

Abstract
Background
Ovarian cancer is the sixth most common cancer and seventh most common cause of cancer death in women world‐wide. Three‐quarters of women present when the disease has spread throughout the abdomen (stage III or IV) and treatment consists of a combination of debulking surgery and platinum‐based chemotherapy. Although initial responses to chemotherapy are good, most women will relapse and require further chemotherapy and will eventually develop resistance to chemotherapy.
PARP (poly (ADP‐ribose) polymerase) inhibitors, are a novel type of medication that works by preventing cancer cells from repairing their DNA once they have been damaged by other chemotherapy agents. It is not clear how PARP inhibitors compare to conventional chemotherapy regimens for the treatment of ovarian cancer, with respect to survival, side effects and quality of life.
Objectives
To determine the benefits and risks of PARP inhibitors for the treatment of epithelial ovarian cancer (EOC).
Search methods
We identified randomised controlled trials (RCTs) by searching the Cochrane Central Register of Controlled Trials (CENTRAL 2015, Issue 3), the Cochrane Gynaecological Cancer Group Trial Register, MEDLINE (1990 to April 2015), EMBASE (1990 to April 2015), ongoing trials on www.controlled‐trials.com/rct, www.clinicaltrials.gov, www.cancer.gov/clinicaltrials and the National Research Register (NRR), the FDA database and pharmaceutical industry biomedical literature.
Selection criteria
Women with histologically proven EOC who were randomised to treatment groups in trials that either compared PARP inhibitors with no treatment, or PARP inhibitors versus conventional chemotherapy, or PARP inhibitors together with conventional chemotherapy versus conventional chemotherapy alone.
Data collection and analysis
We used standard Cochrane methodology. Two review authors independently assessed whether studies met the inclusion criteria. We contacted investigators for additional data, where possible. Outcomes included survival, quality of life and toxicity.
Main results
We included four RCTs involving 599 women with EOC. Data for veliparib were limited and of low quality, due to small numbers (75 women total). Olaparib, on average, improved progression‐free survival (PFS) when added to conventional treatment and when used as maintenance treatment in women with platinum‐sensitive disease compared with placebo (hazard ratio (HR) 0.42, 95% confidence interval (CI) 0.29 to 0.60; 426 participants; two studies), but did not improve overall survival (OS) (HR 1.05, 95% CI 0.79 to 1.39; 426 participants; two studies). We graded this evidence as moderate quality using the GRADE approach. Adverse events of any severity were common in both the PARP inhibitor group and the control group. Olaparib was associated with more severe adverse events (G3/4) during the maintenance phase compared with controls (risk ratio (RR) 1.74, 95% CI 1.22 to 2.49; 385 participants, two studies; high quality evidence). Quality of life data were insufficient for meta‐analysis. We identified four ongoing studies.
Authors' conclusions
PARP inhibitors appear to improve PFS in women with recurrent platinum‐sensitive disease. Ongoing studies are likely to provide more information about whether the improvement in PFS leads to any change in OS in this subgroup of women with EOC. More research is needed to determine whether PARP inhibitors have any role to play in platinum‐resistant disease.
Keywords: Adult; Female; Humans; Poly(ADP‐ribose) Polymerase Inhibitors; Antineoplastic Agents; Antineoplastic Agents/therapeutic use; Benzimidazoles; Benzimidazoles/adverse effects; Benzimidazoles/therapeutic use; DNA Repair; DNA Repair/drug effects; Disease‐Free Survival; Neoplasm Recurrence, Local; Neoplasm Recurrence, Local/drug therapy; Ovarian Neoplasms; Ovarian Neoplasms/drug therapy; Ovarian Neoplasms/genetics; Phthalazines; Phthalazines/adverse effects; Phthalazines/therapeutic use; Piperazines; Piperazines/adverse effects; Piperazines/therapeutic use; Randomized Controlled Trials as Topic
Do PARP inhibitors improve survival in women with ovarian cancer and what are the side effects?
Background Conventional chemotherapy drugs act on dividing cells by damaging cell DNA. As cancer cells divide very rapidly, these drugs affect cancer cells to a greater degree than normal cells. Being able to repair DNA is vital to cell survival and normal cells have more than one DNA repair systems. However, cancer cells often have defects in these repair pathways that makes them harder for them to repair themselves. PARP inhibitors are a new type of medication that works by preventing cancer cells from repairing their DNA once they have been damaged by chemotherapy.
Review question Do PARP inhibitors improve survival in women with epithelial ovarian cancer and what are the side effects?
Main results We searched the literature from 1990 to April 2015 and found four randomised trials of PARP inhibitors versus other treatments or placebo. We also found four ongoing studies. The four completed studies included 599 women with recurrent epithelial ovarian cancer; three included women with platinum‐sensitive disease (return of disease more than 12 months since last chemotherapy treatment), and one included women with platinum‐resistant and partially platinum‐sensitive disease (return of disease less than six months or six to 12 months since last chemotherapy treatment). Three studies all tested a PARP inhibitor known as olaparib and one study with only 75 patients tested veliparib. On average, when added to conventional treatment, olaparib slowed the progression of disease in women with platinum‐sensitive disease compared with placebo or no added treatment, but did not alter the time that patients survived, although there were relatively few women in the studies and larger studies may change this outcome. Adverse events of any severity were common in both the PARP inhibitor group and the control group. However, serious adverse events were more common in the olaparib group than the control group when given as maintenance treatment after a course of chemotherapy. The most common serious adverse events were anaemia and fatigue. Data for veliparib were limited, due to the small number of women included, so we were unable to show if it had any effect on the progression of the disease. Veliparib had few severe side effects, but again the numbers were too small for meaningful conclusions.
Quality of the evidence The evidence is of moderate quality for studies looking at the affects of olaparib and estimates of effect may change with further research. There was low quality evidence for veliparb and we are very uncertain about the effects of the treatment.
Summary of findings
Summary of findings for the main comparison.
Summary of findings: PARP inhibitors versus other monotherapy
| PARP inhibitors compared with other monotherapy drugs for recurrent ovarian cancer | ||||||
|
Patient or population: women with recurrent platinum‐resistant or partially platinum‐sensitive ovarian cancer Settings: specialist hospital Intervention: PARP inhibitor Comparison: other monotherapy (PLD) | ||||||
| Outcomes | Illustrative comparative risks | Relative effect (95% CI) | No of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Overall survival | Due to the way HRs are calculated, the assumed and corresponding risks were not estimated. | HR 0.82 (80% CI 0.52 to 1.31) | 97 (1) | ⊕⊝⊝⊝ very low | Downgraded due to sparseness of data and 80% CIs | |
| Progression‐free survival | Due to the way HRs are calculated, the assumed and corresponding risks were not estimated. | HR 0.88 (0.51 to 1.52) | 97 (1) | ⊕⊕⊝⊝ low | Downgraded due to sparseness of data | |
| CI: confidence interval; RR: risk ratio; HR: hazard ratio; PLD: pegylated liposomal doxorubicin; OLA: olaparib | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
Summary of findings 2.
Summary of findings: PARP inhibitors added to conventional chemotherapy versus no added treatment
| PARP inhibitors in addition to conventional chemotherapy and/or as maintenance treatment for platinum‐sensitive ovarian cancer | ||||||
|
Patient or population: women with recurrent platinum sensitive ovarian cancer Settings: specialist hospital Intervention: PARP inhibitor added to conventional chemotherapy Comparison: placebo or no additional treatment | ||||||
| Outcomes | Illustrative comparative risks | Relative effect (95% CI) | No of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Overall survival | Due to the way HRs are calculated, the assumed and corresponding risks were not estimated. | HR 1.05 (0.79 to 1.39) | 426 (2) | ⊕⊕⊕⊝ moderate | Downgraded due to imprecision | |
| Progression‐free survival | Due to the way HRs are calculated, the assumed and corresponding risks were not estimated. | HR 0.42 (0.29 to 0.60) | 426 (2) | ⊕⊕⊕⊝ moderate | Downgraded due to clinical heterogeneity | |
| CI: confidence interval; RR: risk ratio; HR: hazard ratio; OS: overall survival; PFS: progression‐free survival | ||||||
| The assumed risk was based on the mean control group risk across included studies. GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
Summary of findings 3.
Summary of findings: Adverse Events
| PARP inhibitors versus other treatments or placebo for ovarian cancer | ||||||
|
Patient or population: women with recurrent ovarian cancer Settings: specialist hospital Intervention: PARP inhibitor Comparison: Other treatment or placebo | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Severe adverse events ‐ Nausea (G3‐4) | 20 per 1000 | 25 per 1000 (7 to 92) | RR 1.23 (0.33 to 4.60) | 592 (4) | ⊕⊕⊕⊝ moderate | Downgraded due to imprecision |
| Severe adverse events ‐ Neutropenia (G3‐4) | 270 per 1000 | 159 per 1000 (24 to 1000) | RR 0.59 (0.09 to 3.98) | 220 (2) | ⊕⊕⊝⊝ low | Downgraded due to heterogeneity and imprecision |
| Severe adverse events ‐ Anaemia (G3‐4) | 10 per 1000 | 22 per 1000 (9 to 52) | RR 2.15 (0.89 to 5.21) | 592 (4) | ⊕⊕⊕⊝ moderate | Downgraded due to imprecision |
| Adverse events during maintenance treatment only (grade 3/4) ‐ Nausea | 0 per 1000 | 20 per 10001 | RR 4.21 (0.48 to 36.69) | 385 (2) | ⊕⊕⊕⊝ moderate | Downgraded due to imprecision |
| Adverse events during maintenance treatment only (grade 3/4) ‐ Fatigue | 20 per 1000 | 42 per 1000 (13 to 134) | RR 2.12 (0.67 to 6.71) | 385 (2) | ⊕⊕⊕⊝ moderate | Downgraded due to imprecision |
| Adverse events during maintenance treatment only (grade 3/4) ‐ Anaemia | 10 per 1000 | 53 per 1000 (12 to 230) | RR 5.26 (1.19 to 23.20) | 385 (2) | ⊕⊕⊕⊝ moderate | Downgraded due to imprecision |
| Any severe adverse event during maintenance treatment | 180 per 1000 |
310 per 1000 (220 to 450) |
RR 1.74 (1.22 to 2.49) | 385 (2) |
⊕⊕⊕⊕ high | |
| *The basis for the assumed risk is the median control group risk across studies. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk Ratio | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 Based on the mean experimental group risk across included studies (due to no events in the control group)
Background
This is an updated version of a review first published in 2010 in the Cochrane Database of Systematic Reviews (Issue 6), which had no included studies.
Description of the condition
In 2012, world‐wide, 238,719 women were diagnosed with epithelial ovarian cancer (EOC) and 151,905 died from the disease, corresponding to an annual incidence of 6.1 cases per 100,000 women, an annual mortality rate of 4.3 deaths per 100,000 and a cumulative lifetime risk of 0.5% (GLOBOCAN 2012). In terms of incidence, it is the sixth most common cancer and it is the seventh most common cause of cancer death in women. The onset is often insidious; the symptoms are vague and may mimic other conditions. This may lead to a delay in diagnosis and currently three‐quarters of women with EOC are diagnosed when the disease has spread throughout the abdomen (stage III or IV) (Shepherd 1989), when the five‐year survival is 20% to 30% (Jemal 2008). EOC accounts for 90% of all ovarian cancers and typically presents in post‐menopausal women, with a peak incidence when women are in their early sixties, although it does occur in younger women, often associated with genetic predispositions (Quinn 2001). More recent data suggest that the origin of EOC may often be the lining of the fallopian tubes. Intra‐epithelial precursor lesions (so‐called serous tubal intra‐epithelial carcinoma or STIC) are commonly found in the fimbrial ends of fallopian tubes removed from women at high risk of developing EOC due to BRCA‐mutations (Erickson 2013). These STIC lesions are microscopic and may explain why EOC is difficult to identify at an early stage, since it has immediate access to the abdominal cavity and often does not typically arise from an ovarian cyst, which could be seen on an ultrasound scan.
Description of the intervention
Management of advanced EOC consists of a combination of debulking surgery and platinum‐based chemotherapy, with or without the addition of a taxane (Morrison 2012; Stewart 1999). A randomised controlled trial (RCT) found that there was no difference in survival in women with disease not amenable to surgery to remove all visible (macroscopic) disease, if surgery were performed before or after the first three cycles of chemotherapy (Vergote 2008). However, despite good initial responses to platinum agents and taxanes, most women have disease relapse, require further treatment with chemotherapy, and eventually develop resistance to conventional chemotherapeutic agents.
Conventional chemotherapeutic agents have activity on all rapidly dividing cells, hence the common side effects such as bone marrow suppression and mucositis. Increasing knowledge of the genetic basis for cancer has led to the development of novel reagents, which target cancer‐specific pathways. It is hoped that these reagents will spare normal cells and reduce the toxic side effects of chemotherapy, in addition to having an enhanced therapeutic effect.
How the intervention might work
DNA repair inhibition
Many current therapies for cancer (e.g. cytotoxic chemotherapy and radiotherapy) work by damaging DNA. As this function is fundamental to cell survival there are a number of systems or pathways of DNA repair. Cancer cells are more susceptible to DNA damage than normal cells, because the multiple mutations that have caused cells to become cancerous often affect one or more of these DNA repair pathways.
A number of drugs have been developed, which take advantage of this susceptibility of cancer cells to DNA damage. They work by inhibiting some, but not all, DNA repair pathways. In normal cells other DNA‐repair pathways will compensate. However, cancer cells often have mutations in other DNA‐repair pathways and so DNA damage is not repaired, leading to cell death.
Small‐molecule agents have been identified, which target elements in a number of these pathways, including poly (ADP‐ribose) polymerase (PARP), DNA‐dependent protein kinase (DNA‐PK) and ATM (Bryant 2006). Of these DNA‐repair inhibitors, PARP inhibitors have been most commonly used as anticancer therapy.
PARP inhibitors
PARP inhibitors are a family of related enzymes, which are involved in regulating various cellular processes, including DNA repair, cell death and inflammation. PARP inhibitors therefore have a potentially wide range of applications (Jagtap 2005).
PARP‐1 is the most‐studied of the PARP family. It is a nuclear enzyme, which binds to both single‐stranded and double‐stranded DNA breaks, either facilitating their repair by other enzymes (in the case of mild damage), or triggering cell‐death pathways (in the case of more severe damage) (Curtin 2005; Peralta‐Leal 2008; Ratnam 2007).
Research into the anticancer applications of PARP inhibitors has focused on two main approaches:
Firstly, they can be used in isolation in certain cancers with significant mutations in their DNA‐repair pathways: specifically, those with mutations in the BRCA 1/2 genes (which predispose to inherited forms of breast cancer and some ovarian cancers) (Zaremba 2007). BRCA genes encode for DNA repair enzymes that function independently of the PARP pathway. Cells with BRCA mutations are very susceptible to PARP inhibitors, because both pathways to repair DNA are blocked and so this triggers cell cycle arrest and apoptosis specifically within cells that have the BRCA mutation (Bryant 2005; Farmer 2005). PARP‐1 inhibitors have been shown to be effective, when used alone in cell culture or in mouse models, at killing cells with mutations in the BRCA1 and BRCA2 genes (Bryant 2005; Farmer 2005), and have been used in clinical trials for breast cancer (Fong 2008). BRCA germline (inherited) mutations pre‐dispose women to develop ovarian cancer. In addition, many ovarian cancers, in women who do not have germline BRCA mutations, have developed mutations in the BRCA genes within the tumour ‐ called somatic mutations (Hennessy 2010). 'BRCAness' is when ovarian cancers in women who do not have known BRCA mutations behave similarly to BRCA‐mutated ovarian cancer (Turner 2004). Ovarian cancers with BRCAness have high‐grade serous histology, respond well to platinum‐based chemotherapy and tend to take a relatively long time for disease to relapse (better progression‐free survival). Both somatic BRCA mutation and BRCAness may increase the number of women who may benefit from PARP inhibitors.
Secondly, PARP inhibitors can be used in combination with conventional anticancer agents that act by damaging DNA, such as cytotoxic chemotherapy and radiotherapy, as the PARP inhibitors block the DNA‐repair mechanisms that cancer cells use to resist destruction.
Why it is important to do this review
Novel biological agents that work in different ways to conventional chemotherapy have been developed. It is therefore important to establish whether the addition of these new drugs to conventional chemotherapy regimens is beneficial, in terms of survival and, if so, at what cost, in terms of additional harmful effects. Furthermore, since these compounds may be less toxic compared to conventional chemotherapy agents, it may be feasible to use these new agents in patients who are not currently taking chemotherapy (so called maintenance treatment), to reduce the chance of, or delay, the recurrence of their EOC.
Objectives
To determine the benefits and risks of PARP inhibitors for the treatment of epithelial ovarian cancer (EOC).
Methods
Criteria for considering studies for this review
Types of studies
Randomised controlled trials (RCTs).
Types of participants
Women ≥ 18 years old with histologically proven EOC of any stage. We excluded women with other concurrent malignancies.
Types of interventions
DNA‐repair pathway inhibitors versus no treatment
DNA‐repair pathway inhibitors + conventional chemotherapy versus conventional chemotherapy
DNA‐repair pathway inhibitors versus conventional chemotherapy
Types of outcome measures
Primary outcomes
Overall survival (OS)
Secondary outcomes
Progression‐free survival (PFS)
Objective Response Rate (ORR)
Quality of life, measured by a validated scale, e.g. QLQ‐C30
-
Adverse events: we grouped grades of toxicity (CTEP 2009) as follows:
haematological (leucopenia, anaemia, thrombocytopenia, neutropenia, haemorrhage)
gastrointestinal (nausea, vomiting, anorexia, diarrhoea, liver, proctitis)
genitourinary
skin (stomatitis, mucositis, alopecia, allergy)
neurological (peripheral and central)
other side effects not categorised above
Search methods for identification of studies
We sought papers in all languages and carried out translations where necessary.
Electronic searches
See: Cochrane Gynaecological Cancer Group methods used in reviews.
We searched the following electronic databases:
Cochrane Gynaecological Cancer Group Trial Register;
Cochrane Central Register of Controlled Trials (CENTRAL 2015, Issue 3);
MEDLINE (1990 up to May week 2, 2015);
EMBASE (1990 up to 2015, week 16).
The CENTRAL, MEDLINE and EMBASE search strategies, based on terms related to the review topic, are presented in Appendix 1, Appendix 2 and Appendix 3. We searched the databases from 1990 until April 2015.
We identified all relevant articles found on PubMed using the 'related articles' feature and carried out further searches for newly published articles.
Searching other resources
We searched Physician Data Query, www.controlled‐trials.com/rct, www.clinicaltrials.gov, www.cancer.gov/clinicaltrials and the National Research Register (NRR) for ongoing trials. We also sought details of ongoing or unpublished trials from the Food and Drug Administration (FDA) (www.fda.gov) and the European Medicines Agency (EMA) (www.ema.europa.eu), and from pharmaceutical company sources. We contacted the main investigators of the relevant ongoing trials for further information, as well as the major co‐operative trials groups active in this area. We identified AstraZeneca as the company responsible for ongoing studies and contacted them for preliminary data for these studies.
We searched the reference lists of all included trials for further relevant trials.
Data collection and analysis
Selection of studies
We downloaded all titles and abstracts retrieved by electronic searching to the reference management database, Endnote 2012, and removed duplicates where possible. At least two review authors (IM, KH in the initial version of the review and a combination of AW, GC, JM and TL for the updated review) independently examined the remaining references. We excluded those studies that clearly did not meet the inclusion criteria, and obtained copies of the full text of potentially relevant references. At least two review authors (IM, KH for initial review and a combination of AW, GC, JM and TL for the update) independently assessed the eligibility of retrieved papers. We documented reasons for exclusion.
Data extraction and management
For included studies, we abstracted data as follows:
Author, year of publication and journal citation (including language)
Country
Setting
Inclusion and exclusion criteria
Study design, methodology
-
Study population
Total number enrolled
Patient characteristics
Age
Co‐morbidities
Previous treatment
Total study duration
Total number of intervention groups
-
Ovarian cancer details at diagnosis
FIGO stage
Histological cell type
Tumour grade
Extent of disease
-
Intervention details
Type of DNA‐repair pathway inhibitor
Dose
Duration of treatment
Consolidation treatment or treatment of active disease
-
Comparison details
Type of control: conventional chemotherapy or no treatment
Dose (if appropriate)
Duration (if appropriate)
Deviations from protocol
Risk of bias in study (see below)
Duration of follow‐up
-
Outcomes: overall survival, progression‐free survival, quality of life, toxicity:
for each outcome: outcome definition (with diagnostic criteria if relevant);
unit of measurement (if relevant);
for scales: upper and lower limits, and whether high or low score is good;
results: number of participants allocated to each intervention group;
for each outcome of interest: sample size; missing participants.
We extracted data on outcomes as below:
For time to event (overall and progression‐free survival) data, we extracted the log of the hazard ratio (log(HR)) and its standard error from trial reports; if these were not reported, we attempted to estimate them from other reported statistics using the methods of Parmar 1998.
For dichotomous outcomes (e.g. toxicity or deaths if it was not possible to use a HR), we extracted the number of patients in each treatment arm who experienced the outcome of interest and the number of patients assessed at endpoint, in order to estimate a risk ratio (RR).
For continuous outcomes (e.g. quality of life measures), we extracted the final value and standard deviation of the outcome of interest and the number of patients assessed at endpoint in each treatment arm at the end of follow‐up, in order to estimate the mean difference (if trials measured outcomes on the same scale) or standardised mean difference (if trials measured outcomes on different scales) between treatment arms and its standard error.
We extracted both unadjusted and adjusted statistics, if reported. When we extracted adjusted results, we recorded the variables that were adjusted for.
Where possible, all data extracted were relevant to an intention‐to‐treat analysis, in which participants were analysed in the groups to which they were assigned.
We noted the time points at which outcomes were collected and reported.
At least two review authors (GC, AW and JM) independently extracted data onto a data extraction form specially designed for the review. We resolved differences between review authors by discussion or by appeal to a third review author (TL) if necessary.
Assessment of risk of bias in included studies
We assessed the risk of bias in included RCTs using The Cochrane Collaboration's tool and the criteria specified in Chapter 8 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). This included assessment of:
sequence generation;
allocation concealment;
blinding (of participants, healthcare providers and outcome assessors);
-
incomplete outcome data:
-
we coded the satisfactory level of loss to follow‐up for each outcome as:
yes, if fewer than 20% of patients were lost to follow‐up and reasons for loss to follow‐up were similar in both treatment arms;
no, if more than 20% of patients were lost to follow‐up or reasons for loss to follow‐up differed between treatment arms;
unclear if loss to follow‐up was not reported.
-
selective reporting of outcomes;
other possible sources of bias.
Two review authors (GC, AW) independently applied the risk of bias tool and resolved differences by discussion or by appeal to a third review author (JM). We summarised results in both a 'Risk of bias' graph and a 'Risk of bias' summary. We interpreted the results of meta‐analyses in the light of the findings with respect to risk of bias.
Measures of treatment effect
We used the following measures of the effect of treatment:
For time to event data, we used the HR, if possible.
For dichotomous outcomes, we used the risk ratio (RR).
For continuous outcomes, we used the mean difference (MD) between treatment arms if all trials measured the outcome on the same scale, otherwise we used standardised mean differences (SMD).
Dealing with missing data
We did not impute missing outcome data; if only imputed outcome data were reported, we contacted trial authors to request data on the outcomes only among participants who were assessed.
Assessment of heterogeneity
We assessed heterogeneity between studies by visual inspection of forest plots, by estimation of the percentage of heterogeneity between trials which cannot be ascribed to sampling variation (Higgins 2003), and by a formal statistical test of the significance of the heterogeneity (Deeks 2001). If there was evidence of substantial heterogeneity, we investigated and reported the possible reasons for this.
Assessment of reporting biases
We did not produce funnel plots corresponding to meta‐analysis due to the limited number of included studies. In future versions of this review, we will examine funnel plots for meta‐analysis of the primary outcome to assess the potential for small‐study effects. When there is evidence of small‐study effects, we will consider publication bias as only one of a number of possible explanations. If these plots suggest that treatment effects may not be sampled from a symmetric distribution, as assumed by the random‐effects model, we will perform sensitivity analyses using fixed‐effect models.
Data synthesis
When sufficient clinically similar trials were available we pooled their results in meta‐analyses.
For time‐to‐event data, we pooled HRs using the generic inverse variance facility of RevMan 5.3 (RevMan 2014).
For dichotomous outcomes, we calculated the RR for each study and pooled these.
No continuous data were synthesised for this review. In future versions of this review, for continuous outcomes (e.g. quality of life) we will pool the mean differences between the treatment arms at the end of follow‐up if all trials measured the outcome on the same scale, otherwise we will pool standardised mean differences.
We used random‐effects models with inverse variance weighting for all meta‐analyses (DerSimonian 1986).
Subgroup analysis and investigation of heterogeneity
As we expected to find only a few trials, we did not plan initially any subgroup analyses. However, we considered factors such as type of intervention (e.g. use as early‐stage consolidation therapy in chemo‐sensitive cancers or use in late‐stage chemo‐resistant cancers) and stage of disease in the interpretation of any heterogeneity. Data so far suggest that responses depend on platinum‐sensitivity, BRCA‐mutation status or BRCAness of the tumour. In future updates we will also perform subgroup analysis based on platinum‐sensitivity and BRCA‐mutation status.
Sensitivity analysis
There were too few studies to perform sensitivity analysis. In future versions of the review we will perform sensitivity analysis excluding (i) studies at high risk of bias, and (ii) unadjusted results.
Results
Description of studies
See Characteristics of included studies; Characteristics of excluded studies; Characteristics of ongoing studies.
Results of the search
In 2009, we ran an initial broad search that yielded 473 unique references after deletion of duplicates. Updated searches conducted in October 2013 and May 2014 yielded a further 1062 and136 references respectively, resulting in a total of 1671 references from the combined searches. Two review authors (a combination of AW, GC, JM, TL) independently reviewed the abstracts and we excluded articles that obviously did not meet the inclusion criteria. The original review identified 14 articles and the updated searches identified an additional 10 articles, which we retrieved in full and translated into English where appropriate. The full‐text screening excluded 17 trials for the reasons described in the table Characteristics of excluded studies. Four individual studies (comprising seven citations) met the inclusion criteria. Two ongoing studies in the original review (Assessment of AZD2281a; ICEBERG 3a) from the clinical trials databases were versions of studies that are now included in the updated review (see Characteristics of included studies). For the PRISMA flowchart see Figure 1.
Figure 1.
Study flow diagram.
Searches of clinical trials registries and discussion with reviewers and the National Institute for Clinical Effectiveness Technology Appraisal scoping meeting participants had identified five additional ongoing relevant studies, although we excluded one on further investigation (total 18 records excluded at this stage), as it is an open‐label, non‐randomised trial (see Characteristics of ongoing studies and NCT01891344).
The updated search in April 2015, just prior to publication, found an additional 238 references (making 1909 in total), but did not identify any new studies. Seventeen were retrieved in full text. Five of these were excluded (see Excluded studies). Of the other 12 references: one reference was to the published version of a study to which we had been given data pending publication (Oza 2015); six references were additional references to included studies (Ledermann 2012; Kummar 2015) and another five references were abstracts presented at meetings of previously identified on‐going studies (NCT01844986; NCT01847274; NCT01874353; NCT01968213)
Therefore, we included four studies in total (comprising 14 references), excluded 22 studies (23 references) and classified four studies (nine references) as ongoing.
Included studies
Four studies met our inclusion criteria (Kaye 2012; Kummar 2015; Ledermann 2012; Oza 2015), although data from only two studies had been published in peer‐reviewed journals at the time of the initial search and data extraction (Kaye 2012; Ledermann 2012). Kummar 2015 was initially only published in abstract form; this trial was discontinued early. Limited outcome data for Oza 2015 were published on a clinical trial registry website and at two recent conferences. However, final results were provided by the authors prior to publication of the study. Attempts to obtain additional data/clarification from the investigators of Kummar 2015 were met with limited success, since the authors were reluctant to release further data until their study was published and data were updated just prior to submission of this review. See Characteristics of included studies for further details.
PARP inhibitor versus conventional chemotherapy
One study was included in this comparison. Kaye 2012 compared olaparib to pegylated liposomal doxorubicin (PLD). Ninety‐seven women with EOC who had relapsed within 12 months of platinum‐based chemotherapy (i.e. platinum‐resistant and partially platinum‐sensitive disease) were randomised to one of three treatment arms (olaparib 200 mg, olaparib 400 mg, PLD 50 mg) in a ratio of 1:1:1. There were 32, 32 and 33 women in each arm, respectively. All included women had BRCA mutations; approximately 80% in each group had BRCA1 mutations, although the rate was slightly higher in the olaparib 400 mg group (see Characteristics of included studies). Approximately 50% of women in each group had relapsed within six months of platinum‐based chemotherapy (platinum‐resistant disease). All women had measurable disease according to Response Evaluation Criteria in Solid Tumours (RECIST).
PARP inhibitor versus placebo (as maintenance)
One study was included in this comparison. Ledermann 2012 compared olaparib with placebo as maintenance therapy in women with platinum‐sensitive EOC (relapse after six months of previous platinum‐based chemotherapy). The study enrolled 265 women (136 received olaparib, 129 received placebo, although one woman in the placebo group withdrew consent prior to treatment and evaluations excluded this (non)participant). Participants were required to have received two previous courses of platinum‐based chemotherapy, the most recent of which was to have induced an objective response. All women had normal Ca125 levels and 40% had measurable disease by RECIST. BRCA testing was not mandatory and known BRCA mutation status was similar in the two groups (around 22%), as were other associated factors, e.g. Jewish ancestry.
PARP inhibitor plus conventional chemotherapy versus conventional chemotherapy alone
Two studies were included in this comparison (Kummar 2015; Oza 2015). Oza 2015 compared olaparib with platinum‐based chemotherapy versus platinum‐based chemotherapy alone in 162 women with platinum‐sensitive recurrent serous EOC. BRCA mutation status and BRCA testing was not mandatory, however, 41/107 tested (38%) had BRCA mutations. Randomisation was stratified according to platinum sensitivity. Study interventions comprised olaparib (200 mg bd for 10 days) or placebo added to each conventional platinum‐based chemotherapy cycle and then continued as monotherapy maintenance (400 mg bd continuous) thereafter. Of 162 women randomised, 156 received treatment (81 olaparib versus 75 placebo) and, of these, 121 began the maintenance/no further therapy phase (66 olaparib versus 55 no maintenance).
Kummar 2015 compared veliparib with cyclophosphamide versus cyclophosphamide alone. Data from Kummar 2015 are limited and we were unsuccessful in obtaining significant additional data or clarification from the investigators. The study was closed early due to poor responses observed at interim analysis, when only half the participants had been accrued. The final results of this trial were published after the search date of the review. In total 75 women were recruited (37 cyclophosphamide plus veliparib and 38 cyclophosphamide) and there was no difference in PFS or response rates between the two groups (PFS hazard ratio 1.02, 95% confidence interval 0.69 to 1.50).
Excluded studies
We excluded 23 references after obtaining the full text, for the following reasons:
Four references were non‐randomised, single‐arm, phase I studies of one PARP inhibitor (AZD2281) (Fong 2006; Fong 2008; Fong 2009; Yap 2007);
Ten references were narrative review articles and did not include any other study that met our inclusion criteria (Ashworth 2008; Banerjee 2013; Chen 2013; Drew 2008; Helleday 2008; Lord 2008; Muggia 2009; Shaw 2013; Turner 2005; Yap 2009);
Five references were non‐randomised, phase II cohort studies of PARP inhibitors (Audeh 2009; Audeh 2010; Coleman 2014; Gelmon 2011; NCT01891344);
Two references (Lui 2014) were to an RCT comparing Olaparib plus or minus Cediranib (no randomisation for Olaparib);
One reference (Moore 2014) was an abstract about an on‐going study, but aimed to analyse effects of diet on pharmacokinetics;
One reference was to a biomarker analysis in an excluded RCT (Lee 2014 analysing results from RCT by Lui 2014).
Risk of bias in included studies
We included four studies and evaluated them for risk of bias. We considered three studies to be at a low (Ledermann 2012) to moderate (Kaye 2012; Oza 2015) risk of bias (risk mainly due to lack of blinding). We considered one study to be at a high risk of bias as it closed early and remains unpublished (Kummar 2015). This study could not be included in any meta‐analyses due to insufficient data. See Figure 2.
Figure 2.
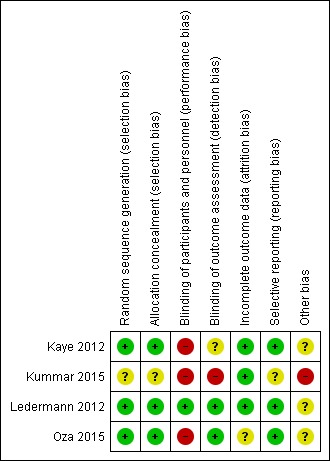
'Risk of bias' summary: review authors' judgements about each risk of bias item for each included study.
Effects of interventions
See: Table 1; Table 2; Table 3
Overall survival (OS)
The included studies were not powered for OS, however there were no differences between PARP inhibitors and any of the control groups in any of the studies individually (Analysis 1.1). Similarly, there was no significant difference in OS when we pooled data from the two studies that included participants with platinum‐sensitive disease (HR 1.05, 95% CI 0.79 to 1.39; 426 participants; I² = 0%; Analysis 1.2). This evidence is of moderate quality and estimates of effect might change with further research.
Analysis 1.1.
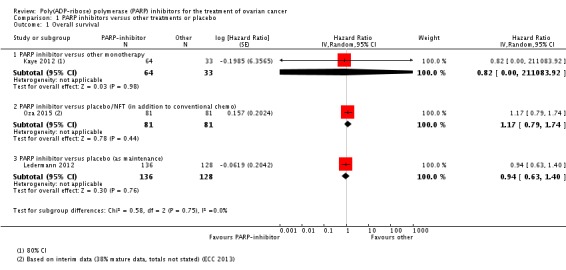
Comparison 1 PARP inhibitors versus other treatments or placebo, Outcome 1 Overall survival.
Analysis 1.2.
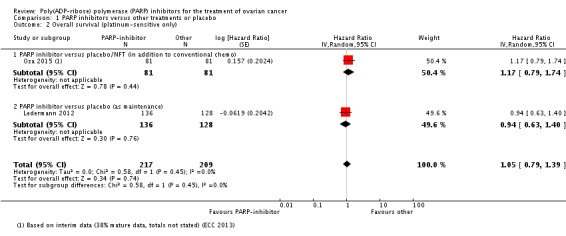
Comparison 1 PARP inhibitors versus other treatments or placebo, Outcome 2 Overall survival (platinum‐sensitive only).
Progression‐free survival (PFS)
One study contributed to the subgroup PARP inhibitors versus other monotherapy (97 participants) (Kaye 2012). In this study, PARP inhibitors (olaparib) resulted in similar PFS compared with pegylated liposomal doxorubicin (PLD) monotherapy in women with platinum‐resistant and partially platinum‐sensitive disease (hazard ratio (HR) 0.88, 95% confidence interval (CI) 0.51 to 1.53; Analysis 1.3). Overall, median PFS was 8.8 months in the olaparib (400 mg) arm (95% CI 5.4 to 9.2 months) and 7.1 months (95% CI 3.7 to 10.7 months) in the PLD arm.
Analysis 1.3.
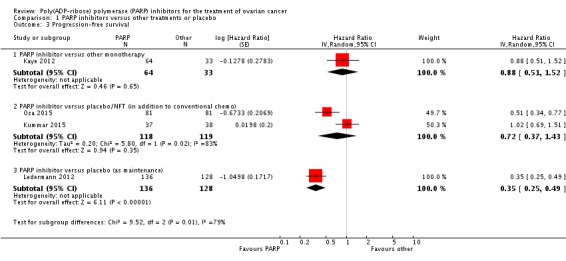
Comparison 1 PARP inhibitors versus other treatments or placebo, Outcome 3 Progression‐free survival.
PARP inhibitors improved PFS when added to conventional treatment in women with platinum‐sensitive disease (one study, Oza 2015, 162 participants), and when used as additional maintenance treatment in women with platinum‐sensitive disease compared with placebo (one study, Ledermann 2012, 264 participants (not including one woman in the placebo group who withdrew consent to the study prior to commencing treatment and for whom no follow‐up data were available)) (Analysis 1.3). Combining data from the latter studies gave an average HR of 0.42 (95% CI 0.29 to 0.60; 426 participants; I² = 49%; Analysis 1.4). Heterogeneity in this analysis was probably due to differences in the types of participants; women in Ledermann 2012 were required to have received and responded to at least two platinum‐based chemotherapy regimens, whereas most women in Oza 2015 had received only one previous platinum‐based regimen and maintenance treatment was administered to women irrespective of response. Median PFS was 8.4 months in the PARP inhibitor group and 4.8 months in the placebo group in Ledermann 2012, whereas in Oza 2015, median PFS was 12.2 months and 9.6 months for PARP inhibitor and placebo groups, respectively. This evidence is of moderate quality and estimates of effect might change with further research. Data from Oza 2015 of 41 patients with BRCA mutations (20 in the olaparib group and 21 in the control group) suggest that olaparib had the greatest benefit in this subgroup of patients (HR 0.21, 95% CI 0.08 to 0.55).
Analysis 1.4.
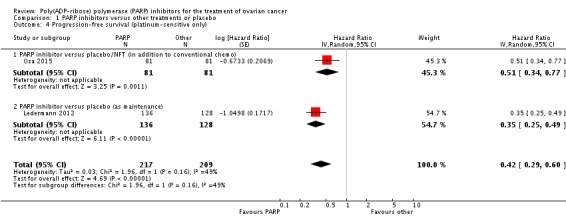
Comparison 1 PARP inhibitors versus other treatments or placebo, Outcome 4 Progression‐free survival (platinum‐sensitive only).
There was no difference in PFS for cyclophosphamide plus veliparib compared to or cyclophosphamide alone (2.1 months compared to 2.3 months; HR 1.02, 95% CI 0.69 to 1.50) (Kummar 2015).
Objective response rate (ORR)
Not all women had Response Evaluation Criteria in Solid Tumours (RECIST) evaluable disease in these studies. In Ledermann 2012 (40% evaluable), the ORR was 12% (7/57 women in the olaparib group) versus 4% (2/48 women in the placebo group). In Kaye 2012 (100% evaluable), the ORR was 28% (18/64) versus 18% (6/33) for the olaparib and placebo groups, respectively. In Oza 2015 (100% evaluable), complete ORR was 10% (8/81) versus 7% (6/81), respectively. Overall, there was a small difference in ORR when we pooled data for non‐response from the four studies (Analysis 1.5: RR 0.90, 95% CI 0.82 to 0.99; I2 = 0%).
Analysis 1.5.
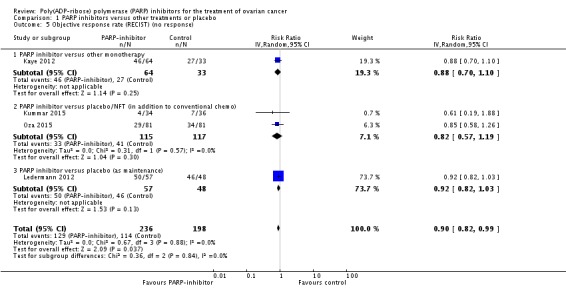
Comparison 1 PARP inhibitors versus other treatments or placebo, Outcome 5 Objective response rate (RECIST) (no response).
Adverse events
There were no differences in gastrointestinal and haematological serious adverse events between the experimental and control groups (Analysis 1.6). Palmar‐plantar erythrodysesthesia was more common in the PLD arm of Kaye 2012 (0/64 versus 12/32 women affected). However, combining adverse event data from Oza 2015 and Ledermann 2012 during the maintenance phases resulted in a trend towards more adverse events in the olaparib group compared with controls, and fatigue of any grade was more common (two studies, 385 participants; RR 1.35, 95% CI 1.02 to 1.78; Analysis 1.7). In addition, there was an increase in the risk of anaemia in the maintenance phase, with more women in the olaparib group experiencing grade 3/4 events (RR 5.26, 95% CI 1.19 to 23.20; Analysis 1.8). There was no difference in adverse events of any grade (Analysis 1.9), reflecting the high level of mild symptoms in women with advanced ovarian cancer. However, when serious adverse events (grade 3/4) were considered, these were more common in the olaparib maintenance arm (RR 1.74, 95% CI 1.22 to 2.49; Analysis 1.10).
Analysis 1.6.

Comparison 1 PARP inhibitors versus other treatments or placebo, Outcome 6 Severe adverse events.
Analysis 1.7.
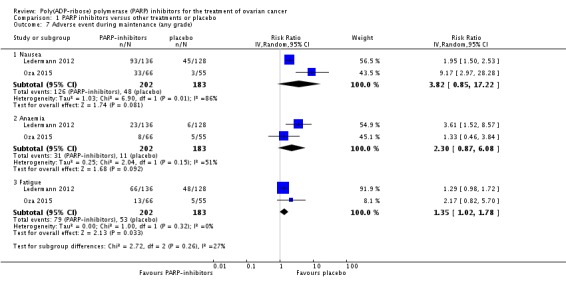
Comparison 1 PARP inhibitors versus other treatments or placebo, Outcome 7 Adverse event during maintenance (any grade).
Analysis 1.8.
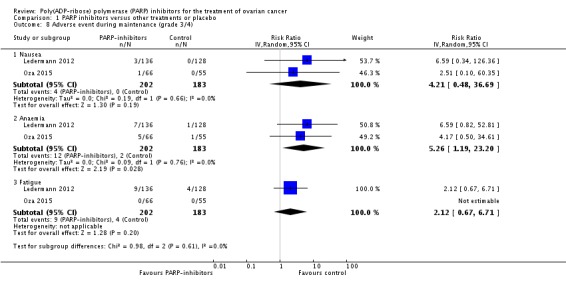
Comparison 1 PARP inhibitors versus other treatments or placebo, Outcome 8 Adverse event during maintenance (grade 3/4).
Analysis 1.9.
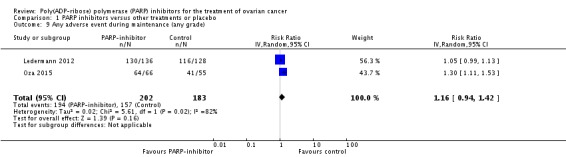
Comparison 1 PARP inhibitors versus other treatments or placebo, Outcome 9 Any adverse event during maintenance (any grade).
Analysis 1.10.
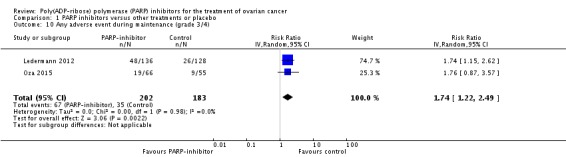
Comparison 1 PARP inhibitors versus other treatments or placebo, Outcome 10 Any adverse event during maintenance (grade 3/4).
Quality of life
Quality of life was reported as not different between treatment groups in Ledermann 2012 and Kaye 2012 (using FACT‐O and Trial Outcome Index), however meta‐analysis was not possible due to insufficient data. Quality of life was not assessed in Oza 2015.
Discussion
Summary of main results
Four studies included 599 women with epithelial ovarian cancer (EOC). One study compared olaparib to pegylated liposomal doxorubicin (PLD) in women with BRCA mutations and platinum‐resistant or partially platinum‐sensitive disease (Kaye 2012). In this study there was no difference in progression‐free survival (PFS) between olaparib and PLD (Table 1), although PFS was longer than expected from historical controls, indicating a survival advantage with both PLD and olaparib in BRCA‐mutation carriers.
Ledermann 2012 and Oza 2015 recruited women with platinum‐sensitive disease, and found an improvement in PFS when olaparib (alongside conventional treatment and/or when used as maintenance treatment) was compared to a placebo or no further treatment. This improvement was statistically significant for the individual studies and when combined in meta‐analysis (hazard ratio (HR) 0.42, 95% confidence interval (CI) 0.29 to 0.60; I² = 49%) (Table 2). We attributed heterogeneity in this analysis to differences in the types of participants involved in these two studies. There was no difference between olaparib and placebo/control arms with regard to overall survival (OS), although the studies were not powered to detect differences in OS, so further data might change this outcome.
Compared with PLD, olaparib had an improved toxicity profile due to lack of palmar‐plantar erythrodysesthesia, which is a known side effect of PLD. Olaparib was associated with a greater risk of severe adverse events (mainly anaemia and fatigue) when given as maintenance treatment.
Overall completeness and applicability of evidence
The review evidence relates primarily to one PARP inhibitor, olaparib. Data regarding veliparib were limited (Kummar 2015), and it is not known whether our findings would apply to this or other new PARP inhibitors, e.g. niraparib and rucaparib, which are currently undergoing evaluation in clinical trials.
In addition, the population for whom PARP inhibitors are most effective has yet to be fully evaluated and it is possible that only a small subset of women may benefit from PARP inhibitors. PARP inhibitors appear to have a better effect in women with platinum‐sensitive disease and ongoing clinical trials are focusing on women undergoing first‐line treatment or with platinum‐sensitive disease who have responded to platinum agents in their most recent course of chemotherapy. Platinum sensitivity is more common in women with germline BRCA mutations, and in those women whose tumours have BRCA mutations. Defining exactly which patients will benefit from PARP inhibitors is therefore a challenge and the need for BRCA‐mutation testing or testing for homologous recombination defects in tumours to define 'BRCA‐ness' will add additional costs to treatment, unless BRCA‐mutation testing becomes standard care for women with high‐grade serous histological subtype of EOC. This may be the case since approximately 16% of women with high‐grade serous EOC (depending on population) have BRCA germline mutations (Risch 2001). This is higher than the 10% risk in breast cancer families when BRCA‐testing should be considered, according to the National Institute of Clinical Excellence (NICE) clinical guidelines for women with a family history of breast cancer (NICE CG164). Participants in the Kaye 2012 trial were restricted to those with BRCA mutations, which may explain the longer survival of women in this study compared with earlier PLD studies. BRCA‐testing was not compulsory in either Ledermann 2012 or Oza 2015, however, these studies recruited women with platinum‐sensitive disease, which is associated with higher BRCA‐mutation rates then the general EOC population. Therefore, these results are currently only applicable to the subgroup of women with BRCA‐mutations or platinum‐sensitive EOC.
From the available data, it is not clear whether PARP inhibitors only delay the onset of recurrent disease, or whether there is an OS benefit for certain subgroups of women. OS endpoints are harder to obtain, since they require longer for the data to mature. In addition, the effects of individual therapeutic agents can be obscured due to the effects of other treatments, especially in EOC where women often have multiple rounds (or lines) of treatment over what can be several years. A more complete picture will emerge with further randomised controlled trials (RCTs) to test their effectiveness and toxicity, and clinicians and eligible women are encouraged to seek out treatment within international RCTs to help answer these questions.
Serious adverse events, which were more common in women receiving olaparib maintenance treatment, may have a significant impact on quality of life. We were unable to evaluate quality of life due to insufficient data and more evidence on this is needed.
Quality of the evidence
Three studies that contributed data appear to be well conducted with pre‐defined outcome criteria and robust randomisation systems. Data from the fourth study, relating to veliparib, were limited and at high risk of bias (Kummar 2015). However, all of the studies were small, open‐label phase II trials and potentially liable to bias. Only one study contributed data to the evidence relating to PARP inhibitor monotherapy in platinum‐resistant and partially platinum‐sensitive disease and we graded this evidence as very low to low quality (see Table 1). With regard to platinum‐sensitive disease, we graded PFS outcomes as moderate quality due to inconsistency (clinical heterogeneity) as per the GRADE criteria (see Table 2) and we graded the quality of the evidence for OS and most serious adverse events as moderate, due to imprecision (see Table 2). Ongoing, appropriately powered, phase III, randomised and blinded studies will have an important impact on our confidence in the estimates of effect and may change the conclusions of this review in the future. We found no good evidence on quality of life.
Potential biases in the review process
We are not aware of any biases in the review process. We conducted this review using standard Cochrane methodology, which aims to reduce bias through double sifting, double data extraction and transparent grading of evidence. None of the authors have any links to drug companies,a financial interest in the prescription of chemotherapeutic agents, nor were they involved in the conduct of any of the included studies.
Agreements and disagreements with other studies or reviews
To date we have not identified any systematic reviews of PARP inhibitors. One review article of PARP inhibitors in gynaecological cancers, including epithelial ovarian cancer, did not identify any additional studies (Reinbolt 2013), and did not include a meta‐analysis of the results.
Authors' conclusions
Women with epithelial ovarian cancer (EOC) high‐grade serous and endometrial serotypes have a relatively high risk of germline BRCA mutation and should be offered genetic screening, if criteria recommended for breast cancer families are applied (NICE CG164; Risch 2001). This is irrespective of whether PARP inhibitors are effective, since it has implications for patients and their families.
These data suggest that there is likely to be a role for PARP inhibitors in the treatment of EOC. Progression‐free survival (PFS) appears to be improved in women with recurrent platinum‐sensitive disease. Limited data suggest that severe adverse effects are uncommon. However, beneficial effects in terms of overall survival (OS) have not been adequately demonstrated and more data are required to determine whether longer PFS translates into an improved (or reduced) OS for subgroups of women with this disease. More data are expected from ongoing phase III clinical trials and at present the use of PARP inhibitors should be encouraged within these studies. However, the European Medicines Agency (EMA) approved olaparib for "monotherapy for the maintenance treatment of adult patients with platinum‑sensitive relapsed BRCA‐mutated (germline and/or somatic) high grade serous epithelial ovarian, fallopian tube, or primary peritoneal cancer who are in response (complete response or partial response) to platinum‐based chemotherapy" in 2014 (EMA 2014). This decision was based on data supplied to the EMA by AstraZeneca. Unpublished data were supplied by AstraZeneca for this review and so it is presumed that the EMA recommendation was based on the results presented here.
Olaparib has been recommended for maintenance treatment after good clinical responses to platinum agents (EMA 2014). This is prior to the publication of phase III studies and OS outcomes. These women may be otherwise well and so it is important to collect good quality of life and adverse event data in ongoing studies to inform women in their risk/benefit decisions. Ongoing studies are limited to women with platinum‐sensitive/responsive disease, either after first‐line treatment or on recurrence, and those with BRCA mutations, either germline or somatic, or likely to behave as if they have BRCA mutations (BRCAness: high‐grade serous tumours with platinum sensitivity and response to a second‐line platinum agent). However, tumours also respond better to conventional chemotherapy, so the challenge remains to improve outcomes for women with poorer prognosis mutations.
Questions remain about how best to use PARP inhibitors, whether to use them in combination with chemotherapy or as maintenance alone and, if used in combination, which drugs to combine with PARP inhibitors. Pre‐clinical studies suggest that PARP inhibitors may work well in combination with chemotherapeutic agents and may sensitise cells to these agents, thereby delaying the onset of drug resistance. Other possibilities for combination treatment with PARP inhibitors include anti‐angiogenic agents or in combination with cyclophosphamide or weekly paclitaxel. Pre‐clinical data suggest that inhibiting vascular endothelial growth factor receptor (VEGFR) may lead to down‐regulation of DNA‐repair activity by DNA‐repair proteins, ERCC1 and XRCC1 (Yadav 2011). This may lead to increased DNA damage and, thereby, increase susceptibility to the effects of PARP inhibition. Clinical studies of PARP inhibitors in combination with chemotherapy agents are ongoing. Future studies should include OS and quality of life as important outcomes. In women with platinum‐resistant EOC objective responses to both PARP inhibitors and pegylated liposomal doxorubicin (PLD) were demonstrated at higher levels than previous studies of women with platinum‐resistant EOC in non‐selected populations (Kaye 2012). PARP inhibitors in combination with other chemotherapeutic agents could be tested in this population, as well as women with platinum‐sensitive‐disease, especially as PARP inhibitors appear to be relatively well tolerated, which is important for women with poorer prognosis, where quality of life issues are even more important.
One argument for using PFS as the primary endpoint in these studies is that they included women with heavily pre‐treated disease, who represent a very heterogenous population. PFS might be a better test of current treatment than OS in this setting. However, it would be important to include OS as a primary outcome measure in future studies, especially those including women at first or second‐line treatment. Many questions remain to be answered regarding optimal drug combinations, scheduling and patient selection for PARP inhibitors, although results so far offer promise.
Acknowledgements
We thank Jane Hayes for help with designing and conducting the searches; Igor Martinek (IM), Krish Haldar (KH), Kezia Gaitskell (KG), Shibani Nicum (SN) and Sean Kehoe (SK) for their work on the original protocol and empty review; Heather Dickinson (HD) for advice on developing the original protocol, Marcia Hall (Contact Editor) for clinical and editorial advice, and Gail Quinn and Clare Jess (Managing Editors) for their contribution to the editorial process. We thank the referees for their many helpful suggestions and Emma Cattell and Michelle Lockley for thought‐provoking discussions that contributed invaluably to the review discussion. We also thank Anitra Fielding and her team from AstraZeneca for helpfully providing additional data for analysis prior to publication and Carol‐Ann Regan and her team from Musgrove Park Hospital Library for their assistance with acquiring full‐text articles.
The National Institute for Health Research (NIHR) is the largest single funder of the Cochrane Gynaecological Cancer Group. The views and opinions expressed therein are those of the authors and do not necessarily reflect those of the NIHR, NHS or the Department of Health.
Appendices
Appendix 1. CENTRAL search strategy
#1 ovar* and (cancer* or carcinom* or neoplasm* or tumor* or tumour* or malignan*) #2 MeSH descriptor Ovarian Neoplasms explode all trees #3 (#1 OR #2) #4 MeSH descriptor DNA Repair Enzymes explode all trees #5 MeSH descriptor DNA Repair explode all trees #6 DNA repair #7 MeSH descriptor Poly(ADP‐ribose) Polymerases explode all trees #8 PARP near/5 inhibit* #9 poly ADP ribose polymerase near/5 inhibit* #10 olaparib or AZD2281 or KU59436 #11 AG014699 #12 ABT‐888 #13 BSI‐201 #14 INO‐1001 #15 MK4827 #16 (#4 OR #5 OR #6 OR #7 OR #8 OR #9 OR #10 OR #11 OR #12 OR #13 OR #14 OR #15) #17 (#3 AND #16)
Appendix 2. MEDLINE search strategy
1 randomized controlled trial.pt. 2 controlled clinical trial.pt. 3 randomized.ab. 4 placebo.ab. 5 drug therapy.fs. 6 randomly.ab. 7 trial.ab. 8 groups.ab. 9 1 or 2 or 3 or 4 or 5 or 6 or 7 or 8 10 (animals not (humans and animals)).sh. 11 9 not 10 12 ovar*.mp. 13 (cancer* or carcinoma*or neoplasm* or tumor*or tumour*or malignan*).mp. 14 12 and 13 15 exp Ovarian Neoplasms/ 16 14 or 15 17 exp DNA Repair Enzymes/ 18 exp DNA Repair/ 19 DNA repair.mp. 20 exp "Poly(ADP‐ribose) Polymerases"/ 21 (PARP adj5 inhibit*).mp. 22 (poly ADP ribose polymerase adj5 inhibit*).mp. 23 (olaparib or AZD2281 or KU59436).mp. 24 AG014699.mp. 25 ABT‐888.mp. 26 BSI‐201.mp. 27 INO‐1001.mp. 28 MK4827.mp. 29 17 or 18 or 19 or 20 or 21 or 22 or 23 or 24 or 25 or 26 or 27 or 28 30 11 and 16 and 29
key: pt=publication type ab=abstract fs=floating subheading mp=title, original title, abstract, name of substance word, subject heading word sh=subject heading
Appendix 3. EMBASE search strategy
1 exp Controlled Clinical Trial/ 2 randomized.ab. 3 placebo.ab. 4 dt.fs. 5 randomly.ab. 6 trial.ab. 7 groups.ab. 8 1 or 2 or 3 or 4 or 5 or 6 or 7 9 (animal not (human and animal)).sh. 10 8 not 9 11 (ovar* and (cancer* or carcinoma* or neoplas* or tumor* or tumour* or malignan*)).mp. 12 exp Ovary Tumor/ 13 11 or 12 14 exp Polydeoxyribonucleotide Synthase/ 15 exp DNA Repair/ 16 DNA repair.mp. 17 exp Nicotinamide Adenine Dinucleotide Adenosine Diphosphate Ribosyltransferase/ 18 (PARP adj5 inhibit*).mp. 19 (poly ADP ribose polymerase adj5 inhibit*).mp. 20 (olaparib or AZD2281 or KU59436).mp. 21 AG014699.mp. 22 ABT‐888.mp. 23 BSI‐201.mp. 24 INO‐1001.mp. 25 MK4827.mp. 26 14 or 15 or 16 or 17 or 18 or 19 or 20 or 21 or 22 or 23 or 24 or 25 27 10 and 13 and 26
key: ab=abstract fs=floating subheading sh=subject heading mp=title, abstract, subject headings, heading word, drug trade name, original title, device manufacturer, drug manufacturer name
Data and analyses
Comparison 1.
PARP inhibitors versus other treatments or placebo
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Overall survival | 3 | Hazard Ratio (Random, 95% CI) | Subtotals only | |
| 1.1 PARP inhibitor versus other monotherapy | 1 | 97 | Hazard Ratio (Random, 95% CI) | 0.82 [0.00, 211083.92] |
| 1.2 PARP inhibitor versus placebo/NFT (in addition to conventional chemo) | 1 | 162 | Hazard Ratio (Random, 95% CI) | 1.17 [0.79, 1.74] |
| 1.3 PARP inhibitor versus placebo (as maintenance) | 1 | 264 | Hazard Ratio (Random, 95% CI) | 0.94 [0.63, 1.40] |
| 2 Overall survival (platinum‐sensitive only) | 2 | 426 | Hazard Ratio (Random, 95% CI) | 1.05 [0.79, 1.39] |
| 2.1 PARP inhibitor versus placebo/NFT (in addition to conventional chemo) | 1 | 162 | Hazard Ratio (Random, 95% CI) | 1.17 [0.79, 1.74] |
| 2.2 PARP inhibitor versus placebo (as maintenance) | 1 | 264 | Hazard Ratio (Random, 95% CI) | 0.94 [0.63, 1.40] |
| 3 Progression‐free survival | 4 | Hazard Ratio (Random, 95% CI) | Subtotals only | |
| 3.1 PARP inhibitor versus other monotherapy | 1 | 97 | Hazard Ratio (Random, 95% CI) | 0.88 [0.51, 1.52] |
| 3.2 PARP inhibitor versus placebo/NFT (in addition to conventional chemo) | 2 | 237 | Hazard Ratio (Random, 95% CI) | 0.72 [0.37, 1.43] |
| 3.3 PARP inhibitor versus placebo (as maintenance) | 1 | 264 | Hazard Ratio (Random, 95% CI) | 0.35 [0.25, 0.49] |
| 4 Progression‐free survival (platinum‐sensitive only) | 2 | 426 | Hazard Ratio (Random, 95% CI) | 0.42 [0.29, 0.60] |
| 4.1 PARP inhibitor versus placebo/NFT (in addition to conventional chemo) | 1 | 162 | Hazard Ratio (Random, 95% CI) | 0.51 [0.34, 0.77] |
| 4.2 PARP inhibitor versus placebo (as maintenance) | 1 | 264 | Hazard Ratio (Random, 95% CI) | 0.35 [0.25, 0.49] |
| 5 Objective response rate (RECIST) (no response) | 4 | 434 | Risk Ratio (IV, Random, 95% CI) | 0.90 [0.82, 0.99] |
| 5.1 PARP inhibitor versus other monotherapy | 1 | 97 | Risk Ratio (IV, Random, 95% CI) | 0.88 [0.70, 1.10] |
| 5.2 PARP inhibitor versus placebo/NFT (in addition to conventional chemo) | 2 | 232 | Risk Ratio (IV, Random, 95% CI) | 0.82 [0.57, 1.19] |
| 5.3 PARP inhibitor versus placebo (as maintenance) | 1 | 105 | Risk Ratio (IV, Random, 95% CI) | 0.92 [0.82, 1.03] |
| 6 Severe adverse events | 4 | Risk Ratio (IV, Random, 95% CI) | Subtotals only | |
| 6.1 Nausea (G3‐4) | 4 | 592 | Risk Ratio (IV, Random, 95% CI) | 1.23 [0.33, 4.60] |
| 6.2 Diarrhoea (G3‐4) | 4 | 592 | Risk Ratio (IV, Random, 95% CI) | 0.53 [0.15, 1.90] |
| 6.3 Vomiting (G3‐4) | 4 | 592 | Risk Ratio (IV, Random, 95% CI) | 1.42 [0.25, 8.10] |
| 6.4 Stomatitis (any grade) | 3 | 503 | Risk Ratio (IV, Random, 95% CI) | 0.44 [0.02, 10.15] |
| 6.5 Anaemia (G3‐4) | 4 | 592 | Risk Ratio (IV, Random, 95% CI) | 2.15 [0.89, 5.21] |
| 6.6 Neutropenia (G3‐4) | 2 | 220 | Risk Ratio (IV, Random, 95% CI) | 0.59 [0.09, 3.98] |
| 6.7 Other (G3‐4) | 3 | 483 | Risk Ratio (IV, Random, 95% CI) | 1.06 [0.16, 6.98] |
| 6.8 Any SAE | 1 | 156 | Risk Ratio (IV, Random, 95% CI) | 1.14 [0.89, 1.47] |
| 7 Adverse event during maintenance (any grade) | 2 | Risk Ratio (IV, Random, 95% CI) | Subtotals only | |
| 7.1 Nausea | 2 | 385 | Risk Ratio (IV, Random, 95% CI) | 3.82 [0.85, 17.22] |
| 7.2 Anaemia | 2 | 385 | Risk Ratio (IV, Random, 95% CI) | 2.30 [0.87, 6.08] |
| 7.3 Fatigue | 2 | 385 | Risk Ratio (IV, Random, 95% CI) | 1.35 [1.02, 1.78] |
| 8 Adverse event during maintenance (grade 3/4) | 2 | Risk Ratio (IV, Random, 95% CI) | Subtotals only | |
| 8.1 Nausea | 2 | 385 | Risk Ratio (IV, Random, 95% CI) | 4.21 [0.48, 36.69] |
| 8.2 Anaemia | 2 | 385 | Risk Ratio (IV, Random, 95% CI) | 5.26 [1.19, 23.20] |
| 8.3 Fatigue | 2 | 385 | Risk Ratio (IV, Random, 95% CI) | 2.12 [0.67, 6.71] |
| 9 Any adverse event during maintenance (any grade) | 2 | 385 | Risk Ratio (IV, Random, 95% CI) | 1.16 [0.94, 1.42] |
| 10 Any adverse event during maintenance (grade 3/4) | 2 | 385 | Risk Ratio (IV, Random, 95% CI) | 1.74 [1.22, 2.49] |
What's new
Last assessed as up‐to‐date: 21 April 2015.
| Date | Event | Description |
|---|---|---|
| 21 September 2016 | Amended | Contact details updated. |
History
Protocol first published: Issue 3, 2009 Review first published: Issue 6, 2010
| Date | Event | Description |
|---|---|---|
| 3 August 2015 | Amended | Typographical error amended. |
| 30 April 2015 | Amended | Literature search text amended |
| 21 April 2015 | New citation required and conclusions have changed | Updated review with four RCTs added. |
| 21 April 2015 | New search has been performed | Searches updated 21 April 2015 |
| 5 October 2013 | New search has been performed | Search updated 5 October 2013. |
Differences between protocol and review
The original review title was changed to limit to PARP inhibitors for clarity.
Another comparison group of PARP inhibitors versus conventional chemotherapy was added following the publication of the original version of the review due to ongoing studies identified in the initial search. We analysed data from studies with women who had EOC sensitive and resistant to platinum treatment separately, since these are heterogeneous populations. Sub‐group analyses were not required since women in each study were limited to either platinum‐resistant or platinum‐sensitive disease. Future up‐dates of the review will contain sub‐group analyses based on platinum‐sensitivity, if appropriate. We will also perform sub‐group analysis based on BRCA‐mutation status. In addition, from on‐going studies identified in the original review, we knew that studies likely to be included were not powered for OS. Objective Response Rate (ORR) was therefore added as a secondary outcome measure at the data extraction stage in this update, since it was identified as a planned outcome measure from published protocols of ongoing studies online in the original review. The outcome 'toxicity' was renamed as 'adverse events' in the update of the review. Future versions of this review should include BRCA mutation status as a subgroup analysis.
Subseqent to the publication of the original protocol, Cochrane methods have changed and it is recommended that quality of evidence should be assessed according to the GRADE system. GRADEPRO software (GRADEpro 2014) was use to import data from Review Manager 5.3 (RevMan 2014) in order to create 'Summary of findings' tables (Table 1; Table 2) according to guidance in the Cochrane Handbook Chapter 11. This allowed us to summarise the overall quality of evidence from studies included in each comparison. The following outcomes were included in the 'Summary of findings' tables by treatment comparisons:
Overall survival;
Progression‐free survival;
Severe adverse effects.
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Kaye 2012
| Methods | Phase II, open‐label, randomised, multicentre study | |
| Participants | 97 women aged 18 years or older with histologically or cytologically confirmed recurrent epithelial ovarian, primary peritoneal or fallopian tube carcinoma Women had confirmed BRCA1/2 mutation (BRCA1 +ve: 81.3% (arm 1); 87.5% (arm 2); 81.8% (arm 3) Recurrence within 12 months of most recent platinum–based chemotherapy regimen (recurrence within 6 months ‐ i.e. platinum‐resistant disease: 56.3% (arm 1); 50.0% (arm 2); 42.4% (arm 3) Performance status (PS) 0 to 2; PS 0: 50.0% (arm 1); 59.4% (arm 2); 57.6% (arm 3) Life expectancy > 16 weeks and one or more measurable lesions according to RECIST No previous exposure to pegylated liposomal doxorubicin (PLD) Mean age: 57.2 (arm 1); 53.8 (arm 2); 54.3 (arm 3) |
|
| Interventions | Arm 1: Olaparib (OLA) 200 mg bd maintenance therapy Arm 2: OLA 400 mg bd maintenance therapy Arm 3: IV pegylated liposomal doxorubicin (PLD) 50 mg/m² every 28 days |
|
| Outcomes | 97 women randomised; 32 women to (33%) OLA 200 mg, 32 women to (33%) OLA 400 mg, and 33 women to (34%) PLD 8 women who progressed on PLD crossed over from PLD to OLA 400 mg group Survival and response outcomes 59 RECIST‐defined progression events were documented (45/63 in arms 1 and 2 combined and 14/28 in arm 3) Median PFS times were 6.5 months (95% CI 5.5 to 10.1 months), 8.8 months (95% CI 5.4 to 9.2 months) and 7.1 months (95% CI 3.7 to 10.7 months) for OLA 200 mg, OLA 400 mg and PLD groups, respectively There was no difference in PFS between OLA (combined or individual doses) and PLD groups (HR 0.88, 95% CI 0.5 to 1.56; P value = 0.66 for arms 1 and 2 combined versus arm 3). OLA 200 mg versus PLD (HR 0.91, 80% CI 0.60 to 1.39; 95% CI 0.48 to 1.74; P value = 0.78); OLA 400 mg versus PLD (HR 0.86, 80% CI 0.56 to 1.30; 95% CI 0.45 to 1.62; Pvalue = 0.63) 9, 11 and 13 deaths in arms 1, 2 and 3, respectively Overall survival of PLD (arm 3) versus OLA 200 mg (arm 1 HR 0.66 (95% CI 0.27 to 1.55) and OLA 400 mg (arm 2 HR 1.01 (95% CI 0.44 to 2.27) Combined response rates (i.e. RECIST and/or GCIG CA125) were 38%, 59% and 39% in the OLA 200 mg, OLA 400 mg and PLD groups, respectively, with odds ratios of OLA 200 mg = 0.98 (P value = 0.97), OLA 400 mg = 2.76 (P value = 0.05) and OLA 200 and 400 mg = 1.64 (P value = 0.27) Quality of life and adverse events outcomes There were no significant differences in improvement or worsening rates between the OLA and PLD group for the FACT‐O Symptom Index and Trial Outcome Index scores. A higher improvement rate was noted for OLA 400 mg compared with PLD for the total FACT‐O score (odds ratio 7.23, 95% CI 1.09 to 143.3; P value = 0.039) Adverse events: Nausea: Grade 3 to 4: 3 (5%) versus 2 (6%) (Arms 1 and 2 versus Arm 3) Fatigue: Grade 3 to 4: 4 (6%) versus 3(9%) (Arms 1 and 2 versus Arm 3) Abdominal pain: Grade 3 to 4: 2 (3%) vs 2 (6%) (Arms 1 and 2 versus Arm 3) Vomiting: Grade 3 to 4: 1 (2%) versus 1 (3%) (Arms 1 and 2 versus Arm 3) |
|
| Notes | Same study as ICEBERG3 study identified as ongoing in initial version of review. Higher response rates in PLD group compared to other studies attributed to high proportion with BRCA mutation, as evidence from other studies that this improves response rate to PLD. Clinical trial identifiers: ICEBERG 3; NCT00628251; D0810C00012; EUCTR2007‐007622‐22‐ GB | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | ''randomisation assignment list was computer‐generated using the Global Randomisation system (DRand)" |
| Allocation concealment (selection bias) | Low risk | "patients were randomly assigned sequentially using an Interactive Voice Response System" |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open‐label |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | "centrally reviewed tumour assessment for all patients with RESIST scans were used for sensitivity analysis" Correspondence with authors confirmed that central reviewers were blinded to treatment groups, which is of low risk, but other outcomes at unclear risk of bias, as open‐label study |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No patients lost to follow‐up and all accounted for in CONSORT flowchart |
| Selective reporting (reporting bias) | Low risk | Outcome measures as declared at trial registration on www.ClinicalTrials.gov |
| Other bias | Unclear risk | Several investigators disclosed financial links to AstraZeneca |
Kummar 2015
| Methods | Open‐label, multicentre, phase II randomised | |
| Participants | 75 women (38 women cyclophosphamide, 37 women veliparib + cyclophosphamide) Women with BRCA mutations and recurrent ovarian or primary peritoneal, fallopian tube or high‐grade serous epithelial ovarian cancer regardless of BRCA mutation status All women had measurable disease by RECIST criteria Women aged 18 years or over (median age 58; range 37 to 79 years) Median 4 (range 1 to 9) previous chemotherapy treatment regimens 2 women had received prior treatment with a PARP inhibitor |
|
| Interventions | Women were randomised to receive either cyclophosphamide (C) alone or veliparib + cyclophosphamide (V+C) administered orally 4x per day (C 50 mg, V 60 mg) at 21‐day intervals until disease progression. At progression those in the C alone arm were able to cross over to combination treatment | |
| Outcomes | 75 women (38 women C, 37 women V+C) Radiological imaging was performed at baseline and every 3 cycles for assessment of response. At interim analysis, 1 complete response was observed in each arm, with a total of 5 partial responses (PR) in the combination arm and 7 PRs in the cyclophosphamide alone arm, so accrual was stopped The study design had an 88% power to detect the difference between a 15% response rate for C alone versus a 35% response rate for V+C; early closure if fewer responses were observed in the combination arm in the first 65 patients enrolled (half of the total projected accrual) These data are different to those published, following limited author response to requests for clarification, since data were inconsistent in the initial meeting abstract Further data published after initial completion of review and review publication delayed to add in. Clarification of data not provided prior to publication, despite requests Data in final publication differ from data in abstract: One complete response was observed in each arm. "PR was seen in six patients in the cyclophosphamide‐only arm [7/36 (19.4%) responses overall; 95% CI: 8.2‐36.0%], three patients in the combination arm [4/34 (11.8%) responses overall; 95% CI: 3.3‐27.5%], with three partial responses (PR) in the combination arm and six PRs in the cyclophosphamide alone arm." 75 enrolled; only 72 had evaluable disease: 1 treatment was discontinued for adverse events; 1 withdrew from the study; and 1 died before the end of the first cycle No improvement in PFS (median 2.3 and 2.1 months for cyclophosphamide alone versus combination treatment; P value = 0.68) Lymphopenia G3 to 4: C = 3/38 (8%) versus V+C = 13/37 (35%) Anaemia G3 to 4: C = 0/38 (0%) versus V+C = 2/37 (5%) Nausea ‐ no G3 to 4 in either arm Vomiting ‐ no G3 to 4 in either arm Abdominal pain ‐ no G3 to 4 in either arm |
|
| Notes | No HR for OS or PFS reported | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "Pts were randomised to receive either C alone or V+C". No additional information provided by authors |
| Allocation concealment (selection bias) | Unclear risk | No additional information provided by authors |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open‐label |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Open‐label ‐ not reported that assessors were blinded. No additional information provided by authors |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | All 75 patients accounted for at end of study |
| Selective reporting (reporting bias) | Unclear risk | Insufficient information to permit judgement |
| Other bias | High risk | Closed early at interim analysis as fewer responses in combination arm than pre‐specified in power calculation but powered to only detect a 20% difference in response rates. Authors did not provide further data/clarification |
Ledermann 2012
| Methods | Randomised, double‐blind, multicentre, international phase 2 study (82 investigational sites in 16 countries) | |
| Participants | 326 women of whom 265 met the eligibility criteria. 136 women were randomly assigned to received OLA and 129 to receive placebo. Women 18 years of age and older with recurrent ovarian or fallopian tube cancer or primary peritoneal cancer (high‐grade (grade 2 or 3) serous features or a serous component) sensitive to platinum (objective response to a previous platinum‐based therapy for more than 6 months). Women had to complete 2 courses of platinum‐based chemotherapy and their most recent regimen induced an objective response (defined by RECIST guidelines or a CA125 response) with a normal CA125 prior to commencement of the study. Median age 58 years (OLA) and 59 years (placebo). Complete response to previous platinum chemotherapy: 57 (41.9%) (OLA); 63 (48.8%) (placebo)). BRCA mutation: 31 (22.8%) (OLA); 28 (21.7%) (placebo). Patients did not have mandatory BRCA1/2 testing as part of eligibility and factors known to affect BRCA status, e.g. Jewish ancestry, were balanced between groups. BRCA1/2 positive: 31 (22.8%) Arm A; 28 (21.7%) Arm B |
|
| Interventions | Arm 1: OLA 400 mg bd maintenance therapy Arm 2: Placebo tablets bd maintenance therapy All women within 8 weeks after completion of the last dose of platinum‐based chemotherapy |
|
| Outcomes | 136 women OLA and 129 women placebo ‐ 1 woman in placebo arm withdrew consent prior to treatment and was not included in the analysis, since there were no follow‐up data available ‐ data based on remaining 264 women Survival and response outcomes 153 progression events (57.7% of women) Median PFS was 8.4 months (OLA) 4.8 months (placebo) HR progression or death 0.35; 95% CI 0.25 to 0.49; P value < 0.001 101 women (38%) had died: 52 (OLA) and 49 (placebo) (OLA HR for death 0.94, 95% CI 0.63 to 1.39; P value = 0.75) Median OS 29.7 months (OLA) and 29.9 months (placebo) 29 women were still receiving OLA after a period of at least 21 months, and 4 women were still receiving placebo Median time to progression (RECIST guidelines or CA‐125 level) 8.3 months (OLA) versus 3.7 months (placebo); HR for progression 0.35, 95% CI 0.25 to 0.47; P value < 0.001) Only 40% of women in the study had measurable disease by RECIST guidelines; the objective response rate (ORR) was 12% (7 of 57 women in the OLA group) versus 4% (2 of 48 women in the placebo group) (OR 3.36, 95% CI 0.75 to 23.72; P value = 0.12) Quality of life and adverse events outcomes 246 of 264 women had 1 or more adverse events, most grade 1 or 2 Adverse events with an incidence 10% higher or more in the OLA group than in the placebo group: nausea; fatigue; vomiting; anaemia. Incidence of grade 3 or 4 adverse events was 35.3% in the OLA group and 20.3% in the placebo group. Seven grade 4 events were reported in the OLA group (5.1% of women), and 2 were reported in the placebo group (1.6% of women) |
|
| Notes | Study sponsored by AstraZeneca: Clinical trial identifiers: NCT01081951; D0810C0041 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "computer generated" |
| Allocation concealment (selection bias) | Low risk | "Randomised by interactive voice response system" |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blinding. Unique identifiers generated during randomisation |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Review of CT scans was blinded. Blinded independent review of data |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 326 patients screened; 61 did not meet inclusion criteria, 265 randomised, 1 withdrew consent, all patients accounted for at end of study and displayed on CONSORT flowchart |
| Selective reporting (reporting bias) | Low risk | Outcomes selected in ClinicalTrials.gov reported |
| Other bias | Unclear risk | Industry‐led study and some authors had documented conflict of interest, but blinding secure and low risk of selective reporting bias as pre‐determined at trial registration. Principle Investigators were not employed by AstraZeneca |
Oza 2015
| Methods | Open‐label, randomised, phase II, multicentre study Randomisation (1:1) stratified by number of platinum treatments and platinum‐free interval |
|
| Participants | 162 women with platinum‐sensitive recurrent high‐grade serous epithelial ovarian cancer Baseline characteristics well balanced between groups. However, 6 randomised to placebo arm withdrew before starting treatment compared to 0 in the OLA group Median age 59.0 (range 27 to 78) (Arm A) 62 (Arm B) (range 31 to 79) |
|
| Interventions |
Arm A ‐ OLA orally (200 mg bd days 1 to 10 of a 21‐day cycle) in combination with paclitaxel (P) intravenous (IV) (175 mg/m2 day 1 of a 21‐day cycle) and carboplatin (C) IV (AUC4 day 1 of a 21‐day cycle) for at least 4 cycles. Followed by OLA monotherapy maintenance (400 mg bd continuous dosing) Arm B ‐ Paclitaxel (P) IV (175 mg/m2 day 1 of a 21‐day cycle) and carboplatin (C) IV (AUC6 day 1 of a 21‐day cycle) for 6 cycles. Followed by a post‐completion phase in which no study treatment was administered |
|
| Outcomes | Primary outcome: progression‐free survival (PFS) by central review (RECIST 1.1) Secondary outcomes: overall survival (OS); objective response rate (ORR); safety 162 women randomised (n = 81 per arm): 156 received treatment (Arm A, n = 81; Arm B, n = 75) and 121 began the maintenance/no further therapy phase (Arm A, n = 66; Arm B, n = 55) Survival and response outcomes OLA + P/C (AUC4) followed by maintenance OLA showed improvement in PFS versus P/C (AUC6) alone (HR 0.51, 95% CI 0.34 to 0.77; P value = 0.0012; median PFS = 12.2 months (95% CI 9.7 to 15.0) versus 9.6 months (95% CI 9.1 to 9.7) OS data (HR 1.17, 95% CI 0.79 to 1.73; P value = 0.4379; median 33.8 versus 37.6 months; 54/81 versus 47/81 deaths in arms A and B respectively; total events = 62%) ORR was similar for Arm A and Arm B (64% versus 58%) Toxicity data (during chemo +/‐ OLA phase) Nausea (G3 to 4): 1/81 (1.2%) (Arm A) and 1/75 (1.3%) (Arm B) Fatigue (G3 to 4): 6/81 (7.4%) (arm A versus 3/75 (4.0%) (Arm B) Abdominal pain: Grade 3 to 4: 0/81 (0%) versus 2/75 (2.67%) (Arm B) Vomiting: Grade 3 to 4: 1/81 (1.23%) versus 0/75 (0%) (Arm B) Anaemia: Grade 3 to 4: 7/82 (8.6%) versus 5/75 (6.7%) Neutropenia: Grade 3 to 4: 35/81 (43.2%) (Arm A) versus 26/75 (34.7%) (Arm B) Toxicity data (during maintenance phase) Nausea (G3 to 4): 1/66 (1.2%) (Arm A) and 0/55 (0%) (Arm B) Fatigue (G3 to 4): 0/66 (0%) (arm A versus 0/55 (0%) (Arm B) Abdominal pain: Grade 3 to 4: 0/66 (0%) versus 0/55 (0%) (Arm B) Vomiting: Grade 3 to 4: 0/66 (1.23%) versus 0/55 (0%) (Arm B) Anaemia: Grade 3 to 4: 5/66 (7.6%) versus 1/55 (1.8%) Neutropenia: Grade 3 to 4: 3/66 (4.5%) (Arm A) versus 0/55 (0%) (Arm B) |
|
| Notes | We contacted authors for additional information and they provided us with the in‐press manuscript | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Patient randomisation was stratified (using an interactive voice response [IVR]system) based on:1) number of prior platinum‐containing treatment lines received(1 or >1) and 2) time to disease progression following completion of the previous platinum‐containing therapy(>6 to <=12 months or >12 months)." |
| Allocation concealment (selection bias) | Low risk | See above |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open‐label |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Assessors for central RECIST review were blinded to treatment groups |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | All patients accounted for from randomisation, although 6 patients in control group withdrew before starting treatment |
| Selective reporting (reporting bias) | Low risk | Outcomes pre‐specified on clinical trial registry website |
| Other bias | Unclear risk | Industry‐sponsored by AstraZeneca with several authors disclosing financial conflict of interest |
bd: twice a day CI: confidence interval CT: computerised tomography GCIG: Gynaecologic Cancer Intergroup HR: hazard ratio IV: intravenous OLA: olaparib OR: odds ratio ORR: objective response rate OS: overall survival PFS: progression‐free survival PLD: pegylated liposomal doxorubicin PS: performance status pts: patients RECIST: Response Evaluation Criteria in Solid Tumours
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Ashworth 2008 | Review article |
| Audeh 2009 | Phase II, single‐arm trial of the oral PARP inhibitor OLA (AZD2281) in BRCA‐deficient advanced ovarian cancer (ASCO 2009 meeting abstract) |
| Audeh 2010 | Non‐randomised, phase II, single‐arm study (update of Audeh 2009) |
| Banerjee 2013 | Review article |
| Chen 2013 | Review article |
| Coleman 2014 | Non‐randomised phase II trial; no control group |
| Drew 2008 | Review article |
| Fong 2006 | Phase I pharmacokinetic (PK) and pharmacodynamic (PD) evaluation of a small molecule inhibitor of Poly(ADP‐ribose) polymerase (PARP), KU‐0059436 (Ku) in patients with advanced tumours (ASCO 2006 meeting abstract) |
| Fong 2008 | Results from a phase I study of AZD2281 (KU‐0059436), a PARP (poly(ADP‐ribose) polymerase) inhibitor with single‐agent anticancer activity in patients with BRCA‐deficient ovarian cancer (ASCO 2008 meeting abstract) |
| Fong 2009 | Non‐randomised phase I clinical trial analysing the pharmacokinetic and pharmacodynamic characteristics of OLA (AZD2281). Selection was aimed at having a study population enriched in carriers of a BRCA1 or BRCA2 mutation |
| Gelmon 2011 | Phase 2, multicentre, open‐label, non‐randomised study |
| Helleday 2008 | Review article |
| Lee 2014 | Biomarker study of RCT comparing olaparib plus/minus cediranib: wrong comparison and wrong outcomes |
| Lord 2008 | Review article |
| Lui 2014 | Comparison of Olaparib versus Olaparib and Cediranib ‐ no randomisation of Olaparib; 2 references to this study. |
| Moore 2014 | On‐going study but study on effects of high fat food on pharmacokinetics |
| Muggia 2009 | Review article |
| NCT01891344 | Ongoing, non‐randomised, open‐label, phase II study (Ariel2) |
| Shaw 2013 | Review article |
| Turner 2005 | Review article |
| Yap 2007 | Phase I pharmacokinetic (PK) and pharmacodynamic (PD) study of KU‐0059436 (Ku), a small molecule inhibitor of poly(ADP‐ribose) polymerase (PARP) in cancer patients, including BRCA1/2 mutation carriers (ASCO 2007 meeting abstract) |
| Yap 2009 | Review article |
OLA: olaparib
Characteristics of ongoing studies [ordered by study ID]
| Trial name or title | Olaparib monotherapy in patients with BRCA mutated ovarian cancer following first line platinum based chemotherapy |
| Methods | A phase III, randomised, double‐blind, placebo‐controlled, multicentre study |
| Participants | Patients with BRCA mutated advanced (FIGO Stage III‐IV) ovarian cancer following first‐line platinum‐based chemotherapy |
| Interventions | Olaparib/placebo tablets po 300 mg twice daily for up to 2 years or until objective radiological disease progression as per RECIST as assessed by the investigator. Patients with evidence of stable disease (or those who have progressed), may continue on treatment beyond 2 years, if in the patient's best interest. Dose reduction to 250 mg and subsequently 200 mg is permitted following confirmation of toxicity |
| Outcomes | Progression‐free survival (PFS) by central review of RECIST data Overall survival Quality of life analysis Safety and tolerability |
| Starting date | August 2013 |
| Contact information | Elizabeth Lowe, AstraZeneca: ClinicalTrialTransparency@astrazeneca.com |
| Notes | Estimated completion: January 2022; estimated enrolment = 2500. Last updated 16 March 2015. Last accessed 7 April 2015. |
| Trial name or title | A maintenance study with niraparib versus placebo in patients with platinum sensitive ovarian cancer |
| Methods | Phase 3, multicentre, randomised, double‐blind, placebo‐controlled study |
| Participants | Platinum‐sensitive ovarian cancer patients who have either gBRCAmut or a tumour with high‐grade serous histology and who have responded to their most recent chemotherapy containing a platinum agent |
| Interventions | 2:1 ratio of niraparib versus placebo Administered once daily continuously during a 28‐day cycle |
| Outcomes | Progression‐free survival overall survival Quality of life Safety and tolerability |
| Starting date | June 2013 |
| Contact information | Shefali Agarwal; Sagarwal@tesarobio.com |
| Notes | Estimated completion date October 2016. Last updated 23 Feb 2015. Last accessed 7 April 2015. |
| Trial name or title | Olaparib treatment in BRCA mutated ovarian cancer patients after complete or partial response to platinum chemotherapy |
| Methods | A phase III, randomised, double‐blind, placebo‐controlled, multicentre study to assess the efficacy of olaparib maintenance monotherapy |
| Participants | Women with relapsed high‐grade serous ovarian cancer (HGSOC) (including patients with primary peritoneal and/or fallopian tube cancer) or high‐grade endometrioid cancer with BRCA mutations (documented mutation in BRCA1 or BRCA2 that is predicted to be deleterious or suspected deleterious (known or predicted to be detrimental/lead to loss of function)) who have responded following platinum‐based chemotherapy |
| Interventions | 300 mg olaparib or placebo tablets taken orally twice daily until objective radiological disease progression as per RECIST as assessed by the investigator. Dose reduction to 250 mg and subsequently 200 mg is permitted following confirmation of toxicity |
| Outcomes | Progression‐free survival (PFS) by central review of RECIST data Overall survival Quality of life analysis Safety and tolerability |
| Starting date | September 2013 |
| Contact information | Elizabeth Lowe, AstraZeneca: ClinicalTrialTransparency@astrazeneca.com |
| Notes | Estimated completion date June 2020; estimated enrolment = 440; Last updated 2 Feb 2015. Last accessed 7 April 2015. |
| Trial name or title | A study of rucaparib as switch maintenance following platinum‐based chemotherapy in patients with platinum‐sensitive, high‐grade serous or endometrioid epithelial ovarian, primary peritoneal or fallopian tube cancer (ARIEL3) |
| Methods | Phase 3 study of rucaparib as switch maintenance after platinum in relapsed high‐grade serous and endometrioid ovarian cancer |
| Participants | Women with high‐grade serous or endometrioid epithelial ovarian, primary peritoneal or fallopian tube cancer Received ≥ 2 prior platinum‐based treatment regimens Received no more than 1 non‐platinum regimen Must have had at least a 6‐month disease‐free period following prior treatment with platinum‐based chemotherapy and achieved a response Have sufficient archival tumour tissue for analysis |
| Interventions | Rucaparib Oral tablets or placebo administered twice daily with 28‐day cycles of treatment until evidence of recurrence |
| Outcomes | Disease progression according to RECIST version 1.1 Disease progression according to RECIST version 1.1, as assessed by Independent Radiology Review (IRR), or death from any cause (irrPFS), in molecularly defined subgroups Quality of life: time to a specified decrease in the DSR P subscale of the FOSI‐18 patient‐reported outcome questionnaire; time to a specified decrease in the total score of the FOSI‐18 patient‐reported outcome questionnaire Overall survival (OS) Incidence of adverse events (AEs), clinical laboratory abnormalities and dose modifications Individual model parameter estimates of rucaparib and covariates identification (PK) |
| Starting date | January 2014 |
| Contact information | clovistrials@emergingmed.com |
| Notes | estimated completion November 2016; Last updated 23 March 2015; Date last accessed 7 April 2015 |
po: orally RECIST: Response Evaluation Criteria in Solid Tumours
Contributions of authors
GC and AW contributed equally to the review and are joint first authors.
SK and JM had the initial concept for the original title. The original protocol was written by JM and KG, with significant input from HD and AB. The original searching was performed by IM and HD and in the updated review GC, AW, TL and JM analysed the results of the searches, extracted data in pairs, with discussion with a third author where there were disagreements. GC, AW, JM and TL contacted authors and pharmaceutical companies for additional information. AW, GC, TL and JM wrote the final review.
Sources of support
Internal sources
-
Taunton and Somerset NHS Trust NHS Supporting Programmed Activity, UK.
JM (1 hr per/week)
External sources
No sources of support supplied
Declarations of interest
AW ‐ no conflict of interest declared. GC ‐ no conflict of interest declared. AB ‐ no conflict of interest declared. TL ‐ no conflict of interest declared. JM ‐ no conflict of interest declared.
Edited (no change to conclusions)
References
References to studies included in this review
- Kaye SB. ICEBERG3. http://clinicaltrials.gov/show/NCT00628251. ClinicalTrials.gov, 26 February 2008. ; Kaye SB, Lubinski J, Matulonis U, Ang J E, Gourley C, Karlan BY, et al. Phase II, open‐label, randomized, multicenter study comparing the efficacy and safety of olaparib, a poly (ADP‐ribose) polymerase inhibitor, and pegylated liposomal doxorubicin in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer. Journal of Clinical Oncology 2012;30(4):372‐9. [DOI] [PubMed] [Google Scholar]
- Kummar S, Fleming GF, Oza AM, Sullivan DM, Gandara DR, Naughton M, et al. Randomized trial of oral cyclophosphamide and veliparib in high‐grade serous ovarian, primary peritoneal, or fallopian tube cancers, or BRCA‐mutant ovarian cancer. Clinical Cancer Research 2015;14:2562. [DOI: 10.1158/1078-0432.CCR-14-2565] [DOI] [PMC free article] [PubMed] [Google Scholar]; Kummar S, Oza A, Fleming G, Sullivan D, Gandara D, Erlichman C, et al. Randomized trial of oral cyclophosphamide © with or without veliparib (V), an oral poly (ADP‐ribose) polymerase (PARP) inhibitor, in patients with recurrent BRCA‐positive ovarian, or primary peritoneal or high‐grade serous ovarian carcinoma. 2012 ASCO Annual Meeting. Journal of Clinical Oncology 2012;30(15 Suppl):5020. [http://meetinglibrary.asco.org/content/98169‐114] [Google Scholar]
- Ledermann J. Assessment of efficacy of AZD2281 in platinum sensitive relapsed serous ovarian cancer. http://clinicaltrials.gov/show/NCT00753545 12 September 2008. ; Ledermann J, Harter P, Gourley C, Friedlander M, Vergote I, Rustin G, Scott C, Meier W, Shapira Frommer R, Safra T, Matei D, Macpherson E, Watkins C, Carmichael J, Matulonis U. Phase 2 randomized placebo‐controlled study of olaparib (AZD2281) in patients with platinum‐sensitive relapsed serous ovarian cancer (PSR SOC). International journal of gynecological cancer. 2011; Vol. 21 (S13). [Google Scholar]; Ledermann J, Harter P, Gourley C, Friedlander M, Vergote I, Rustin G, Scott CL, Meier W, Shapira‐Frommer R, Safra T, Matei D, Fielding A, Spencer S, Dougherty B, Orr M, Hodgson D, Barrett JC, Matulonis U. Olaparib maintenance therapy in patients with platinum‐sensitive relapsed serous ovarian cancer: A preplanned retrospective analysis of outcomes by BRCA status in a randomised phase 2 trial. Lancet oncology 2014;15(8):852‐861. [DOI] [PubMed] [Google Scholar]; Ledermann J, Harter P, Gourley C, Friedlander M, Vergote I, Rustin G, et al. Olaparib maintenance therapy in platinum‐sensitive relapsed ovarian cancer. New England Journal of Medicine 2012;366(15):1382‐92. [DOI] [PubMed] [Google Scholar]; Ledermann J, Harter P, Gourley C, Friedlander M, Vergote I, Rustin G, Scott CL, Meier W, Shapira‐Frommer R, Safra T, Matei D, Fielding A, Spencer S, Dougherty B, Orr M, Hodgson D, Barrett JC, Matulonis U. Olaparib maintenance therapy in patients with platinum‐sensitive relapsed serous ovarian cancer: A preplanned retrospective analysis of outcomes by BRCA status in a randomised phase 2 trial. Obstetrical & gynecological survey 2015;69((10)):594‐596. [DOI] [PubMed] [Google Scholar]; Ledermann JA, Harter P, Gourley C, Friedlander M, Vergote IB, Rustin GJS, Scott C, Meier W, Shapira‐Frommer R, Safra T, Matei D, Macpherson E, Watkins C, Carmichael J, Matulonis U. Phase II randomized placebo‐controlled study of olaparib (AZD2281) in patients with platinum‐sensitive relapsed serous ovarian cancer (PSR SOC). Journal of clinical oncology. 2011; Vol. 29. [Google Scholar]; Matulonis UA, Harter P, Gourley C, Friedlander M, Vergote IB, Rustin GJS, Fielding A, Spencer S, Ho TW, Ledermann JA. Analysis of intermediate clinical endpoints from a Phase II trial of olaparib maintenance therapy in patients with platinum‐sensitive relapsed serous ovarian cancer (PSR SOC). Gynecologic oncology 2014;Conference: 45th Annual Meeting on Women's Cancer of the Society of Gynecologic Oncology, SGO 2014 Tampa, FL United States. Conference Start: 20140322 Conference End: 20140325. Conference Publication:(var.pagings):54‐55. 2014. [Google Scholar]
- Oza A, Cibula D, Oaknin Benzaquen A, Poole C, Mathijssen RHJ, Sonke GS, et al. Olaparib combined with chemotherapy for recurrent platinum‐sensitive ovarian cancer: a randomised phase II trial. Lancet Oncology 2015;; Vol. 16, issue 1:87‐97. [DOI] [PubMed]; Oza AM, Cibula D, Oaknin A, Poole CJ, Mathijssen RHJ, Sonke GS, et al. Olaparib plus paclitaxel plus carboplatin (P/C) followed by olaparib maintenance treatment in patients (pts) with platinum‐sensitive recurrent serous ovarian cancer (PSR SOC): A randomized, open‐label phase II study. Journal of Clinical Oncology 2012;30(15 Suppl):5001. [http://meeting.ascopubs.org/cgi/content/abstract/30/15_suppl/5001] [Google Scholar]; Oza AM, Cibula D, Oaknin Benzaquen A, Poole CJ, Mathijssen RHJ, et al. Olaparib plus chemotherapy, followed by maintenance monotherapy, in women with platinum‐sensitive recurrent serous ovarian cancer (PSR SOC): BRCA1/2 mutation (BRCAm) and interim overall survival analyses. European Journal of Cancer 2013;49 Suppl 2:S712‐S3. [Google Scholar]
References to studies excluded from this review
- Ashworth A. A synthetic lethal therapeutic approach: Poly(ADP) ribose polymerase inhibitors for the treatment of cancers deficient in DNA double‐strand break repair. Journal of Clinical Oncology 2008;26(22):3785‐90. [DOI] [PubMed] [Google Scholar]
- Audeh MW, Penson RT, Friedlander M. Phase II trial of the oral PARP inhibitor olaparib (AZD2281) in BRCA‐deficient advanced ovarian cancer. Journal of Clinical Oncology (Meeting Abstracts) 2009;27(15S):5500. [Google Scholar]
- Audeh MW, Carmichael J, Penson RT, Friedlander M, Powell B, Bell‐McGuinn KM, et al. Oral poly(ADP‐ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer: a proof‐of‐concept trial. Lancet 2010;376(9737):245‐51. [DOI] [PubMed] [Google Scholar]
- Banerjee S, Kaye SB. New strategies in the treatment of ovarian cancer: current clinical perspectives and future potential. Clinical Cancer Research 2013;19(5):961‐8. [DOI] [PubMed] [Google Scholar]
- Chen Y, Zhang L, Hao Q. Olaparib: a promising PARP inhibitor in ovarian cancer therapy. Archives of Gynecology and Obstetrics 2013;288(2):367‐74. [DOI] [PubMed] [Google Scholar]
- Coleman RL, Sill M, Aghajanian C, Gray HJ, Tewari KS, Rubin SC, Rutherford TJ, Chan JK, Chen HX, Swisher EM. A phase II evaluation of the potent, highly selective PARP inhibitor veliparib in the treatment of persistent or recurrent epithelial ovarian, fallopian tube, or primary peritoneal cancer in patients who carry a germline BRCA1 or BRCA2 mutation ‐ A Gynecologic Oncology Group study. Gynecologic oncology. 2014; Vol. 45th Annual Meeting on Women's Cancer of the Society of Gynecologic Oncology, SGO 2014 Tampa, FL United States. Conference Start: 20140322 Conference End: 20140325. Conference Publication:56‐57. 24680592 [Google Scholar]
- Drew Y, Calvert H. The potential of PARP inhibitors in genetic breast and ovarian cancers. Annals of the New York Academy of Science 2008;1138:136‐45. [DOI] [PubMed] [Google Scholar]
- Fong PC, Spicer J, Reade S. Phase I pharmacokinetic (PK) and pharmacodynamic (PD) evaluation of a small molecule inhibitor of Poly ADP‐Ribose Polymerase (PARP), KU‐0059436 (Ku) in patients (p) with advanced tumours. Journal of Clinical Oncology (Meeting Abstracts) 2006;24(18 Suppl):3022. [Google Scholar]
- Fong PC, Boss DS, Carden CP. AZD2281 (KU‐0059436), a PARP (poly ADP‐ribose polymerase) inhibitor with single agent anticancer activity in patients with BRCA deficient ovarian cancer: Results from a phase I study. Journal of Clinical Oncology (Meeting Abstracts) 2008;26(15 Suppl):5510. [Google Scholar]
- Fong PC, Boss DS, Yap TA. Inhibition of poly(ADP‐ribose) polymerase in tumors from BRCA mutation carriers. New England Journal of Medicine 2009;361(2):123‐34. [DOI] [PubMed] [Google Scholar]
- Gelmon KA, Tischkowitz M, Mackay H, Swenerton K, Robidoux A, Tonkin K, et al. Olaparib in patients with recurrent high‐grade serous or poorly differentiated ovarian carcinoma or triple‐negative breast cancer: A phase 2, multicentre, open‐label, non‐randomised study. Lancet Oncology 2011;12(9):852‐61. [DOI] [PubMed] [Google Scholar]
- Helleday T, Petermann E, Lundin C, Hodgson B, Sharma RA. DNA repair pathways as targets for cancer therapy. Nature Reviews Cancer 2008;8(3):193‐204. [DOI] [PubMed] [Google Scholar]
- Lee J‐M, Liu J, Choyke PL, Elbuluk O, Turkbey IB, Trepel JB, Lee M‐J, Cao L, Houston ND, Gordon N, Figg WD, Barry WT, Matulonis U, Birrer MJ, Ivy P, Kohn EC. Biomarker correlates from the randomized phase 2 trial of the PARP inhibitor olaparib (O) with or without the antiangiogenic TKI cediranib (C) in recurrent platinum‐sensitive ovarian cancer (NCT01116648). Journal of clinical oncology. 2014; Vol. 32:5s:(suppl; abstr 5535). [Google Scholar]
- Lord CJ, Ashworth A. Targeted therapy for cancer using PARP inhibitors. Current Opinion in Pharmacology 2008;8(4):363‐9. [DOI] [PubMed] [Google Scholar]
- Liu J, Barry WT, Birrer MJ, Lee J‐M, Buckanovich RJ, Fleming GF, Rimel B, Buss MK, Nattam SR, Hurteau J, Luo W, Quy P, Obermayer E, Whalen C, Lee H, Winer EP, Kohn EC, Ivy SP, Matulonis U. A randomized phase 2 trial comparing efficacy of the combination of the PARP inhibitor olaparib and the antiangiogenic cediranib against olaparib alone in recurrent platinum‐sensitive ovarian cancer.. Journal of Clinical Oncology, 2014 ASCO Annual Meeting Abstracts. 32(15_suppl); Vol. LBA550. [Google Scholar]; Liu JF, Barry WT, Birrer M, Lee JM, Buckanovich RJ, Fleming GF, Rimel B, Buss MK, Nattam S, Hurteau J, Luo W, Quy P, Whalen C, Obermayer L, Lee H, Winer EP, Kohn EC, Ivy SP, Matulonis UA. Combination cediranib and olaparib versus olaparib alone for women with recurrent platinum‐sensitive ovarian cancer: a randomised phase 2 study. Lancet oncology 2014;15(11):1207‐1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore KN, Zhang X‐Y, Agarwal S, Patel MR, Burris HA, Martell RE, Kansra, V. Food effect substudy of a phase 3 randomized double‐blind trial of maintenance with niraparib (MK4827), a poly(ADP)ribose polymerase (PARP) inhibitor versus placebo in patients with platinum‐sensitive ovarian cancer.. Journal of Clinical Oncology, 2014 ASCO Annual Meeting Abstracts.. May 2014; Vol. 32(15_suppl):e16531. [Google Scholar]
- Muggia F. Platinum compounds 30 years after the introduction of cisplatin: implications for the treatment of ovarian cancer. Gynecologic Oncology 2009;112(1):275‐81. [DOI] [PubMed] [Google Scholar]
- McNeish I, Coleman RL, Oza A, Konecny G, O'Malley DM, Kichenadasse G, et al. Preliminary results of ARIEL2, a phase 2 open‐label study to identify ovarian cancer patients likely to respond to rucaparib. Annals of Oncology 2014;25(Suppl 4):iv305‐26. [clovistrials@emergingmed.com; https://clinicaltrials.gov/ct2/show/NCT01891344] [Google Scholar]
- Shaw HM, Hall M. Emerging treatment options for recurrent ovarian cancer: the potential role of olaparib. OncoTargets and Therapy 2013;6:1197‐206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner N, Tutt A, Ashworth A. Targeting the DNA repair defect of BRCA tumours. Current Opinion in Pharmacology 2005;5(4):388‐93. [DOI] [PubMed] [Google Scholar]
- Yap TA, Boss DS, Fong PC. First in human phase I pharmacokinetic (PK) and pharmacodynamic (PD) study of KU‐0059436 (Ku), a small molecule inhibitor of poly ADP‐ribose polymerase (PARP) in cancer patients (p), including BRCA1/2 mutation carriers. Journal of Clinical Oncology (Meeting Abstracts) 2007;25(18 Suppl):3529. [Google Scholar]
- Yap TA, Carden CP, Kaye SB. Beyond chemotherapy: targeted therapies in ovarian cancer. Nature Reviews Cancer 2009;9(3):167‐81. [DOI] [PubMed] [Google Scholar]
References to ongoing studies
- DiSilvestro P, Moore K. A phase III, randomised, double blind, placebo controlled, multicentre study of olaparib maintenance monotherapy in patients with BRCA mutated advanced (FIGO stage III‐IV) ovarian cancer following first line platinum based chemotherapy. https://clinicaltrials.gov/ct2/show/NCT018449862013. ; Moore KN, DiSilvestro P, Lowe ES, Garnett S Pujade‐Lauraine E. SOLO1 and SOLO2: Randomized phase III trials of olaparib in patients (pts) with ovarian cancer and a BRCA1/2 mutation (BRCAm).. J Clin Oncol (Meeting Abstracts). May 2014; Vol. 32 (15‐suppl):TPS5616. [Google Scholar]
- Matulonis U, Mahner S, Wenham RM, Ledermann JA, Monk BJ, Campo JM, Berek JS, Vergote I, Fabbro M, Katsaros D, Marth C, Lorusso D, Herrstedt J, Agarwal S, RMartell RE, Mirza MR. A phase 3 randomized double‐blind trial of maintenance with niraparib versus placebo in patients with platinum‐sensitive ovarian cancer (ENGOT‐OV16/NOVA trial).. Journal of Clinical Oncology, 2014 ASCO Annual Meeting Abstracts. 32(15_sppl); Vol. TPS5625. [Google Scholar]; Mirza M, Berek JS, Vergote I, Wenham RM, Campo JM, Oza AM, Mahner S, Monk BJ, Fabbro M, Ledermann JA, Marth C, Bruchim I, Katsaros D, Lorusso D, Malander S, Dorum A, Agarwal S, Martell RE, Matulonis U. Engot‐ov16/nova: A phase 3 randomized double‐blind trial of maintenance with PARP‐inhibitor niraparib versus placebo in patients with platinum‐sensitive ovarian cancer. International journal of gynecological cancer 2014;Conference: 15th Biennial Meeting of the International Gynecologic Cancer Society Melbourne, VIC Australia. Conference Start: 20141108 Conference End: 20141111. Conference Publication. 2014:33‐34. [Google Scholar]; Mirza MR, Matulonis U. A maintenance study with niraparib versus placebo in patients with platinum sensitive ovarian cancer. https://clinicaltrials.gov/ct2/show/NCT018472742013.
- Moore KN, DiSilvestro P, Lowe ES, Garnett S Pujade‐Lauraine E. SOLO1 and SOLO2: Randomized phase III trials of olaparib in patients (pts) with ovarian cancer and a BRCA1/2 mutation (BRCAm).. Journal of Clinical Oncology, 2014 ASCO Annual Meeting Abstracts.Vol 32, No 15_suppl (May 20 Supplement), 2014: TPS5616. 2014. [Google Scholar]; Pujade‐Lauraine E. Phase III randomised, double blind, placebo controlled study of olaparib maintenance monotherapy in platinum sensitive relapsed BRCA mutated ovarian cancer patients with a complete or partial response following platinum based chemotherapy. https://clinicaltrials.gov/NCT018743532013.
- A study of rucaparib as switch maintenance following platinum‐based chemotherapy in patients with platinum‐sensitive, high‐grade serous or endometrioid epithelial ovarian, primary peritoneal or fallopian tube cancer (ARIEL3). ClinicalTrials.gov website 6 April 2015. [https://clinicaltrials.gov/ct2/show/NCT01968213?term=NCT01968213&rank=1] ; Swisher EM, McNeish IA, ColemanRL, Brenton J, Kaufmann SH, A Allen AR, Raponi M, Giordano H, Maloney L, JIsaacson J, aLedermann JA. ARIEL 2/3: An integrated clinical trial program to assess activity of rucaparib in ovarian cancer and to identify tumor molecular characteristics predictive of response.. J Clin Oncol (Meeting Abstracts) vol. 32 no. 15_suppl TPS5619. May 2014; Vol. 32 (15_suppl):TPS5619. [Google Scholar]
Additional references
- Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, et al. Specific killing of BRCA2‐deficient tumours with inhibitors of poly(ADP‐ribose) polymerase. Nature 2005;434(7035):913‐7. [DOI] [PubMed] [Google Scholar]
- Bryant HE, Helleday T. Inhibition of poly (ADP‐ribose) polymerase activates ATM which is required for subsequent homologous recombination repair. Nucleic Acids Research 2006;34(6):1685‐91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Therapy Evaluation Program (CTEP), Common Terminology Criteria for Adverse Events, Version 4.03, DCTD, NCI, NIH, DHHS. US Department of Health and Human Services (http://ctep.cancer.gov) May 2009; updated June 2010, issue NIH Publication No. 09‐5410.
- Curtin NJ. PARP inhibitors for cancer therapy. Expert Reviews in Molecular Medicine 2005;7(4):1‐20. [DOI] [PubMed] [Google Scholar]
- Deeks JJ, Altman DG, Bradburn MJ. Statistical methods for examining heterogeneity and combining results from several studies in meta‐analysis. In: Egger M, Davey Smith G, Altman DG editor(s). Systematic Reviews in Health Care: Meta‐Analysis in Context. 2nd Edition. London: BMJ Publication Group, 2001. [Google Scholar]
- DerSimonian R, Laird N. Meta‐analysis in clinical trials. Controlled Clinical Trials 1986;7(3):177‐88. [DOI] [PubMed] [Google Scholar]
- Committee for Medicinal Products for Human Use (CHMP). European Medicines Agency. Summary of opinion (initial authorisation): Lynparza (olaparib). European Medicines Agency, London 23 October 2014.
- Adept Scientific Ltd.. Endnote X6. Letchworth,UK: Adept Scientific Ltd., 2012.
- Erickson BK, Conner MC, Landen CN. The role of the fallopian tube in the origin of ovarian cancer. American Journal of Obstetrics and Gynecology Nov 2013;209(5):409‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005;434(7035):917‐21. [DOI] [PubMed] [Google Scholar]
- Ferlay J, Soerjomataram I, Ervik M, Dikshit R, Eser S, Mathers C, et al. GLOBOCAN 2012v1.0, Cancer Incidence and Mortality Worldwide. IARC CancerBase2013, issue No. 11 [Internet].
- McMaster University. GRADEpro[Computer program on www.gradepro.org].. Version Version [Jan 2015]. McMaster University, 2014.
- Hennessy BT, Timms KM, Carey MS, Gutin A, Meyer LA, Flake DD 2nd, et al. Somatic mutations in BRCA1 and BRCA2 could expand the number of patients that benefit from poly (ADP ribose) polymerase inhibitors in ovarian cancer. Journal of Clinical Oncology 2010;28(22):3570‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgins JP, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta‐analyses. BMJ 2003;327(7414):557‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
- Jagtap P, Szabo C. Poly(ADP‐ribose) polymerase and the therapeutic effects of its inhibitors. National Reviews of Drug Discovery 2005;4(5):421‐40. [DOI] [PubMed] [Google Scholar]
- Jemal A, Siegel R, Ward E, Hao Y, Xu J, Murray T, et al. Cancer statistics. CA: A Cancer Journal for Clinicians 2008;58(2):71‐96. [DOI] [PubMed] [Google Scholar]
- Morrison J, Haldar K, Kehoe S, Lawrie TA. Chemotherapy versus surgery for initial treatment in advanced ovarian epithelial cancer. Cochrane Database of Systematic Reviews 2012, Issue 8. [DOI: 10.1002/14651858.CD005343] [DOI] [PMC free article] [PubMed] [Google Scholar]
- National Institute for Clinical Excellence. Familial breast cancer: Classification and care of people at risk of familial breast cancer and management of breast cancer and related risks in people with a family history of breast cancer ‐ guidance (CG164). London: National Institute for Clinical Excellence, 2013. [Google Scholar]
- Parmar MK, Torri V, Stewart L. Extracting summary statistics to perform meta‐analyses of the published literature for survival endpoints. Statistics in Medicine 1998;17(24):2815‐34. [DOI] [PubMed] [Google Scholar]
- Peralta‐Leal A, Rodriguez MI, Oliver FJ. Poly(ADP‐ribose)polymerase‐1 (PARP‐1) in carcinogenesis: potential role of PARP inhibitors in cancer treatment. Clinical Translations in Oncology 2008;10(6):318‐23. [DOI] [PubMed] [Google Scholar]
- Quinn M, Babb B, Brock A, Jones J. Cancer Trends in England and Wales. London: The Stationery Office, 2001. [Google Scholar]
- Ratnam K, Low JA. Current development of clinical inhibitors of poly(ADP‐ribose) polymerase in oncology. Clinical Cancer Research 2007;13(5):1383‐8. [DOI] [PubMed] [Google Scholar]
- Reinbolt RE, Hays JL. The role of PARP inhibitors in the treatment of gynecologic malignancies. Frontiers in Oncology 2013;3(237):1‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Nordic Cochrane Centre, The Cochrane Collaboration. Review Manager (RevMan). Version 5.3. Copenhagen: The Nordic Cochrane Centre, The Cochrane Collaboration, 2014.
- Risch HA, McLaughlin JR, Cole DE, Rosen B, Bradley L, Kwan E, et al. Prevalence and penetrance of germline BRCA1 and BRCA2 mutations in a population series of 649 women with ovarian cancer. American Journal of Human Genetics 2001;68(3):700‐10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepherd JH. Revised FIGO staging for gynaecological cancer. British Journal of Obstetrics and Gynaecology 1989;96(8):889‐92. [DOI] [PubMed] [Google Scholar]
- Stewart L, Advanced Ovarian Cancer Trialists Group. Chemotherapy for advanced ovarian cancer. Cochrane Database of Systematic Reviews 1999, Issue 1. [DOI: 10.1002/14651858.CD001418] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner N, Tutt A, Ashworth A. Hallmarks of 'BRCAness' in sporadic cancers. Nature Reviews Cancer 2004;4:814‐9. [DOI] [PubMed] [Google Scholar]
- Vergote I, Tropé CG, Amant F, Kristensen GB, Sardi JE, Ehlen T, et al. EORTC‐GCG/NCIC‐CTG Randomised trial comparing primary debulking surgery with neoadjuvant chemotherapy in stage IIIC‐IV ovarian, fallopian tube and peritoneal cancer (OVCA). Proceedings of the 12th Biennial Meeting of the International Gynecologic Cancer Society ‐ IGCS. Bangkok, 2008. [Google Scholar]
- Yadav A, Kumar B, Teknos TN, Kumar P. Sorafenib enhances the anti‐tumor effects of chemo‐radiation treatment by down‐regulating DNA repair proteins. Molecular Cancer Therapeutics 2011;10:1241‐51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaremba T, Curtin NJ. PARP inhibitor development for systemic cancer targeting. Anti‐cancer Agents in Medicinal Chemistry 2007;7(5):515‐23. [DOI] [PubMed] [Google Scholar]
References to other published versions of this review
- Martinek I, Haldar K, Gaitskell K, Nicum S, Kehoe S, Morrison J. DNA‐repair pathway inhibitors for the treatment of ovarian cancer. Cochrane Database of Systematic Reviews 2009, Issue 4. [DOI: 10.1002/14651858.CD007929] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinek I, Haldar K, Gaitskell K, Bryant A, Nicum S, Kehoe S, Morrison J. DNA‐repair pathway inhibitors for the treatment of ovarian cancer. Cochrane Database of Systematic Reviews 2010, Issue 6. [DOI: 10.1002/14651858.CD007929.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]