and solutes. Also, note that in contrast to the non-permeable solid pipes in our house, the vessels in our body can also contract or dilate and therefore changes in vascular wall tension will modify the volume of the “closed” circuit.
2.2. The kidney and blood pressure
The human kidney is central to fluid and salt homeostasis. Each kidney is composed of around one million nephrons, each independently operating as a filtration, reabsorption, and secretion unit. Individual nephrons adjust blood composition and form urine beginning with the glomerulus (filtration unit), followed by proximal convoluted tubule, loop of Henle, distal convoluted tubule (reabsorption units), and the common collecting duct (secretion unit). Numerous collecting ducts merge into the renal pelvis that ultimately becomes the ureter connecting the kidneys to the bladder. Unfiltered blood enters the glomerulus via afferent arterioles where small molecular weight substances pass through fenestrations in glomerular capillaries and filtration slits formed by the foot processes of podocyte cells wrapped around these capillaries. Larger molecular weight proteins remain in the plasma and exit the glomerulus via efferent arterioles. The principal role of the renal tubule is to reabsorb water and key solutes (e.g. sugars, amino acids, and ions) while eliminating waste products and toxins in the urine. In the opening paragraph, we indicated that salt and water were interconnected and they directly affected whole body water homeostasis. While the bulk of Na+ reabsorption occurs in early segments (e.g. proximal tubule and thick ascending limb), finely tuned reabsorption occurs more distally by the sodium-chloride cotransporter (NCC) in the distal convoluted tubule and the epithelial Na+ channel (ENaC) in the connecting segment and the cortical collecting duct. Because NCC and ENaC are the last mechanisms participating in Na+ reabsorption, they each play critical roles in fine-tuning plasma Na+ levels and systemic blood pressure.
Multiple observations support the role of NCC and ENaC functions in regulating blood pressure. First, thiazides and amiloride are compounds that inhibit with great specificity NCC and ENaC, respectively (Garty & Benos, 1988; Stokes, Lee & D’Amico, 1984). These drugs are widely used in clinical medicine, often in combination, to treat volume expansion (e.g. congestive heart failure) and high blood pressure (Padilla, Armas-Hernández, Hernández, Israili & Valasco, 2007). Second, loss-of-function mutations in NCC results in Gitelman’s syndrome, a renal salt wasting disorder with many patients suffering from arterial hypotension (Simon, et al., 1996), whereas loss-of function-mutations in ENaC results in pseudohypoaldosteronism type I (PHA-I), a disease that also involves the loss of salts leading to hyponatremia (Chang, et al., 1996). The disease is called pseudohypoaldosteronism, because it mimics the absence of aldosterone production (or hypo-aldosteronism). Aldosterone is a key hormone produced by the adrenal gland that enhances Na+ reabsorption through NCC and ENaC in the distal tubule. Absence of aldosterone causes urinary Na+ wasting, leading to volume depletion and hypotension (Merriam & Baer, 1980). In contrast, there are genetic conditions that lead to increased salt reabsorption, volume retention, and high blood pressure. Liddle syndrome is due to a mutation in ENaC that prevents its recycling from the plasma membrane and degradation (Schild, et al., 1996; Tamura, et al., 1996). The consequence of this lack of recycling is increased channel density at the plasma membrane and increased ENaC function. Type II pseudohypoaldosteronism (PHA-II) is caused by mutations in two “with no lysine kinases” (WNK1 or WNK4) leading to increased phosphorylation and activity of NCC (Wilson, et al., 2001). Mutations in Kelch-3 or CUL3, genes products that are part of the ubiquitination and degradation of NCC, also manifest as PHA-II (Boyden, et al., 2012).
In summary, decreased NCC or ENaC function results in volume depletion and hypotension, whereas increased NCC or ENaC function lead to volume retention and hypertension. In the preceding paragraph, we focused on inhibitors and genetic disorders that affect salt transport and blood pressure. In a normal situation, however, how exactly does the body regulate “physiological” changes in blood volume?
2.3. Hypovolemia and Tubuloglomerular feedback mechanism.
Afferent and efferent arterioles carry blood into and out of the glomerulus, respectively. Hypovolemia, or low blood volume, leads to a decrease in glomerular filtration, which in turn, leads to decreased flow of Cl− at the transition between the end portion of the loop of Henle and the beginning of the distal convoluted tubule. Specialized epithelial cells (macula densa) in this transition zone sense the reduced flow of Cl− and send signals to the adjacent afferent arteriole to vasodilate and increase glomerular filtration. In addition, the decreased flow of Cl− stimulates the release of renin (Schnermann & Briggs, 2013) from other specialized cells that are located in the wall of the afferent arteriole (Figure 2). Renin, in turn, stimulates the conversion of plasma angiotensinogen to angiotensin II, which is a potent vasoconstrictor. Thus, angiotensin II leads to increased vascular resistance and increased blood pressure. In addition to its vasoconstrictive effect, angiotensin II activates the release of the mineralocorticoid hormone aldosterone from the adrenal gland. Aldosterone as we discussed before stimulates salt and water reabsorption in the distal portion of the nephron.
Figure 2. The kidney nephron is a rheostat for plasma volume.
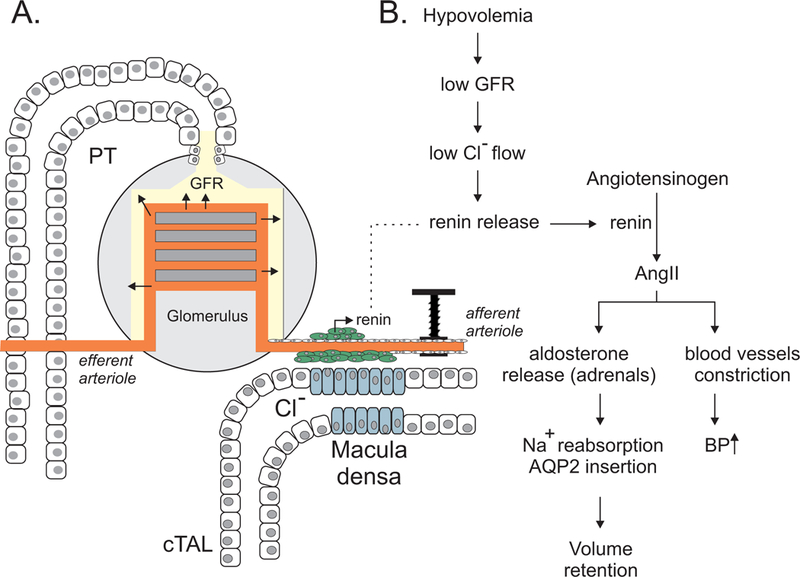
A, The ultrafiltration of blood into urine that begins in the glomerulus depends upon the wall pressure of afferent and efferent blood vessels. The rate of filtration at the glomerulus is one component that defines the amount of Cl− that flows at the macula densa. Macula densa cells serve as “sensors” of the amount of Cl−-containing filtrate that passes along the cortical thick ascending limb (cTAL). These cells relay information to specialized renin cells located in the wall of the afferent arteriole to release renin when necessary. B, Low blood volume (hypovolemia) ultimately results in renin release that acts on the renin-angiotensin-aldosterone system. Angiotensin II and aldosterone restore blood volume (and blood pressure) by affecting Na+ and water reabsorption (increased AQP2 insertion) and constriction of blood vessels.
2.4. Urinary concentrating mechanism
The human body, like the bodies of all mammals and birds, concentrates its urine. This important function stems from the fact that the body needs to eliminate the excess salts it consumes, but not at the expense of losing too much water. The combined activity of the apical Na-K-2Cl cotransporter, NKCC2, and basolateral Na+/K+ ATPase in the thick ascending limb of Henle (TALH) moves Na+ from the tubular fluid to the renal interstitium (Castrop & Schießl, 2014). Because the TALH is impermeable to water, this transport of Na+ contributes to a hyperosmotic interstitium. The juxtaposition of descending limb (permeable to water but not ions) and ascending limb (pumping ions and impermeable to water) creates a countercurrent multiplier mechanism that maintains high osmolarity in the medulla, while keeping normal the osmolarity in the cortex (Gottschalk & Mylle, 1958; Gottschalk & Mylle, 1959; Hargitay & Kuhn, 1951). Comparative physiologists have observed that in species that typically survive with minimal water and produce a very concentrated urine, the majority of their nephrons possess very long loops of Henle (Jamison, 1987). Along with Na+, urea is another key osmolyte that contributes to a hyperosmotic medullary interstitium (Sands, 2004). The urea cycle converts ammonia (NH3), a very toxic compound, into urea, a water-soluble non-toxic compound easily excreted. Some of the urea present in urine is re-absorbed in the inner medullary collecting ducts thereby raising the osmolarity of the medullary interstitium. Anti-diuretic hormone, also called vasopressin or arginine vasopressin (AVP), controls this re-absorptive mechanism (Nielsen & Knepper, 1993 3586).
2.5. Circumventricular Organ, hypothalamus and vasopressin
The vascular organ of lamina terminalis (VOLT) and the subfornical organ are highly vascularized circumventricular organs that sense changes as small as 1% (3 mOsmol/L) in plasma osmolarity. They project to hypothalamic neurons that produce AVP when increased osmolarity is detected (Prager-Khoutorsky, 2017). They also send signals to the cortex where conscious water craving is processed. AVP produced in the somata of magnocellular neurosecretory cells (located in the hypothalamic supraoptic (SON) and paraventricular nuclei) then travels down the axons of these neurons towards the posterior pituitary and is released into the circulation. AVP has multiple sites of action. The primary role of AVP is to increase the number of water channels (aquaporin 2) on the apical membrane of collecting duct principal cells, leading to increased water reabsorption and conservation. Second, AVP also increases the number of urea transporters located in the inner medullary collecting duct, increasing interstitial osmolarity and the capacity to concentrate urine, thereby also contributing to water conservation. Third, AVP stimulates Na+ reabsorption in the TALH, adding to the countercurrent multiplication phenomenon further helping in water conservation. Fourth, AVP constricts blood vessels, leading to increase in arterial blood pressure. Thus, in conditions of dehydration when plasma osmolarity increases, release of AVP into the circulation results in several water conservation mechanisms to maintain whole body water homeostasis.
2.6. Back to the relationship between Na+ and water
Arthur Guyton proposed that under normal (i.e. steady-state) conditions, dietary Na+ increase is matched by urinary Na+ excretion with no net consequences on volume and blood pressure (Guyton, 1987). However, if the individual is unable to secrete the excess dietary Na+, extracellular fluid volume will expand leading to an increase in blood pressure (Guyton, 1987). Natriuresis lowers the sodium concentration in the blood by excreting sodium in the urine. Osmotic forces draw water out of the body’s blood circulation decreasing blood volume and restoring blood pressure to normal values. As stated by Alan Pao “high blood pressure is adaptive from the standpoint of maintaining Na+ homeostasis, but this adaptation comes at the price of exposing end organs to the damaging effects of high blood pressure (Pao, 2014). Recent studies indicates that Na+ and water are not always linked, as Na+ can be stored in organs without increasing body fluid content (Kopp, et al., 2013). Furthermore, evidence has shown that the body can vary its Na+ content while maintaining constant its water content (Rakova, et al., 2017). It is indeed clear than in healthy individuals, the kidney concentrating mechanism accommodates the disposal of any extra dietary Na+, without the loss of any additional water.
3. Cell Volume Homeostasis
The activities that exist within a cell are intimately linked to the spatial organization of internal macromolecular structures. In this respect, any change to the volume of a cell potentially risks major disorganization of these structures and consequently perturbation of the many key activities of the cell.
The composition of the lipid bilayer greatly influences the movement of water and ions across the plasma membrane. Proteins embedded in the membrane provide pathways for the movement of water (e.g. aquaporins or water channels) and ions (e.g. ion channels and transporters). Even in the absence of these integral membrane proteins, the plasma membrane still possesses some intrinsic water permeability. Asymmetry between the nature and composition of the lipids between the inner and outer leaflets of the bilayer has a significant effect on this water permeability (Hill & Zeidel, 2000; Krylov, Pohl, Zeidel & Hill, 2001). Indeed, membranes containing sphingolipids, glycosphingolipids, phospholipids, and cholesterol have much lower water permeabilities (3.4 × 10−4 cm/s) than membranes containing only phospholipids and cholesterol (34.4 × 10−4 cm/s).
Irrespective of its lipid composition, the permeability of a membrane to water will always be greater than the membrane permeability to ions and other solutes. Under these conditions, the cell is subjected to osmotic pressure differences that arise between the outside environment and the interior of the cell. If we exclude bacteria and plants that have a rigid membrane, all other cells have flexible membranes and thus undergo significant changes in cell volume proportional to the intensity of the osmotic gradient. Before we consider the behavior of cells under anisosmotic conditions, we will examine the challenge of cells in maintaining their volume under isosmotic conditions.
3.1. Isosmotic cell volume maintenance
Cells can grow and change their size while still maintaining their water content constant. In this section, we will not be considering cell growth, but instead, the volume of a cell occupied by water. Cells contain in their cytoplasm macromolecules that are negatively charged. The presence of these macromolecules creates a colloido-osmotic disequilibrium first described by British chemist Frederick G. Donnan in 1911 (Donnan, 1911). While some ionic species can cross cellular membranes, larger anionic proteins are generally not permeable. These larger anions attract smaller cations while simultaneously repelling smaller anions creating an uneven electric potential called the Donnan potential.
Consider the following two simple scenarios: in the first scenario, consider a system composed of two compartments separated by a membrane that is permeable to small cations (Na+, and K+) but impermeable to large anions (A−) (Figure 3A). If we add NaA and KA salts to these compartments, respectively, and the salt concentration is different between the two compartments, the dissociative ions will re-distribute to create a new equilibrium (Figure 3B-C). At equilibrium, the equations in Box 1 describe the work represented by the movement of Na+ and K+.
Figure 3. Physicochemical system of two compartments separated by a membrane permeable only to small cations.
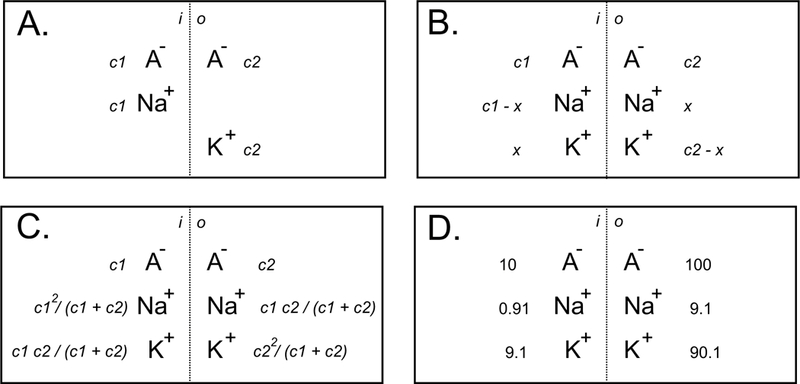
A, Starting conditions with c1 and c2 concentrations of NaA and KA salts in their respective compartments. B, Conditions after salt dissociation and cationic redistribution (impermeant anions remained in their respective compartments). C, Concentrations obtained after x has been solved. D, Example of ionic redistribution when starting with a 10-fold gradient difference between NaA and KA.
Box 1. Donnan Equilibrium for scenario 1 of Figure 3.
| {1} |
| {2} |
| {3} |
| {4} |
Which leads at equilibrium to the concentrations written in Figure 3C.
One can play with the numbers and dissolve 10 mM NaA in the first compartment and 100 mM KA in the second compartment and calculate x to be 9.1 mM. At equilibrium, most of the Na+ (91%) will have traveled to the second compartment, while only 9.1% of the K+ will have moved to the first compartment (Figure 3D). Thus, very different ion distributions across the membrane can exist due to the presence of the large impermeable anions. However, equal concentrations would be reached if either the system starts with equal concentrations of the large anion, or the anion can freely cross the membrane.
In a second scenario, consider a similar system composed of two compartments separated by a semi-permeable membrane where the large macromolecules are present only in the first compartment (called “i” for internal) with K+ at a concentration of c1 and NaCl is only present in the second compartment (called “e” for external) at a concentration of c2 (Figure 4A). At equilibrium, the amount of Cl−, K+ and Na+ that moved are x, z, and y, respectively (Figure 4B). At equilibrium, the values will be as shown in Figure 4C. If we examine the case of c1 = c2, one can calculate that [Cl−]e = 2 [Cl−]i, [K+]i = 2 [K+]o, and [Na+]i = 2 [Na+]o. Note that the majority of the Na+, which started on the outside is now on the inside, while Cl− is mostly prevented to move to the inside compartment. The calculated osmolalities will be 250 mOsM inside versus 200 mOsM outside, if the non-diffusible anions are osmotically inactive (i.e. large negatively charged macromolecules). If the non-diffusible charges are osmotically active then the calculated osmolalities will be 400 mOsM versus 200 mOsM. Thus, this scenario creates an osmotic gradient or “Gibbs-Donnan disequilibrium” that creates a driving force for the movement of water into the internal compartment.
Figure 4. Physicochemical system closer to the situation of a cell.

A, Impermeant anions (A−) were placed in one compartment with K+, NaCl salt was placed in the other compartment. B, Conditions after salt dissociation and ionic redistribution. C, Concentrations obtained after x, y, and z have been solved. D, Example of resulting osmolality differential between the two compartments after ionic redistribution when 150 mM NaCl and KA salts were placed in their respective compartments.
Why is this physicochemical system relevant to a cell? As mentioned earlier, the membrane of a cell is permeable to water and far less permeable to ions. The cells also contain in their cytoplasm many macromolecules with anionic charges. Indeed, the majority of cytosolic proteins have amino and carboxyl moieties that are negatively charged at physiological pH (Gianazza & Righetti, 1980). Similarly, nucleic acids have an acidic isoelectric point making them also carry negative charges at physiological pH (Sherbet, Lakshmi & Cajone, 1983). Thus, cells observe all the requirements for Gibbs-Donnan disequilibrium and they are under pressure to swell. The reason they maintain their volume is because they use cellular energy to fuel the Na+/K+-pump to constantly fight the leak of ions and the influx of water (Kay, 2017; Tosteson & Hoffman, 1960). The pump basically renders the membrane “impermeable” to Na+ by actively extruding the Na+ that enters the cell through leak pathways. The presence of non-diffusible anionic charges in cells is likely a key factor in the measured low intracellular Cl− concentration in most cells, including central neurons which have the lowest intracellular Cl− concentrations (Delpire & Staley, 2014; Glykys, et al., 2014).
3.1.2. Epithelial Transport
Epithelia are specialized barriers that keep compartments separate within the body. The compartments can be both internal (e.g. choroid plexus separating the blood from CNS) or external (e.g. airway, intestinal, bladder epithelia separating the blood from the outside). In addition to forming a barrier, epithelial cells actively participate to the movement of solutes from one side to the other. This requires coordinated movement at both poles (apical and basolateral) of the membrane. Since transport is a dynamic process, any disruption at one membrane will create a disruption that will last until the second membrane responds proportionally. As water obligatorily follows the movement of solutes, any disruption in one membrane only will tend to disrupt cell volume.
As an example, we will consider the choroid plexus, a cuboidal epithelium that participates to CSF fluid secretion and K+ reabsorption (Johanson, et al., 2008). As shown in Figure 5A, cells isolated with mild collagenase treatment dissociate and keep their polarity with an apical pole of light density due to brush borders (white arrows), and a rough basolateral pole (black arrows) (Wu, Delpire, Hebert & Strange, 1998). Staining with anti-NKCC1 antibody reveals staining on the apical membrane only (Figure 5B) – consistent with staining in intact tissue (Plotkin, et al., 1997). When isolated cells were exposed to bumetanide as a NKCC1-specific inhibitor (see Figure 5E), they demonstrated cell volume reduction, consistent with ion fluxes on the apical membrane not being balanced by ion movement on the basolateral side. If, however, the entry of salts and water on the apical pole is not balanced by a reduction of efflux on the basolateral pole, the net loss of ions and water will result in cell shrinkage. As the effect of bumetanide is reversible, when the inhibitor was removed, the cells started to regain their volume (Figure 5E). The addition of bumetanide to the choroid plexus epithelial cells was a manipulation that disrupted the balance of transport between the two membranes. Under normal conditions, if an epithelium uptakes solute on the basolateral side, and extrudes them on the apical side, it just facilitates their trans-epithelial movement while keeping in check (homeostasis) the internal milieu. This is the case for Cl− secreting epithelia (e.g. salivary gland, sweat gland, stomach), K+ secreting epithelia (inner ear), and Na+ reabsorbing epithelia (intestine, distal convoluted tubule, collecting duct).
Figure 5. Ion and water transport in choroid plexus.
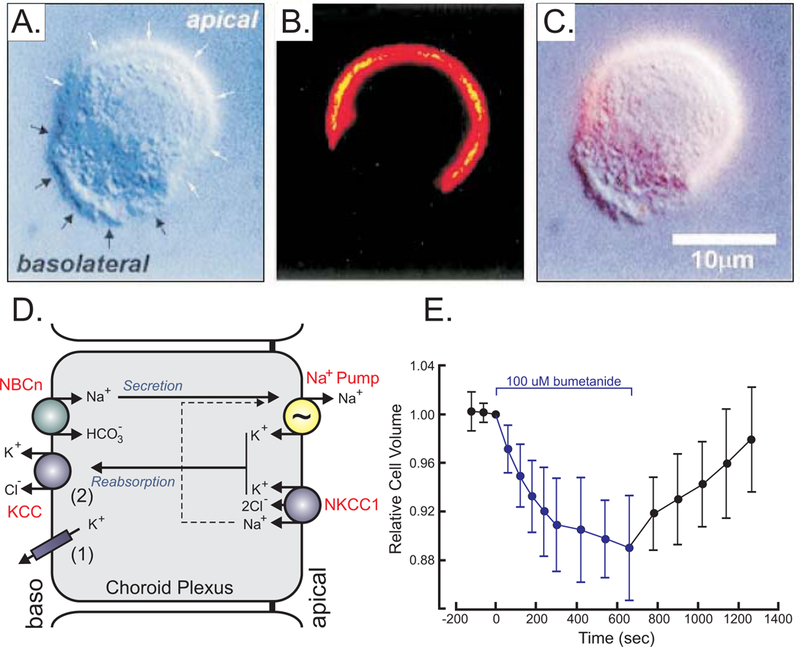
A, Mouse choroid plexus epithelial cell observed by Differential interference contrast (DIC) microscopy with white arrows identifying the apical and black arrows identifying the basolateral membrane, respectively. B, Fluorescent microscopy image of NKCC1 staining of the same cell using a secondary Cy3-conjugated antibody targeting a primary anti-NKCC1 antibody. C, Overlay of A & B showing presence of NKCC1 in apical membrane. D, Model of choroid plexus epithelial cell with apical Na+/K+-ATPase (pump) and NKCC1 and basolateral Na-bicarbonate cotransporter (NBCn) and K+ efflux mechanisms (KCC and K+ channels). E, Effect of bumetanide on relative cell volume, as measured by microscopy. Materials were taken with permission from (Wu, Delpire, Hebert & Strange, 1998).
3.1.3. Neurotransmission
Charges move in and out of a neuron during synaptic transmission. Glutamate-induced excitation involves the movement of Na+ and Ca2+ into a neuron. An action potential involves the entrance of Na+, followed by the exit of K+. GABA (γ-aminobutyric acid)-mediated inhibition involves the inward movement of Cl− in central neurons (Rivera, et al., 1999; Zhu, Lovinger & Delpire, 2005) or outward movement of Cl− in the terminals of peripheral sensory fibers (Sung, Kirby, McDonald, Lovinger & Delpire, 2000). Compared to the amount of charges (ions) existing in and out of cells, the amount of ions moving during these processes is relatively small and thus, the ion concentrations are not likely to be affected that much. If for instance during an action potential the membrane of a cell moves from −70 mV to +40 mV due to the inward movement of Na+, this translates to a 110 mV difference. The charge associated with that change can be calculated to be 6,875 ions per μm2. If we hypothetically consider a round cell with a diameter of 10 μm, the calculated surface area is approximately 314 μm2 and the amount of ions would be 6,875 × 314 = 2.16 × 106 ions. If the intracellular Na+ concentration of a neuron is 10 mM, the cytoplasm of the cell contains 3.2 × 109 Na+. Adding 2.6 × 106 Na+ ions is equivalent to adding 0.08% to the total, which is negligible. This calculation is based on the overall volume of a cell, and not micro-domains like dendritic spines which can be as small as 0.01 μm3 or 2 × 105 smaller than a 10 μm cell. In addition, a cell or a dendrite can experience repeated stimulation which could conceivably alter the ion concentrations. Using gramicidin-perforated patch, we demonstrated that long (6 seconds) application of GABA or repeated applications of GABA to a neuron in culture could lead to measurable changes in the Cl− concentration (Zhu, Lovinger & Delpire, 2005). These changes if not regulated, are likely to lead to changes in cell volume.
3.1.4. Cell Cycle
Cell volume is a major determinant of the timing of cell division in unicellular algae like Chlamydomonas (Craigie & Cavalier-Smith, 1982). The algae grow to a critical volume of 140 μm3, which seems to be the factor that triggers cell division. In mammalian cells, cell volume changes during the different phases of the cell cycle. When Ehrlich Ascites tumor cells are synchronized, their average mean corpuscular cell volume increases during the S phase and plateau in the G2 phase (DuPre & Hempling, 1978). When mitosis is initiated the volume decreases significantly and this cell volume reduction continues for several hours after the mitosis phase has been completed (DuPre & Hempling, 1978). Interestingly the volume change seems to be related to an increase in osmotically inactive cell volume (Figure 6) that we define as “b” or the intercept on the Y axis of the relationship between volume ratios over osmotic pressure ratios (see below).
Figure 6. Cell volume changes with cell cycle phases.
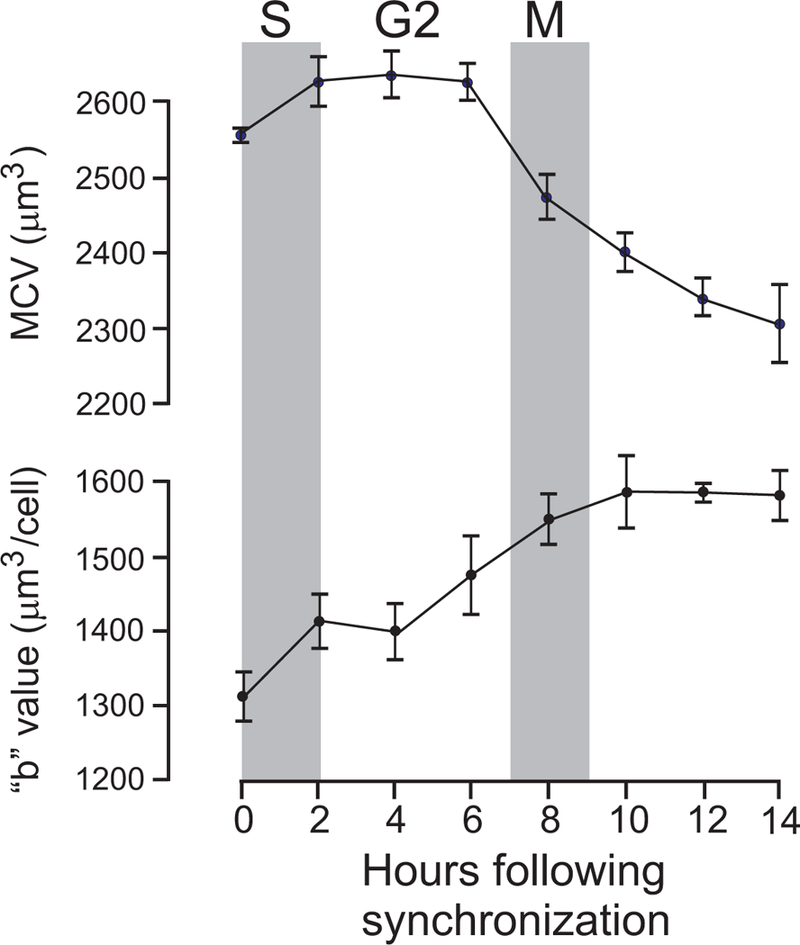
Measurement of mean corpuscular volume (MCV) and osmotically-inactive volume “b” in synchronized Ehrlich ascites tumor cells maintained in suspension culture during S, G2, and M phases of the cell cycle. Figure re-drawn (DuPre & Hempling, 1978).
3.1.5. Apoptosis
Cells die through the distinct processes of necrosis or apoptosis. In necrotic cell death, cells originally undergo ATP depletion, cell swelling, increased membrane permeability and lysis (Nieminen, 2003). In contrast, apoptotic cell death is associated with isosmotic cell shrinkage (Kerr, Wyllie & Currie, 1972). As K+ is the major osmolytes in the cell, loss of K+ is a major component of apoptotic cell volume decrease (Bortner & Cidlowski, 2007; Bortner, Hughes & Cidlowski, 1997). When mouse and human lymphoma cells are exposed to dexamethasone, A23187, thaspigargin, or staurosporine, they demonstrate a significant reduction in cell volume that is accompanied by a reduction in K+ content. While the Na+ content of a cell is much lower than that of K+, there is also a significant reduction in Na+ content (Bortner, Hughes & Cidlowski, 1997). Note that increase in Na+ (in contrast to decrease) has also been observed in apoptotic cells (Yurinskaya, Rubashkin & Vereninov, 2011). It is important to stress that loss of K+ by itself is not a factor in the decision of a cell to undergo apoptotic cell death. Indeed, cells exposed to hypotonic solution, lose a sizable fraction of their K+ content, but do not undergo apoptotic cell death. That being said, the K+ loss is an important component of apoptosis, as cells induced to undergo apoptosis failed to follow through when they were exposed to high external K+ environments (Bortner, Hughes & Cidlowski, 1997).
Interestingly, apoptosis can also be triggered by exposing cells to hypertonic cell shrinkage. Their ability to proceed through regulatory volume decrease (RVI) then determines their resistance to apoptosis (Bortner & Cidlowski, 2007; Numata, Sato, Okada & Wehner, 2008). In 1985, we published two papers showing that rat pheochromocytoma (PC12) cells exposed to hypertonicity demonstrated condensation of chromatin (a hallmark of apoptotic cells (Kerr, Wyllie & Currie, 1972)), ruffling of the nuclear envelope with loosening of condensed chromatin from the lamina, and apparent loss of nucleolar fibrillar component which disappears in a background of diffuse granular material (Delpire, Duchêne, Gilles & Goessens, 1985; Delpire, Duchêne, Goessens & Gilles, 1985). These cells were unable to undergo RVI (Delpire, 1989). We did not observe the formation of small cytoplasmic fragments with or without pyknotic remnants of nuclei, indicating that if the cells were on the path to apoptosis, they were captured in the very early stages (Delpire, Duchêne, Gilles & Goessens, 1985; Delpire, Duchêne, Goessens & Gilles, 1985). Several studies have attempted to model apoptotic cell volume decrease (Model, 2014; Yurinskaya, Rubashkin & Vereninov, 2011). Whether the loss of soluble inorganic (K+ and Cl−) and organic (taurine, etc.) osmolytes is enough to account for the extent of shrinkage during apoptosis is still a matter of debate. The participation of non-soluble negative (Donnan) charges and osmotically-inactive water molecules might also need to be considered (Model, 2014).
3.1.6. Cell migration
Cell migration is a key component in the formation and maintenance of multicellular organisms. While this phenomenon occurs predominantly during development, it does continue to occur throughout life, as it is associated with processes such as inflammation and tissue repair. Unfortunately, cell migration is also largely facilitated by malignant cancer cells invading neighboring tissues. Cell migration involves the formation of membrane protrusions such as lamellipodia and membrane ruffles at the leading edge of the cell. These protrusions or localized cell swelling require the rapid movement of ions and water into the protrusion (Condeelis, 1993; Jakab & Ritter, 2006; Lauffenburger & Horwitz, 1996). Alan Verkman (Verkman, 2005) hypothesized that “actin cleavage and ion uptake at the tip of the lamellipodium could create local osmotic gradients that drive the influx of water” across the membrane at the leading edge. Water channels have also been shown to be essential for T cell function and macrophage migration (Rump & Adamzik, 2018). In addition, aortic endothelial cells isolated from aquaporin-1 knockout mice demonstrated a deficit in cell migration (Saadoun, Papadopoulos, Hara-Chikuma & Verkman, 2005). Interestingly, this deficit could be rescued by expressing non-endothelial cell aquaporin 1 or unrelated aquaporin-4 into these aortic endothelial cells (Saadoun, Papadopoulos, Hara-Chikuma & Verkman, 2005). Aquaporin inhibition is currently being investigated in animal models to reduce metastatic cancer cell invasion (Simone, et al., 2018) or in culture to prevent cell migration (Chen, et al., 2017). The ion transport mechanisms at play in the swelling of the leading edge are multiple. They include the Na+/H+ and Cl−/HCO3 exchangers (Schwab, 2001), and the Na-K-2Cl cotransporter (Schwab, Wojnowski, Gabriel & Oberleithner, 1994). Interestingly, these same mechanisms are involved in RVI (see below). Cell migration also involves cell retraction at the trailing edge of the cell (Jakab & Ritter, 2006). Transport mechanisms involved in regulatory volume decrease or RVD are thought to be at play for this process. This is the case of the K-Cl cotransporter (KCC) (Gagnon, 2012; Shen, Chou & Ellory, 2000; Shen, et al., 2004), the volume activated Cl− channel (Ransom, O’Neal & Sontheimer, 2001; Soroceanu, Manning & Sontheimer, 1999), as well as K+ channels (Soroceanu, Manning & Sontheimer, 1999).
3.2. Anisosmotic Cell Volume Regulation
In the previous section we considered the movement of water and changes to cell volume under conditions of constant external osmotic pressure. In this section, we will consider the behavior of cells following osmotic challenges.
3.2.1. The osmotic equilibration phase
When two solutions of different osmotic pressures are separated by a semi-permeable membrane, water will diffuse from the least concentrated solution to the more concentrated solution in order to equilibrate the pressures. Such a physical system was first described in 1748 by Jean-Antoine Nollet, a catholic priest, who used a pig bladder to close a vial containing spirit of wine that he placed in water. After several hours, he observed a bulged membrane that he pierced with a pin to witness a column of water shooting out (Nollet, 1748). The term osmosis (or endosmosis) was later introduced by French physiologist Henri Dutrochet (Dutrochet, 1826). In his book, Dutrochet relates experiments done with chicken intestines filled with different liquid contents (milk, egg white, etc.) placed in buckets filled with rain water. In each case, the weight of the intestinal bag increased over time (see box 2). He called this process endosmosis as the water moved from the bucket into the intestinal bag. He was also clever enough to repeat the experiment in reverse by placing the intestinal bag in buckets containing higher solute concentrations and observe exosmosis. Similar experiments were later performed by the German botanist, Wilhelm Pfeffer (Pfeffer, 1877). He created a device (the Pfeffer Cell) that measured the osmotic pressure of a solution. Many studies in the latter part of the 19th century and early part of the 20th century described swelling and shrinking of cells, both vegetal and animal, when exposed to hypotonic or hypertonic solutions, respectively (de Vries, 1884; de Vries, 1888; Gough, 1924; Koeppe, 1895; Nägeli, 1855; Overton, 1907; Pringsheim, 1854).
Box 2. Experiment performed by Dutrochet and described on pages 123–124 of his book.
Je pris in coecum de poulet qui, dans l’état de distenstion, avait 12 millimètres de largeur, et qui, courbe en arc, avait 10 centimètres de longeur. Je le remplis à peu près à moitié avec de l’albumen. Dans cet état, il pesait 58 grains. Le coecum fut plongé dand de l’eau de pluie... Huit heures et demi après le commencement de l’expérience, le coecum pesait 130 grains. Il avait gagné 72 grains, et était devenu tres-turgide… Il me fut pleinement demontré que l’introduction de l’eau dand la cavité organique dépendait entièrement de la nature du fluide, plus dense que l’eau que contenait cette cavité. Tant que ce fluide conservait son integrité de composition, l’endosmose avait lieu.
Translation: “I took a chicken caecum which when distended measured 12 mm in diameter and 10 cm in length. I filled it half with egg white. In this state, it weighed 3.08 g (1 grain = 53.114 mg). The caecum was placed in rain water… Eight and a half hour after the beginning of the experiment, the caecum weighed 6.9 g. It had gained 3.8 g and became very turgid… It was fully demonstrated to me that the introduction of water in the cavity depended on the nature of the fluid, more dense than water existing in this cavity. As long as this fluid kept its integrity [these experiments had tendency to lead to putrefaction], endosmosis occurred.”
The Dutch chemist Jacobus H. van’t Hoff, the recipient of the first (1901) Nobel Prize in chemistry, postulated that solutes in dilute solutions obey the ideal gas laws, which led to the osmotic pressure formula π = nRT/V where R is the gas constant and T the absolute temperature.
The definition of the osmotically inactive volume (called “b”) in Figure 7 depends upon the method of cell volume measurement. If the cell volume measurement does not involve the measurement of cell water, b is the combination of the volume occupied by “dry” (or non-water) material added to the volume of water that does not participate in intracellular osmolality. If, however, the amount of water represents cell volume, b then represents the osmotically inactive water content. The ratio of osmotically active water to total water is sometimes referred to as Ponder’s R factor (Ponder, 1948). In the muscle fiber of the giant barnacle, Balanus nubilis, solvent water was estimated to be 64–72% of total water (Hinke, 1970). Osmotically inactive water is believed to be associated or bound to macromolecules. Note that if experiments with cells are performed with an infinite extracellular volume, in vivo, the extracellular space is often limited and tends to buffer volume changes. Let us consider a rigid system with two compartments designated in and out. At equilibrium, each compartment has an osmotic pressure π0. If we modify the osmotic pressure of the “out” compartment (π0 becomes π1), a new equilibrium will develop leading to new volumes in’ and out’ with a new osmotic pressure π’.
Figure 7. Relationship between cell volume and osmolarity.
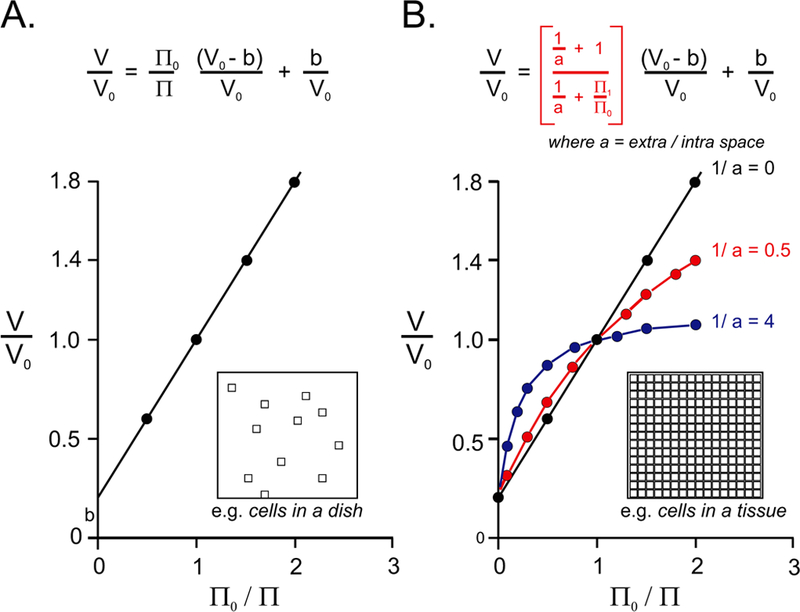
A, Linear relationship of volume ratio divided by osmotic ratio is based on modified ideal gas law for dilute solutions. Note that the slope is always less than 1 and the intercept on the Y axis has a positive value “b”. B, Same relationship but considering the ratio of extracellular versus intracellular space (called “a”). Note that as “a” decreases (or 1/a increases), the relationship deviates from a linear relationship and volume changes are minimized or buffered. The insets visually represents cells in an infinite volume (in A) compared to cells packed in a tissue (in B).
If the extracellular volume (out) is infinite, 1/a is equal to 0 and equation 11 becomes equation 7 (straight line in Figure 7B). If, however the ratio of extracellular to intracellular volume is small, the relationship is no longer linear and extracellular space plays the role of a buffer. Figure 7B shows the relationship for intracellular to extracellular ratios of 0.5 and 4. Thus, in vivo, a confined extracellular volume buffers the effect of osmotic pressure changes. Note that in the mathematical model described in Box 4 above, we postulated that the sum of the “in” and “out” compartments was constant, which is not entirely accurate as tissues and organs are not fully rigid.
Box 4. Role of the extracellular space in the relationship between cell volume and osmolality.
First, we can stipulate in + out = in’ + out’
in’ = in . π0/π’
out = out . π1/π’
| {8} |
| {9} |
| {10} |
Inserting equation 10 into equation 8, we obtain:
| {11} |
where a = extracellular space / intracellular space
The behavior of isolated cells varies from cell type to cell type. While some cells at the peak response closely follow the van’t Hoff linear relationship (Dellasega & Grantham, 1973; Olson & Holtzman, 1982), others (e.g. chicken lymphocytes, Hela cells, and murine hepatocytes) demonstrate resistance to swelling during large hypotonic shocks (similar to channel rectification) (Doljanski, 1974; Howard & Wondergem, 1987; Tivey, Simmons & Aiton, 1985). This likely indicates some participation of the cytoskeleton in creating resistance to cell swelling.
3.2.2. Cell Response to Hypotonicity
When cells are exposed to hypotonic conditions, their swelling induces the activation of separate K+ and Cl− channels, leading to a loss of K+, Cl−, and obligatory water, leading the restoration of cell volume (Figure 8) (Hoffmann, Lambert & Simonsen, 1986; Hoffmann, Simonsen & Lambert, 1984). Because the conductive Cl− permeability of cells is typically low, an increase in in conductive K+ permeability alone cannot account for an observed net loss of KCI during RVD, an increase in conductive Cl− permeability must also occur. We will now discuss the participation of K+ channels and Cl− channels in regulatory volume decrease.
Figure 8. Behavior of mammalian cells exposed to a change in osmolalities from 300 mOsM to 240 mOsM.

A, separation between the osmotic (swelling) phase and the regulatory phase. As water enters the cell to equilibrate osmotic pressure, the cell swells. The loss of osmolytes such as K+, Cl−, and taurine lead to water loss and cell volume recovery. Note that the inside of the cell after RVD is at 240 mOsM. B, Example of a cell (PC12 cells) undergoing cell swelling and regulatory volume decrease upon exposure to hypotonic conditions. Close symbols are cells exposed to hypotonic conditions, open symbols are cells remaining under isosmotic conditions. Redrawn from (Delpire, 1989; Delpire, Cornet & Gilles, 1991).
A. Volume-activated K+ channels
Early work has shown that in Ehrlich ascites tumor cells, the K+ loss that is activated by cell swelling (Hendil & Hoffmann, 1974) is mediated by a Ca2+-dependent K+ channel sensitive to quinine (Hoffmann, Simonsen & Lambert, 1984). In these cells, the loss of K+ exceeds the loss of Cl− loss by a factor of about 1.6 (Hendil & Hoffmann, 1974). Interestingly, while the permeability of the cells to Cl− increases some 60-fold, the permeability of a cell for K+ increases only 2-fold upon cell swelling (Hoffmann & Simonsen, 1989). This indicates that the Cl− permeability is likely to be a greater limiting factor to RVD than the K+ permeability (Hendil & Hoffmann, 1974). The conductance of the Ca2+-activated K+ channel in Ehrlich cells was measured under normal external K+ concentration at 7 pS (Christensen & Hoffmann, 1992). Arachidonic acid metabolites such as leukotriene D4 (LTD4) have been shown to enhance KCl loss and RVD following hypotonic shock (Jorgensen, Lambert & Hoffmann, 1996). While there is a concomitant increase in intracellular Ca2+, the LTD4 effect seemed to be independent of the transient rise in the divalent cation. The K+ current activated by cell swelling was shown to be insensitive to charybdotoxin as well as to apamin, inhibitors of calcium-activated voltage-gated shaker K+ channels and small conductance SK channels, respectively (Riquelme, Sepúlveda, Jørgensen, Pedersen & Hoffmann, 1998). In contrast, clofilium, a class III antiarrhythmic drug blocked the volume-sensitive K+ current in a voltage independent manner and acted as a strong inhibitor of the regulatory volume decrease response of Ehrlich cells (Niemeyer, et al., 2000). Because the volume-sensitive K+ channel is pH-sensitive and has a small ion conductance, TASK-1 and TASK-2 channels expressed in Ehrlich cells (Niemeyer, et al., 2000) were proposed as candidates (Hougaard, Jørgensen & Hoffmann, 2001). The TASK-2 channel was later shown to be a major component of the K+ conductance activated by cell swelling (Kirkegaard, Lambert, Gammeltoft & Hoffmann, 2010; Kirkegaard, Strøm, Gammeltoft, Hansen & Hoffmann, 2016). KCNQ4, but not KCNQ2/3, were also shown to be involved in regulatory volume decrease in HEK293 cells overexpressing these channels (Hougaard, Klaerke, Hoffmann, Olesen & Jorgensen, 2004).
B. Volume-activated Cl− channels
Work from Else Hoffmann’s group in Ehrlich Ascites tumor cells and of Sergio Grinstein and colleagues in human lymphocytes showed activation of a Cl− conductance during RVD (Grinstein, Clarke, Dupre & Rothstein, 1982; Grinstein, Clarke, Rothstein & Gelfand, 1983; Hoffmann, Simonsen & Sjøholm, 1979). Because ionophores, which produce increases in K+ permeability far greater than a hypotonic shock, failed to produce any changes in cell volume, Grinstein argued that the anion conductance was the rate-limiting factor in salt and water transport (Grinstein, Clarke, Dupre & Rothstein, 1982). However, because the gramicidin ionophore greatly accelerated RVD in Ehrlich cells, Hoffmann argued that the K+ conductance was also limiting (Hoffmann, Lambert & Simonsen, 1986). Thus, it can be concluded that both conductance are low under isotonic conditions, and the movement of one ion through one conductance is limited unless there is movement of the oppositely charged ion in the other conductance. In Ehrlich cells the Cl− conductance was described as unselective carrying CI−, Br−, NO3−, and SCN− (Hoffmann, Lambert & Simonsen, 1986), while in lymphocytes the channel had the following anion series: SCN− = I− > NO3− > Br− >= Cl− > acetate− > SO42− = gluconate−.
In 1983, Else Hoffman and Ian Lambert demonstrated that Ehrlich Ascites tumor cells exposed to a hypotonic solution lose significant amount of amino acids (Hoffmann & Lambert, 1983). In these cells, taurine (53 mM) is definitely the most abundant amino acid, followed by glycine (9 mM), and other amino acids at 1–2 mM each. During a decrease in osmotic pressure from 300 mOsM to 225, the vast majority (90%) of taurine is lost, whereas only 30% of glycine is lost, making taurine the most significant organic osmolytes in cells. The permeability (loss) of taurine is proportional to the amount of cell swelling (Hoffmann & Lambert, 1983). Ten years later, the authors demonstrated that the swelling-induced activation of the taurine leak involved arachidonic acid and leukotrienes, as did the activation of K+ and Cl− conductances (Lambert & Hoffmann, 1993). The authors indicated for the first time the possibility that the taurine efflux was directly linked to the activation of the Cl− channels. While the Cl− channel blocker MK196 inhibited both Cl− efflux and taurine efflux pathways, the fact that 4,4’-Diisothiocyano-2,2’-stilbenedisulfonic acid (DIDS) minimally affected Cl− efflux in their cells, prompted them to later conclude that the swelling-activated Cl− channel and the taurine channel in the Ehrlich cells represented two distinct types of channels (Hoffmann & Lambert, 1994; Lambert & Hoffmann, 1994).
Demonstration that amino acid efflux and Cl− efflux were mediated by one single channel was made by Kevin Strange (Jackson & Strange, 1993; Strange, Morrison, Shrode & Putnam, 1992). They demonstrated that Ptaurine/PCl was ~ 0.2 meaning that the channel was preferentially permeable to Cl−, but the permeability to taurine was not negligible. They called the channel Volume Sensitive Organic Anion Channel or VSOAC. In C6 cells, the channel has a unitary conductance of 35–40 pS (Jackson, Morrison & Strange, 1994). Many attempts have been made to identify the molecular makup of this functional channel unit. Proteins like P-glycoprotein (Gill, et al., 1992), ICln (Paulmichl, et al., 1992), CLC2 (Gründer, Thiemann, Pusch & Jentsch, 1992), and ClC3 (Duan, Winter, Cowley, Hume & Horowitz, 1997) were all initially considered as candidates. Following these first attempts at identifying the Cl− channels involved in RVD, came the demonstration that TMEM16, a Ca2+-activated Cl− channel is active during apoptosis and cell swelling (Martins, et al., 2011). Jurkat cells activated by Fas ligand express an outwardly rectifying Cl− current, sensitive to NPPB (5-Nitro-2-(3-phenylpropylamino) benzoic acid) and to the anoctamin-blocker CaCCinh-AO1 that was completely abrogated when TMEM16F was knocked down. Outwardly rectifying Cl− currents, which were NPPB-, DIDS-, and CaCCinh-AO1-sensitive, were also eliminated by TMEM16F siRNA in airway epithelial cells (Martins, et al., 2011). The authors demonstrated that regulatory volume decrease after hypotonic cell swelling was augmented in HEK293 cells overexpressing TMEM16F. Another Ca2+-activated Cl− channel, drosophila Bestrophin-1 (or dBest1) was proposed to mediate volume-stimulated Cl− currents. Knockdown of dBest 1 in Drosophila S2 cells abolished volume-regulated Cl− currents and significantly reduced regulatory volume decrease (Chien & Hartzell, 2007).
It took another few years for the molecular identification of the volume-activated Cl− channel associated with amino acid efflux to be revealed in two landmark papers (Qiu, et al., 2014; Voss, et al., 2014). In the two papers, the authors used an RNA interference screen and swelling-induced iodine uptake and quenching of a yellow fluorescent protein, to identify LRRC8A (Leucine Rich Repeat Containing 8 VRAC Subunit A). The first paper showed that knockdown of LRRC8A protein strongly suppressed ICl-swell in follow-up patch clamping experiments. When LRRC8A was overexpressed in HEK293 cells, the ICl-swell current decreased, but when LRRC8A and LRRC8C were co-transfected, a significant increase in ICl-swell current was observed. These data indicated that multiple subunits were needed to form a functional channel that traffics properly to the plasma membrane (Voss, et al., 2014). The authors also demonstrated that cas9-mediated inactivation of LRRC8A resulted in cells that could no longer efflux taurine upon hypotonic treatment. The specific participation of TMEMs, bestrophins, and LRRC8 channels to chloride loss during RVD will still need to be individually resolved from cell type to cell type. Whether these channels also regulate cell volume under isosmotic conditions is currently unknown.
C. K-Cl cotransporters
A volume- and N-ethylmaleimide-stimulated Cl−-dependent K+ flux was first described in human and sheep red blood cells (Dunham & Ellory, 1981; Dunham, Steward & Ellory, 1980; Lauf & Theg, 1980). This Cl−-dependent K+ transport was attributed to a K-Cl transport mechanism (now K-Cl cotransporter). Coincidentally, the function of a Na-K-2Cl cotransport mechanism was also described the same year (see below and (Geck, Pietrzyk, Burckhardt, Pfeiffer & Heinz, 1980)). The red blood cell K-Cl cotransporter is functionally silent under control conditions (Lauf & Theg, 1980) and activated by suflhydryl reagents as well as cell swelling. In his 1980 paper, Peter Lauf indicated that red blood cell precursors had 10 fold higher K+ flux than mature red blood cells (Lauf & Theg, 1980). Similarly, reticulocytes which are larger young red blood cells, demonstrate greater Cl−-sensitive K+ fluxes than mature erythrocytes (Lauf, 1983). The cells are thus able to reduce their volume by losing K+, Cl−, and water (Lauf & J., 1987). K-Cl cotransport, through a still unknown signaling mechanism, is activated in red blood cell from sickle cell patients, contributing to the dehydration and sickling process (Berkowitz & Orringer, 1987; Brugnara, 1989; Quarmyne, Risinger, Linkugel, Frazier & Joiner, 2011).
When healthy red blood cells are exposed to a hypotonic solution, they rapidly swell and then slowly recover their volume. This behavior was first demonstrated in erythrocytes isolated from Pleuronectes flesus, although this study investigated the role of amino acids loss and not the role of ions (Fugelli, 1967). It was later shown in duck red blood cells, that a loss of K+ was associated with the “volume regulatory phase” (Kregenow, 1971), and this loss of K+ was Cl−-dependent in marine teleost Opsanus tau erytrhrocytes (Lauf, 1982). KCC activation has also been shown in rat (Haas & Harrison, 1989), rabbit (Brugnara, Van Ha & Tosteson, 1989; Jennings & Schultz, 1990), dog (Fujise, et al., 1991; Parker, Colclasure & McManus, 1991), sheep (Bergh, Kelly & Dunham, 1990), pig (Kim, Sergeant, Forte, Sohn & Im, 1989) and human (Brugnara, Van Ha & Tosteson, 1989; Dunham & Ellory, 1981; Kaji, 1986; Sachs & Martin, 1993) erythrocytes.
Molecular cloning in the late 1990s led to the identification of four genes (SLC12A4–7) in mammals that encode four K-Cl cotransporters: KCC1–4 (Gillen, Brill, Payne & Forbush, 1996; Hiki, et al., 1999; Mount, et al., 1999; Payne, Stevenson & Donaldson, 1996; Race, et al., 1999). Interestingly, only KCC2 displays some basal transport activity under isotonic conditions (Mercado, Broumand, Zandi-Nejad, Enck & Mount, 2006). Activation of all the other K-Cl cotransporters could only be demonstrated following cell swelling. After observing a lag phase of activation following cell swelling, Michael Jennings proposed in 1990 a two-state kinetic model, where KCC is inactivated by the action of a protein kinase and activated by the action of a phosphatase (Jennings & al-Rohil, 1990). The model and the data indicated that cell swelling inhibited a kinase with no effect on the conjugate phosphatase thereby resulting in an increase in the rate of dephosphorylation concurrent with a decrease in the rate of phosphorylation (Figure 9). While we always assume that swelling is the factor that leads to KCC activation (because we utilize this manipulation to activate the transporter), any physiological factor that decreases transporter phosphorylation or increases transporter de-phosphorylation will lead to an active K-Cl cotransporter.
Figure 9. Diagram showing the two-state model of K-Cl cotransport activation/inactivation.
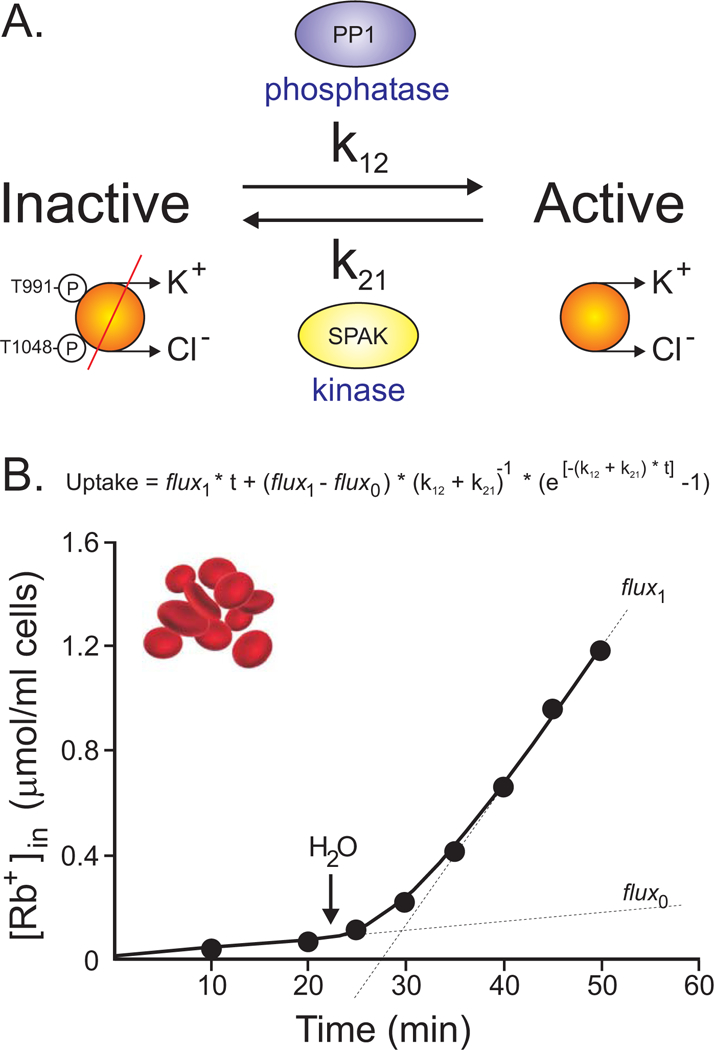
A, A protein phosphatase (PP1) mediates a de-phosphorylation (activation) reaction of specific threonine residues (T991 & T1048) with a rate constant of k12, whereas a kinase (SPAK) mediates the opposite phosphorylation (inhibition) reaction of these same threonine residues with a different rate constant (k21). B, Measurement of 86Rb uptake versus time in rabbit red blood cells illustrates a lag phase occurs in the stimulation of the flux from flux0 to flux1. Water is added at arrow to decrease the medium’s osmolarity. Figure adapted/redrawn from (Jennings & al-Rohil, 1990).
A role for KCC3 in cell volume maintenance of peripheral nerve fibers was evidenced by genetic disruptions of the human SLC12A6 gene. Loss of function mutations in the gene are found in a few sporadic families throughout the world (Uyanik, et al., 2006) as well as in many Canadian families living in two regions around Quebec City (Howard, et al., 2002). Axonal swelling was found in KCC3-deficient patients (Auer, et al., 2016; Dupré, et al., 2003), consistent with this cotransporter isoform playing a role in regulatory volume decrease. Analysis of peripheral nerves in a knockout mouse model also found significant swelling of peripheral nerve fibers (Byun & Delpire, 2007). Interestingly, a single individual that carries a gain of function mutation in KCC3 was also identified (Kahle, et al., 2016). The mutated residue (Thr991) is one of the two key threonine (Thr991 and Thr1048), involved in shutting off KCC3 function (Figure 9). Mutation of Thr991 into an alanine prevents phosphorylation of the residue, leading to a constitutively active transporter (Rinehart, et al., 2009). Our laboratory also created a KCC3-T991A knock-in mouse (Kahle, et al., 2016) and demonstrated the shrinkage of peripheral nerve fibers (Flores, Schornack & Delpire, 2018). Thus, when KCC3 function is abolished the nerve fibers are swollen, and when KCC3 function is enhanced, the nerve fibers are shrunken. These data clearly establish a key role for KCC3 in cell volume maintenance.
There are two recently published cell volume experiments that provide puzzling data worth discussing in this section. A 2015 paper reported the behavior of HEK293 cells overexpressing a human KCC3a T991A-T1048A double mutant protein (Adragna, et al., 2015). Cell volume was measured using a well-established microscopic method based on fluorescent dye dilution (Blanco, Márquez & Alvarez-Leefmans, 2013; Lenart, Kintner, Shull & Sun, 2004). When wild-type HEK293 cells were exposed to a large 150 mOsM/Kg water osmotic shock, the cells increased their volume 3–4 times, then recovered their volume with a slope of 12% cell water volume recovery per min. When HEK293 cells overexpressing KCC3-T991A-T1048A were exposed to the same hypotonic shock, cell swelling was significantly attenuated (up to 2-fold) and the rate of recovery was also dampened (4% recovery/min). The authors did not attempt to decrease the extent of the osmotic shock to match the swelling. Such an experiment would have determined if the rate of regulatory volume decrease would have matched that of the mutant cells. The significant reduction in peak swelling observed in the KCC3-T991A-T1048A expressing cells was totally unexpected. Because the cells might have changed their phenotype (e.g. attachment to substrate), furosemide was also added to inhibit the overactive cotransporter. When treated acutely with furosemide, the cells expressing the constitutively active transporter showed significantly increased swelling (3-fold) followed by minimal cell volume recovery. Note that native HEK293 cells treated with furosemide also showed a small increase in peak swelling. These data clearly indicated that the activity of the transporter was responsible in preventing cell swelling.
Note that the data presented in that paper did not comply with the van’t Hoff relationship. If the osmotically inactive volume of the cells is negligible (or equal to zero), the van’t Hoff relationship passes through the graph origin (X = 0 and Y = 0), and at best, a 300 mOsM to 150 mOsM (2 fold) osmotic shock should lead to a 2 fold increase in cell volume. Why was a 4-fold increase in cell volume, measured with wild-type HEK293 cells? This error could be related to the methodology that overestimated the changes in cell volume. Irrespective of this discrepancy with the van’ Hoff relationship, the fact remains that cells overexpressing the mutant KCC3 did not swell as much as wild-type cells. This could only be explained by cells dumping K+, Cl−, and water during the swelling phase. In contrast to a wild-type transporter which demonstrates a lag-phase of activation upon swelling (see above), the mutant transporter is already fully active when the swelling occurs. The data therefore suggest that with an active KCC3 transporter, the membrane can no longer be regarded as semi-permeable, i.e. far more permeable to water than solutes.
This unusual behavior was also observed with human fibroblasts isolated from the patient carrying the single T991A mutation in KCC3 (Kahle, et al., 2016). Fibroblasts from the patient as well as control human fibroblasts were exposed to the same hypotonic shock and cell volume was measured using the same methodology. In fact, the experiments were performed in the same laboratory. Wild-type fibroblasts increased their volume by 2.5 fold, whereas the patient fibroblasts swelled only marginally (Kahle, et al., 2016). No regulatory volume decrease was observed in the wild-type cells.
Along with red blood cells and peripheral nerve fibers, KCC has been implicated in cell volume regulation in mouse sperm (Klein, Cooper & Yeung, 2006), and glial cells (Ringel & Plesnila, 2008). Interestingly, as with nerve fibers, specific inhibition of KCCs caused glial cell swelling under isotonic conditions indicating that either the cotransporter is active in vivo under isotonic conditions, or that the cotransporter responds to small increases in cell volume. Cell volume increase could be due to Donnan forces (see above) or to cell transport activities that leads to small water intake and swelling. In this regard, a study published in 1986 provides evidence that cells can maintain their volume constant under anisosmotic conditions provided that the changes in osmolarities are slow and gradual. Indeed, Lohr and Grantham exposed kidney tubules to a solution with an osmolarity that changed from 293 mOsM/Kg to 110 mOsM/Kg at a rate of 1.5 mOsM decrease per min. They observe no changes in cell volume over a large range of osmolarities (293 – 170 mOsM) (Lohr & Grantham, 1986). They named the portion of the study in which cell volume did not change as the osmolality decreased as the isovolumetric regulation phase (Lohr & Grantham, 1986). They determined that cells that did remain isovolumetric during a 100 mOsM change had lost significant amount of osmolytes, as these cells shrank abruptly when re-exposed to isosmotic conditions. A similar isovolumetric behavior was also observed in the direction of increased osmolarities.
3.2.3. Cell Response to Hypertonicity
A. Short term – Regulatory Volume Increase
When cells are exposed to a hypertonic solution, they first lose water to equilibrate internal and external osmotic pressures (Figure 10A). Following this initial cell volume reduction, some cells undergo a regulatory volume increase process (called RVI) to restore their volume, while other cells do not volume regulate and remain shrunken (Figure 10B, Box 5). Several transport mechanisms, that include the Na+/H+ exchanger and the Na-K-2Cl cotransporter, have been implicated in RVI. If the cells remain in the hypertonic environment, they will adapt by accumulating organic osmolytes. This will be covered in more detail in the long-term accumulation of organic osmolytes section.
Figure 10. Behavior of mammalian cells exposed to changes in osmolality.
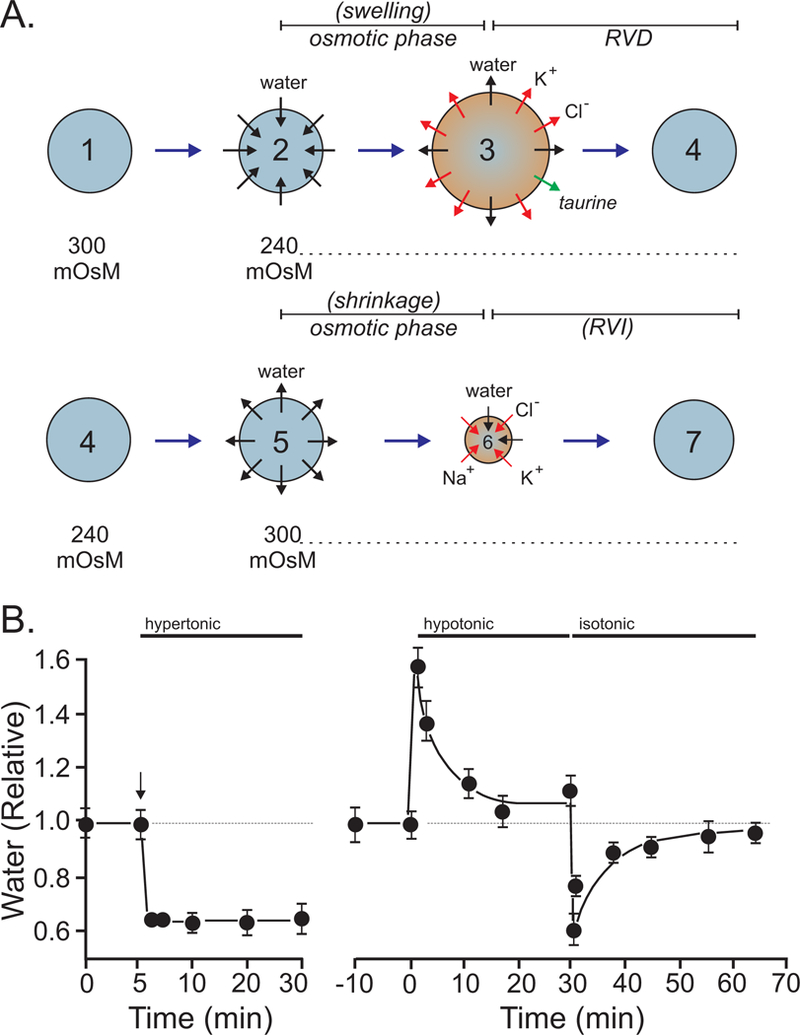
A, After the hypotonic (240 mOsM) swelling (2 to 3) and regulatory volume decrease (3 to 4) phases, the cells are re-exposed to isotonic (300 mOsM) conditions. They then lose water and shrink (5 to 6). This treatment triggers a secondary regulatory volume increase (6 to 7). B, Example of a cell (PC12 cells) undergoing cell shrinkage with no volume recovery when exposed to hypertonic conditions. Upon return to isotonic conditions after regulatory volume decrease, the same cells now demonstrate post-RVD RVI. Redrawn from (Delpire, 1989).
Box 5. Behavior of different cell types when exposed to a hypertonic solution.
| Cell Type | RVI – Yes | NKCC-mediated | RVI – No |
|---|---|---|---|
| Guinea pig jejunal villus cells | (MacLeod & Hamilton, 1990) | Yes | |
| Duck red blood cells | (Schmidt & McManus, 1977) | Yes | |
| Ehrlich Ascites cells | (Geck & Pfeiffer, 1985) | Yes | |
| Ehrlich Ascites cells | (Hendil & Hoffmann, 1974) | Yes | |
| (Hoffmann & Lambert, 1983) | |||
| Amphiuma red blood cells | (Cala, 1980) | No | |
| Human lymphoid cells | No | (Roti Roti & Rothstein, 1973) | |
| Human PBM | No | (Grinstein, Clarke & Rothstein, 1983) |
The Na+/H+ and Cl−/HCO3− exchangers
When Amphiuma red blood cells are transferred from a 245 mOsM isosmotic saline to a 320 mOsM hypertonic saline, they first shrink, then undergo regulatory volume increase in response to net cellular uptake of Na+ and Cl− (Cala, 1980). The net influx of Na+ associated with RVI is completely prevented by the addition of 1 mM amiloride (Cala, 1980; Siebens & Kregenow, 1978). Because the accumulation of Na+ is pH-dependent and amiloride-sensitive, a Na+/H+ exchanger is involved. Due to the stimulation of Na+ uptake by high HCO3− medium, it was postulated that Na+/H+ exchange is functionally coupled to Cl−/HCO3− exchange. The net result is accumulation of NaCl and obligatory water (Cala, 1980). Similar transport systems are activated by hypertonicity if dog red blood cells (Parker, 1983), Necturus gallbladder (Ericson & Spring, 1982) and human lymphocytes (Grinstein, Clarke & Rothstein, 1983). In Amphiuma red blood cells, the accumulation of Na+ triggers the exchange of some of that Na+ with K+ through activation of the Na+/K+-ATPase. This phenomenon is prevented by the application of ouabain, while the overall amount of Na+ plus K+ that enters during RVI is ouabain-independent (Siebens & Kregenow, 1985).
The Na-K-2Cl cotransporter
Na-K-2Cl cotransport was functionally defined by Peter Geck in a landmark 1980 paper (Geck, Pietrzyk, Burckhardt, Pfeiffer & Heinz, 1980). The molecular identification was done 14 years later with the cloning of a kidney-specific isoform, NKCC2 (Gamba, et al., 1994; Igarashi, Vanden Heuvel, Payne & Forbush, 1995; Payne & Forbush, 1994), and a more widely-distributed isoform, NKCC1 (Delpire, Rauchman, Beier, Hebert & Gullans, 1994; Payne, et al., 1995; Xu, et al., 1994). The Na-K-2Cl cotransporters are integral transmembrane proteins activated (in opposition to K-Cl cotransporters) by phosphorylation and inactivated by dephosphorylation (Lytle & Forbush, 1992; Palfrey & Pewitt, 1993). Several key regulatory phospho-residues are located in the cytosolic N-terminal of the cotransporter (Darman & Forbush, 2002; Flemmer, Gimenez, Dowd, Darman & Forbush, 2002; Gimenez & Forbush, 2005). The two Ste20p-kinases, SPAK and OSR1, which constitute the germinal center kinase 6 subfamily, were identified as the kinases that bind and phosphorylate the cotransporter (Gagnon & Delpire, 2010; Gagnon, England & Delpire, 2007; Piechotta, Lu & Delpire, 2002). These terminal kinases are themselves under the control of upstream kinases, such as WNKs (Gagnon, England & Delpire, 2006; Moriguchi, et al., 2005; Piechotta, Garbarini, England & Delpire, 2003; Vitari, Deak, Morrice & Alessi, 2005).
Cells that fail to undergo RVI typically do express the Na-K-2Cl cotransporter, but the mechanism is functionally silent. This is evidenced by the ability of these cells to demonstrate RVI after they have been first exposed to hypotonicity and allowed to volume regulate, and then returned to the isosmotic solution (Grinstein, Clarke & Rothstein, 1983; Grinstein, Rothstein, Sarkadi & Gelfand, 1984; Hoffmann, Sjoholm & Simonsen, 1983). This secondary RVI is now possible because of the loss of Cl− that occurred during the RVD, allowing the activation of NKCC1 by the shrinkage induced by the return to the isosmotic solution (Figure 10). Alternatively, the rate of RVI greatly increased in simian eccrine clear cells exposed to a hypertonic solution when cells are co-treated with forskolin (Toyomoto, Knutsen, Soos & Sato, 1997) (Figure 11). This observation can be simply explained by forskolin inducing the opening of Cl− channels (such as CFTR), the loss of intracellular Cl−, and the activation of the WNK-SPAK signaling cascade, leading to stimulation of the Na-K-2Cl cotransport, and ultimately cell volume recovery (Figure 11C). The reverse situation is also true. When eccrine cells were returned to isosmoticity after a hypotonic challenge, they readily underwent secondary RVI. This regulatory volume phase was inhibited with co-treatment with phorbol ester 12-O-Tetradecanoylphorbol-13-acetate (TPA) which activates protein kinase C and inhibits NKCC1 (Figure 11B)(Toyomoto, Knutsen, Soos & Sato, 1997).
Figure 11. Intracellular Cl− and NKCC1 account for RVI in cells.
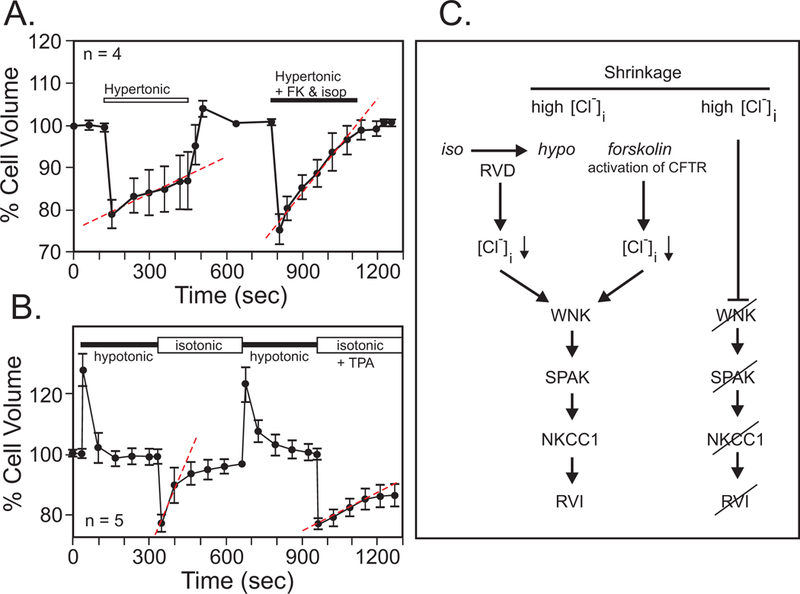
A, Behavior of simian eccrine clear cells exposed to hypertonic solution. Minimal RVI is observed unless the cells were treated with forskolin (FK) and isoproterenol (isop). B, Post-RVD RVI is strong in these cells but inhibited by phorbol ester (TPA). Data were redrawn and modified from (Toyomoto, Knutsen, Soos & Sato, 1997). C, Both RVD and forskolin through activation of Cl− channels cause a loss of intracellular Cl−. Through the activation of the protein kinases WNK and SPAK, this leads to an active NKCC1, which helps the cells recover their volume. Under non-stimulated conditions, the high intracellular Cl− prevents activation of Na-K-2Cl cotransport and the cells remain shrunken.
The Na-K-2Cl cotransporter is often cited as a mechanism for regulatory volume increase (Hoffmann, Lambert & Pedersen, 2009; Hoffmann & Simonsen, 1989) and indeed, its activity seems to be activated by hypertonicity. It is, however, important to note that the transporter in some conditions mediates K+/K+ exchange instead of Na-K-2Cl cotransport (Canessa, Brugnara, Cusi & Tosteson, 1986; Gagnon & Delpire, 2010; Haas, Starke & McManus, 1984; Lauf, et al., 1987; Orlov, Tremblay & Hamet, 1996). This is important because the preferred method to assay “cotransporter” function is unidirectional rubidium (Rb+) uptake, where cold (85Rb) or radiolabeled (86Rb) rubidium is used as a tracer for K+ movement. Many studies have demonstrated activation of Rb+ influx under hypertonicity. This is the case for vascular endothelial cells (Klein & O’Neill, 1995; O’Donnell, Martinez & Sun, 1995), guinea pig jenual villus cells (MacLeod & Hamilton, 1990), HeLa cells (Ikehara, Yamaguschi, Hosokawa & Miyamoto, 1993), turkey erythrocytes (Ueberschär & Bakker-Grunwald, 1985), duck red blood cells (Lytle, 1998), rat vascular smooth muscle cells (Orlov, Tremblay & Hamet, 1996), and osteoblasts (Whisenant, Zhang, Khademazad, Loessberg & Muallem, 1991). Whether increased 86Rb uptake signifies in all cases activation of Na-K-2Cl cotransport is unclear. When NKCC1 mediates Na-K-2Cl cotransport, unidirectional K+ uptake is proportional to the uptake of one Na+ and 2 Cl− ions. The entry of ions is followed by obligatory water movement. If, however, NKCC1 functions in K+/K+ exchange mode, the unidirectional movement of K+ is not accompanied by the movement of Na+, Cl−, and obligatory water. In Xenopus laevis oocytes over-expressing mouse NKCC1 and subjected to hypertonicity, the protein functions preferentially in K+/K+ exchange mode with no net water movement (Delpire & Gagnon, 2011; Gagnon & Delpire, 2010).
While shrinkage itself tends to increase the concentration of ions inside the cells, when cells undergo RVI, they also accumulate salt and water. Because salts are “toxic” to proteins (Arakawa & Timasheff, 1982; Arakawa & Timasheff, 1982; Gekko & Timasheff, 1981; Gekko & Timasheff, 1981), long-term adaptation to high osmolality conditions involves accumulation of organic osmolytes and replacement of high intracellular salts by such neutral compounds.
B. Long term – Accumulation of organic osmolytes
Early studies examining the inner medulla of the kidney demonstrated that mammalian cells accumulate osmolytes when exposed to hypertonic environments. These studies demonstrated that cells which live in a high urea environment accumulate organic osmolytes like trimethylamine (Balaban & Knepper, 1983), glycerylphosphorylcholine and inositol (Bagnasco, Balaban, Fales, Yang & Burg, 1986), sorbitol (Bagnasco, Uchida, Balaban, Kador & Burg, 1987), and amino acids (Law & Turner, 1987). High urea and salts environments perturb the structure of proteins (Arakawa & Timasheff, 1982) and accumulation of organic osmolytes not only allow for respective decrease in intracellular ions, but also for the protection of proteins from denaturation (Arakawa & Timasheff, 1982; Arakawa & Timasheff, 1985; Gekko & Timasheff, 1981; Gekko & Timasheff, 1981). Amino acids also stabilize chromatin from salt denaturation (Buche, et al., 1989). Accumulation of organic osmolytes is the result of uptake through membrane transport mechanisms (e.g. myo-inositol, betaine transporters) as well as synthesis (e.g. sorbitol). Transporters and enzymes are up-regulated at the transcriptional level by hypertonicity, due to the presence of a tonicity-responsive element (TonE) in the promoter of these genes (Ferraris & Burg, 2006; Takenaka, Preston, Kwon & Handler, 1994).
The demonstration that marine organisms accumulate organic osmolytes was first made at the end of the 19th century by Leon Fredericq (1851–1935) at the University of Liège, Belgium, and expanded by work from distinguished physiologists/biochemists such as Marcel Florquin (1900–1979), Ernest Schoffeniels (1927–1992), and Raymond Gilles (1940–2018). The latter 3 individuals spent summers working in marine biology stations both in France (Roscoff and Concarneau in Brittany) and in the United States (Hopkins Marine Biology station in Pacific Grove, CA, and Duke marine biology station in Beaufort, NC). In several seminal papers, they demonstrated that in euryhaline species, or species that are able to adapt to a wide range of salinities, organic osmolytes represent a very sizable portion of the intracellular osmolarity (Gilles, 1979; Gilles, 1987; Gilles & Delpire, 1997). For example, in the muscle of sea water acclimated Callinectes sapidus, amino acids account to 70% of the intracellular osmolality (Gilles, 1979). I (first author) was extremely fortunate to have been taught biochemistry and physiology by Ernest Schoffeniels and Raymond Gilles, respectively. I also completed my graduate work under the mentorship of Raymond Gilles. While I made my career in the United States, I do feel privileged to belong to this lineage of renowned Liège physiologists.
Box 3. Van’Hoff’s cell volume versus osmotic pressure relationship.
| {5} |
If we consider that the osmotically active component of a cell does not occupy the entire volume of a cell, the equation becomes:
| {6} |
If we then divide both terms by the initial volume (v0), one obtains the linear relationship:
| {7} |
The intercept on the Y axis does not cross 0 but results in a positive value that is equivalent to b, the osmotically inactive volume. As the osmolarity of the solution decreases, the volume of the cell increases proportionally (Figure 7A).
Acknowledgments
E. Delpire is funded by NIH grants DK93501, DK110375, GM118944, a Transatlantic Network of Excellence grant from the Leducq Foundation (17CVD05), and by an endowed directorship from Vanderbilt University Medical Center. K.B. Gagnon is supported by the University of Louisville, School of Medicine—Division of Nephrology and Hypertension.
REFERENCES
- Adragna NC, Ravilla NB, Lauf PK, Begum G, Khanna AR, Sun D, & Kahle KT (2015). Regulated phosphorylation of the K-Cl cotransporter KCC3 is a molecular switch of intracellular potassium content and cell volume homeostasis. Front. Cell. Neurosci 9, 255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arakawa T, & Timasheff SN (1982). Preferential interactions of protein with salts in concentrated solutions. Biochemistry 21, 6545–6552. [DOI] [PubMed] [Google Scholar]
- Arakawa T, & Timasheff SN (1982). Stabilization of protein structure by sugars. Biochemistry 21, 6536–6544. [DOI] [PubMed] [Google Scholar]
- Arakawa T, & Timasheff SN (1985). The stabilization of proteins by osmolytes. Biophys. J 47, 411–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auer RN, Laganière JL, Robitaille YO, Richardson J, Dion PA, Rouleau GA, & Shekarabi M (2016). KCC3 axonopathy: neuropathological features in the central and peripheral nervous system. Mod. Pathol 29, 962–976. [DOI] [PubMed] [Google Scholar]
- Bach W, Paulick H, Garrido CJ, Ildefonse B, Meurer WP, & Humphris SE (2006). Unraveling the sequence of serpentinization reactions: petrography, mineral chemistry, and petrophysics of serpentinites from MAR 15N (ODP Leg 209, Site 1274)`. Geophys. Res. Lett 33, L13306. [Google Scholar]
- Bada JL (2004). How life began on Earth: a status report. Earth Planet. Sci. Lett 226, 1–15. [Google Scholar]
- Bagnasco SM, Balaban RS, Fales HM, Yang YM, & Burg MD (1986). Predominant osmotically active organic solutes in rat and rabbit renal medulla. J. Biol. Chem 261, 5872–5877. [PubMed] [Google Scholar]
- Bagnasco SM, Uchida S, Balaban RS, Kador PF, & Burg MD (1987). Induction of aldose reductase and sorbitol in renal inner medullary cells by elevated extracellular NaCl. Proc. Natl. Acad. Sci. U.S.A 84, 1718–1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balaban RS, & Knepper MA (1983). Nitrogen-14 nuclear magnetic resonance spectroscopy of mammalian tissues. In Am. J. Physiol pp. C439–C444. [DOI] [PubMed]
- Bergh C, Kelly SJ, & Dunham PB (1990). K-Cl cotransport in LK sheep erythrocytes: Kinetics of stimulation by cell swelling. J. Membr. Biol 117, 177–188. [DOI] [PubMed] [Google Scholar]
- Berkowitz LR, & Orringer EP (1987). Cell volume regulation in hemoglobin CC and AA erythrocytes. Am. J. Physiol 252, C300–C306. [DOI] [PubMed] [Google Scholar]
- Blanco VM, Márquez MS, & Alvarez-Leefmans FJ (2013). Parallel changes in intracellular water volume and pH induced by NH(3)/NH(4)(+) exposure in single neuroblastoma cells. Cell Physiol. Biochem 32, 57–76. [DOI] [PubMed] [Google Scholar]
- Bortner CD, & Cidlowski JA (2007). Cell shrinkage and monovalent cation fluxes: role in apoptosis. Arch. Biochem. Biophys 462, 176–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bortner CD, Hughes FMJ, & Cidlowski JA (1997). A primary role for K+ and Na+ efflux in the activation of apoptosis. J. Biol. Chem 272, 32436–32442. [DOI] [PubMed] [Google Scholar]
- Boyden LM, Choi M, Choate KA, Nelson-Williams CJ, Farhi A, Toka HR, Tikhonova IR, Bjornson R, Mane SM, Colussi G, Lebel M, Gordon RD, Semmekrot BA, Poujol A, Välimäki MJ, De Ferrari ME, Sanjad SA, Gutkin M, Karet FE, Tucci JR, Stockigt JR, Keppler-Noreuil KM, Porter CC, Anand SK, Whiteford ML, Davis ID, Dewar SB, Bettinelli A, Fadrowski JJ, Belsha CW, Hunley TE, Nelson RD, Trachtman H, Cole TR, Pinsk M, Bockenhauer D, Shenoy M, Vaidyanathan P, Foreman JW, Rasoulpour M, Thameem F, Al-Shahrouri HZ, Radhakrishnan J, Gharavi AG, Goilav B, & Lifton RP (2012). Mutations in kelch-like 3 and cullin 3 cause hypertension and electrolyte abnormalities. Nature 482, 98–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brugnara C (1989). Characteristics of the volume- and chloride-dependent K transport in human erythrocytes homozygous for hemoglobin C. J. Membr. Biol 111, 69–81. [DOI] [PubMed] [Google Scholar]
- Brugnara C, Van Ha T, & Tosteson DC (1989). Role of chloride in potassium transport through a K-Cl cotransport system in human red cells. Am. J. Physiol 256, C994–C1103. [DOI] [PubMed] [Google Scholar]
- Buche A, Ouassaidi A, Hacha R, Delpire E, Gilles R, & Houssier C (1989). Glycine and other amino compounds prevent chromatin precipitation at physiological ionic strength. FEBS Lett 247, 367–370. [DOI] [PubMed] [Google Scholar]
- Byun N, & Delpire E (2007). Axonal and periaxonal swelling precede peripheral neurodegeneration in KCC3 knockout mice. Neurobiol. Dis 28, 39–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cala PM (1980). Volume regulation by Amphiuma red blood cells: the membrane potential and its implications regarding the nature of the ion-flux pathways. J. Gen. Physiol 76, 683–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canessa M, Brugnara C, Cusi D, & Tosteson DC (1986). Modes of operation and variable stoichiometry of the furosemide- sensitive Na and K fluxes in human red cells. J Gen Physiol 87, 113–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castrop H, & Schießl IM (2014). Physiology and pathophysiology of the renal Na-K-2Cl cotransporter (NKCC2). Am. J. Physiol. Renal Physiol 307, F991–F1002. [DOI] [PubMed] [Google Scholar]
- Chang SS, Grunder S, Hanukoglu A, Rosler A, Mathew PM, Hanukoglu I, Schild L, Lu Y, Shimkets RA, Nelson-Williams C, Rossier BC, & Lifton RP (1996). Mutations in subunits of the epithelial sodium channel cause salt wasting with hyperkalaemic acidosis, pseudohypoaldosteronism type 1. Nat. Genet 12, 248–253. [DOI] [PubMed] [Google Scholar]
- Chen Y, Gao F, Jiang R, Liu H, Hou J, Yi Y, Kang L, Liu X, Li Y, & Yang M (2017). Down-Regulation of AQP4 Expression via p38 MAPK Signaling in Temozolomide-Induced Glioma Cells Growth Inhibition and Invasion Impairment. J. Cell Biochem 118, 4905–4913. [DOI] [PubMed] [Google Scholar]
- Chien LT, & Hartzell HC (2007). Drosophila bestrophin-1 chloride current is dually regulated by calcium and cell volume. J. Gen. Physiol 130, 513–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen O, & Hoffmann E,K.(1992). Cell swelling activates K+ and Cl- channels as well as nonselective, stretch-activated cation channels in Ehrlich ascites tumor cells. J. Membr. Biol 129, 13–36. [DOI] [PubMed] [Google Scholar]
- Condeelis J (1993). Life at the leading edge: the formation of cell protrusions. Annu. Rev. Cell Biol 9, 411–444. [DOI] [PubMed] [Google Scholar]
- Cook SF, Cramer CF, & Kenyon K (1952). A rapid titrimetric method for determining the water content of animal tissues. Science 115, 353–354. [DOI] [PubMed] [Google Scholar]
- Corliss JB, Dymond J, Gordon LI, Edmond JM, von Herzen RP, Ballard RD, Green K, Williams D, Bainbridge A, Crane K, & van Andel TH (1979). Submarine thermal sprirngs on the galapagos rift. Science 203, 1073–1083. [DOI] [PubMed] [Google Scholar]
- Craigie RA, & Cavalier-Smith T (1982). Cell Volume and the Control of the Chlamydomonas Cell Cycle. J. Cell Science
- Daniel I, Oger P, & Winter R (2006). Origins of life and biochemistry under high-pressure conditions. Chem. Soc. Rev 35, 858–875. [DOI] [PubMed] [Google Scholar]
- Darman RB, & Forbush B (2002). A regulatory locus of phosphorylation in the N terminus of the Na-K-Cl cotransporter, NKCC1. J. Biol. Chem 277, 37542–37550. [DOI] [PubMed] [Google Scholar]
- de Vries H (1884). Jahrb. f. wissenschaft. Bot XIV, 427. [Google Scholar]
- de Vries H (1888). Zeitschr. f. physik. Chemie II, 415. [Google Scholar]
- Dellasega M, & Grantham JJ (1973). Regulation of renal tubule cell volume in hypotonic media. Am. J. Physiol 224, 1288–1294. [DOI] [PubMed] [Google Scholar]
- Delpire E (1989). Controle du volume cellulaire: Etudes au niveau de cellules PC12 en culture, Université de Liège, Liège. [Google Scholar]
- Delpire E, Cornet M, & Gilles R (1991). Volume regulation in rat pheochromocytoma cultured cells submitted to hypoosmotic conditions. Arch. Inter. Physiol. Biochem 99, 71–76. [DOI] [PubMed] [Google Scholar]
- Delpire E, Duchêne C, Gilles R, & Goessens G (1985). Effects of osmotic shocks on the ions content and ultrastructure of rat pheochromocytoma cells of line PC12. Molecular Physiology 8, 293–305. [Google Scholar]
- Delpire E, Duchêne C, Goessens G, & Gilles R (1985). Effects of osmotic shocks on the ultrastructure of different tissues and cell types. Exp. Cell Res 160, 106–116. [DOI] [PubMed] [Google Scholar]
- Delpire E, & Gagnon KB (2011). Kinetics of hyperosmotically-stimulated Na-K-2Cl cotransporter in Xenopus laevis oocytes. Am. J. Physiol. Cell Physiol 301, C1074–C1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delpire E, Rauchman MI, Beier DR, Hebert SC, & Gullans SR (1994). Molecular cloning and chromosome localization of a putative basolateral Na-K-2Cl cotransporter from mouse inner medullary collecting duct (mIMCD-3) cells. J. Biol. Chem 269, 25677–25683. [PubMed] [Google Scholar]
- Delpire E, & Staley KJ (2014). Novel determinants of the neuronal Cl- concentration. J. Physiol 592, 4099–4114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doljanski F, Ben-Sasson S, Reich M, Grover NB (1974). Dynamic osmotic behavior of chick blood lymphocytes. J. Cell Physiol 84, 215–224. [DOI] [PubMed] [Google Scholar]
- Donnan FG (1911). Theorie der Membrangleichgewichte und Membranpotentiale bei Vorhandensein von nicht dialysierenden Elektrolyten. Ein Beitrag zur physikalisch-chemischen Physiologie” (The theory of membrane equilibrium and membrane potential in the presence of a non-dialyzable electrolyte. A contribution to physical-chemical physiology). Zeitschrift für Elektrochemie und angewandte physikalische Chemie 17, 572–581. [Google Scholar]
- Duan D, Winter C, Cowley S, Hume JR, & Horowitz B (1997). Molecular identification of a volume-regulated chloride channel. Nature 390, 417–421. [DOI] [PubMed] [Google Scholar]
- Dunham PB, & Ellory JC (1981). Passive potassium transport in low potassium sheep red cells: Dependence upon cell volume and chloride. J. Physiol. (London) 318, 511–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunham PB, Steward GW, & Ellory JC (1980). Chloride-activated passive potassium transport in human erythrocytes. Proc. Natl. Acad Sci. USA 77, 1711–1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DuPre AM, & Hempling HG (1978). Osmotic properties of Ehrlich ascites tumor cells during the cell cycle. J. Cell Physiol 93, 381–396. [DOI] [PubMed] [Google Scholar]
- Dupré N, Howard HC, Mathieu J, Karpati G, Vanasse M, Bouchard JP, Carpenter S, & Rouleau GA (2003). Hereditary motor and sensory neuropathy with agenesis of the corpus callosum. Ann. Neurol 54, 9–18. [DOI] [PubMed] [Google Scholar]
- Dutrochet MH (1826). L’agent immédiat du mouvement vital dévoilé dans sa nature et dans son monde d’action chez les végétaux et chez les animaux (Paris: Chez Dentu; ). [Google Scholar]
- Ericson AC, & Spring KR (1982). Volume regulation by Necturus gallbladder: apical Na+-H+ and Cl−-HCO−3 exchange. Am. J. Physiol 243, C146–C150. [DOI] [PubMed] [Google Scholar]
- Ferraris JD, & Burg MB (2006). Tonicity-dependent regulation of osmoprotective genes in mammalian cells. Contrib. Nephrol 2006 152, 125–141. [DOI] [PubMed] [Google Scholar]
- Flemmer AW, Gimenez I, Dowd BF, Darman RB, & Forbush B (2002). Activation of the Na-K-Cl cotransporter NKCC1 detected with a phospho-specific antibody. J. Biol. Chem 277, 37551–37558. [DOI] [PubMed] [Google Scholar]
- Flores B, Schornack CC, & Delpire E (2018). A role for KCC3 in maintaining cell volume of peripheral nerve fibers. Neurochemistry Int [DOI] [PMC free article] [PubMed]
- Fredericq L (1885). Influence du milieu ambiant sur la composition du sang des animaux aquatiques. Arch. Zool. Exp. & Gen. Série 2, XXXIV–XXXVIII. [Google Scholar]
- Fugelli K (1967). Regulation of cell volume in flounder (Pleuronectes flesus) erythrocytes accompanying a decrease in plasma osmolarity. Comp. Biochem. Physiol 22, 253–260. [DOI] [PubMed] [Google Scholar]
- Fujise H, Yamada I, Masuda M, Miyazawa Y, Ogawa E, & Takahashi R (1991). Several cation transporters and volume regulation in high-K dog red blood cells. Am. J. Physiol 260, C589–C597. [DOI] [PubMed] [Google Scholar]
- Gagnon KB (2012). High-grade glioma motility reduced by genetic knockdown of KCC3. Cell Physiol. Biochem 30, 466–472. [DOI] [PubMed] [Google Scholar]
- Gagnon KB, & Delpire E (2010). Molecular determinants of hyperosmotically activated NKCC1-mediated K+/K+ exchange. J. Physiol. (Lond) 588, 3385–3396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gagnon KB, & Delpire E (2010). On the substrate recognition and autoinhibitory domain of SPAK. FASEB J 44, 609–611. [Google Scholar]
- Gagnon KB, England R, & Delpire E (2006). Volume sensitivity of cation-chloride cotransporters is modulated by the interaction of two kinases: SPAK and WNK4. Am. J. Physiol. Cell Physiol 290, C134–C142. [DOI] [PubMed] [Google Scholar]
- Gagnon KB, England R, & Delpire E (2007). A single binding motif is required for SPAK activation of the Na-K-2Cl cotransporter. Cell Physiol. Biochem 20, 131–142. [DOI] [PubMed] [Google Scholar]
- Gamba G, Miyanoshita A, Lombardi M, Lytton J, Lee W-S, Hediger M, & Hebert SC (1994). Molecular cloning, primary structure, and characterization of two members of the mammalian electroneutral sodium-(potassium)-chloride cotransporter family expressed in kidney. J. Biol. Chem 269, 17713–17722. [PubMed] [Google Scholar]
- Garty H, & Benos DJ (1988). Characteristics and regulatory mechanisms of the amiloride-blockable Na+ channel. Physiol. Rev 68, 309–373. [DOI] [PubMed] [Google Scholar]
- Geck P, & Pfeiffer B (1985). Na+ +K+ +2Cl− cotransport in animal cells - its role in volume regulation. Ann. NY Acad. Sci 456, 166–182. [DOI] [PubMed] [Google Scholar]
- Geck P, Pietrzyk C, Burckhardt B-C, Pfeiffer B, & Heinz E (1980). Electrically silent cotransport of Na+, K+ and Cl− in Ehrlich cells. Biochim. Biophys. Acta 600, 432–447. [DOI] [PubMed] [Google Scholar]
- Gekko K, & Timasheff SN (1981). Mechanism of protein stabilization by glycerol: preferential hydration in glycerol-water mixtures. Biochemistry 20, 4667–4676. [DOI] [PubMed] [Google Scholar]
- Gekko K, & Timasheff SN (1981). Thermodynamic and kinetic examination of protein stabilization by glycerol. Biochemistry 20, 4677–4686. [DOI] [PubMed] [Google Scholar]
- Gianazza E, & Righetti PG (1980). Size and charge distribution of macromolecules in living systems. J. Chromatogr 193, 1–8. [Google Scholar]
- Gill DR, Hyde SC, Higgins CF, Valverde MA, Mintenig GM, & Sepúlveda FV (1992). Separation of drug transport and chloride channel functions of the human multidrug resistance P-glycoprotein. Cell 71, 23–32. [DOI] [PubMed] [Google Scholar]
- Gillen CM, Brill S, Payne JA, & Forbush BI (1996). Molecular cloning and functional expression of the K-Cl cotransporter from rabbit, rat, and human. A new member of the cation-chloride cotransporter family. J. Biol. Chem 271, 16237–16244. [DOI] [PubMed] [Google Scholar]
- Gilles R (1979). Intracellular organic osmotic effectors. In Mechanisms of Osmoregulation in Animals, Gilles R, ed. (London, New York: Wiley Interscience; ), pp. 111–153. [Google Scholar]
- Gilles R (1987). Volume regulation in cells of euryhaline invertebrates. Curr. Top. Membr. Transp 30, 205–247. [Google Scholar]
- Gilles R, & Delpire E (1997). Variations in salinity, osmolarity, and water availability: vertebrates and invertebrates. In Handbook of Physiology: Section 13, Comparative Physiology, Volume 2, Danzler WH, ed. (New York: American Physiological Society; ), pp. 1523–1586. [Google Scholar]
- Gimenez I, & Forbush B (2005). Regulatory phosphorylation sites in the NH2 terminus of the renal Na-K-Cl cotransporter (NKCC2). Am. J. Physiol. Renal Physiol 289, F1341–F1345. [DOI] [PubMed] [Google Scholar]
- Glykys J, Dzhala V, Egawa K, Balena T, Kuchibhotla KV, Bacskai BJ, Kahle KT, Zeuthen T, & Staley KJ (2014). Local impermeant anions set the GABAA reversal potential. Science 343, 670–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottschalk CW, & Mylle M (1958). Evidence that the mammalian nephron functions as a countercurrent multiplier system. Science 128, 594. [DOI] [PubMed] [Google Scholar]
- Gottschalk CW, & Mylle M (1959). Micropuncture study of the mammalian urinary concentrating mechanism: evidence for the countercurrent hypothesis. Am. J. Physiol 196, 927–936. [DOI] [PubMed] [Google Scholar]
- Gough A (1924). The nature of the red blood corpuscle. Biochem. J 18, 202–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grinstein S, Clarke CA, Dupre A, & Rothstein A (1982). Volume-induced increase of anion permeability in human lymphocytes. J. Gen. Physiol 80, 801–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grinstein S, Clarke CA, & Rothstein A (1983). Activation of Na+/H+ exchange in lymphocytes by osmotically induced volume changes and by cytoplasmic acidification. J. Gen. Physiol 82, 619–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grinstein S, Clarke CA, Rothstein A, & Gelfand EW (1983). Volume-induced anion conductance in human B lymphocytes is cation independant. Am. J. Physiol 245, C160–C163. [DOI] [PubMed] [Google Scholar]
- Grinstein S, Rothstein A, Sarkadi B, & Gelfand EW (1984). Responses of lymphocytes to anisotonic media: volume-regulating behavior. Am. J. Physiol 246, C204–C215. [DOI] [PubMed] [Google Scholar]
- Gründer S, Thiemann A, Pusch M, & Jentsch TJ (1992). Regions involved in the opening of CIC-2 chloride channel by voltage and cell volume. Nature 360, 759–762. [DOI] [PubMed] [Google Scholar]
- Guyton AC (1987). Renal function curve--a key to understanding the pathogenesis of hypertension. Hypertension 10, 1–6. [DOI] [PubMed] [Google Scholar]
- Haas M, & Harrison JH Jr(1989). Stimulation of K-Cl cotransport in rat red cells by a hemolytic anemia-producing metabolite of dapsone. Am. J. Physiol 256, C265–272. [DOI] [PubMed] [Google Scholar]
- Haas M, Starke LC, & McManus TJ (1984). Soium modulation of cathecholamine-stimulated K/K (K/Rb) exchange in duck red blood cells. J. Gen. Physiol 82, 10a–11a. [Google Scholar]
- Haldane JBS (1929). Origin of life. Ration. Annu 148, 3–10. [Google Scholar]
- Hargitay B, & Kuhn W (1951). Das multiplikationsprinzip als Grudnlage der harkonzentrier in der Niere. Zeitshrift fur Electrochemie und angewandte physikalische Chemie 55, 539–558. [Google Scholar]
- Hendil KB, & Hoffmann EK (1974). Cell volume regulation in Ehrlich ascites tumor cells. J. Cell Physiol 84, 115–125. [DOI] [PubMed] [Google Scholar]
- Hiki K, D’Andrea RJ, Furze J, Crawford J, Woollatt E, Sutherland GR, Vadas MA, & Gamble JR (1999). Cloning, characterization, and chromosomal location of a novel human K+-Cl− cotransporter. J. Biol. Chem 274, 10661–10667. [DOI] [PubMed] [Google Scholar]
- Hill WG, & Zeidel ML (2000). Reconstituting the barrier properties of a water-tight epithelial membrane by design of leaflet-specific liposomes. J. Biol. Chem 275, 30176–30185. [DOI] [PubMed] [Google Scholar]
- Hinke JAM (1970). Solvant water for electrolytes in the muscle fiber of the giant barnacle. J. Gen. Physiol 56, 521–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann EK, & Lambert IH (1983). Amino acid transport and cell volume regulation in Ehrlich ascites tumour cells. J. Physiol 338, 613–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann EK, & Lambert IH (1994). On the similarity between the small Cl- channel and the taurine channel activated after cell swelling in Ehrlich ascites tumor cells. Jpn. J. Physiol 44, S49–S53. [PubMed] [Google Scholar]
- Hoffmann EK, Lambert IH, & Pedersen SF (2009). Physiology of cell volume regulation in vertebrates. Physiol Rev 89, 193–277. [DOI] [PubMed] [Google Scholar]
- Hoffmann EK, Lambert IH, & Simonsen LO (1986). Separate, Ca2+-activated K+ and Cl− transport pathways in Ehrlich ascites tumor cells. J. Membrane Biol 91, 227–244. [DOI] [PubMed] [Google Scholar]
- Hoffmann EK, & Simonsen EK (1989). Membrane mechanisms in volume and pH regulation in vertebrate cells. Physiol. Rev 69, 315–382. [DOI] [PubMed] [Google Scholar]
- Hoffmann EK, Simonsen LO, & Lambert IH (1984). Volume-induced increase of K+ and Cl− permeabilities in Ehrlich ascites tumor cells. Role of internal Ca2+. J. Membrane Biol 78, 211–222. [DOI] [PubMed] [Google Scholar]
- Hoffmann EK, Simonsen LO, & Sjøholm C (1979). Membrane potential, chloride exchange, and chloride conductance in Ehrlich mouse ascites tumour cells. J. Physiol 296, 61–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann EK, Sjoholm C, & Simonsen LO (1983). Na+, Cl− cotransport in Ehrlich asites tumor cells activated during volume regulation (Regulatory Volume Increase). J. Membrane Biol 76, 269–280. [DOI] [PubMed] [Google Scholar]
- Hougaard C, Jørgensen F, & Hoffmann EK (2001). Modulation of the volume-sensitive K+ current in Ehrlich ascites tumour cells by pH. Pflugers Arch 442, 622–633. [DOI] [PubMed] [Google Scholar]
- Hougaard C, Klaerke DA, Hoffmann EK, Olesen SP, & Jorgensen NK (2004). Modulation of KCNQ4 channel activity by changes in cell volume. Biochim. Biophys. Acta 1660, 1–6. [DOI] [PubMed] [Google Scholar]
- Howard HC, Mount DB, Rochefort D, Byun N, Dupré N, Lu J, Fan X, Song L, Rivière J-B, Prévost C, Welch R, England R, Zhan FQ, Mercado A, Siesser WB, George AL, Horst J, Simonati A, McDonald MP, Bouchard J-P, Mathieu J, Delpire E, & Rouleau GA (2002). Mutations in the K-Cl cotransporter KCC3 cause a severe peripheral neuropathy associated with agenesis of the corpus callosum. Nat. Genet 32, 384–392. [DOI] [PubMed] [Google Scholar]
- Howard LD, & Wondergem R (1987). Effects of anisosmotic medium on cell volume, transmembrane potential and intracellular K+ activity in mouse hepatocytes. J. Membr. Biol 100, 53–61. [DOI] [PubMed] [Google Scholar]
- Igarashi P, Vanden Heuvel G.B., Payne JA, & Forbush BI (1995). Cloning, embryonic expression and alternative splicing of a murine kidney specific Na-K-Cl cotransporter. Am. J. Physiol. (Renal Fluid Electrolyte Physiol.) 269, F405–F418. [DOI] [PubMed] [Google Scholar]
- Ikehara T, Yamaguschi H, Hosokawa K, & Miyamoto H (1993). Kinetic study non the effects [DOI] [PubMed]
- Jackson PS, Morrison R, & Strange K (1994). The volume-sensitive organic osmolyte-anion channel VSOAC is regulated by nonhydrolytic ATP binding. Am. J. Physiol 267, C1203–C1209. [DOI] [PubMed] [Google Scholar]
- Jackson PS, & Strange K (1993). Volume-sensitive anion channels mediate swelling-activated inositol and taurine efflux. Am J. Physiol 265, C1489–C1500. [DOI] [PubMed] [Google Scholar]
- Jakab M, & Ritter M (2006). Cell volume regulatory ion transport in the regulation of cell migration. Contrib. Nephrol 152, 151–180. [DOI] [PubMed] [Google Scholar]
- Jamison RL (1987). Short and long ioop nephrons. Kidney Int 31, 597–605. [DOI] [PubMed] [Google Scholar]
- Jennings ML, & al-Rohil N (1990). Kinetics of activation and inactivation of swelling-stimulated K+/Cl− transport. The volume-sensitive parameter is the rate constant for inactivation. J. Gen. Physiol 95, 1021–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jennings ML, & Schultz RK (1990). Swelling-activated KCl cotransport in rabbit red cells: flux is determined mainly by cell volume rather than shape. Am. J. Physiol 259, C960–967. [DOI] [PubMed] [Google Scholar]
- Johanson CE, Duncan JA 3rd, Klinge PM, Brinker T, Stopa EG, & Silverberg GD (2008). Multiplicity of cerebrospinal fluid functions: New challenges in health and disease. Cerebrospinal Fluid Res 5, 1–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen NK, Lambert IH, & Hoffmann EK (1996). Role of LTD4 in the regulatory volume decrease response in Ehrlich ascites tumor cells. J. Membr. Biol 151, 159–173. [DOI] [PubMed] [Google Scholar]
- Kahle KT, Flores B, Bharucha-Goebel D, Zhang J, Donkervoort S, Hegde M, Hussain G, Duran D, Liang B, Sun D, Bönnemann CG, & Delpire E (2016). Peripheral motor neuropathy is associated with defective kinase regulation of the KCC3 cotransporter. Sci. Signaling 9, ra77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaji D (1986). Volume-sensitive K transport in human erythrocytes. J. Gen. Physiol 88, 719–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kay AR (2017). How Cells Can Control Their Size by Pumping Ions. Front. Cell Dev. Biol 5, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley DS, Karson JA, Blackman DK, Früh-Green GL, Butterfield DA, Lilley MD, Olson EJ, Schrenk MO, Roe KK, Lebon GT, Rivizzigno P, & Partyq A-S (2001). An off-axis hydrothermal vent field near the Mid-Atlantic Ridge at 30 degrees N. Nature 412, 145–149. [DOI] [PubMed] [Google Scholar]
- Kerr JF, Wyllie AH, & Currie AR (1972). Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer 26, 239–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HD, Sergeant S, Forte LR, Sohn DK, & Im JH (1989). Activation of a Cl-dependent K flux by cAMP in pig red cells. Am. J. Physiol 256, C772–C778. [DOI] [PubMed] [Google Scholar]
- Kirkegaard SS, Lambert IH, Gammeltoft S, & Hoffmann EK (2010). Activation of the TASK-2 channel after cell swelling is dependent on tyrosine phosphorylation. Am. J. Physiol. Cell Physiol 299, C844–C853. [DOI] [PubMed] [Google Scholar]
- Kirkegaard SS, Strøm PD, Gammeltoft S, Hansen AJ, & Hoffmann EK (2016). The Volume Activated Potassium Channel KCNK5 is Up-Regulated in Activated Human T Cells, but Volume Regulation is Impaired. Cell. Physiol. Biochem 38, 883–892. [DOI] [PubMed] [Google Scholar]
- Klein JD, & O’Neill WC (1995). Volume-sensitive myosin phosphorylation in vascular endothelial cells: correlation with Na-K-2Cl cotransport. Am. J. Physiol. (Cell Physiol.) 269, C1524–C1531. [DOI] [PubMed] [Google Scholar]
- Klein T, Cooper TG, & Yeung CH (2006). The role of potassium chloride cotransporters in murine and human sperm volume regulation. Biol. Reprod 75, 853–858. [DOI] [PubMed] [Google Scholar]
- Koeppe H (1895). Ueber den Quellungsgrad der rothen Blutscheiben durch aequimoleculare Salzlösungen und über den osmotischen Druck des Blutplasmas. Arch. F. Phsyiol, 154–184.
- Kopp C, Linz P, Dahlmann A, Hammon M, Jantsch J, Müller DN, Schmieder RE, Cavallaro A, Eckardt KU, Uder M, Luft FC, & Titze J (2013). 23Na magnetic resonance imaging-determined tissue sodium in healthy subjects and hypertensive patients. Hypertension 61, 635–640. [DOI] [PubMed] [Google Scholar]
- Kregenow FM (1971). The response of duck erythrocytes to nonhemolytic hypotonic media. Evidence for a volume-controlling mechanism. J. Gen. Physiol 58, 372–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krylov AV, Pohl P, Zeidel ML, & Hill WG (2001). Water permeability of asymmetric planar lipid bilayers: leaflets of different composition offer independent and additive resistances to permeation. J. Gen. Physiol 118, 333–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert IH, & Hoffmann EK (1993). Regulation of taurine transport in Ehrlich ascites tumor cells. J. Membr. Biol 131, 67–69. [DOI] [PubMed] [Google Scholar]
- Lambert IH, & Hoffmann EK (1994). Cell swelling activates separate taurine and chloride channels in Ehrlich mouse ascites tumor cells. J. Membr. Biol 142, 289–298. [DOI] [PubMed] [Google Scholar]
- Lane N, & Martin WF (2012). The origin of membrane bioenergetics. Cell 151, 1406–1416. [DOI] [PubMed] [Google Scholar]
- Lauf PK (1982). Evidence for a chloride-dependent potassium and water transport induced by hypo-osmotic stress in erythrocytes of the marine teleost Opsanus tau. J. Comp. Physiol 146, 9–16. [Google Scholar]
- Lauf PK (1983). Thiol-dependent passive K/Cl transport in sheep red cells. II. Loss of Cl- and N-ethylmaleimide sensitivity in maturing high K+ cells. J. Membrane Biol 73, 247–256. [DOI] [PubMed] [Google Scholar]
- Lauf PK, & J.B. (1987). Direct evidence for chloride-dependent volume reduction in macrocytic sheep reticulocytes. Biochem. Biophys. Res. Commun 144, 849–855. [DOI] [PubMed] [Google Scholar]
- Lauf PK, McManus TJ, Haas M, Forbush B 3rd, Duhm J, Flatman PW, Saier MH Jr., & Russell JM (1987). Physiology and biophysics of chloride and cation cotransport across cell membranes. Fed. Proc 46, 2377–2394. [PubMed] [Google Scholar]
- Lauf PK, & Theg BE (1980). A chloride dependent K+ flux induced by N-ethylmaleimide in genetically low K+ sheep and goat erythrocytes. Biochem. Biophys. Res. Commun 70, 221–242. [DOI] [PubMed] [Google Scholar]
- Lauffenburger DA, & Horwitz AF (1996). Cell migration: a physically integrated molecular process. Cell 84, 359–369. [DOI] [PubMed] [Google Scholar]
- Law RO, & Turner DP (1987). Are ninhydrin-positive substances volume-regulatory osmolytes in rat renal papillary cells? J. Physiol 386, 45–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenart B, Kintner DB, Shull GE, & Sun D (2004). Na-K-Cl cotransporter-mediated intracellular Na+ accumulation affects Ca2+ signaling in astrocytes in an in vitro ischemic model. J. Neurosci 24, 9585–9587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohr JW, & Grantham JJ (1986). Isovolumetric regulation of isolated S2 proximal tubules in anisosmotic media. J. Clin. Invest 78, 1165–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lytle C (1998). A volume-sensitive protein kinase regulates the Na-K-2Cl cotransporter in duck red blood cells. Am. J. Physiol. (Cell Physiol.) 274, C1002–C1010. [DOI] [PubMed] [Google Scholar]
- Lytle C, & Forbush BI (1992). The Na-K-Cl cotransport protein of shark rectal gland. II. regulation by direct phosphorylation. J. Biol. Chem 267, 25438–25443. [PubMed] [Google Scholar]
- MacLeod RJ, & Hamilton J, R. (1990). Regulatory volume increase in mammalian jejunal villus cells is due to bumetanide-sensitive NaKCl2 cotransport. Am. J. Physiol. Gastrointest. Liver Physiol 285, G665–G674. [DOI] [PubMed] [Google Scholar]
- Martin W, & Russell MJ (2007). On the origin of biochemistry at an alkaline hydrothermal vent. Phil. Trans. Roy. Soc. Lond. B 367, 1887–1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martins JR, Faria D, Kongsuphol P, Reisch B, Schreiber R, & Kunzelmann K (2011). Anoctamin 6 is an essential component of the outwardly rectifying chloride channel. Proc. Natl. Acad. Sci. USA 108, 18168–18172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercado A, Broumand V, Zandi-Nejad K, Enck AH, & Mount DB (2006). A C-terminal domain in KCC2 confers constitutive K+-Cl− cotransport. J. Biol. Chem 281, 1016–1026. [DOI] [PubMed] [Google Scholar]
- Merriam GR, & Baer L (1980). Adrenocorticotropin deficiency: correction of hyponatremia and hypoaldosteronism with chronic glucocorticoid therapy. J. Clin. Endocrinol. Metab 50, 10–14. [DOI] [PubMed] [Google Scholar]
- Model MA (2014). Possible causes of apoptotic volume decrease: an attempt at quantitative review. Am. J. Physiol. Cell Physiol 306, C417–C424. [DOI] [PubMed] [Google Scholar]
- Moriguchi T, Urushiyama S, Hisamoto N, Iemura SI, Uchida S, Natsume T, Matsumoto K, & Shibuya H (2005). WNK1 regulates phosphorylation of cation-chloride-coupled cotransporters via the STE20-related kinases, SPAK and OSR1. J. Biol. Chem 280, 42685–42693. [DOI] [PubMed] [Google Scholar]
- Mount DB, Mercado A, Song L, Xu J, George JAL, Delpire E, & Gamba G (1999). Cloning and Characterization of KCC3 and KCC4, new members of the cation-chloride cotransporter gene family. J. Biol. Chem 274, 16355–16362. [DOI] [PubMed] [Google Scholar]
- Nägeli C (1855). Pflanzenphysiologische Untersuchungen von Carl NÄgeli und Carl Cramer, Volume I (Zürich: Friedrich Schulthness; ). [Google Scholar]
- Nielsen S, & Knepper MA (1993). Vasopressin activates collecting duct urea transporters and water channels by distinct physical processes. Am. J. Physiol 265, F204–F213. [DOI] [PubMed] [Google Scholar]
- Niemeyer MI, Hougaard C, Hoffmann EK, Jorgensen F, Stutzin A, & Sepúlveda FV (2000). Characterisation of a cell swelling-activated K+-selective conductance of ehrlich mouse ascites tumour cells. J. Physiol 524, 757–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieminen AL (2003). Apoptosis and necrosis in health and disease: role of mitochondria. Int. Rev. Cytol 224, 29–55. [DOI] [PubMed] [Google Scholar]
- Nollet A (1748). Lecons de Physique Experimentale (Paris: Frères Guérin; ). [Google Scholar]
- Numata T, Sato K, Okada Y, & Wehner F (2008). Hypertonicity-induced cation channels rescue cells from staurosporine-elicited apoptosis. Apoptosis 13, 895–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Donnell ME, Martinez A, & Sun D (1995). Endothelial Na-K-Cl cotransport regulation by tonicity and hormones: phosphorylation of cotransport protein. Am. J. Physiol. (Cell Physiol.) 269, C1513–C1523. [DOI] [PubMed] [Google Scholar]
- Olson J, & Holtzman D (1982). Respiration and cell volume of primary cultured cerebral astrocytes in media of various osmolarities. Brain Res 246, 273–279. [DOI] [PubMed] [Google Scholar]
- Orgel LE (2008). The implausibility of metabolic cycles on the prebiotic Earth. PLoS Biol 6, 5–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orlov SN, Tremblay J, & Hamet P (1996). Bumetanide-sensitive ion fluxes in vascular smooth muscle cells: lack of functional Na+, K+, 2 Cl− cotransport. J. Membr. Biol 153, 125–135. [DOI] [PubMed] [Google Scholar]
- Overton E (1907). Uber den Mechanismus der Resorption und der Secretion. In Handbuch der Physiologie des Menschen, Volume II, pt. 2, Nagel W, ed. (Braunschweig: Vieweg u. Sohn; ). [Google Scholar]
- Padilla MC, Armas-Hernández MJ, Hernández RH, Israili ZH, & Valasco M (2007). Update of diuretics in the treatment of hypertension. Am. J. Ther 14, 154–160. [DOI] [PubMed] [Google Scholar]
- Palfrey HC, & Pewitt EB (1993). The ATP and Mg2+ dependence of Na(+)-K(+)-2Cl− cotransport reflects a requirement for protein phosphorylation: studies using calyculin A. Pflugers Archiv 425, 321–328. [DOI] [PubMed] [Google Scholar]
- Pao AC (2014). Update on the Guytonian view of hypertension. Curr. Opin. Nephrol. Hypertens 23, 391–398. [DOI] [PubMed] [Google Scholar]
- Parker JC (1983). Volume-responsive sodium movements in dog red cells. Am. J. Physiol 244, C324–C330. [DOI] [PubMed] [Google Scholar]
- Parker JC, Colclasure GG, & McManus TJ (1991). Coordinated regulation of shrinkage-induced Na/H exchange and swelling-induced [K-Cl] cotransport in dog red cells. Further evidence from activation kinetics and phosphatase inhibition. J. Gen. Physiol 98, 869–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulmichl M, Li Y, Wickman K, Ackerman M, Peralta E, & Clapham D (1992). New mammalian chloride channel identified by expression cloning. Nature 356, 238–241. [DOI] [PubMed] [Google Scholar]
- Payne JA, & Forbush BI (1994). Alternatively spliced isoforms of the putative renal Na-K-Cl cotransporter are differentially distributed within the rabbit kidney. Proc. Natl. Acad. Sci. USA 91, 4544–4548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne JA, Stevenson TJ, & Donaldson LF (1996). Molecular characterization of a putative K-Cl cotransporter in rat brain. A neuronal-specific isoform. J. Biol. Chem 271, 16245–16252. [DOI] [PubMed] [Google Scholar]
- Payne JA, Xu J-C, Haas M, Lytle CY, Ward D, & Forbush BI (1995). Primary structure, functional expression, and chromosome localization of the bumetanide sensitive Na-K-Cl cotransporter in human colon. J. Biol. Chem 270, 17977–17985. [DOI] [PubMed] [Google Scholar]
- Pfeffer W (1877). Osmotische Untersuchungen – Studien zur Zellmechanik (Leipzig: Engelmann, W.). [Google Scholar]
- Piechotta K, Garbarini NJ, England R, & Delpire E (2003). Characterization of the interaction of the stress kinase SPAK with the Na+-K+−2Cl− cotransporter in the nervous system: Evidence for a scaffolding role of the kinase. J. Biol. Chem 278, 52848–52856. [DOI] [PubMed] [Google Scholar]
- Piechotta K, Lu J, & Delpire E (2002). Cation-chloride cotransporters interact with the stress-related kinases SPAK and OSR1. J. Biol. Chem 277, 50812–50819. [DOI] [PubMed] [Google Scholar]
- Plotkin MD, Kaplan MR, Peterson LN, Gullans SR, Hebert SC, & Delpire E (1997). Expression of the Na+-K+−2Cl− cotransporter BSC2 in the nervous system. Am. J. Physiol. Cell Physiol 272, C173–C183. [DOI] [PubMed] [Google Scholar]
- Ponder E (1948). Hemolysis and Related Phenomena (New York: Grune and Stratton; ), pp. 83–114. [Google Scholar]
- Prager-Khoutorsky M (2017). Mechanosensing in hypothalamic osmosensory neurons. Semin. Cell Dev. Biol 71, 13021. [DOI] [PubMed] [Google Scholar]
- Pringsheim EG (1854). Untersuchungen über den Bau und die Bildung der Pflanzenzellen (Leipzig: ). [Google Scholar]
- Qiu Z, Dubin AE, Mathur J, Tu B, Reddy K, Miraglia LJ, Reinhardt J, Orth AP, & Patapoutian A (2014). SWELL1, a plasma membrane protein, is an essential component of volume-regulated anion channel. Cell 157, 447–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quarmyne MO, Risinger M, Linkugel A, Frazier A, & Joiner C (2011). Volume regulation and KCl cotransport in reticulocyte populations of sickle and normal red blood cells. Blood Cells Mol. Dis 47, 95–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Race JE, Makhlouf FN, Logue PJ, Wilson FH, Dunham PB, & Holtzman EJ (1999). Molecular cloning and functional characterization of KCC3, a new K-Cl cotransporter. Am. J. Physiol. (Cell Physiol.) 277, C1210–C1219. [DOI] [PubMed] [Google Scholar]
- Rakova N, Kitada K, Lerchl K, Dahlmann A, Birukov A, Daub S, Kopp C, Pedchenko T, Zhang Y, Beck L, Johannes B, Marton A, Müller DN, Rauh M, Luft FC, & Titze J (2017). Increased salt consumption induces body water conservation and decreases fluid intake. J. Clin. Invest 127, 1932–1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ransom CB, O’Neal JT, & Sontheimer H (2001). Volume-activated chloride currents contribute to the resting conductance and invasive migration of human glioma cells. J. Neurosci 21, 7674–7683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinehart J, Maksimova YD, Tanis JE, Stone KL, Hodson CA, Zhang J, Risinger M, Pan W, Wu D, Colangelo CM, Forbush B, Joiner CH, Gulcicek EE, Gallagher PG, & Lifton RP (2009). Sites of regulated phosphorylation that control K-Cl cotransporter activity. Cell 138, 525–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ringel F, & Plesnila N (2008). Expression and functional role of potassium-chloride cotransporters (KCC) in astrocytes and C6 glioma cells. Neurosci. Lett 442, 219–223. [DOI] [PubMed] [Google Scholar]
- Riquelme G, Sepúlveda FV, Jørgensen F, Pedersen S, & Hoffmann EK (1998). Swelling-activated potassium currents of Ehrlich ascites tumour cells. Biochim. Biophys. Acta 1371, 101–106. [DOI] [PubMed] [Google Scholar]
- Rivera C, Voipio J, Payne JA, Ruusuvuori E, Lahtinen H, Lamsa K, Pirvola U, Saarma M, & Kaila K (1999). The K+/Cl− co-transporter KCC2 renders GABA hyperpolarizing during neuronal maturation. Nature 397, 251–255. [DOI] [PubMed] [Google Scholar]
- Roti Roti LW, & Rothstein A (1973). Adaptation of mouse leukemic cells (L5178Y) to anisotonic media. I. Cell volume regulation. Exp. Cell Res 79, 295–310. [DOI] [PubMed] [Google Scholar]
- Rump K, & Adamzik M (2018). Function of aquaporins in sepsis: a systematic review. Cell. Biosci 8, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell MJ, Daniel RM, & Hall A (1993). On the emergence of life via catalytic iron-sulphide mem branes Terra Nova 5, 343–345. [Google Scholar]
- Russell MJ, Daniel RM, Hall A, & Sherringham JA (1994). A Hydrothermally Precipitated Catalytic Iron Sulphide Membrane as a First Step Toward Life J. Mol. Evol 39, 231–243. [Google Scholar]
- Russell MJ, & Martin W (2004). The rocky roots of the acetyl-CoA pathway. Trends Biochem. Sci 29, 358–363. [DOI] [PubMed] [Google Scholar]
- Saadoun S, Papadopoulos MC, Hara-Chikuma M, & Verkman AS (2005). Impairment of angiogenesis and cell migration by targeted aquaporin-1 gene disruption. Nature 434, 786–792. [DOI] [PubMed] [Google Scholar]
- Sachs JR, & Martin DW (1993). The role of ATP in swelling-stimulated K-Cl cotransport in human red cell ghosts. J. Gen. Physiol 102, 551–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sands JM (2004). Renal urea transporters. Curr. Opin. Nephrol. Hypertens 13, 525–532. [DOI] [PubMed] [Google Scholar]
- Schild L, Lu Y, Gautschi I, Schneeberger E, Lifton RP, & Rossier BC (1996). Identification of a PY motif in the epithelial Na channel subunits as a target sequence for mutations causing channel activation found in Liddle syndrome. EMBO J 15, 2381–2387. [PMC free article] [PubMed] [Google Scholar]
- Schmidt WF 3rd, & McManus TJ (1977). Ouabain-insensitive salt and water movements in duck red cells. I. Kinetics of cation transport under hypertonic conditions. J Gen Physiol 70, 59–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnermann J, & Briggs JP (2013). Tubular control of renin synthesis and secretion. Pflugers Arch 465, 39–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab A (2001). Function and spatial distribution of ion channels and transporters in cell migration. Am. J. Physiol. Renal Physiol 280, F739–F747. [DOI] [PubMed] [Google Scholar]
- Schwab A, Wojnowski L, Gabriel K, & Oberleithner H (1994). Oscillating activity of a Ca(2+)-sensitive K+ channel. A prerequisite for migration of transformed Madin-Darby canine kidney focus cells. J. Clin. Invest 93, 1631–1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen MR, Chou CY,, & Ellory JC (2000). Volume-sensitive KCI cotransport associated with human cervical carcinogenesis. Pflugers Arch 440, 751–760. [DOI] [PubMed] [Google Scholar]
- Shen MR, Lin AC, Hsu YM, Chang TJ, Tang MJ, Alper SL, Ellory JC, & Chou CY (2004). Insulin-like growth factor 1 stimulates KCl cotransport, which is necessary for invasion and proliferation of cervical cancer and ovarian cancer cells. J. Biol. Chem 279, 40017–40025. [DOI] [PubMed] [Google Scholar]
- Sherbet GV, Lakshmi MS, & Cajone F (1983). Isoelectric characteristics and the secondary structure of some nucleic acids. Biophys. Struct. Mech 10, 121–128. [DOI] [PubMed] [Google Scholar]
- Shock EL, & Schulte MD (1998). Organic synthesis during fluid mixing in hydrothermal systems. J. Geophys. Res. E: Planets 103, 28513–28527. [Google Scholar]
- Siebens AW, & Kregenow FM (1978). Volume regulatory responses of salamander red cells incubated in anisotonic media. Effect of amiloride. Physiologist 21, 110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siebens AW, & Kregenow FM (1985). Volume-regulatory responses of Amphiuma red cells in anisotonic media. The effect of amiloride. J. Gen. Physiol 86, 527–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon DB, Nelson-Williams C, Johnson Bia M., Ellison D, Karet FE, Morey Molina A., Vaara I, Iwata F, Cushner HM, Koolen M, Gainza FJ, Gitelman HJ, & Lifton RP (1996). Gitelman’s variant of Bartter’s syndrome, inherited hypokalaemic alkalosis, is caused by mutations in the thiazide-sensitive Na-Cl cotransporter. Nature Genetics 12, 24–30. [DOI] [PubMed] [Google Scholar]
- Simone L, Gargano CD, Pisani F, Cibelli A, Mola MG, Frigeri A, Svelto M, & Nicchia GP (2018). Aquaporin-1 inhibition reduces metastatic formation in a mouse model of melanoma. J. Cell Mol. Med 22, 904–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sleep NH, Meibom A, Fridriksson T, Coleman RG, & Bird DK (2004). H2-rich fluids from serpentinization: geochemical and biotic implications. Proc. Natl. Acad. Sci. USA 101, 12818–12823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soroceanu L, Manning TJJ, & Sontheimer H (1999). Modulation of glioma cell migration and invasion using Cl(−) and K(+) ion channel blockers. J. Neurosci 19, 5942–5954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stokes JB, Lee I, & D’Amico M (1984). Sodium chloride absorption by the urinary bladder of the winter flounder: A thiazide-sensitive, electrically neutral transport system. J. Clin. Invest 74, 7–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strange K, Morrison R, Shrode L, & Putnam R (1992). Mechanism and regulation of swelling-activated inositol efflux in brain glial cells. Am. J. Physiol 265, C244–C256. [DOI] [PubMed] [Google Scholar]
- Sung K-W, Kirby M, McDonald MP, Lovinger DM, & Delpire E (2000). Abnormal GABAA-receptor mediated currents in dorsal root ganglion neurons isolated from Na-K-2Cl cotransporter null mice. J. Neurosci 20, 7531–7538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takenaka M, Preston AS, Kwon HM, & Handler JS (1994). The tonicity-sensitive element that mediates increased transcription of the betaine transporter gene in response to hypertonic stress. J. Biol. Chem 269, 29379–29381. [PubMed] [Google Scholar]
- Tamura H, Schild L, Enomoto N, Matsui N, Marumo F, & Rossier BC (1996). Liddle disease caused by a missense mutation of beta subunit of the epithelial sodium channel gene. J. Clin. Invest 97, 1780–1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tivey DR, Simmons NL, & Aiton JF (1985). Role of passive potassium fluxes in cell volume regulation in cultured HeLa cells. J. Membr. Biol 87, 93–95. [DOI] [PubMed] [Google Scholar]
- Tosteson DC, & Hoffman JF (1960). Regulation of cell volume by active cation transport in high and low potassium sheep red cells. J Gen Physiol 44, 169–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyomoto T, Knutsen D, Soos G, & Sato K (1997). Na-K-2Cl cotransporters are present and regulated in simian eccrine clear cells. Am. J. Physiol 273, R270–R277. [DOI] [PubMed] [Google Scholar]
- Ueberschär S, & Bakker-Grunwald T (1985). Effects of ATP and cAMP on the (Na++K++2Cl-)-cotransport system in turkey erythrocytes. Biochim. Biophys. Acta 818, 260–266. [DOI] [PubMed] [Google Scholar]
- Uyanik G, Elcioglu N, Penzien J, Gross C, Yilmaz Y, Olmez A, Demir E, Wahl D, Scheglmann K, Winner B, Bogdahn U, Topaloglu H, Hehr U, & Winkler J (2006). Novel truncating and missense mutations of the KCC3 gene associated with Andermann syndrome. Neurology 66, 1044–1048. [DOI] [PubMed] [Google Scholar]
- Verkman AS (2005). More than just water channels: unexpected cellular roles of aquaporins. J. Cell Sci 118, 3225–3232. [DOI] [PubMed] [Google Scholar]
- Vitari AC, Deak M, Morrice NA, & Alessi DR (2005). The WNK1 and WNK4 protein kinases that are mutated in Gordon’s hypertension syndrome, phosphorylate and active SPAK and OSR1 protein kinases. Biochem. J 391, 17–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voss FK, Ullrich F, Münch J, Lazarow K, Lutter D, Mah N, Andrade-Navarro MA, von Kries JP, Stauber T, & Jentsch TJ (2014). Identification of LRRC8 heteromers as an essential component of the volume-regulated anion channel VRAC. Science 344, 634–638. [DOI] [PubMed] [Google Scholar]
- Whisenant N, Zhang B-X, Khademazad M, Loessberg P, & Muallem S (1991). Regulation of Na-K-2Cl cotransport in osteoblasts. Am. J. Physiol 261, C433–C440. [DOI] [PubMed] [Google Scholar]
- Wilson FH, Disse-Nicodeme S, Choate KA, Ishikawa K, Nelson-Williams C, Desitter I, Gunel M, Milford DV, Lipkin GW, Achard JM, Feely MP, Dussol B, Berland Y, Unwin RJ, Mayan H, Simon DB, Farfel Z, Jeunemaitre X, & Lifton RP (2001). Human hypertension caused by mutations in WNK kinases. Science 293, 1107–1112. [DOI] [PubMed] [Google Scholar]
- Wu Q, Delpire E, Hebert SC, & Strange K (1998). Functional demonstration of Na-K-2Cl cotransporter activity in isolated, polarized choroid plexus cells. Am. J. Physiol. Cell Physiol 275, C1565–C1572. [DOI] [PubMed] [Google Scholar]
- Xu J-C, Lytle C, Zhu TT, Payne JA, Benz EJ, & Forbush BI (1994). Molecular cloning and functional expression of the bumetanide-sensitive Na-K-2Cl cotransporter. Proc. Natl. Acad. Sci. U.S.A 91, 2201–2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yurinskaya VE, Rubashkin AA, & Vereninov AA (2011). Balance of unidirectional monovalent ion fluxes in cells undergoing apoptosis: why does Na+/K+ pump suppression not cause cell swelling? J. Physiol 589, 2197–2211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu L, Lovinger D, & Delpire E (2005). Cortical neurons lacking KCC2 expression show impaired regulation of intracellular chloride. J. Neurophysiol 93, 1557–1568. [DOI] [PubMed] [Google Scholar]