

Abstract
Background
Bipolar disorder is a common recurrent illness with high levels of chronicity. Previous trials have suggested that the anticonvulsant topiramate may be efficacious in bipolar disorder. This is an update of a previous Cochrane review (last published 2006) on the role of topiramate in bipolar disorder.
Objectives
To assess the effects of topiramate for acute mood episodes in bipolar disorder in adults compared to placebo, alternative pharmacological treatment, and combination pharmacological treatment as measured by treatment of symptoms on specific rating scales for individual episodes.
Search methods
We searched the Cochrane Depression, Anxiety and Neurosis Controlled Trials Register to 13 October 2015, which includes records from the Cochrane Central Register of Controlled Trials (CENTRAL) all years; MEDLINE 1950‐; EMBASE 1974‐; and PsycINFO 1967‐.
We performed handsearching, reviewing of grey literature and reference lists, and correspondence with authors and pharmaceutical companies.
Selection criteria
Randomised controlled trials comparing topiramate with placebo or with active agents in the treatment of acute mood episodes in adult male and female patients with bipolar disorder.
Data collection and analysis
Two review authors independently performed data extraction and methodological quality assessment. For analysis, we used odds ratio (OR) for binary efficacy outcomes and mean difference (MD) for continuously distributed outcomes.
Main results
This review included six studies with a total of 1638 male and female participants, of all ethnic backgrounds in both inpatient and outpatient settings. In five studies, participants were experiencing a manic or mixed episode, and in the other study the participants met the criteria for a depressive phase. Topiramate was compared with placebo and alternative pharmacological treatment as both monotherapy and as adjunctive treatment.
Moderate‐quality evidence showed topiramate to be no more or less efficacious than placebo as monotherapy, in terms of mean change on Young Mania Rating Scale (YMRS) (range 0 to 60), at endpoint 3 weeks (MD 1.17, 95% confidence interval (CI) ‐0.52 to 2.86; participants = 664; studies = 3; P = 0.17) and at endpoint 12 weeks (MD ‐0.58, 95% CI ‐3.45 to 2.29; participants = 212; studies = 1; P = 0.69; low‐quality evidence). For the same outcome, low‐quality evidence also showed topiramate to be no more or less efficacious than placebo as add‐on therapy (endpoint 12 weeks) (MD ‐0.14, 95% CI ‐2.10 to 1.82; participants = 287; studies = 1; P = 0.89) in the treatment of manic and mixed episodes. We found high‐quality evidence that lithium was more efficacious than topiramate as monotherapy in the treatment of manic and mixed episodes in terms of mean change on YMRS (range 0 to 60) (endpoint 12 weeks) (MD 8.46, 95% CI 5.86 to 11.06; participants = 449; studies = 2; P < 0.00001).
For troublesome side effects experienced of any nature, we found no difference between topiramate and placebo as monotherapy (endpoint 12 weeks) (OR 0.68, 95% CI 0.33 to 1.40; participants = 212; studies = 1; P = 0.30; low‐quality evidence) or as add‐on therapy (endpoint 12 weeks) (OR 1.10, 95% CI 0.58 to 2.10; participants = 287; studies = 1; P = 0.76; low‐quality evidence). In terms of participants experiencing side effects of any nature, we found no difference between topiramate and an alternative drug as monotherapy (endpoint 12 weeks) (OR 0.87, 95% CI 0.50 to 1.52; participants = 230; studies = 1; P = 0.63; low‐quality evidence) or as add‐on therapy (endpoint 8 weeks) (OR 1.57, 95% CI 0.42 to 5.90; participants = 36; studies = 1; P = 0.50; very low‐quality evidence).
We considered five of the studies to be at low risk of selection bias for random sequence generation, performance, detection, attrition, and reporting biases, and at unclear risk for allocation concealment and other potential sources of bias. We considered the McIntyre 2000 study to be at high risk of performance bias; unclear risk of bias for random sequence generation, allocation concealment, blinding of outcome assessment, and other potential sources of bias; and at low risk for attrition bias and reporting bias.
Authors' conclusions
It is not possible to draw any firm conclusions about the use of topiramate in clinical practice from this evidence. The only high‐quality evidence found was that lithium is more efficacious than topiramate when used as monotherapy in the treatment of acute affective episodes in bipolar disorder, and we note that this evidence came from only two studies. Moderate‐quality evidence showed that topiramate was no more or less efficacious than placebo as monotherapy when a 3‐week endpoint was used, but the quality of the evidence for this outcome at a 12‐week endpoint dropped to low. As we graded the quality of the evidence for the other findings as low and very low, it was not possible to draw any conclusions from the results.
To best address this research question, if investigators see the indication in so doing, more double‐blind randomised controlled trials could be conducted that are more explicit with regard to methodological issues. In particular, investigators could compare placebo, alternative, and combination treatments (including a wide range of mood stabilisers), atypical antipsychotics for manic and mixed episodes, and antidepressants in combination with mood stabilisers or atypical antipsychotics for depressive episodes.
Keywords: Humans; Affective Disorders, Psychotic; Affective Disorders, Psychotic/drug therapy; Antimanic Agents; Antimanic Agents/therapeutic use; Bipolar Disorder; Bipolar Disorder/psychology; Bupropion; Bupropion/therapeutic use; Depressive Disorder; Depressive Disorder/drug therapy; Fructose; Fructose/analogs & derivatives; Fructose/therapeutic use; Randomized Controlled Trials as Topic; Topiramate
Plain language summary
Topiramate for acute affective episodes in bipolar disorder
Who may be interested in this review?
People with bipolar disorder and their healthcare providers.
Why is this review important?
Bipolar disorder is a mood disorder that is a common mental health problem. Patients may experience recurrent symptoms of elevated or irritable mood, depression, or a combination of both. Treatment is usually with psychiatric medication; commonly used medications include mood stabilisers, antidepressants, and antipsychotics. Topiramate is a drug used in epilepsy, however it may have a role in the treatment of bipolar disorder.
What questions does this review aim to answer?
This review investigated the effectiveness and acceptability of topiramate compared to placebo and other agents in the treatment of acute affective episodes in bipolar disorder.
Which studies were included in the review?
We searched medical databases to find reports of clinical trials (specifically randomised controlled trials) published up to 13 October 2015. We identified six studies that involved 1638 people. The studies compared topiramate with placebo or conventional medication such as lithium, both on its own or in combination with other treatments such as sodium valproate or atypical antipsychotics.
Five of the studies were at low risk of bias in the majority of the domains, and at unclear risk for allocation concealment (because of insufficient details regarding how those who admitted participants to a study were shielded from knowing the assignments) and other potential sources of bias (because they were industry funded). The McIntyre 2000 study was at high risk of performance bias (because participants and personnel knew who was taking each medication); at low risk for attrition bias and reporting bias; and at unclear risk of bias for the other domains.
What does the evidence from the review tell us?
We found moderate‐quality evidence showing that topiramate is no more or less effective than placebo when used alone, and low‐quality evidence that topiramate is no more or less effective than placebo when added to other drugs in the treatment of manic and mixed episodes. We found high‐quality evidence that lithium is more effective than topiramate when used alone in the treatment of manic and mixed episodes. Low‐ and very low‐quality evidence showed no difference in side effect profiles when topiramate was compared with placebo or alternative drugs when used alone or in combination with other treatments.
Limitations of the review were that the quality of the evidence was generally low, therefore there is a need for further research. Future research could involve more controlled studies with clearly detailed methods comparing topiramate with placebo and alternative or combination bipolar treatments for manic, mixed, and depressive episodes.
Summary of findings
Summary of findings for the main comparison. Topiramate compared to placebo as monotherapy.
| Topiramate compared to placebo as monotherapy | ||||||
| Patient or population: adults with acute affective episodes in bipolar disorder Settings: inpatients Intervention: topiramate as monotherapy Comparison: placebo as monotherapy | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Placebo as monotherapy | Topiramate as monotherapy | |||||
| Efficacy: change in scores from baseline to endpoint (3 weeks) on symptom rating scales in the treatment of manic and mixed episodes. Lower scores indicate improvement Follow‐up: 3 weeks | ‐ | The mean change in scores from baseline to endpoint (3 weeks) on symptom rating scales in the treatment of manic and mixed episodes in the intervention groups was 1.17 higher (0.52 lower to 2.86 higher) | ‐ | 664 (3 studies) | ⊕⊕⊕⊝ moderate1 | There's no clinical difference between topiramate and placebo as the confidence interval crosses 0 |
| Efficacy: change in scores from baseline to endpoint (12 weeks) on symptom rating scales in the treatment of manic and mixed episodes Lower scores indicate improvement Follow‐up: 12 weeks | ‐ | The mean change in scores from baseline to endpoint (12 weeks) on symptom rating scales in the treatment of manic and mixed episodes in the intervention groups was 0.58 lower (3.45 lower to 2.29 higher) | ‐ | 212 (1 study) | ⊕⊕⊝⊝ low1,2 | There's no clinical difference between topiramate and placebo as the confidence interval crosses 0 |
| Efficacy: change in scores from baseline to endpoint on symptom rating scales in the treatment of depressive episodes | No data available | ‐ | ‐ | ‐ | ‐ | ‐ |
| Acceptability with topiramate (endpoint: 12 weeks) Number of participants experiencing troublesome side effects of any nature Follow‐up: 12 weeks |
Study population mean age (SD) [topiramate]: 40 (12) [placebo]: 37(10) |
OR 0.68 (0.33 to 1.4) | 212 (1 study) | ⊕⊕⊝⊝ low3,4 | ‐ | |
| 85 per 100 | 79 per 100 (65 to 89) | |||||
| Moderate | ||||||
| 85 per 100 | 79 per 100 (65 to 89) | |||||
| Response to treatment with topiramate. Number of participants experiencing a 50% reduction or greater in mean score from baseline to end of treatment on symptom rating scales in manic and mixed episodes | No data available | ‐ | ‐ | ‐ | ‐ | ‐ |
| Response to treatment with topiramate. Number of participants experiencing a 50% reduction or greater in mean score from baseline to end of treatment on symptom rating scales in depressive episodes | No data available | ‐ | ‐ | ‐ | ‐ | ‐ |
| Remission with topiramate. Number of participants presenting with a rating scale score within the normal range at the endpoint in manic and mixed episodes | No data available | ‐ | ‐ | ‐ | ‐ | ‐ |
| Remission with topiramate. Number of participants presenting with a rating scale score within the normal range at the endpoint in depressive episodes | No data available | ‐ | ‐ | ‐ | ‐ | ‐ |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; OR: odds ratio; SD: standard deviation | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1Downgraded one point because of imprecision caused by a 95% confidence interval that includes 1) no effect and 2) the upper and lower confidence limits cross an effect size of 0.5 in either direction. 2Downgraded one point because of imprecision caused by a total population size of less than 400. 3Downgraded one point because of imprecision caused by a total number of events of less than 300. 4Downgraded one point because of imprecision caused by a 95% confidence interval that includes both 1) no effect and 2) relative risk reduction and relative risk increase greater than 25%.
Summary of findings 2. Topiramate as add‐on therapy compared to placebo as add‐on therapy for acute affective episodes in bipolar disorder.
| Topiramate as add‐on therapy compared to placebo as add‐on therapy for acute affective episodes in bipolar disorder | ||||||
| Patient or population: adults with acute affective episodes in bipolar disorder Settings: outpatients Intervention: topiramate as add‐on therapy Comparison: placebo as add‐on therapy | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Placebo as add‐on therapy | Topiramate as add‐on therapy | |||||
| Efficacy: change in scores from baseline to endpoint (12 weeks) on symptom rating scales in the treatment of manic and mixed episodes Lower scores indicate improvement Follow‐up: 12 weeks | ‐ | The mean change in scores from baseline to endpoint (12 weeks) on symptom rating scales in the treatment of manic and mixed episodes in the intervention groups was 0.14 lower (2.1 lower to 1.82 higher) | ‐ | 287 (1 study) | ⊕⊕⊝⊝ low1,2 | There was no clinical difference between topiramate and placebo as the confidence interval crosses 0 |
| Efficacy: change in scores from baseline to endpoint on symptom rating scales in the treatment of depressive episodes | No data available | ‐ | ‐ | ‐ | ‐ | ‐ |
| Acceptability with topiramate (endpoint: 12 weeks) Number of participants experiencing troublesome side effects of any nature Follow‐up: 12 weeks |
Study population mean age (SD) [topiramate]: 41 (12.2) [placebo]: 39 (11.9) |
OR 1.1 (0.58 to 2.1) | 287 (1 study) | ⊕⊕⊝⊝ low3,4 | ‐ | |
| 84 per 100 | 85 per 100 (75 to 92) | |||||
| Moderate | ||||||
| 84 per 100 | 85 per 100 (75 to 92) | |||||
| Response to treatment with topiramate. Number of participants experiencing a 50% reduction or greater in mean score from baseline to end of treatment on symptom rating scales in manic and mixed episodes (endpoint: 12 weeks) Follow‐up: 12 weeks | Study population | OR 0.98 (0.61 to 1.58) | 287 (1 study) | ⊕⊕⊝⊝ low3,4 | ‐ | |
| 38 per 100 | 37 per 100 (27 to 49) | |||||
| Moderate | ||||||
| 38 per 100 | 37 per 100 (27 to 49) | |||||
| Response to treatment with topiramate. Number of participants experiencing a 50% reduction or greater in mean score from baseline to end of treatment on symptom rating scales in depressive episodes | No data available | ‐ | ‐ | ‐ | ‐ | ‐ |
| Remission with topiramate. Number of participants presenting with a rating scale score within the normal range at the endpoint in manic and mixed episodes | No data available | ‐ | ‐ | ‐ | ‐ | ‐ |
| Remission with topiramate. Number of participants presenting with a rating scale score within the normal range at the endpoint in depressive episodes | No data available | ‐ | ‐ | ‐ | ‐ | ‐ |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; OR: odds ratio; SD: standard deviation | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1Downgraded one point because of imprecision caused by a total population size of less than 400. 2Downgraded one point because of imprecision caused by 95% confidence interval that includes 1) no effect and 2) upper and lower confidence limits cross an effect size of 0.5 in either direction. 3Downgraded one point because of imprecision caused by a total number of events of less than 300. 4Downgraded one point because of imprecision caused by 95% confidence interval that includes both 1) no effect and 2) relative risk reduction and relative risk increase greater than 25%.
Summary of findings 3. Topiramate as monotherapy compared to an alternative drug as monotherapy for acute affective episodes in bipolar disorder.
| Topiramate as monotherapy compared to an alternative drug as monotherapy for acute affective episodes in bipolar disorder | ||||||
| Patient or population: adults with acute affective episodes in bipolar disorder Settings: inpatients Intervention: topiramate as monotherapy Comparison: an alternative drug as monotherapy (lithium) | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Lithium as monotherapy | Topiramate as monotherapy | |||||
| Efficacy: change in scores from baseline to endpoint (12 weeks) on symptom rating scales in the treatment of manic and mixed episodes Lower scores indicate improvement Follow‐up: 12 weeks | ‐ | The mean change in scores from baseline to endpoint (12 weeks) on symptom rating scales in the treatment of manic and mixed episodes in the intervention groups was 8.46 higher (5.86 to 11.06 higher) | ‐ | 449 (2 studies) | ⊕⊕⊕⊕ high | The Young Mania Rating Scale score was on average 8.46 higher in participants treated with topiramate at 12 weeks compared to the lithium group |
| Efficacy: change in scores from baseline to endpoint on symptom rating scales in the treatment of depressive episodes | No data available | ‐ | ‐ | ‐ | ‐ | ‐ |
| Acceptability with topiramate (12 weeks) Number of participants experiencing troublesome side effects of any nature Follow‐up: 12 weeks |
Study population mean age (SD) [topiramate]: 40 (12) [lithium]: 42 (11) |
OR 0.87 (0.5 to 1.52) | 230 (1 study) | ⊕⊕⊝⊝ low1,2 | ‐ | |
| 70 per 100 | 67 per 100 (54 to 78) | |||||
| Moderate | ||||||
| 70 per 100 | 67 per 100 (54 to 78) | |||||
| Response to treatment with topiramate. Number of participants experiencing a 50% reduction or greater in mean score from baseline to end of treatment on symptom rating scales in manic and mixed episodes | No data available | ‐ | ‐ | ‐ | ‐ | ‐ |
| Response to treatment with topiramate. Number of participants experiencing a 50% reduction or greater in mean score from baseline to end of treatment on symptom rating scales in depressive episodes | No data available | ‐ | ‐ | ‐ | ‐ | ‐ |
| Remission with topiramate. Number of participants presenting with a rating scale score within the normal range at the endpoint in manic and mixed episodes | No data available | ‐ | ‐ | ‐ | ‐ | ‐ |
| Remission with topiramate. Number of participants presenting with a rating scale score within the normal range at the endpoint in depressive episodes | No data available | ‐ | ‐ | ‐ | ‐ | ‐ |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; OR: odds ratio; SD: standard deviation | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1Downgraded one point because of imprecision caused by a total number of events of less than 300. 2Downgraded one point because of imprecision caused by 95% confidence interval that includes both 1) no effect and 2) relative risk reduction and relative risk increase greater than 25%.
Summary of findings 4. Topiramate as add‐on therapy compared to alternative drug as add‐on therapy for acute affective episodes in bipolar disorder.
| Topiramate as add‐on therapy compared to alternative drug as add‐on therapy for acute affective episodes in bipolar disorder | ||||||
| Patient or population: people with acute affective episodes in bipolar disorder Settings: unspecified Intervention: topiramate as add‐on therapy Comparison: alternative drug as add‐on therapy | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Alternative drug as add‐on therapy | Topiramate as add‐on therapy | |||||
| Efficacy: change in scores from baseline to endpoint on symptom rating scales in the treatment of manic and mixed episodes | No data available | ‐ | ‐ | ‐ | ‐ | ‐ |
| Efficacy: change in scores from baseline to endpoint on symptom rating scales in the treatment of depressive episodes | No data available | ‐ | ‐ | ‐ | ‐ | ‐ |
| Acceptability with topiramate (8 weeks) Number of participants experiencing troublesome side effects of any nature Follow‐up: 8 weeks |
Study population mean age [topiramate]: 39 [bupropion SR]: 43 |
OR 1.57 (0.42 to 5.9) | 36 (1 study) | ⊕⊝⊝⊝ very low1,2,3 | ‐ | |
| 50 per 100 | 61 per 100 (30 to 86) | |||||
| Moderate | ||||||
| 50 per 100 | 61 per 100 (30 to 86) | |||||
| Response to treatment with topiramate. Number of participants experiencing a 50% reduction or greater in mean score from baseline to end of treatment on symptom rating scales in manic and mixed episodes | No data available | ‐ | ‐ | ‐ | ‐ | ‐ |
| Response to treatment with topiramate. Number of participants experiencing a 50% reduction or greater in mean score from baseline to end of treatment on symptom rating scales in depressive episodes (8 weeks) Follow‐up: 8 weeks | Study population | OR 0.8 (0.21 to 3) | 36 (1 study) | ⊕⊝⊝⊝ very low1,2,3 | ‐ | |
| 61 per 100 | 56 per 100 (25 to 83) | |||||
| Moderate | ||||||
| 61 per 100 | 56 per 100 (25 to 82) | |||||
| Remission with topiramate as add‐on therapy. Number of participants presenting with a rating scale score within the normal range at the endpoint in manic and mixed episodes | No data available | ‐ | ‐ | ‐ | ‐ | ‐ |
| Remission with topiramate. Number of participants presenting with a rating scale score within the normal range at the endpoint in depressive episodes (8 weeks) Follow‐up: 8 weeks | Study population | OR 1 (0.23 to 4.3) | 36 (1 study) | ⊕⊝⊝⊝ very low2,3,4 | ‐ | |
| 28 per 100 | 28 per 100 (8 to 62) | |||||
| Moderate | ||||||
| 28 per 100 | 28 per 100 (8 to 62) | |||||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; OR: odds ratio | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1Downgraded one point because of risk of bias. The study was at high risk of performance bias as it was single blind. 2Downgraded one point because of imprecision caused by a total number of events of less than 300. 3Downgraded one point because of imprecision caused by 95% confidence interval that includes both 1) no effect and 2) relative risk reduction and relative risk increase greater than 25%.
Background
Description of the condition
Bipolar disorder is a common recurrent illness with high rates of chronicity. Although it is classically manifested in recurrent manic, depressed, or mixed episodes with complete interepisode recovery, one‐third of patients suffer chronic symptoms, which affect their social and occupational development.
In present classification systems, bipolar disorder now refers to a group of affective (mood) disorders in which patients experience acute episodes of depression for at least two weeks, characterised by low mood and related symptoms (for example loss of pleasure and reduced energy), and acute episodes of either mania for at least one week, characterised by elated or irritable mood or both, and related symptoms such as increased energy and reduced need for sleep, or hypomania, for at least four days, the symptoms of which are less severe or less protracted than are those of mania (Phillips 2013).
In a combined sample of 61,392 adults from 11 countries, the total lifetime prevalences were 0.6% for bipolar disorder type I and 0.4% for bipolar disorder type II (Merikangas 2011).
According to the World Health Organization’s 2004 Global Burden of Disease report, along with other psychiatric illnesses, bipolar disorder is one of the 10 most debilitating of all non‐communicable diseases (WHO 2004). In addition, bipolar disorder is among the mental disorders that contribute the most disability‐adjusted life‐years, which is the sum of years lived with disability and years of life lost (Prince 2007).
Description of the intervention
According to the National Institute for Health and Care Excellence (NICE) guidance (NICE 2006), lithium, olanzapine, and valproate should be considered as first‐line treatments for the long‐term management of bipolar disorder. For the treatment of acute affective episodes, an antipsychotic (normally olanzapine, quetiapine, or risperidone), valproate, or lithium should be considered for managing episodes of mania or hypomania. For moderate or severe depressive symptoms, prescribing a selective serotonin reuptake inhibitor (SSRI) should be considered in addition to an antimanic drug, taking into account the risk of manic switching. Lamotrigine is considered to be a second‐line treatment for bipolar depression (Taylor 2015). Patients suffering from acute mixed episodes should be treated as if they were having an acute manic episode, and prescribing antidepressants should be avoided.
Despite available treatments, there is a high prevalence of incomplete or unsatisfactory treatment responses in bipolar disorder (Poon 2012). The characteristics of bipolar disorder that are associated with inferior treatment responses include: very early onset age, rapid cycling, prominent psychotic features, and comorbidity (Goodwin 2007).
There is ongoing interest in the efficacy of anticonvulsants in bipolar disorder. Topiramate is an anticonvulsant used in the adjunctive treatment of partial seizures and primary generalised tonic‐clonic seizures. Retrospective and prospective uncontrolled open trials have supported the efficacy of topiramate in refractory bipolar disorder both as monotherapy (Sachs 2000), and as an adjunctive treatment (Marcotte 1998; McElroy 2000b; Vieta 2000; Vieta 2003). The most common side effects of topiramate include sedation, word‐finding difficulties, and impaired concentration. Unlike established agents, topiramate is associated with decreased appetite and weight loss (Bourgeois 1998; Meldrum 1996). A large randomised, placebo‐controlled trial over one year supported use of the drug in the treatment of obese participants, with significant improvements in blood pressure and plasma glucose levels (Wilding 2004). There is evidence in the form of a review that topiramate‐treated patients with affective (more commonly known as mood) disorders may also experience weight reduction (Woods 2004).
How the intervention might work
Topiramate has several actions that are potentially therapeutic in bipolar disorder. For example, it can stabilise the membranes of neurons by blocking sodium channels, block glutamate receptors, and increase the effect of gamma‐aminobutyric acid (GABA) receptors (Bourgeois 1998; Meldrum 1996). Indeed, people with bipolar disorder have been found to have decreased plasma levels of GABA during both depressive and manic episodes (Petty 1995) manic episodes.
Why it is important to do this review
Treatment resistance in bipolar disorder remains a problem, and new medications that are both efficacious and well‐tolerated are required. The Cochrane Depression, Anxiety and Neurosis Review Group has already carried out several reviews on a variety of anticonvulsants in the treatment of bipolar disorder (Macritchie 2009; Vasudev 2009; Young 2006). This is an update of a Cochrane review first published in 2006 (Vasudev 2006), which studied the evidence for the use of topiramate in the treatment of acute episodes of this disorder. The review authors concluded that there was insufficient evidence on which to base any recommendations regarding the use of topiramate in any phase of bipolar illness, either in monotherapy or as an adjunctive treatment. At the time of the previous review several unpublished trials were identified, therefore it is important to update the review in order to include them as well as other randomised controlled trials.
Objectives
To assess the effects of topiramate for acute mood episodes in bipolar disorder in adults compared to placebo, alternative pharmacological treatment, and combination pharmacological treatment as measured by treatment of symptoms on specific rating scales for individual episodes.
Methods
Criteria for considering studies for this review
Types of studies
Randomised trials comparing topiramate in the treatment of acute mood episodes (manic, mixed‐mood, or depressive episodes) with placebo or alternative drug treatments in bipolar disorder.
Cross‐over studies were eligible for inclusion, although we planned to include data only from the first phase of randomisation. We also planned to include cluster‐randomised controlled trials, with assessment of their potential for unit‐of‐analysis errors (Higgins 2011).
We considered both published and unpublished trials.
Types of participants
Participant characteristics
Males and females aged 18 and over of any ethnicity.
We included studies with different age ranges if the majority of participants were 18 and over, and investigated the impact of including such studies through sensitivity analyses.
Diagnosis
We included studies when participants had a diagnosis of bipolar disorder corresponding to the International Classification of Diseases, 10th Revision (ICD‐10), code F31*, WHO 1992, and the Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition (DSM‐IV) code 296* (APA 1994).
We included people with all subtypes of bipolar disorder (types I and II and other, and rapid cycling disorder); we excluded cyclothymia.
We included patients with acute mood episodes with:
depressive episodes, with or without psychotic symptoms, approximating to ICD‐10 codes F31.3‐31.5* and DSM‐IV codes 296.21‐4 and 296.31‐4*;
a diagnosis of mixed mood disorder, with or without psychotic symptoms, approximating to ICD‐10 code F31.6* and DSM‐IV code 296.61‐4*;
a diagnosis of hypomania or mania, with or without psychotic symptoms, approximating to ICD‐10 codes F30.0* and F31.0‐31.2* and DSM‐IV code 296.40* or 296.41‐4*.
*We included trials with ICD‐9 and DSM‐III/DSM‐IIIR diagnoses approximating to these codes.
Where available, we noted previous mood stabiliser treatment and planned to perform subgroup analyses to examine the efficacy of topiramate in those who failed to respond to previous mood stabiliser treatment. Some trials might potentially involve heterogenous groups of participants, in particular schizoaffective disorder and recurrent unipolar depression (diagnoses approximating to ICD‐10 F25 and DSM‐IV 295.70, and ICD‐10 F33 and DSM‐IV 296.3, respectively). If possible, we would separate data from these studies into diagnostic groups.
Comorbidities
We excluded people with DSM‐IV Axis I and II and physical comorbidities from the review.
Setting
We included studies from all settings, for example primary or secondary care, outpatients or inpatients.
Types of interventions
Experimental intervention
Topiramate in the treatment of acute manic, mixed mood, or depressive episodes in the context of bipolar disorder.
We defined acute treatment as treatment instituted specifically to alleviate symptoms of an existing acute episode. We would analyse discontinuation trials, in which participants received topiramate prior to randomisation (other than for short periods of stabilisation), separately. When trials combined acute treatment and maintenance phases, we would analyse data separately.
Comparator intervention
Placebo
Alternative pharmacological treatment
Combination pharmacological treatment
We would consider studies where topiramate was used as an adjunctive treatment in combination with another agent separately from studies where it was used in monotherapy.
Types of outcome measures
Primary outcomes
Efficacy of topiramate in the treatment of acute mood episodes in bipolar disorder:
For manic and mixed episodes, efficacy of treatment was measured by change in mean scores from baseline to end of treatment on symptom rating scales, for example Young Mania Rating Scale, Young 1978, and Cavanagh scale for mixed states (Cavanagh 2009).
For depressive episodes, efficacy of treatment was measured by change in mean scores from baseline to end of treatment on depressive symptom rating scale, such as Hamilton Depression Rating Scale (HAMD‐17) (Hamilton 1980), Montgomery‐Åsberg Depression Rating Scale (MADRS) (Montgomery 1979).
Acceptability:
Number of participants experiencing troublesome side effects of any nature.
Secondary outcomes
Response to treatment:
For manic and mixed episodes, we defined response as a 50% reduction or greater in mean score from baseline to end of treatment on symptom rating scales like Young Mania Rating Scale, Young 1978, and Cavanagh scale for mixed states (Cavanagh 2009).
For depressive episodes, we defined response as a 50% reduction or greater in mean score from baseline to end of treatment on depressive symptom rating scales like HAMD‐17 (Hamilton 1980).
Remission:
For manic and mixed episodes, we defined remission as a mood rating scale score within the normal range at the end of the study.
For depressive episodes, we defined remission as a mood rating scale score within the normal range at the end of the study.
Hierarchy of outcome measures
If data on more than one efficacy of treatment were provided for a trial, we would extract data according to the following hierarchy.
For manic episodes:
Young Mania Rating Scale
Other outcome measure of efficacy of treatment with manic symptom rating scales.
For mixed mood episodes:
Young Mania Rating Scale
Cavanagh scale for mixed states
Hamilton Depression Rating Scale
Other outcome measure of efficacy of treatment with mixed symptom rating scales.
For depressive episodes:
Hamilton Depression Rating Scale
Montgomery‐Åsberg Depression Rating Scale
Other outcome measure of efficacy of treatment with depressive symptom rating scales
Timing of outcome assessment
All outcomes were short term, which we defined as acute‐phase treatment which normally would last up to six months.
When studies reported response rates at different time points within the six months, we gave the time point closest to 12 weeks preference.
Search methods for identification of studies
We used a comprehensive search strategy to identify all relevant studies regardless of language or publication status (published, unpublished, in press, and in progress).
Electronic searches
Cochrane Depression, Anxiety and Neurosis Review Group's Specialised Register (CCDANCTR)
The Cochrane Depression, Anxiety and Neurosis Group (CCDAN) maintains two clinical trials registers at their editorial base in Bristol, UK: a references register and a studies‐based register. The CCDANCTR‐References Register contains over 39,000 reports of randomised controlled trials (RCTs) in depression, anxiety, bipolar disorder, eating disorders, self harm, and other mental disorders within the scope of this Group. Approximately 60% of these references have been tagged to individual, coded trials. The coded trials are held in the CCDANCTR‐Studies Register, and records are linked between the two registers through the use of unique study ID tags. Coding of trials is based on the EU‐Psi coding manual, using a controlled vocabulary; please contact the CCDAN Trials Search Co‐ordinator (TSC) for further details. Reports of trials for inclusion in the CCDAN's registers are collated from routine (quarterly) searches of the Cochrane Central Register of Controlled Trials (CENTRAL); weekly generic searches of MEDLINE (1950‐), EMBASE (1974‐), and PsycINFO (1967‐); quarterly searches of the Cochrane Central Register of Controlled Trials (CENTRAL); and review‐specific searches of additional databases. Reports of trials are also sourced from international trials registers c/o the World Health Organization's trials portal (the International Clinical Trials Registry Platform (ICTRP)) and ClinicalTrials.gov, pharmaceutical companies, the handsearching of key journals, conference proceedings, and other (non‐Cochrane) systematic reviews and meta‐analyses. Details of CCDAN's generic search strategies (used to identify RCTs) can be found on the Group's website.
The Group's TSC searched the CCDANCTR to 13 October 2015 using the following terms: (Topiramate or Topamax) AND (“affective disorder*” or bipolar or mania* or manic* or hypomani* or psychos* or psychotic or postpsychotic or post‐psychotic or “rapid cycling” or schizoaffective).
International trial registers
We ran an additional search of ClinicalTrials.gov and the ICTRP to 13 October 2015 to identify unpublished or ongoing studies, search terms: Topiramate or Topamax.
Searching other resources
Grey literature
We also searched the grey literature using the terms Topiramate and Topamax. We searched the website www.greylit.org/ on 21 May 2014.
Handsearching
We reviewed the latest versions of: The Maudsley Prescribing Guidelines in Psychiatry; the annual conference proceedings and guidelines of the British Association of Psychopharmacology, the World Federation of Societies of Biological Psychiatry (WFSBP), and the Canadian Network for Mood and Anxiety Treatments (CANMAT).
Reference lists
We checked the reference lists of all identified RCTs, review articles, and other relevant papers.
Correspondence
We would identify the authors of significant papers over the last five years from the authorship of trials and review articles found in the search and contact them and other experts in the field to ask of their knowledge of other published or unpublished studies relevant to the review. We asked pharmaceutical companies marketing topiramate products to provide relevant published and unpublished data (see CCDAN group policy).
Data collection and analysis
Selection of studies
Two review authors (KP and IG) independently screened titles and abstracts of all the potential studies identified as a result of the search for inclusion and coded them as 'retrieve' (eligible or potentially eligible/unclear) or 'do not retrieve'. We retrieved the full‐text study reports/publication, and two review authors (KP and IG) independently screened the full‐text and identified studies for inclusion, and identified and recorded reasons for exclusion of the ineligible studies. Any disagreements were resolved through discussion or, if required, by consulting a third person (AY).
We identified and excluded duplicate records and collated multiple reports that related to the same study so that each study, rather than each report, was the unit of interest in the review.
We recorded the selection process in sufficient detail to complete a PRISMA flow diagram and 'Characteristics of excluded studies' table.
Data extraction and management
We used a data collection form to extract study characteristics and outcome data that had been piloted on at least one study in the review. Two review authors (KP and IG) extracted study characteristics and outcome data from included studies. We extracted the following study characteristics.
Eligibility: confirm eligibility for review, reason for exclusion.
Methods: study design, total duration of study, study setting, withdrawals, date of study, sequence generation, allocation sequence concealment, blinding, and other concerns about bias.
Participants: total number, age range, gender, diagnostic criteria, country.
Interventions: intervention, comparison, concomitant medications, and excluded medications.
Outcomes: primary and secondary outcomes specified and collected, and time points reported.
Notes: funding for trial and notable conflicts of interest of trial authors.
We noted in the 'Characteristics of included studies' table if outcome data were not reported in a useable way. Disagreements were resolved by consensus or by involving a third person (AY). One review author (IG) transferred data into the Review Manager file (RevMan 2012). We double‐checked that data were entered correctly by comparing the data presented in the systematic review with the study reports. A second review author (KP) spot‐checked study characteristics for accuracy against the trial report.
Main comparisons
We analysed: 1) acute depressive and 2) acute mixed and manic episodes. For each episode, the main comparisons were as follows.
Topiramate versus placebo as monotherapy
Topiramate versus alternative pharmacological treatment as monotherapy
Topiramate versus placebo as adjunctive treatment
Topiramate versus an alternative pharmacological agent as adjunctive treatment
We would include discontinuation trials but analyse them separately.
Assessment of risk of bias in included studies
Two review authors (SW and KV) independently assessed risk of bias for each study using the criteria outlined in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). Any disagreements were resolved by discussion or by involving another review author (JG). We assessed the risk of bias according to the following domains.
Random sequence generation
Allocation concealment
Blinding of participants and personnel
Blinding of outcome assessment
Incomplete outcome data
Selective outcome reporting
Other bias
We judged each potential source of bias as high, low, or unclear and provided a supporting quotation from the study report together with a justification for our judgement in the 'Risk of bias' table. The 'Risk of bias' judgements were summarised across different studies for each of the domains listed. Blinding was considered separately for different key outcomes where necessary (for example for unblinded outcome assessment, risk of bias for all‐cause mortality may be very different than for a patient‐reported pain scale). Where information on risk of bias related to unpublished data or correspondence with an author, we would note this in the 'Risk of bias' table.
When considering treatment effects, we took into account the risk of bias for the studies that contributed to that outcome.
Measures of treatment effect
Dichotomous data
We analysed dichotomous data as odds ratios with 95% confidence intervals (CIs).
Continuous data
We analysed continuous data as mean difference with 95% CIs or standardised mean difference with 95% CIs. We presented data as a scale with a consistent direction of effect.
We undertook meta‐analyses only where this was meaningful, that is if the treatments, participants, and underlying clinical question were similar enough for pooling to make sense.
We would report a narrative description of skewed data as medians and interquartile ranges.
Where a single trial reported multiple trial arms, we included only the relevant arms.
Unit of analysis issues
Cluster‐randomised trials
In cluster‐randomised trials, groups of individuals rather than individuals are randomised to different interventions. Cluster‐randomised trials would need to account for intraclass correlation. To adjust for cluster effects, we would use the generic inverse variance technique, provided that cluster‐randomised trials have been appropriately analysed taking into account intraclass correlation coefficients (ICC). If the necessary summary statistics were not reported, we would contact the authors; otherwise the data cannot be re‐analysed.
Cross‐over trials
In cross‐over trials, each participant is allocated to a sequence of interventions and each participant acts as his/her own control. We used only data from the first phase of cross‐over trials because the effect of treatment in the first period can affect the outcome in the second period.
Studies with multiple treatment groups
In trials with multiple treatment groups, we selected one pair of interventions and excluded the others to avoid including the same group of participants twice in the same meta‐analysis.
Dealing with missing data
We contacted investigators or study sponsors in order to verify key study characteristics and to obtain missing numerical outcome data where possible (for example when a study was identified as abstract only). We documented all correspondence with authors and reported which authors responded in the full review.
We used intention‐to‐treat (ITT) data when available. Missing data would be imputed with replacement values and treated as if they were observed (last observation carried forward). Where this was not possible, in order to carry out an ITT analysis including all randomised participants, we imputed the missing data as follows.
For continuous efficacy outcomes, we imputed the missing data assuming that these participants had no change in their mean score on the Young Mania Rating Scale from baseline to the endpoint. As access to the raw participant data was not possible for their baseline score, we used the mean baseline provided by the other participants. For the imputed means, we used the same standard deviation of the original mean reported in the studies. We carried out sensitivity analyses to assess the robustness of the assumptions. We assumed these participants to have had the same mean change as the other participants.
For dichotomous outcomes, we used missing data based on a consideration of a 'worst‐case' scenario. To assess the robustness of the assumptions, we carried out sensitivity analyses based on a 'best‐case' scenario.
Assessment of heterogeneity
We assessed heterogeneity between studies using the I2 statistic (Higgins 2003), and by visual inspection of the forest plot. As per recommendations in the Cochrane Handbook for Systematic Reviews of Interventions, we interpreted I2 values as follows: 0% to 40% might not be important; 30% to 60% may represent moderate heterogeneity; 50% to 90% may represent substantial heterogeneity; 75% to 100% considerable heterogeneity. If we identified significant heterogeneity, we investigated the sources.
Assessment of reporting biases
No analysis in this review contained more than 10 studies, however should this be the case in future versions of the review, we plan to contruct funnel plots to examine for small‐study effects (Higgins 2011). In addition to publication bias, such effects may be due to selective reporting, poor methodological quality leading to spuriously inflated effects in smaller studies, true heterogeneity of effect, artefact, and chance (Higgins 2011).
Data synthesis
We presented non‐quantitative data descriptively.
We analysed outcomes concerning relapse/recurrence of mood disorder excluding data from studies of discontinuation design. We analysed data from these studies separately, to assess the effects of topiramate discontinuation.
We used a random‐effects method (DerSimonian 1986), as it assumes that studies estimate different but related treatment effects. We considered a random‐effects model to be appropriate because changes in the symptom rating scales may measure similar but different effects. For example, a change in total score in a manic symptom rating scale could reflect improvements in physical symptoms of mania (for example sleep disturbance, motor activity), whilst in another study it may reflect a change in psychological symptoms (for example thought disorder, insight). We carried out a sensitivity analysis using a fixed‐effect instead of a random‐effects model to see if this affected the results.
When we could not combine studies, we explained the reasons for this and provided a narrative summary documenting the principal findings.
Subgroup analysis and investigation of heterogeneity
Participants with different characteristics may respond differently to topiramate and to other treatments. We were unable to conduct subgroup analyses in this version of the review due to insufficient data, however in future we plan the following subgroup analyses. We will interpret results with caution because multiple comparisons can lead to false‐positive conclusions (Oxman 1992).
Mixed mood episodes: the coexistence of depressive and manic symptoms during the same time period has been considered to be a more severe form of mood episode than either one of these states alone and may respond differently to trial medications.
Mood disorder with psychotic features: psychotic features are associated with more severe episodes of mood disorder and may respond differently to mild‐moderate mood disorder or to non‐psychotic episodes of similar severity.
Rapid cycling disorder: this is a variant of bipolar disorder characterised by four or more affective episodes per year. It is associated with greater morbidity and mortality than its classic form, including greater risk of suicide, and a high incidence of inadequate response to lithium. Therefore participants with this variant may respond differently to trial medications.
Previous failure to treatment: participants who had previously failed to respond to treatment may respond differently to trial medications.
We would separate data from trials including participants with schizoaffective and recurrent unipolar depression into diagnostic groups.
Sensitivity analysis
We performed sensitivity analyses as follows to assess the robustness of the results.
Excluding trials with high risk of bias in one of the domains (i.e. trials with inadequate allocation concealment and blinding, with incomplete data reporting and/or with high probability of selective reporting). We assessed the risk of bias for each study as per the guidelines in the Cochrane Handbook (Higgins 2011).
Excluding trials with high levels of missing data (more than 30%). In studies with high drop‐out rates, the assumptions involved in the use of the last‐observation‐carried‐forward approach may introduce considerable bias.
Fixed‐effect instead of random‐effects model. The random‐effects model, rather than the fixed‐effect model, allows for heterogeneity, however the estimate of distribution of studies may not be accurate if biases are present.
Excluding cluster‐randomised trials in order to investigate the strength of their conclusions, as these trials can introduce the risk of bias in several ways, for example recruitment bias, baseline imbalance, and incorrect analysis (Higgins 2011), and also when ICCs have been borrowed from external sources.
'Summary of findings' table
We prepared 'Summary of findings' tables as per methods in the Cochrane Handbook (Higgins 2011). We included the following elements.
-
A list of all important outcomes, both desirable and undesirable:
change in mean scores from baseline to end of treatment on manic and mixed symptom rating scales for manic and mixed episodes;
change in mean scores from baseline to end of treatment on depression symptom rating scales for depressive episodes;
participants experiencing troublesome side effects of any nature;
response to treatment, defined as a 50% reduction or greater in mean score from baseline to end of treatment on manic and mixed symptom rating scales for manic and mixed episodes;
response to treatment, defined as a 50% reduction or greater in mean score from baseline to end of treatment on depression symptom rating scales for depressive episodes;
remission, for manic and mixed episodes, defined as a mood rating scale score within the normal range at the end of the study;
remission, for depressive episodes, defined as a mood rating scale score within the normal range at the end of the study.
A measure of the typical burden of these outcomes (e.g. illustrative risk on control intervention). According to the Systematic Treatment Enhancement Program for Bipolar Disorder (STEP‐BD) cohort (n = 1469), 58% of people with bipolar disorder types I and II achieved recovery, but 49% had recurrences in a two‐year interval (Perlis 2006).
Absolute and relative magnitude of effect (if both are appropriate).
Numbers of participants and studies addressing these outcomes.
A grade of the overall quality of the body of evidence for each outcome (which may vary by outcome).
Space for comments.
We used the GRADE approach to assessing the quality of the body of evidence. We adhered to the standard methods for the preparation and presentation of results outlined in the Cochrane Handbook (Higgins 2011).
Results
Description of studies
See Characteristics of included studies; Characteristics of excluded studies; Characteristics of studies awaiting classification.
Results of the search
We identified 43 references in total: 38 from the CCDANCTR, 1 from search of the reference lists, and 4 following a handsearch. The number of records screened after de‐duplication remained unchanged. Following a review of the abstracts, we excluded 20 references (10 for study type, 6 for population, 4 for intervention). We assessed 23 full‐text articles for eligibility excluding 3 records for study type and categorising 5 as awaiting classification. The number of studies included in the qualitative analysis and in the meta‐analysis was 6 (15 references). Of these 15 references, the 3 identified from handsearching were secondary reports of studies already identified from the CCDANCTR. See PRISMA flow diagram for details (Figure 1). We last updated the literature search in October 2015.
1.
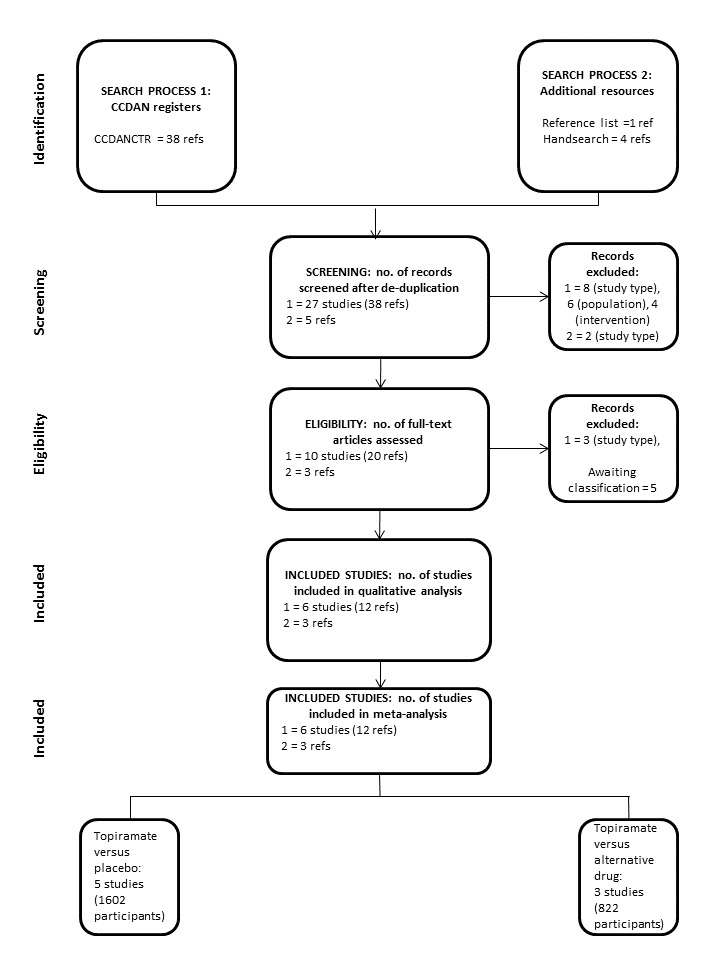
PRISMA flow diagram
We liaised with Professor MP DelBello to obtain all available published versions of her clinical trials (DelBello 2004); she confirmed they were not yet published.
We liaised with Dr J Wozniak and obtained a copy of her paper presented at the Stanley Foundation Research awards (Wozniak 2000).
We liaised with Professor R McIntyre to request the original data from his study (McIntyre 2000); he was not able to retrieve archived information.
We liaised with Dr Z Mirsepassi to obtain original unpublished data from his study (Mirsepassi 2013); he was not able to provide detailed records.
We contacted Professor PS Power, Professor G Sachs, and Janssen and obtained clarification on their conference abstract (Power 2004).
We contacted the Korean Journal of Psychopharmacology to retrieve an English version of the trial Yoon 2003a, but were unsuccessful. The King's College library was also not able to retrieve an English version.
Attempts to contact the following authors were unsuccessful: Professor J Calabrese (to clarify which study was cited in his conference abstract Calabrese 2000); Professor S Kushner and Janssen (to identify which placebo‐controlled trials they made reference to in their article Kushner 2006).
Included studies
See Characteristics of included studies.
We included a total of six studies in the systematic review. We contacted the authors regarding missing data but did not obtain any additional information.
Study design
All six studies were RCTs. One was a single‐centre and single‐blind trial (McIntyre 2000), while the remaining were all multicentre and double‐blind trials.
Three trials were two‐armed, with topiramate versus placebo in Chengappa 2006 and PDMD‐006 Kushner 2006, and topiramate versus an alternative pharmacological treatment in McIntyre 2000.
Two trials were three‐armed, with topiramate in two different doses and placebo in PDMD‐005 Kushner 2006, and topiramate, lithium, and placebo in PDMD‐008 Kushner 2006; the latter trial had an arm that crossed over.
One trial was four‐armed, with topiramate in two different doses, lithium, and placebo (PDMD‐004 Kushner 2006); one of these arms crossed over.
Four studies used topiramate as monotherapy (PDMD‐004 Kushner 2006; PDMD‐005 Kushner 2006; PDMD‐006 Kushner 2006; PDMD‐008 Kushner 2006), and two studies used topiramate as adjunctive therapy, in combination with valproate or lithium (Chengappa 2006), or as adjunctive therapy with divalproex sodium, lithium, or atypical antipsychotics (McIntyre 2000).
Two trials included a cross‐over phase (PDMD‐004 Kushner 2006; PDMD‐008 Kushner 2006).
Sample size
We included a total of 1638 participants. One study contained 36 participants (McIntyre 2000), while each of the others contained over 200 participants.
The mean sample size per arm was 102 participants (range of 18 to 144).
Participants
All studies included male and female patients of all ethnic backgrounds.
Two studies Chengappa 2006; McIntyre 2000 included participants aged over 18 years. In four studies PDMD‐004 Kushner 2006; PDMD‐005 Kushner 2006; PDMD‐006 Kushner 2006; PDMD‐008 Kushner 2006 participants were eligible if they were 16 years of age or older. These 4 studies PDMD‐004 Kushner 2006; PDMD‐005 Kushner 2006; PDMD‐006 Kushner 2006; PDMD‐008 Kushner 2006 had mean ages between 37 and 43. We decided to include the data from these 4 studies and undertook a sensitivity analysis to assess robustness of the results.
In five studies participants met the criteria for bipolar 1 disorder (DSM‐IV) experiencing a manic or mixed episode (Chengappa 2006; PDMD‐004 Kushner 2006; PDMD‐005 Kushner 2006; PDMD‐006 Kushner 2006; PDMD‐008 Kushner 2006). In one study participants met the criteria for bipolar disorder 1 or 2 depressive phase (DSM‐IV) (McIntyre 2000). Five of the six studies included mixed‐mood episodes (Chengappa 2006; PDMD‐004 Kushner 2006; PDMD‐005 Kushner 2006; PDMD‐006 Kushner 2006; PDMD‐008 Kushner 2006). In four studies patients with a mood disorder with psychotic features were included (PDMD‐004 Kushner 2006; PDMD‐005 Kushner 2006; PDMD‐006 Kushner 2006; PDMD‐008 Kushner 2006), and in one study they were excluded (McIntyre 2000). The remaining study did not comment on mood disorders with psychotic features (Chengappa 2006). Rapid cyclers were included in two studies (Chengappa 2006; McIntyre 2000), and excluded in four studies (PDMD‐004 Kushner 2006; PDMD‐005 Kushner 2006; PDMD‐006 Kushner 2006; PDMD‐008 Kushner 2006). None of the studies commented on previous failure to treatment. We were not able to conduct subgroup analysis with regard to mixed‐mood episodes, mood disorders with psychotic features, rapid cycling disorder, or previous failure to treatment because no individual data was provided.
In all six studies the exclusion criteria regarding comorbidities were not fully explicit. However, we included the studies as baseline clinical characteristics were generally similar between the treatment groups and any significant comorbidities were not reported.
Two studies were conducted in an outpatient setting (Chengappa 2006; McIntyre 2000), while the remaining four studies were conducted in an inpatient setting (PDMD‐004 Kushner 2006; PDMD‐005 Kushner 2006; PDMD‐006 Kushner 2006; PDMD‐008 Kushner 2006).
Interventions/comparison
In two studies topiramate was used as an adjunctive treatment. In one study topiramate (target dose 400 mg/day) was added to valproate or lithium versus placebo (Chengappa 2006). In the other study topiramate (titrated until clinical response from 50 mg up to 300 mg/day) was added to divalproex sodium, lithium, or an atypical antipsychotic versus bupropion sustained‐release (McIntyre 2000). In four studies topiramate was administered as a monotherapy versus placebo. Two studies used topiramate in two different doses (200 mg/day and 400 mg/day in PDMD‐004 Kushner 2006 and 400 mg/day and 600 mg/day in PDMD‐005 Kushner 2006). In two studies lithium (1500 mg/day) was an active comparator (PDMD‐004 Kushner 2006; PDMD‐008 Kushner 2006). Where there were multiple treatment groups (PDMD‐004 Kushner 2006; PDMD‐005 Kushner 2006; PDMD‐008 Kushner 2006), we selected one pair of interventions (topiramate doses of 400 mg/day versus placebo) and excluded the others (200 mg/day and 600 mg/day) to avoid including the same group of participants twice in the same meta‐analysis.
In one study participants were permitted to continue taking a stable dose of an oral antipsychotic agent (Chengappa 2006), as well as subtherapeutic doses of antidepressants. In addition, the use of short‐acting benzodiazepines for sleep or agitation was permitted only during the first four weeks. There was no significant difference in the treatment response of participants receiving concomitant antipsychotics versus those who did not. In one study participants were not permitted to receive concomitant antidepressant treatment (McIntyre 2000); there was no mention of use of benzodiazepines. In four studies (PDMD‐004 Kushner 2006; PDMD‐005 Kushner 2006; PDMD‐006 Kushner 2006; PDMD‐008 Kushner 2006), during the first 14 days chloral hydrate, non‐benzodiazepine short‐acting sedative hypnotics, and short‐acting benzodiazepine anxiolytics could be used as rescue medication. Participants requiring antipsychotics or mood stabilisers were discontinued. Non‐pharmacological interventions other than supportive or educational psychotherapy were prohibited.
In the Chengappa et al trial, participants received the interventions for 12 weeks. The McIntyre et al trial was an eight‐week trial. Three of the Kushner trials were 12 weeks' long (PDMD‐004 Kushner 2006; PDMD‐005 Kushner 2006; PDMD‐008 Kushner 2006); two of these included a nine‐week cross‐over phase (PDMD‐004 Kushner 2006; PDMD‐008 Kushner 2006). One trial was three weeks' long (PDMD‐006 Kushner 2006).
Outcomes
Primary outcomes
In five studies the primary efficacy measure was the change in Young Mania Rating Score (YMRS) from baseline to endpoint. In the remaining study (McIntyre 2000), the primary efficacy measure was the percentage of participants responding to treatment as measured by: (a) 50% reduction from baseline in Hamilton Depression Rating Scale (HDRS‐17); and (b) remission defined as endpoint HDRS‐17 score of equal to or less than 7. As the primary efficacy outcome in this review was change in mean score from baseline to endpoint in rating scale, and as per previous Cochrane review Vasudev 2006, we attempted to contact the author to obtain original data but were unsuccessful.
All studies reported side effects. In five studies safety measures also included clinical laboratory values and vital signs. Rehospitalisation was considered in four of the studies (PDMD‐004 Kushner 2006; PDMD‐005 Kushner 2006; PDMD‐006 Kushner 2006; PDMD‐008 Kushner 2006). The safety evaluation in McIntyre 2000 also looked at concomitant medications and weight change.
We were able to use data regarding side effects of any nature from four studies: Chengappa 2006, McIntyre 2000, PDMD‐005 Kushner 2006, and in the topiramate and lithium arms of PDMD‐008 Kushner 2006.
Secondary outcomes
In five studies secondary efficacy assessments included the Clinical Global Impressions‐Severity of Illness scale (CGI‐S), the Brief Psychiatric Rating Scale (BPRS), the Montgomery‐Åsberg Depression Rating Scale (MADRS), and the Global Assessment Scale (GAS) (Chengappa 2006; PDMD‐004 Kushner 2006; PDMD‐005 Kushner 2006; PDMD‐006 Kushner 2006; PDMD‐008 Kushner 2006). In addition, four studies included mean per cent change in baseline body weight, the percentage of participants with ≥ 50% change in YMRS, YMRS ≤ 12 at final visit, ≥ 10% increase from baseline YMRS, participants who no longer met DSM‐IV criteria for manic or mixed episodes of bipolar I disorder as secondary efficacy measures. These studies also determined participants with treatment‐emergent depression (PDMD‐004 Kushner 2006; PDMD‐005 Kushner 2006; PDMD‐006 Kushner 2006; PDMD‐008 Kushner 2006). In the McIntyre 2000 study the secondary efficacy measures were MADRS, CGI‐S, and CGI‐I (Improvement scale).
We were able to retrieve data on response to treatment from two studies (Chengappa 2006; McIntyre 2000), and on remission from one study (McIntyre 2000).
We used data regarding specific side effects from one study (Chengappa 2006).
Timing of outcome assessment
The endpoint was 12 weeks for one study (Chengappa 2006), eight weeks for one study (McIntyre 2000), and three weeks for one study (PDMD‐006 Kushner 2006). Regarding the other three studies, there was a three‐week core and a nine‐week extension period (PDMD‐004 Kushner 2006; PDMD‐005 Kushner 2006PDMD‐008 Kushner 2006). Our protocol stated that we would use the time point closest to 12 weeks. Due to a cross‐over phase in the placebo arm of two studies (PDMD‐004 Kushner 2006; PDMD‐008 Kushner 2006), we were only able to use data from the three‐week core phase in the placebo arm. Otherwise we used a 12‐week endpoint.
Excluded studies
See Characteristics of excluded studies.
We excluded 10 studies for several reasons, such as the absence of a control group, inappropriate age ranges of participants, and inappropriate outcome (Bahk 2005; DelBello 2004; Hebrani 2009; Mahmoudi‐Gharaei 2012; McIntyre 2005; Mirsepassi 2014;Sahraian 2014; Vieta 2004; Wozniak 2000; Wozniak 2009).
Studies awaiting classification
See Characteristics of studies awaiting classification.
We identified three studies that are awaiting classification (Calabrese 2000; Mirsepassi 2013; Yoon 2003). We were unable to obtain the full text of the Calabrese study. We have requested and are awaiting original data from the Mirsepassi trial. We were unable to retrieve an English version of the Yoon study despite our efforts to contact the Korean Journal of Psychopharmacology and the King's College library.
Ongoing studies
We identified no ongoing studies.
Risk of bias in included studies
We considered five of the studies to be at low risk of selection bias for random sequence generation, performance, detection, attrition, and reporting biases, and the risk was unclear for allocation concealment and for other potential sources of bias as the available information was insufficient to make an assessment (Chengappa 2006; PDMD‐004 Kushner 2006; PDMD‐005 Kushner 2006; PDMD‐006 Kushner 2006; PDMD‐008 Kushner 2006).
We considered the McIntyre 2000 study to have a high risk of performance bias because it was a single‐blind study and participants and personnel had knowledge of the allocated interventions. Also, the risk of bias was unclear for random sequence generation, allocation concealment, blinding of outcome assessment, and other potential sources of bias due to insufficient information to make a judgement. We considered the risk of attrition bias and reporting bias to be low.
See 'Risk of bias' tables in the Characteristics of included studies and Figure 2; Figure 3.
2.
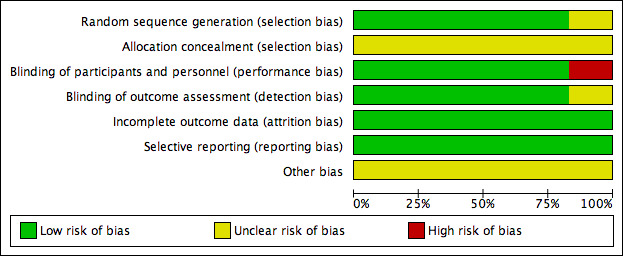
Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
3.

Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
Allocation
The risk of bias for random sequence generation was low for all of the studies except McIntyre 2000. We were unable to judge the risk of bias for allocation concealment for any of the studies due to insufficient information.
Blinding
The risk of performance and detection bias was low for five studies (Chengappa 2006; PDMD‐004 Kushner 2006; PDMD‐005 Kushner 2006; PDMD‐006 Kushner 2006; PDMD‐008 Kushner 2006), as they maintained blinding of participants, personnel, and outcome assessment. We considered McIntyre 2000 to have a high risk of performance bias due to single blinding; the risk of detection bias was unclear.
Incomplete outcome data
Overall, we considered risk of attrition bias for all studies to be low. All studies reported attrition rates and reasons for withdrawals. They all used intention‐to‐treat analysis including all randomised participants with at least one postbaseline data. They accounted for missing data using the last‐observation‐carried‐forward approach.
Selective reporting
We considered the risk of reporting bias to be low across all the studies. We gave this careful consideration, as no study protocols were available. However, all expected outcomes, including those that were prespecified in the protocol section of the final reports, were included in the results.
Other potential sources of bias
Five of the studies were industry‐funded (Chengappa 2006; PDMD‐004 Kushner 2006; PDMD‐005 Kushner 2006; PDMD‐006 Kushner 2006; PDMD‐008 Kushner 2006). It was not specified whether or not the McIntyre 2000 trial was industry‐funded. Taking into account this information, we judged the risk of other potential sources of bias to be unclear.
Effects of interventions
See: Table 1; Table 2; Table 3; Table 4
Comparison 1: Topiramate versus placebo as monotherapy
The four studies that contributed to this comparison were PDMD‐004 Kushner 2006, PDMD‐005 Kushner 2006, PDMD‐006 Kushner 2006, and PDMD‐008 Kushner 2006. The endpoint was three weeks for one study (PDMD‐006 Kushner 2006). In the other three studies there was a three‐week core and a nine‐week extension period (PDMD‐004 Kushner 2006; PDMD‐005 Kushner 2006PDMD‐008 Kushner 2006). Our protocol stated we would use the time point closest to 12 weeks. Due to a cross‐over phase in the placebo arm of two studies (PDMD‐004 Kushner 2006; PDMD‐008 Kushner 2006), we were only able to use data from the three‐week core phase in the placebo arm. Otherwise, we used a 12‐week endpoint.
Primary outcomes
1.1 Efficacy of treatment for manic and mixed episodes: change in mean scores from baseline to end of treatment on symptom rating scales (negative numbers indicate improvement)
There was no evidence that topiramate was more efficacious than placebo as monotherapy in the treatment of manic and mixed episodes in terms of mean change (endpoint: 3 weeks) on YMRS (YMRS range 0‐60) (mean difference (MD) 1.17, 95% confidence interval (CI) ‐0.52 to 2.86; participants = 664; studies = 3; P = 0.17) (Analysis 1.1). We assessed the evidence contributing to this outcome as moderate quality.
1.1. Analysis.
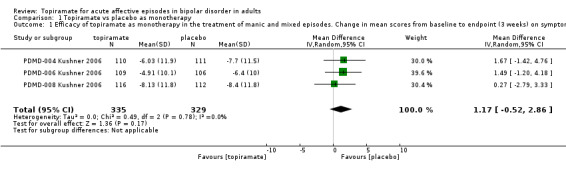
Comparison 1 Topiramate vs placebo as monotherapy, Outcome 1 Efficacy of topiramate as monotherapy in the treatment of manic and mixed episodes. Change in mean scores from baseline to endpoint (3 weeks) on symptom rating scales (negative numbers indicate improvement).
There was no evidence to suggest that topiramate as monotherapy is more efficacious than placebo at endpoint 12 weeks (MD ‐0.58, 95% CI ‐3.45 to 2.29; participants = 212; studies = 1; P = 0.69) (Analysis 1.2). We assessed the evidence contributing to this outcome as low quality.
1.2. Analysis.
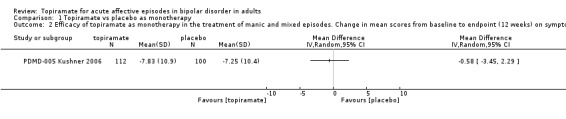
Comparison 1 Topiramate vs placebo as monotherapy, Outcome 2 Efficacy of topiramate as monotherapy in the treatment of manic and mixed episodes. Change in mean scores from baseline to endpoint (12 weeks) on symptom rating scales (negative numbers indicate improvement).
1.2 Efficacy of treatment for depressive episodes: change in mean scores from baseline to end of treatment on symptom rating scales (negative numbers indicate improvement)
No data were available.
1.3 Acceptability: number of participants experiencing troublesome side effects of any nature
There was no difference between the groups with regard to experiencing troublesome side effects of any nature (endpoint: 12 weeks) (odds ratio (OR) 0.68, 95% CI 0.33 to 1.40; participants = 212; studies = 1; P = 0.30) (Analysis 1.3). We assessed the evidence contributing to this outcome as low quality.
1.3. Analysis.
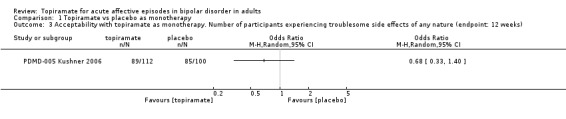
Comparison 1 Topiramate vs placebo as monotherapy, Outcome 3 Acceptability with topiramate as monotherapy. Number of participants experiencing troublesome side effects of any nature (endpoint: 12 weeks).
Secondary outcomes
1.4 Response to treatment for manic and mixed episodes: number of participants experiencing a 50% reduction or greater in mean score from baseline to end of treatment on symptom rating scales
No data were available.
1.5 Response to treatment for depressive episodes: number of participants experiencing a 50% reduction or greater in mean score from baseline to end of treatment on symptom rating scales
No data were available.
1.6 Remission for manic and mixed episodes: number of participants presenting with a score within the normal range on symptom rating scales
No data were available.
1.7 Remission for depressive episodes: number of participants presenting with a score within the normal range on symptom rating scales
No data were available.
Comparison 2: Topiramate versus placebo as add‐on therapy
One study contributed to this comparison (Chengappa 2006). In this study topiramate was used at a target dose of 400 mg/day and was added to valproate or lithium.
Primary outcomes
2.1 Efficacy of treatment for manic and mixed episodes: change in mean scores from baseline to end of treatment on symptom rating scales (negative numbers indicate improvement)
There was no evidence to show that topiramate as add‐on therapy is superior to placebo in the treatment of manic and mixed episodes (endpoint: 12 weeks) (YMRS range 0 to 60) (MD ‐0.14, 95% CI ‐2.10 to 1.82; participants = 287; studies = 1; P = 0.89) (Analysis 2.1). We assessed the evidence contributing to this outcome as low quality.
2.1. Analysis.

Comparison 2 Topiramate vs placebo as add‐on therapy, Outcome 1 Efficacy of topiramate as add‐on therapy in the treatment of manic and mixed episodes. Change in mean scores from baseline to endpoint (12 weeks) on symptom rating scales (negative numbers indicate improvement).
2.2 Efficacy of treatment for depressive episodes: change in mean scores from baseline to end of treatment on symptom rating scales (negative numbers indicate improvement)
No data were available.
2.3 Acceptability: number of participants experiencing troublesome side effects of any nature
We identified no difference in number of participants experiencing troublesome side effects of any nature (endpoint: 12 weeks) (OR 1.10, 95% CI 0.58 to 2.10; participants = 287; studies = 1; P = 0.76) (Analysis 2.2). We assessed the evidence contributing to this outcome as low quality.
2.2. Analysis.
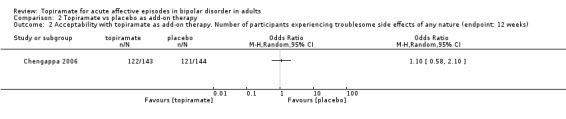
Comparison 2 Topiramate vs placebo as add‐on therapy, Outcome 2 Acceptability with topiramate as add‐on therapy. Number of participants experiencing troublesome side effects of any nature (endpoint: 12 weeks).
Secondary outcomes
2.4 Response to treatment for manic and mixed episodes: number of participants experiencing a 50% reduction or greater in mean score from baseline to end of treatment on symptom rating scales
There was no evidence that topiramate is superior to placebo as an add‐on therapy (endpoint: 12 weeks) (OR 0.98, 95% CI 0.61 to 1.58; participants = 287; studies = 1; P = 0.94) (Analysis 2.3). We assessed the evidence contributing to this outcome as low quality.
2.3. Analysis.
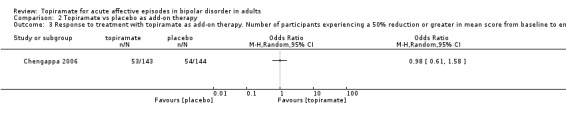
Comparison 2 Topiramate vs placebo as add‐on therapy, Outcome 3 Response to treatment with topiramate as add‐on therapy. Number of participants experiencing a 50% reduction or greater in mean score from baseline to end of treatment on symptom rating scales in manic and mixed episodes (endpoint: 12 weeks).
2.5 Response to treatment for depressive episodes: number of participants experiencing a 50% reduction or greater in mean score from baseline to end of treatment on symptom rating scales
No data were available.
2.6 Remission for manic and mixed episodes: number of participants presenting with a score within the normal range on symptom rating scales
No data were available.
2.7 Remission for depressive episodes: number of participants presenting with a score within the normal range on symptom rating scales
No data were available.
Comparison 3: Topiramate versus an alternative drug as monotherapy
Two studies were eligible for this comparison (PDMD‐004 Kushner 2006; PDMD‐008 Kushner 2006). Topiramate was compared to lithium (1500 mg/day) as an active comparator.
Primary outcomes
3.1 Efficacy of treatment for manic and mixed episodes: change in mean scores from baseline to end of treatment on symptom rating scales (negative numbers indicate improvement)
There was evidence that topiramate is less efficacious than an alternative drug as monotherapy in the treatment of manic and mixed episodes in terms of mean change on YMRS (endpoint: 12 weeks) (YMRS range 0 to 60) (MD 8.46, 95% CI 5.86 to 11.06; participants = 449; studies = 2; P < 0.00001) (Analysis 3.1). We assessed the evidence contributing to this outcome as high quality, although it should be noted that only two studies contributed.
3.1. Analysis.
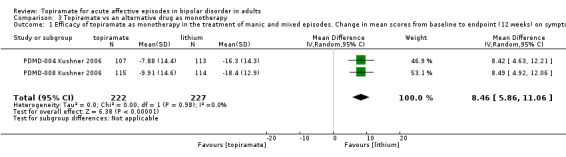
Comparison 3 Topiramate vs an alternative drug as monotherapy, Outcome 1 Efficacy of topiramate as monotherapy in the treatment of manic and mixed episodes. Change in mean scores from baseline to endpoint (12 weeks) on symptom rating scales (negative numbers indicate improvement).
3.2 Efficacy of treatment for depressive episodes: change in mean scores from baseline to end of treatment on symptom rating scales (negative numbers indicate improvement)
No data were available.
3.3 Acceptability: number of participants experiencing troublesome side effects of any nature
There was no difference between the groups in terms of side effects (endpoint: 12 weeks) (OR 0.87, 95% CI 0.50 to 1.52; participants = 230; studies = 1; P = 0.63) (Analysis 3.2). We assessed the evidence contributing to this outcome as low quality.
3.2. Analysis.
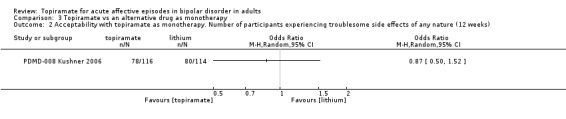
Comparison 3 Topiramate vs an alternative drug as monotherapy, Outcome 2 Acceptability with topiramate as monotherapy. Number of participants experiencing troublesome side effects of any nature (12 weeks).
Secondary outcomes
3.4 Response to treatment for manic and mixed episodes: number of participants experiencing a 50% reduction or greater in mean score from baseline to end of treatment on symptom rating scales
No data were available.
3.5 Response to treatment for depressive episodes: number of participants experiencing a 50% reduction or greater in mean score from baseline to end of treatment on symptom rating scales
No data were available.
3.6 Remission for manic and mixed episodes: number of participants presenting with a score within the normal range on symptom rating scales
No data were available.
3.7 Remission for depressive episodes: number of participants presenting with a score within the normal range on symptom rating scales
No data were available.
Comparison 4: Topiramate versus an alternative drug as add‐on therapy
One study was eligible for this comparison (McIntyre 2000). Topiramate (titrated until clinical response from 50 mg up to 300 mg/day) was added to divalproex sodium, lithium, or an atypical antipsychotic versus bupropion sustained‐release.
Primary outcomes
3.1 Efficacy of treatment for manic and mixed episodes: change in mean scores from baseline to end of treatment on symptom rating scales (negative numbers indicate improvement)
No data were available.
3.2 Efficacy of treatment for depressive episodes: change in mean scores from baseline to end of treatment on symptom rating scales (negative numbers indicate improvement)
No data were available.
3.3 Acceptability: number of participants experiencing troublesome side effects of any nature
There was no difference between the groups in terms of side effects (endpoint: 8 weeks) (OR 1.57, 95% CI 0.42 to 5.90; participants = 36; studies = 1; P = 0.50) (Analysis 4.1). We assessed the evidence contributing to this outcome as very low quality.
4.1. Analysis.
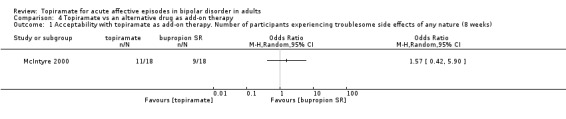
Comparison 4 Topiramate vs an alternative drug as add‐on therapy, Outcome 1 Acceptability with topiramate as add‐on therapy. Number of participants experiencing troublesome side effects of any nature (8 weeks).
Secondary outcomes
3.4 Response to treatment for manic and mixed episodes: number of participants experiencing a 50% reduction or greater in mean score from baseline to end of treatment on symptom rating scales
No data were available.
3.5 Response to treatment for depressive episodes: number of participants experiencing a 50% reduction or greater in mean score from baseline to end of treatment on symptom rating scales
There was no difference between the groups in terms of response to treatment (endpoint: 8 weeks) (OR 0.80, 95% CI 0.21 to 3.00; participants = 36; studies = 1; P = 0.74) (Analysis 4.2). We assessed the evidence contributing to this outcome as very low quality.
4.2. Analysis.
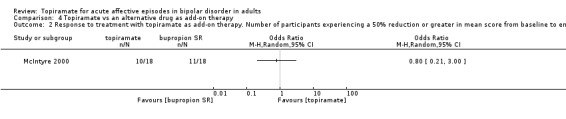
Comparison 4 Topiramate vs an alternative drug as add‐on therapy, Outcome 2 Response to treatment with topiramate as add‐on therapy. Number of participants experiencing a 50% reduction or greater in mean score from baseline to end of treatment on symptom rating scales in depressive episodes (8 weeks).
3.6 Remission for manic and mixed episodes: number of participants presenting with a score within the normal range on symptom rating scales
No data were available.
3.7 Remission for depressive episodes: number of participants presenting with a score within the normal range on symptom rating scales
There was no difference between the groups in terms of remission (endpoint: 8 weeks) (OR 1.00, 95% CI 0.23 to 4.30; participants = 36; studies = 1; P = 1.00) (Analysis 4.3). We assessed the evidence contributing to this outcome as very low quality.
4.3. Analysis.
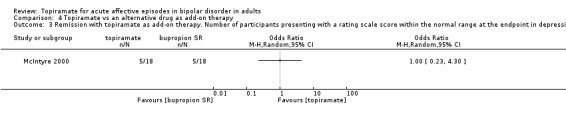
Comparison 4 Topiramate vs an alternative drug as add‐on therapy, Outcome 3 Remission with topiramate as add‐on therapy. Number of participants presenting with a rating scale score within the normal range at the endpoint in depressive episodes (8 weeks).
Subgroup analyses
We were not able to conduct subgroup analyses with regard to mixed‐mood episodes, mood disorders with psychotic features, rapid cycling disorder, or previous failure to treatment because no individual data was provided.
Assessment of heterogeneity
We identified no significant heterogeneity during the analysis.
Sensitivity analyses
We conducted a sensitivity analysis comparing random‐effects and fixed‐effect models. This sensitivity analysis did not affect the results, and the confidence interval and the P value remained not significant.
Where possible, we conducted a sensitivity analysis excluding trials with high levels of missing data, that is more than 30%. The sensitivity analysis did not affect the results.
To assess the robustness of the assumptions made for the imputation of missing data, we performed a sensitivity analysis as follows.
For continuous efficacy outcomes, the missing participants were assumed to have had the same mean change as the other participants.
For dichotomous outcomes, we conducted sensitivity analyses based on a 'best‐case' scenario.
The above sensitivity analyses did not affect the results.
We found one study to have a high risk of performance bias (McIntyre 2000). It was not possible to perform a sensitivity analysis because it was the only study included in that comparison.
Reporting bias
This review included six studies, therefore we were unable to construct a funnel plot to examine for small‐study effects (Higgins 2011).
Discussion
Summary of main results
This systematic review found six RCTs evaluating the use of topiramate in the treatment of acute affective episodes in bipolar disorder. The 'Summary of findings' tables (Table 1; Table 2; Table 3; Table 4) show high‐quality evidence that topiramate is less efficacious than lithium when used as monotherapy in the treatment of manic and mixed episodes, although this evidence was provided by two studies with a total of 449 participants. Moderate‐quality evidence suggests no difference in efficacy between topiramate and placebo as monotherapy in the treatment of manic and mixed episodes. The evidence for topiramate for the other outcomes was generally of low quality. The evidence for the use of topiramate in depressive episodes was of very low quality, so we are uncertain about the estimate.
Overall completeness and applicability of evidence
Of the studies identified in this review, five examined manic and mixed episodes, and one looked at depressive episodes. Overall, the objectives of the review (Objectives) were addressed, although the studies did not provide appropriate data for some of the outcomes. We investigated all relevant types of participants in terms of their characteristics, diagnosis, and the setting in which they were treated. However, patients were excluded if they had DSM‐IV Axis I and II and physical comorbidities which would limit the applicability of the evidence. We examined relevant types of intervention, however the only alternative drug topiramate was compared to was lithium. In addition, the add‐on drugs used were limited to atypical antipsychotics. This would limit the applicability of the findings when compared to current clinical practice.
Cognitive side effects were not specifically elicited with cognitive testing with pre‐ and post‐neuropsychological tests, which are one of the main types of side effects limiting the use of topiramate. Also, in the 'Summary of findings' tables the reader will note that there was no data available for many outcomes. However, the studies were worthy of inclusion in the meta‐analysis because they met the inclusion criteria and addressed the primary outcome. We are not clear why remission and response to treatment topiramate were not reported for many of the studies, as they are important in terms of clinical practice and prognosis.
Quality of the evidence
In summary, we included six studies with a total of 1638 participants, and the overall quality of the evidence was low.
We rated the quality of body of evidence for the outcomes taking into account the following factors.
Risk of bias
The quality of the evidence was downgraded if the studies were at high risk of bias.
Inconsistency
None of the evidence was downgraded due to inconsistency.
Indirectness
None of the evidence was downgraded due to indirectness.
Imprecision
The quality of the evidence was downgraded due to imprecision if there was a small sample size or small number of events and if 95% confidence intervals included no effect and were wide.
Publication bias
None of the evidence was downgraded due to publication bias.
Potential biases in the review process
Strengths of the review include the likelihood that all relevant studies were identified by searching electronic databases and grey literature, handsearching, and checking reference lists. Limitations of the review were that not all the relevant data could be obtained despite efforts to contact the authors. We did not believe the methods used in the review process to have introduced bias.
Agreements and disagreements with other studies or reviews
This systematic review is an update of the previous systematic Cochrane intervention review on topiramate for acute affective episodes in bipolar disorder conducted in 2006 (Vasudev 2006). The previous review included only one study, which was also included in this review (McIntyre 2000), along with more RCTs. This review was in agreement with other reviews that evaluated topiramate in the treatment of bipolar disorder which did not find evidence of efficacy for topiramate in the short term or when used to supplement standard mood‐stabilising treatments (Poon 2012), and did not support the efficacy of topiramate as monotherapy in acute mania and mixed episodes (Rosa 2011).
Authors' conclusions
Implications for practice.
It is not possible to draw any firm conclusions about the use of topiramate in clinical practice from this evidence. The only high‐quality evidence found was that lithium was more efficacious than topiramate when used as monotherapy in the treatment of acute affective episodes in bipolar disorder, and we note that this evidence came from only two studies. Moderate‐quality evidence did not find topiramate to be more efficacious than placebo when a 3‐week endpoint was used, however the evidence contributing to the 12‐week time point for this outcome was of only low quality. It was not possible to draw conclusions about the use of topiramate as an adjunctive treatment in acute affective episodes or to comment on the acceptability of topiramate in a clinical context, as overall the quality of the evidence was low. The practical usefulness of topiramate in the treatment of acute affective episodes in bipolar disorder therefore remains questionable.
Implications for research.
As the evidence base for the use of topiramate in acute affective episodes in bipolar is limited by the quality of the research, for example imprecision (small population size, small number of events, wide confidence intervals which also include no effect) or high risk of bias (single‐blind study), further research could address this. For example, more double‐blind RCTs would be appropriate that are more explicit with regard to methodological issues and comparing placebo, alternative and combination treatments (including a wide range of mood stabilisers), atypical antipsychotics for manic and mixed episodes, and also antidepressants in combination with mood stabilisers or atypical antipsychotics for depressive episodes.
To help people make well‐informed decisions about future healthcare research, we propose the following research suggestions.
Population: patients with all subtypes of bipolar disorder, acute affective episodes, both sexes, 18 and over, all ethnicities, all settings.
Intervention: monotherapy, adjunctive therapy.
Comparison: placebo, alternative and combination treatments including a wide range of mood stabilisers, atypical antipsychotics for manic and mixed episodes, and also antidepressants in combination with mood stabilisers or atypical antipsychotics for depressive episodes.
Outcome: rating scale scores, remission and response rates, dropouts and reasons why, and detailed side effects. It would be desirable if these were documented for each individual.
To best address this research question, if investigators see the indication in so doing, future studies should be RCTs as well as more explicit with regard to methodological issues such as allocation concealment.
What's new
| Date | Event | Description |
|---|---|---|
| 7 September 2015 | New citation required but conclusions have not changed | Review updated |
| 7 September 2015 | New search has been performed | Review methodology updated and new studies incorporated |
History
Protocol first published: Issue 4, 2001 Review first published: Issue 1, 2006
| Date | Event | Description |
|---|---|---|
| 25 August 2005 | New citation required and conclusions have changed | Substantive amendment |
Acknowledgements
The review authors wish to gratefully acknowledge the help of Sarah Dawson of the Cochrane Depression, Anxiety and Neurosis Group (CCDAN) in conducting the database searches and CCDAN Managing Editor Chris Champion for his important contributions.
We would also like to thank Karine Macritchie for the work she has undertaken thus far, including cowriting the previous protocol and review.
Data and analyses
Comparison 1. Topiramate vs placebo as monotherapy.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Efficacy of topiramate as monotherapy in the treatment of manic and mixed episodes. Change in mean scores from baseline to endpoint (3 weeks) on symptom rating scales (negative numbers indicate improvement) | 3 | 664 | Mean Difference (IV, Random, 95% CI) | 1.17 [‐0.52, 2.86] |
| 2 Efficacy of topiramate as monotherapy in the treatment of manic and mixed episodes. Change in mean scores from baseline to endpoint (12 weeks) on symptom rating scales (negative numbers indicate improvement) | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 3 Acceptability with topiramate as monotherapy. Number of participants experiencing troublesome side effects of any nature (endpoint: 12 weeks) | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 2. Topiramate vs placebo as add‐on therapy.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Efficacy of topiramate as add‐on therapy in the treatment of manic and mixed episodes. Change in mean scores from baseline to endpoint (12 weeks) on symptom rating scales (negative numbers indicate improvement) | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 2 Acceptability with topiramate as add‐on therapy. Number of participants experiencing troublesome side effects of any nature (endpoint: 12 weeks) | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 3 Response to treatment with topiramate as add‐on therapy. Number of participants experiencing a 50% reduction or greater in mean score from baseline to end of treatment on symptom rating scales in manic and mixed episodes (endpoint: 12 weeks) | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 3. Topiramate vs an alternative drug as monotherapy.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Efficacy of topiramate as monotherapy in the treatment of manic and mixed episodes. Change in mean scores from baseline to endpoint (12 weeks) on symptom rating scales (negative numbers indicate improvement) | 2 | 449 | Mean Difference (IV, Random, 95% CI) | 8.46 [5.86, 11.06] |
| 2 Acceptability with topiramate as monotherapy. Number of participants experiencing troublesome side effects of any nature (12 weeks) | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 4. Topiramate vs an alternative drug as add‐on therapy.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Acceptability with topiramate as add‐on therapy. Number of participants experiencing troublesome side effects of any nature (8 weeks) | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2 Response to treatment with topiramate as add‐on therapy. Number of participants experiencing a 50% reduction or greater in mean score from baseline to end of treatment on symptom rating scales in depressive episodes (8 weeks) | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 3 Remission with topiramate as add‐on therapy. Number of participants presenting with a rating scale score within the normal range at the endpoint in depressive episodes (8 weeks) | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Chengappa 2006.
| Methods | DESIGN Description: randomised, double‐blind, placebo‐controlled, parallel‐group, multicentre trial. Study visits occurred weekly during an 8‐week titration period and then biweekly for the remaining 4 weeks. BLINDING Participants: yes Assessors: yes Administrators: yes ALLOCATION CONCEALMENT Method: study medication was packaged by the sponsor according to the randomisation schedule and provided to the study sites with identification numbers RANDOMISATION: Method: a computer‐generated randomisation schedule was prepared before the study and was balanced using randomly permuted blocks |
|
| Participants | SAMPLE Description: 287 adult outpatients with bipolar 1 disorder (DSM‐IV criteria) experiencing a manic or a mixed episode with a YMRS score of ≥ 18 while taking therapeutic levels of valproate or lithium. Participants were required to have received either lithium or valproate for 6 weeks or more including a stable dose during the 2 weeks before the screening visit. Serum levels of mood stabiliser at the screening visit were required to be between 0.5 and 1.2 mEq/L for lithium or between 45 and 100 mg/L for valproate when drawn 8 to 12 hours after the last dose. Male and female adults aged between 18 and 70 years. Exclusion criteria included substance abuse or dependence (except alcohol or marijuana) in the previous 3 months; mania requiring hospitalisation; an organic mental disorder; mental retardation or a developmental disability; treatment‐emergent mania due to antidepressant use; use of an antidepressant or stimulant within the previous 2 weeks (4 weeks for fluoxetine) unless the antidepressant was given at a subtherapeutic dose; use of carbamazepine within the previous 2 weeks or another anticonvulsant within the previous 3 weeks; initiation of nutraceutical treatment (e.g. St John's wort) within the previous 4 weeks; long‐acting antipsychotic medication or oral antipsychotic medicine that was given above the maximum recommended dose or was not given at a stable dose during the previous 4 weeks; use of opiates or barbiturates within the previous 3 months; any clinically unstable comorbid disease; liver disease; untreated hypothyroidism; history of nephrolithiasis, seizures, or any contraindication or precaution that would preclude use of topiramate; and pregnancy, lactation, or inadequate contraception in women. SCREENING Primary diagnosis: SCID |
|
| Interventions | Interventions: 143 participants received adjunctive topiramate (titrated from 25 to 400 mg/day over 8 weeks and was continued for 4 additional weeks), and 144 participants received placebo, while taking therapeutic levels of valproate or lithium, for 12 weeks. The initial dose of topiramate was 25 mg once daily. The total daily dose was then titrated at weekly study visits, using the following sequence and a twice‐daily dosing schedule: 50, 75, 100, 150, 200, 300, and 400 mg. The investigator could stop titrating study medication due to adverse events, but the minimum allowed dose after the first week was 50 mg daily. After the 8‐week titration period, the same dose was continued for the remaining 4 weeks |
|
| Outcomes | Primary outcomes: measure was the change in YMRS score from baseline to last study visit during the double‐blind phase. YMRS ratings were assessed during screening and then at every visit during the double‐blind phase. Secondary outcomes: efficacy assessments were measured at screening and baseline (day 1) and then biweekly and included the CGI‐S, BPRS, MADRS, and GAS. Adverse events were reported at every visit. Data estimation: SAS version 9.1 was used for statistical analyses. Efficacy analyses were performed in the ITT population of participants who received at least 1 dose of study medication and completed at least 1 postbaseline assessment. Missing efficacy data were imputed with LOCF |
|
| Notes | INDUSTRY SUPPORT Industry funded: this study was supported by Ortho‐McNeil Neurologics, Inc. Medication provided by industry: yes Any of the authors work for industry: yes ADDITIONAL INFORMATION The study was conducted from October 2001 through October 2003. In addition to study medication and a stable dose of divalproex sodium or lithium, participants were permitted to continue taking a stable dose of an oral antipsychotic agent. The use of a short‐acting benzodiazepine (lorazepam) for sleep or agitation was permitted only during the first 4 weeks of the titration period. 49 participants (26 topiramate treated and 23 placebo treated) were included in the ITT population for efficacy analysis despite violating the study protocol by starting lithium or valproate fewer than 6 weeks before the study |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | A computer‐generated randomisation schedule was prepared before the study and was balanced using randomly permuted blocks |
| Allocation concealment (selection bias) | Unclear risk | No information available to make judgement |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double blind |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Double blind |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Figure 1 summarises the numbers screened, randomised, intent to treat, and discontinued with reasons. Both efficacy and safety analyses included all randomised participants with a least 1 postbaseline data |
| Selective reporting (reporting bias) | Low risk | All expected outcomes in terms of efficacy and safety are reported. All the study's prespecified primary outcomes are reported |
| Other bias | Unclear risk | The study was industry funded |
McIntyre 2000.
| Methods | DESIGN Description: randomised, single‐blind trial (raters were blinded). BLINDING Participants: no Assessors: yes Administrators: no ALLOCATION CONCEALMENT No information available RANDOMISATION Method: participants were randomised (no other information provided) |
|
| Participants | SAMPLE Description: 36 patients with DSM‐IV diagnosis of bipolar I or II (depressive phase). Participants were 18 to 70 years of age. Exclusion criteria: prior exposure to trial drugs, substance dependence in past 30 days, ECT in preceding 4 weeks prior to entry or at any time during the trial, high suicide risk, history of nephrolithiasis, history of seizures, active neurological or medical problems, incapacity to provide consent, psychotic symptoms. SCREENING Primary diagnosis: SCID‐I/P version 2.0 |
|
| Interventions | Interventions: 8‐week trial of topiramate vs bupropion SR adjunctive therapy with 18 participants in each arm. Topiramate 50 mg/day or bupropion SR 100 mg/day. Titrated every 2 weeks until clinical response. Final dose range: topiramate 50 to 300 mg/day; bupropion SR 100 to 400 mg/day. Participants were allowed to titrate to lower doses to enhance tolerability. Interventions added to existing treatments. For those participants randomised to topiramate, 5 received lithium, 13 received divalproex sodium, and 3 received atypical antipsychotics. For those participants randomised to bupropion SR, 8 received lithium, 10 received divalproex sodium, and 3 received atypical antipsychotics |
|
| Outcomes | Primary outcomes: the primary efficacy measure was the percentage of participants responding to treatment as measured by:
Secondary outcomes: the secondary efficacy measures were MADRS, CGI‐S, and CGI‐I results, but these were not detailed in the published account. Safety evaluation was assessed by report of adverse events, concomitant medications, vital signs, weight change, and laboratory tests. Data estimation: data were analysed on an intent‐to‐treat basis using LOCF. Independent t tests and analysis of variance (ANOVA) repeated measures design were used to compare baseline and 8 weeks of single‐blind treatment with topiramate and bupropion SR |
|
| Notes | INDUSTRY SUPPORT Industry funded: not specified Medication provided by industry: not specified Any of the authors work for industry: not specified |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Insufficient information available to make judgement, e.g. "subjects were randomised to receive either." |
| Allocation concealment (selection bias) | Unclear risk | No information available to make judgement |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | This was a single‐blind study. Performance bias due to knowledge of the allocated interventions by participants and personnel |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Insufficient information to make judgement, e.g. “single‐blind (rater‐blind)” |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Attrition and reason for attrition are reported, attrition rate is balanced in numbers across intervention groups. LOCF used to account for missing data |
| Selective reporting (reporting bias) | Low risk | All expected outcomes in terms of efficacy and safety are reported. All the study's prespecified outcomes are reported |
| Other bias | Unclear risk | It is not specified if this study was industry funded |
PDMD‐004 Kushner 2006.
| Methods | DESIGN Description: multicentre, randomised, double‐blind, placebo‐controlled, parallel‐group, conducted in Eastern and Western Europe, Argentina, India, Israel, and Australia. The trial included a screening phase during which previous psychotropic medications were discontinued, followed by randomisation to double‐blind treatment for 3 weeks (core study); double‐blind treatment was continued for a total of 12 weeks (3‐week core study + 9‐week double‐blind extension). The screening‐phase duration varied according to the time needed for medication washout. Following randomisation, participants were hospitalised for at least 4 days and remained hospitalised as clinically warranted during the core 3‐week study. BLINDING Participants: yes Assessors: yes Administrators: yes ALLOCATION CONCEALMENT Method: the study drug was packaged and labelled for each participant, with participant numbers pre‐printed on study drug labels RANDOMISATION Method: computer‐generated randomisation code that was balanced by using permuted blocks |
|
| Participants | SAMPLE Description: the randomised population included 444 patients, who were eligible if they were ≥ 16 years of age with a primary DSM‐IV diagnosis of bipolar I disorder and hospitalised with an acute manic or mixed episode, confirmed by a Structured Clinical Interview for DSM‐IV Axis I Disorders. Patients had to have a history of at least 1 previous manic or mixed episode. Patients with comorbid diagnoses were included unless the primary diagnosis was schizoaffective or impulse control disorder or the patient had antisocial or borderline personality disorder. A YMRS score ≥ 20 at screening and randomisation was required. Women of childbearing age were eligible if they were surgically incapable of bearing children or practicing a medically acceptable method of birth control, were not lactating, and had negative pregnancy tests at screening and baseline. Other exclusion criteria included rapid cycling bipolar disorder, DSM‐IV‐defined alcohol or substance abuse/dependence within the previous 3 months, and symptoms associated with recent antidepressant/psychostimulant treatment or with intoxication or withdrawal from psychostimulants such as alcohol, (meth)amphetamines, cocaine, or hallucinogens. Patients were excluded if they were at high risk for suicide, violence, or alcohol/substance abuse, or were likely to require acute intervention with psychotropic medications that could not be discontinued. Patients with unstable or serious medical conditions, seizure disorders, history of nephrolithiasis, or patients taking medication associated with nephrolithiasis were excluded. Exclusion criteria also included the use of substances with potentially confounding psychotropic effects (e.g. St. John’s wort, calcium channel blockers administered for mania, or antidepressants) within the previous 4 weeks (5 weeks if fluoxetine), clozapine use within the previous 3 months, previous use of topiramate monotherapy for acute mania or mixed episodes for > 4 weeks, previous participation in a topiramate trial, history of severe drug allergy, known hypersensitivity to topiramate, treatment with an experimental drug or medical device within 30 days before screening, known or suspected intolerance to lithium and low‐salt diet. SCREENING Primary diagnosis: SCID |
|
| Interventions | Interventions: 108 participants were randomised to topiramate 200 mg/day, 107 to topiramate 400 mg/day, 113 to lithium 1500 mg/day, and 111 to placebo. The starting dose of 300 mg/day lithium was increased daily in 300 mg increments to 1200 mg/day at day 4 and 1500 mg/day at day 6; lithium dosage could be reduced a maximum of 600 mg/day. Lithium dosage was individualised based on target serum levels (titration, 0.8 to 1.2 mEq/L; stabilisation, 0.6 to 1.2 mEq/L; maximum 1800 mg/day). The starting dose of 50 mg/day topiramate was increased to 100 mg/day at day 2 and in 100 mg increments each day for the next 1 to 5 days until the target dose (200, 400, or 600 mg/day) or the maximally tolerated dose was achieved. Participants were crossed over from placebo to lithium (1500 mg/day) during the double‐blind extension. Dose titration and adjustments for lithium followed the same schedule as in the core double‐blind study |
|
| Outcomes | Primary outcomes: the primary efficacy endpoint was mean YMRS change from baseline in the core 3‐week double‐blind study. Secondary outcomes: secondary efficacy measures included mean YMRS change from baseline after 12 weeks and mean per cent change in baseline body weight after 3 and 12 weeks. Other planned efficacy measures were changes from baseline in CGI‐S, GAS, BPRS, and MADRS. The percentage of participants with ≥ 50% change in YMRS, YMRS ≤ 12 at final visit, ≥ 10% increase from baseline YMRS, participants who no longer met DSM‐IV diagnostic criteria for manic or mixed episode of bipolar I disorder (DSM‐IV Responder), and participants with treatment‐emergent depression (MADRS ≥ 18 and a change of ≥ 4 points from baseline on at least 2 consecutive visits or at the last visit) were also determined. Tolerability/safety measures included treatment‐emergent adverse events, clinical laboratory values, vital signs, and re‐hospitalisation. Data estimation: missing values were imputed using the LOCF. Analysis of covariance (ANCOVA) was used to compare between‐group differences |
|
| Notes | INDUSTRY SUPPORT Industry funded: yes Medication provided by industry: not specified Any of the authors work for industry: yes ADDITIONAL INFORMATION During washout, short‐acting benzodiazepine anxiolytics such as lorazepam (maximum 8 mg/day) were allowed for agitation and insomnia. Investigators could slow down the topiramate titration by withholding doses or could reduce the dosage, with a maximum reduction of 2 tablets or capsules (100 mg/day topiramate) to improve tolerability. During the first 14 days of double‐blind treatment, chloral hydrate, non‐benzodiazepine short‐acting sedative hypnotics, and short‐acting benzodiazepine anxiolytics could be used as rescue medication. Participants requiring antipsychotics or mood stabilisers were discontinued. Non‐pharmacologic interventions other than supportive or educational psychotherapy were prohibited |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated randomisation code |
| Allocation concealment (selection bias) | Unclear risk | No information available to make judgement |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "study treatment was blinded to patients, investigators, and clinical staff, study monitors, data reviewers, and data‐entry personnel until the double‐blind phase was completed and database finalised." "Based on the randomisation code, the study drug was packaged and labelled for each patient, with patient numbers pre printed on study drug labels." "The central lab had access to the randomisation code to identify blood samples for lithium." |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | As above. Also, "to maintain study blinding, investigators received instructions from central laboratory to increase/decrease the mid‐day lithium dose to achieve target levels, with sham adjustments in the mid‐day (inactive) dose for patients in topiramate and placebo groups." |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Table 2 summarises the numbers randomised, intent to treat, completed and withdrawals, reasons for withdrawal. Both efficacy and safety analyses included all randomised participants with at least 1 postbaseline data during double‐blind treatment |
| Selective reporting (reporting bias) | Low risk | All expected outcomes in terms of efficacy and safety are reported. All the study’s prespecified primary outcomes are reported |
| Other bias | Unclear risk | The study was industry funded |
PDMD‐005 Kushner 2006.
| Methods | DESIGN Description: multicentre, randomised, double‐blind, placebo‐controlled, parallel‐group, conducted in the United States. The trial included a screening phase during which previous psychotropic medications were discontinued, followed by randomisation to double‐blind treatment for 3 weeks (core study); double‐blind treatment was continued for a total of 12 weeks (3‐week core study + 9‐week double‐blind extension). The duration of the screening phase varied according to the time needed for medication washout. Following randomisation, participants were hospitalised for at least 4 days and remained hospitalised as clinically warranted during the core 3‐week study. BLINDING Participants: yes Assessors: yes Administrators: yes ALLOCATION CONCEALMENT Method: the study drug was packaged and labelled for each participant, with participant numbers pre‐printed on study drug labels RANDOMISATION Method: computer‐generated randomisation code that was balanced by using permuted blocks |
|
| Participants | SAMPLE Description: the randomised population included 314 participants, who were eligible if they were ≥ 16 years of age with a primary DSM‐IV diagnosis of bipolar I disorder and hospitalised with an acute manic or mixed episode, confirmed by a Structured Clinical Interview for DSM‐IV Axis I Disorders. Patients had to have a history of at least 1 previous manic or mixed episode. Patients with comorbid diagnoses were included unless the primary diagnosis was schizoaffective or impulse control disorder or the patient had antisocial or borderline personality disorder. A YMRS score ≥ 20 at screening and randomisation was required. Women of childbearing age were eligible if they were surgically incapable of bearing children or practicing a medically acceptable method of birth control, were not lactating, and had negative pregnancy tests at screening and baseline. Other exclusion criteria included rapid cycling bipolar disorder, DSM‐IV‐defined alcohol or substance abuse/dependence within the previous 3 months, and symptoms associated with recent antidepressant/psychostimulant treatment or with intoxication or withdrawal from psychostimulants such as alcohol, (meth)amphetamines, cocaine, or hallucinogens. Patients were excluded if they were at high risk for suicide, violence, or alcohol/substance abuse, or were likely to require acute intervention with psychotropic medications that could not be discontinued. Patients with unstable or serious medical conditions, seizure disorders, history of nephrolithiasis, or patients taking medication associated with nephrolithiasis were excluded. Exclusion criteria also included the use of substances with potentially confounding psychotropic effects (e.g. St. John’s wort, calcium channel blockers administered for mania, or antidepressants) within the previous 4 weeks (5 weeks if fluoxetine), clozapine use within the previous 3 months, previous use of topiramate monotherapy for acute mania or mixed episodes for > 4 weeks, previous participation in a topiramate trial, history of severe drug allergy, known hypersensitivity to topiramate, and treatment with an experimental drug or medical device within 30 days before screening. SCREENING Primary diagnosis: SCID |
|
| Interventions | Interventions: 108 participants were randomised to topiramate 400 mg/day, 101 to topiramate 600 mg/day, 99 to placebo. The starting dose of 50 mg/day topiramate was increased to 100 mg/day at day 2 and in 100 mg increments each day for the next 1 to 5 days until the target dose (200, 400, or 600 mg/day) or the maximally tolerated dose was achieved |
|
| Outcomes | Primary outcomes: the primary efficacy endpoint was mean YMRS change from baseline in the core 3‐week double‐blind study. Secondary outcomes: secondary efficacy measures included mean YMRS change from baseline after 12 weeks and mean per cent change in baseline body weight after 3 and 12 weeks. Other planned efficacy measures were changes from baseline in CGI‐S, GAS, BPRS, and MADRS. The percentage of participants with ≥ 50% change in YMRS, YMRS ≤ 12 at final visit, ≥ 10% increase from baseline YMRS, participants who no longer met DSM‐IV diagnostic criteria for manic or mixed episode of bipolar I disorder (DSM‐IV Responder), and participants with treatment‐emergent depression (MADRS ≥ 18 and a change of ≥ 4 points from baseline on at least 2 consecutive visits or at the last visit) were also determined. Tolerability/safety measures included treatment‐emergent adverse events, clinical laboratory values, vital signs, and re‐hospitalisation. Data estimation: missing values were imputed using the LOCF. Analysis of covariance (ANCOVA) was used to compare between‐group differences |
|
| Notes | INDUSTRY SUPPORT Industry funded: yes Medication provided by industry: not specified Any of the authors work for industry: yes ADDITIONAL INFORMATION During washout, short‐acting benzodiazepine anxiolytics such as lorazepam (maximum 8 mg/day) were allowed for agitation and insomnia. Investigators could slow down the topiramate titration by withholding doses or could reduce the dosage, with a maximum reduction of 2 tablets or capsules (100 mg/day topiramate) to improve tolerability. During the first 14 days of double‐blind treatment, chloral hydrate, non‐benzodiazepine short‐acting sedative hypnotics, and short‐acting benzodiazepine anxiolytics could be used as rescue medication. Participants requiring antipsychotics or mood stabilisers were discontinued. Non‐pharmacologic interventions other than supportive or educational psychotherapy were prohibited |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated randomisation code |
| Allocation concealment (selection bias) | Unclear risk | No information available to make judgement |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "study treatment was blinded to patients, investigators, and clinical staff, study monitors, data reviewers, and data‐entry personnel until the double‐blind phase was completed and database finalised." "Based on the randomisation code, the study drug was packaged and labelled for each patient, with patient numbers pre printed on study drug labels." "The central lab had access to the randomisation code to identify blood samples for lithium." |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | As above. Also, "to maintain study blinding, investigators received instructions from central laboratory to increase/decrease the mid‐day lithium dose to achieve target levels, with sham adjustments in the mid‐day (inactive) dose for patients in topiramate and placebo groups." |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Table 2 summarises the numbers randomised, intent to treat, completed and withdrawals, reasons for withdrawal. Both efficacy and safety analyses included all randomised participants with at least 1 postbaseline data during double‐blind treatment |
| Selective reporting (reporting bias) | Low risk | All expected outcomes in terms of efficacy and safety are reported. All the study’s prespecified primary outcomes are reported |
| Other bias | Unclear risk | The study was industry funded |
PDMD‐006 Kushner 2006.
| Methods | DESIGN Description: multicentre, randomised, double‐blind, placebo‐controlled, parallel‐group, conducted in the United States. The trial included a screening phase during which previous psychotropic medications were discontinued, followed by randomisation to double‐blind treatment for 3 weeks (core study). The duration of the screening phase varied according to the time needed for medication washout. Following randomisation, participants were hospitalised for at least 4 days and remained hospitalised as clinically warranted during the core 3‐week study. BLINDING Participants: yes Assessors: yes Administrators: yes ALLOCATION CONCEALMENT Method: the study drug was packaged and labelled for each participant, with participant numbers pre‐printed on study drug labels RANDOMISATION Method: computer‐generated randomisation code that was balanced by using permuted blocks |
|
| Participants | SAMPLE Description: the randomised population included 215 participants, who were eligible if they were ≥ 16 years of age with a primary DSM‐IV diagnosis of bipolar I disorder and hospitalised with an acute manic or mixed episode, confirmed by a Structured Clinical Interview for DSM‐IV Axis I Disorders. Patients had to have a history of at least 1 previous manic or mixed episode. Patients with comorbid diagnoses were included unless the primary diagnosis was schizoaffective or impulse control disorder or the patient had antisocial or borderline personality disorder. A YMRS score ≥ 20 at screening and randomisation was required. Women of childbearing age were eligible if they were surgically incapable of bearing children or practicing a medically acceptable method of birth control, were not lactating, and had negative pregnancy tests at screening and baseline. Other exclusion criteria included rapid cycling bipolar disorder, DSM‐IV‐defined alcohol or substance abuse/dependence within the previous 3 months, and symptoms associated with recent antidepressant/psychostimulant treatment or with intoxication or withdrawal from psychostimulants such as alcohol, (meth)amphetamines, cocaine, or hallucinogens. Patients were excluded if they were at high risk for suicide, violence, or alcohol/substance abuse, or were likely to require acute intervention with psychotropic medications that could not be discontinued. Patients with unstable or serious medical conditions, seizure disorders, history of nephrolithiasis, or patients taking medication associated with nephrolithiasis were excluded. Exclusion criteria also included the use of substances with potentially confounding psychotropic effects (e.g. St. John’s wort, calcium channel blockers administered for mania, or antidepressants) within the previous 4 weeks (5 weeks if fluoxetine), clozapine use within the previous 3 months, previous use of topiramate monotherapy for acute mania or mixed episodes for > 4 weeks, previous participation in a topiramate trial, history of severe drug allergy, known hypersensitivity to topiramate, and treatment with an experimental drug or medical device within 30 days before screening. SCREENING Primary diagnosis: SCID |
|
| Interventions | Interventions: 107 participants were randomised to topiramate 400 mg/day and 106 to placebo. The starting dose of 50 mg/day topiramate was increased to 100 mg/day at day 2 and in 100 mg increments each day for the next 1 to 5 days until the target dose (200, 400, or 600 mg/day) or the maximally tolerated dose was achieved. |
|
| Outcomes | Primary outcomes: the primary efficacy endpoint was mean YMRS change from baseline in the core 3‐week double‐blind study. Secondary outcomes: efficacy measures included mean YMRS change from baseline after 12 weeks and mean per cent change in baseline body weight after 3 and 12 weeks. Other planned efficacy measures were changes from baseline in CGI‐S, GAS, BPRS, and MADRS. The percentage of participants with ≥ 50% change in YMRS, YMRS ≤ 12 at final visit, ≥ 10% increase from baseline YMRS, participants who no longer met DSM‐IV diagnostic criteria for manic or mixed episode of bipolar I disorder (DSM‐IV Responder), and participants with treatment‐emergent depression (MADRS ≥ 18 and a change of ≥ 4 points from baseline on at least 2 consecutive visits or at the last visit) were also determined. Tolerability/safety measures included treatment‐emergent adverse events, clinical laboratory values, vital signs, and re‐hospitalisation. Data estimation: missing values were imputed using the LOCF. Analysis of covariance (ANCOVA) was used to compare between‐group differences |
|
| Notes | INDUSTRY SUPPORT Industry funded: yes Medication provided by industry: not specified Any of the authors work for industry: yes ADDITIONAL INFORMATION During washout, short‐acting benzodiazepine anxiolytics such as lorazepam (maximum 8 mg/day) were allowed for agitation and insomnia. Investigators could slow down the topiramate titration by withholding doses or could reduce the dosage, with a maximum reduction of 2 tablets or capsules (100 mg/day topiramate) to improve tolerability. During the first 14 days of double‐blind treatment, chloral hydrate, non‐benzodiazepine short‐acting sedative hypnotics, and short‐acting benzodiazepine anxiolytics could be used as rescue medication. Participants requiring antipsychotics or mood stabilisers were discontinued. Non‐pharmacologic interventions other than supportive or educational psychotherapy were prohibited |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated randomisation code |
| Allocation concealment (selection bias) | Unclear risk | No information available to make judgement |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "study treatment was blinded to patients, investigators, and clinical staff, study monitors, data reviewers, and data‐entry personnel until the double‐blind phase was completed and database finalised." "Based on the randomisation code, the study drug was packaged and labelled for each patient, with patient numbers pre printed on study drug labels." "The central lab had access to the randomisation code to identify blood samples for lithium." |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | As above. Also, "to maintain study blinding, investigators received instructions from central laboratory to increase/decrease the mid‐day lithium dose to achieve target levels, with sham adjustments in the mid‐day (inactive) dose for patients in topiramate and placebo groups." |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Table 2 summarises the numbers randomised, intent to treat, completed and withdrawals, reasons for withdrawal. Both efficacy and safety analyses included all randomised participants with at least 1 postbaseline data during double‐blind treatment |
| Selective reporting (reporting bias) | Low risk | All expected outcomes in terms of efficacy and safety are reported. All the study’s prespecified primary outcomes are reported |
| Other bias | Unclear risk | The study was industry funded |
PDMD‐008 Kushner 2006.
| Methods | DESIGN Description: multicentre, randomised, double‐blind, placebo‐controlled, parallel‐group, conducted in Eastern Europe, South Africa, Latin America, and India. The trial included a screening phase during which previous psychotropic medications were discontinued, followed by randomisation to double‐blind treatment for 3 weeks (core study); double‐blind treatment was continued for a total of 12 weeks (3‐week core study + 9‐week double‐blind extension). The duration of the screening phase varied according to the time needed for medication washout. Following randomisation, participants were hospitalised for at least 4 days and remained hospitalised as clinically warranted during the core 3‐week study. BLINDING Participants: yes Assessors: yes Administrators: yes ALLOCATION CONCEALMENT Method: the study drug was packaged and labelled for each participant, with participant numbers pre‐printed on study drug labels RANDOMISATION Method: computer‐generated randomisation code that was balanced by using permuted blocks |
|
| Participants | SAMPLE Description: the randomised population included 342 participants, who were eligible if they were ≥ 16 years of age with a primary DSM‐IV diagnosis of bipolar I disorder and hospitalised with an acute manic or mixed episode, confirmed by a Structured Clinical Interview for DSM‐IV Axis I Disorders. Patients had to have a history of at least 1 previous manic or mixed episode. Patients with comorbid diagnoses were included unless the primary diagnosis was schizoaffective or impulse control disorder or the patient had antisocial or borderline personality disorder. A YMRS score ≥ 20 at screening and randomisation was required. Women of childbearing age were eligible if they were surgically incapable of bearing children or practicing a medically acceptable method of birth control, were not lactating, and had negative pregnancy tests at screening and baseline. Other exclusion criteria included rapid cycling bipolar disorder, DSM‐IV‐defined alcohol or substance abuse/dependence within the previous 3 months, and symptoms associated with recent antidepressant/psychostimulant treatment or with intoxication or withdrawal from psychostimulants such as alcohol, (meth)amphetamines, cocaine, or hallucinogens. Patients were excluded if they were at high risk for suicide, violence, or alcohol/substance abuse, or were likely to require acute intervention with psychotropic medications that could not be discontinued. Patients with unstable or serious medical conditions, seizure disorders, history of nephrolithiasis, or patients taking medication associated with nephrolithiasis were excluded. Exclusion criteria also included the use of substances with potentially confounding psychotropic effects (e.g. St. John’s wort, calcium channel blockers administered for mania, or antidepressants) within the previous 4 weeks (5 weeks if fluoxetine), clozapine use within the previous 3 months, previous use of topiramate monotherapy for acute mania or mixed episodes for > 4 weeks, previous participation in a topiramate trial, history of severe drug allergy, known hypersensitivity to topiramate, treatment with an experimental drug or medical device within 30 days before screening, known or suspected intolerance to lithium and low‐salt diet. SCREENING Primary diagnosis: SCID |
|
| Interventions | Interventions: 115 participants were randomised to topiramate 400 mg/day, 114 to lithium 1500 mg/day, and 112 to placebo. The starting dose of 300 mg/day lithium was increased daily in 300 mg increments to 1200 mg/day at day 4 and 1500 mg/day at day 6; lithium dosage could be reduced a maximum of 600 mg/day. Lithium dosage was individualised based on target serum levels (titration, 0.8 to 1.2 mEq/L; stabilisation, 0.6 to 1.2 mEq/L; maximum 1800 mg/day). Participants were converted from placebo to lithium (1500 mg/day) during the double‐blind extension. The starting dose of 50 mg/day topiramate was increased to 100 mg/day at day 2 and in 100 mg increments each day for the next 1 to 5 days until the target dose (200, 400, or 600 mg/day) or the maximally tolerated dose was achieved. Participants were crossed over from placebo to topiramate (150 mg/day) during the double‐blind extension. For participants converted to topiramate, the starting dose of topiramate was 50 mg/day increased in 50 mg increments on days 2 and 3 |
|
| Outcomes | Primary outcomes: the primary efficacy endpoint was mean YMRS change from baseline in the core 3‐week double‐blind study. Secondary outcomes: secondary efficacy measures included mean YMRS change from baseline after 12 weeks and mean per cent change in baseline body weight after 3 and 12 weeks. Other planned efficacy measures were changes from baseline in CGI‐S, GAS, BPRS, and MADRS. The percentage of participants with ≥ 50% change in YMRS, YMRS ≤ 12 at final visit, ≥ 10% increase from baseline YMRS, participants who no longer met DSM‐IV diagnostic criteria for manic or mixed episode of bipolar I disorder (DSM‐IV Responder), and participants with treatment‐emergent depression (MADRS ≥ 18 and a change of ≥ 4 points from baseline on at least 2 consecutive visits or at the last visit) were also determined. Tolerability/safety measures included treatment‐emergent adverse events, clinical laboratory values, vital signs, and re‐hospitalisation. Data estimation: missing values were imputed using the LOCF. Analysis of covariance (ANCOVA) was used to compare between‐group differences |
|
| Notes | INDUSTRY SUPPORT Industry funded: yes Medication provided by industry: not specified Any of the authors work for industry: yes ADDITIONAL INFORMATION During washout, short‐acting benzodiazepine anxiolytics such as lorazepam (maximum 8 mg/day) were allowed for agitation and insomnia. Investigators could slow down the topiramate titration by withholding doses or could reduce the dosage, with a maximum reduction of 2 tablets or capsules (100 mg/day topiramate) to improve tolerability. During the first 14 days of double‐blind treatment, chloral hydrate, non‐benzodiazepine short‐acting sedative hypnotics, and short‐acting benzodiazepine anxiolytics could be used as rescue medication. Participants requiring antipsychotics or mood stabilisers were discontinued. Non‐pharmacologic interventions other than supportive or educational psychotherapy were prohibited |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated randomisation code |
| Allocation concealment (selection bias) | Unclear risk | No information available to make judgement |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "study treatment was blinded to patients, investigators, and clinical staff, study monitors, data reviewers, and data‐entry personnel until the double‐blind phase was completed and database finalised." "Based on the randomisation code, the study drug was packaged and labelled for each patient, with patient numbers pre printed on study drug labels." "The central lab had access to the randomisation code to identify blood samples for lithium." |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | As above. Also, "to maintain study blinding, investigators received instructions from central laboratory to increase/decrease the mid‐day lithium dose to achieve target levels, with sham adjustments in the mid‐day (inactive) dose for patients in topiramate and placebo groups." |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Table 2 summarises the numbers randomised, intent to treat, completed and withdrawals, reasons for withdrawal. Both efficacy and safety analyses included all randomised participants with at least 1 postbaseline data during double‐blind treatment |
| Selective reporting (reporting bias) | Low risk | All expected outcomes in terms of efficacy and safety are reported. All the study’s prespecified primary outcomes are reported |
| Other bias | Unclear risk | The study was industry funded |
BPRS: Brief Psychiatric Rating Scale CGI‐I: Clinical Global Impressions‐Improvement scale CGI‐S: Clinical Global Impressions‐Severity of Illness scale DSM‐IV: Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition ECT: electroconvulsive therapy GAS: Global Assessment Scale HDRS‐17: Hamilton Depression Rating Scale ITT: intention‐to‐treat LOCF: last observation carried forward MADRS: Montgomery‐Åsberg Depression Rating Scale SCID: Structured Clinical Interview for DSM‐IV Axis I disorders SCID‐I/P: Structured Clinical Interview for DSM‐IV Axis I disorders Patient Edition SR: sustained‐release YMRS: Young Mania Rating Scale
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Bahk 2005 | The study does not include a control group |
| DelBello 2004 | The participants were children and adolescents |
| Hebrani 2009 | The participants were adolescents |
| Mahmoudi‐Gharaei 2012 | The participants were adolescents |
| McIntyre 2005 | The study does not include a control group |
| Mirsepassi 2014 | This study evaluated weight control in people with bipolar type I receiving lithium and antipsychotics during manic episodes |
| Sahraian 2014 | This study looked at controlling the symptoms of obsessive‐compulsive disorder in people with bipolar disorder |
| Vieta 2004 | The study does not include a control group |
| Wozniak 2000 | The participants were children and adolescents |
| Wozniak 2009 | The participants were children and adolescents |
Characteristics of studies awaiting assessment [ordered by study ID]
Calabrese 2000.
| Methods | Multicentre, double‐blind, placebo‐controlled study |
| Participants | |
| Interventions | |
| Outcomes | The primary outcome measure was improvement in people with bipolar I on the Young Mania Rating Scale Secondary outcome measures included the Brief Psychiatric Rating Scale, Montgomery‐Åsberg Depression Rating Scale, Clinical Global Impression, and Global Assessment Scale. The results of safety evaluations will also be addressed |
| Notes |
Mirsepassi 2013.
| Methods | Double‐blind, randomised, 8‐week clinical trial |
| Participants | 46 participants, aged 18 to 65, suffering from bipolar I disorder most recent manic episode hospitalised were included. The exclusion criteria were pregnant and lactating women, people with disabling medical or neurological disorders, substance‐related disorders, history of kidney disease or renal stone, and history of hypersensitivity to topiramate |
| Interventions | In the intervention group, participants received 900 mg of lithium carbonate and 2 mg of risperidone per day (increased to 8 mg per day if needed) or equivalent dosage of other antipsychotics plus topiramate starting with 50 mg per day and increasing the dosage 50 mg each 3 days to reach the maximum dosage of 200 mg per day. In the control group, participants received 900 mg of lithium carbonate and 2 mg of risperidone per day (increased to 8 mg per day if needed) or equivalent dosage of other antipsychotics plus placebo. Duration of the study took 6 weeks after reaching the optimal dosage of topiramate, which was 200 mg per day (8 weeks after onset of the study) |
| Outcomes | Participants were interviewed weekly, and the response to treatment was assessed at baseline and weekly thereafter, using the Young Mania Rating Scale. Medication compliance, appetite, weight, suicidal thoughts, side effects of topiramate, and symptoms and signs of lithium toxicity were assessed in every interview. At the first visit, height and weight were measured and accordingly body mass index was recorded |
| Notes |
Yoon 2003.
| Methods | |
| Participants | |
| Interventions | |
| Outcomes | |
| Notes | English version not retrievable despite efforts |
Differences between protocol and review
The Cochrane Handbook for Systematic Reviews of Interventions recommends a maximum of seven outcomes, therefore we reduced the number of outcomes in the protocol for the review. We added extra information to the section Dealing with missing data to clarify the methods used. We added headings for 'Timing of outcome assessment', 'Hierarchy of outcome measures', and 'Summary of findings' tables, as per current Cochrane guidance.
In the last version of the review we used a fixed‐effect model (using random‐effects models to investigate the sensitivity of results to the choice of statistical method). We believed it to be more appropriate in this version of the review to use a random‐effects model for analyses, as it assumes that studies estimate different but related treatment effects.
We slightly reworded the Objectives to fit the recommended Cochrane template.
Contributions of authors
Dr Katie Pigott and Dr Ilaria Galizia cowrote the protocol, performed the literature search, data extraction, data analysis, and selection of trials, and drafted the final review. Professor Allan Young supervised the selection of trials and the data analysis and contributed to the writing of the final review. Dr Kamini Vasudev and Dr Stuart Watson performed the 'Risk of bias' assessment and resolved any disagreements between themselves. Professor John Geddes reviewed the final draft.
Sources of support
Internal sources
No sources of support supplied
External sources
Stanley Medical Research Institute, USA.
Declarations of interest
In the past, Allan Young has undertaken consultancy work and paid lectures for both Johnson & Johnson and GlaxoSmithKline, who manufacture topiramate and bupropion sustained‐release.
Katie Pigott, Ilaria Galizia, Kamini Vasudev, Stuart Watson, and John Geddes do not have any conflicts of interest.
New search for studies and content updated (no change to conclusions)
References
References to studies included in this review
Chengappa 2006 {published data only}
- Chengappa KN, Schwarzmann LK, Hulihan JF, Xiang J, Rosenthal NR. Adjunctive topiramate therapy in patients receiving a mood stabilizer for bipolar I disorder: A randomized, placebo‐controlled trial. Neuropsychopharmacology 2005;30(Suppl 1):S235. [DOI] [PubMed] [Google Scholar]
- Chengappa KNR, Schwarzman LK, Hulihan JF, Xiang J, Rosenthal NR. Adjunctive topiramate therapy in patients receiving a mood stabilizer for bipolar I disorder: a randomized, placebo‐controlled trial. Journal of Clinical Psychiatry 2006;67(11):1698‐1706. [DOI] [PubMed] [Google Scholar]
- NCT00237289. Topiramate versus placebo as add‐on treatment in patients with bipolar disorder in the outpatient setting. http://www.clinicaltrials.gov/ct2/show/NCT00237289 2006, issue accessed 1 January 2014.
McIntyre 2000 {published and unpublished data}
- Chengappa KN, Gershon S, Levine J. The evolving role of topiramate among other mood stabilizers in the management of bipolar disorder. Bipolar Disorders 2001;3(5):215‐32. [PubMed] [Google Scholar]
- McIntyre R, Mancini DA, McCann SM, Srinavasan J, Sagman D, Kennedy SH. Randomised, single‐blind comparison of topiramate and bupropion sr in bipolar depression. International Journal of Neuropsychopharmacology 2002;5(Suppl 1):56. [Google Scholar]
- McIntyre RS, Mancini DA, McCann S, Srinivasan J, Sagman D, Kennedy SH. Topiramate versus bupropion SR when added to mood stabilizer therapy for the depressive phase of bipolar disorder: a preliminary single‐blind study. Bipolar Disorder 2002;4:207‐13. [DOI] [PubMed] [Google Scholar]
- McIntyre RS, Mancini DA, McCann SM, Srinivasan J, Sagman D, Kennedy SH. Randomized, single‐blind comparison of topiramate and bupropion SR as add‐on therapy in bipolar depression. 39th Annual Meeting of the American College of Neuropsychopharmacology. 2000 Dec 10‐14; San Juan, Puerto Rico. 149.
- McIntyre RS, Mancini DA, McCann SM, Srinivasan J, Sagman D, Kennedy SH. Randomized, single‐blind comparison of topiramate and bupropion sustained release as add‐on therapy in bipolar depression. 154th Annual Meeting of the American Psychiatric Association; 2001 May 5‐10; New Orleans. NR104.
- McIntyre RS, Mancini DA, McCann SM, Srinivasan J, Sagman D, Kennedy SH. Randomized, single‐blind comparison of topiramate and bupropion sustained release as add‐on therapy in bipolar depression. 155th Annual Meeting of the American Psychiatric Association; 2002 May 18‐23; Philadelphia.
- McIntyre RS, McCann SM, Mancini DA, Kennedy SH. Randomized, single‐blind comparison of topiramate and bupropion SR added to mood stabilizer in bipolar disorder. Acta Neuropsychiatrica 2000;12(3):163. [Google Scholar]
PDMD‐004 Kushner 2006 {published data only}
- Kushner SF, Khan A, Lane R, Olson WH. Topiramate monotherapy in the managment of acute mania: results of four double‐blind placebo‐controlled trials. Bipolar Disorder 2006;8:15‐27. [DOI] [PubMed] [Google Scholar]
- NCT00037674. A randomized, double‐blind, multicenter, placebo‐controlled 12‐week study of the safety and efficacy of two doses of topiramate for the treatment of acute manic or mixed episodes in patients with bipolar I disorder with an optional open‐label extension. https://clinicaltrials.gov/ct2/show/NCT00037674 2002, issue accessed 1 January 2014.
- Power PS, Sachs GS, Kushner SF, Wang D, Olson W, Capece J, et al. Topiramate in adults with acute bipolar I mania. New Research Abstracts, American Psychiatric Association 2004, 157th Annual Meeting, New York, NY, May 1‐6, 2004 (Abstract NR 365).
PDMD‐005 Kushner 2006 {published data only}
- Kushner SF, Khan A, Lane R, Olson WH. Topiramate monotherapy in the managment of acute mania: results of four double‐blind placebo‐controlled trials. Bipolar Disorders 2006;8:15‐27. [DOI] [PubMed] [Google Scholar]
- NCT00240721. A randomized, double‐blind, multicenter, placebo‐controlled 12‐week study of the safety and efficacy of two doses of topiramate for the treatment of acute manic or mixed episodes in subjects with bipolar I disorder with an optional open‐label extension. https://clinicaltrials.gov/ct2/show/NCT00240721 2005, issue accessed 1 January 2014.
- Power PS, Sachs GS, Kushner SF, Wang D, Olson W, Capece J, et al. Topiramate in adults with acute bipolar I mania. New Research Abstracts, American Psychiatric Association 2004, 157th Annual Meeting, New York, NY, May 1‐6, 2004 (Abstract NR 365).
PDMD‐006 Kushner 2006 {published data only}
- Kushner SF, Khan A, Lane R, Olson WH. Topiramate monotherapy in the managment of acute mania: results of four double‐blind placebo‐controlled trials. Bipolar Disorders 2006;8:15‐27. [DOI] [PubMed] [Google Scholar]
- Power PS, Sachs GS, Kushner SF, Wang D, Olson W, Capece J, et al. Topiramate in adults with acute bipolar I mania. New Research Abstracts, American Psychiatric Association 2004, 157th Annual Meeting, New York, NY, May 1‐6, 2004 (Abstract NR 365).
PDMD‐008 Kushner 2006 {published data only}
- Kushner SF, Khan A, Lane R, Olson WH. Topiramate monotherapy in the managment of acute mania: results of four double‐blind placebo‐controlled trials. Bipolar Disorders 2006;8:15‐27. [DOI] [PubMed] [Google Scholar]
- NCT00035230. A randomized, double‐blind, multicenter, placebo‐controlled 12‐week study of the safety and efficacy of topiramate in patients with acute manic or mixed episodes of bipolar I disorder with an optional open‐label extension. https://clinicaltrials.gov/ct2/show/NCT00035230 2002, issue accessed 1 January 2014.
- Power PS, Sachs GS, Kushner SF, Wang D, Olson W, Capece J, et al. Topiramate in adults with acute bipolar I mania. New Research Abstracts, American Psychiatric Association 2004, 157th Annual Meeting, New York, NY, May 1‐6, 2004 (Abstract NR 365).
References to studies excluded from this review
Bahk 2005 {published data only}
- Bahk WM, Shin YC, Woo JM, Yoon BH, Lee JS, Jon DI, et al. Effectiveness and tolerability of topiramate versus divalproex in bipolar mania. Korean Journal of Psychopharmacology 2004;15(4):425‐32. [Google Scholar]
- Bahk WM, Shin YC, Woo JM, Yoon BH, Lee JS, Jon DI, et al. Topiramate and divalproex in combination with risperidone for acute mania: a randomized open‐label study. Progress in Neuro‐Psychopharmacology and Biological Psychiatry 2005;29(1):115‐21. [DOI] [PubMed] [Google Scholar]
DelBello 2004 {published data only}
- DelBello MP, Kushner SF, Wang D, Olson W, Capece J, Fazzio L, et al. Topiramate for acute mania in adolescent patients with bipolar I disorder. 157th Annual Meeting of the American Psychiatric Association; 2004 May 1‐6; New York, NY. NR363.
- Delbello MP, Findling RL, Kushner S, Wang D, Olson WH, Capece JA, et al. A pilot controlled trial of topiramate for mania in children and adolescents with bipolar disorder. Journal of the American Academy of Child & Adolescent Psychiatry 2005;44(6):539‐47. [DOI] [PubMed] [Google Scholar]
Hebrani 2009 {published data only}
- Hebrani P, Behdani F, Manteghi AA. Double‐blind, randomized, clinical trial of topiramate versus sodium valproate for the treatment of bipolar disorder in adolescents. Pakistan Journal of Medical Sciences 2009;25(2):247‐52. [Google Scholar]
Mahmoudi‐Gharaei 2012 {published data only}
- Mahmoudi‐Gharaei J, Shahrivar Z, Faghihi T, Mohammadi MR, Tehrani‐Doost M, Alaghband‐Rad J, et al. Topiramate versus valproate sodium as adjunctive therapies to a combination of lithium and risperidone for adolescents with bipolar I disorder: effects on weight and serum lipid profile. Iranian Journal of Psychiatry 2012;7(1):1‐10. [http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3395968/ ] [PMC free article] [PubMed] [Google Scholar]
McIntyre 2005 {published data only}
- McIntyre RS, Riccardelli R, Binder C, Kusumakar V. Open‐label adjunctive topiramate in the treatment of unstable bipolar disorder. The Canadian Journal of Psychiatry 2005;50(7):415‐22. [DOI] [PubMed] [Google Scholar]
Mirsepassi 2014 {published data only}
- Mirsepassi Z, Mazinani R, Fadai F, Beigi NA, Astaneh AN. Effect of topiramate on weight control in bipolar type I patients receiving lithium and antipsychotics during manic episode. Iranian Journal of Psychiatry and Clinical Psychology 2014;19(3):195‐201. [Google Scholar]
Sahraian 2014 {published data only}
- IRCT2012061310013N1. Comparison of treatment of bipolar disorder (lithium + olanzapine + clonazepam) and placebo with a treatment regimen containing topiramate in controlling the OCD symptoms of bipolar patients. http://apps.who.int/trialsearch/Trial2.aspx?TrialID=IRCT2012061310013N1 issue accessed 1 January 2014.
- Sahraian A, Bigdeli M, Ghanizadeh A, Rezaee V. Topiramate as an adjuvant treatment for obsessive compulsive symptoms in patients with bipolar disorder, a randomized double blind placebo controlled clinical trial [abstract]. Bipolar Disorders 2014;96. [IRCT2012061310013N1] [DOI] [PubMed] [Google Scholar]
- Sahraian A, Bigdeli M, Ghanizadeha A, Akhondzadeh S. Topiramate as an adjuvant treatment for obsessive compulsive symptoms in patients with bipolar disorder: a randomized double blind placebo controlled clinical trial. Journal of Affective Disorders 2014;166:201–5. [DOI] [PubMed] [Google Scholar]
Vieta 2004 {published data only}
- Vieta E, Sánchez‐Moreno J, Goikolea JM, Colom F, Martínez‐Arán A, Benabarre A, et al. Effects on weight and outcome of long‐term olanzapine‐topiramate combination treatment in bipolar disorder. Journal of Clinical Psychopharmacology 2004;24(4):374‐8. [DOI] [PubMed] [Google Scholar]
Wozniak 2000 {published data only}
- Wozniak J. Olanzapine plus topiramate versus olanzapine plus omega‐3 fatty acids for bipolar disorder. http://www.stanleyresearch.org/ 2000, issue accessed 1 January 2014.
Wozniak 2009 {published data only}
- Wozniak J, Mick E, Waxmonsky J, Kotarski M, Hantsoo L, Biederman J. Comparison of open‐label, 8‐week trials of olanzapine monotherapy and topiramate augmentation of olanzapine for the treatment of paediatric bipolar disorder. Journal of Child and Adolescent Psychopharmacology 2009;19(5):539‐45. [DOI] [PubMed] [Google Scholar]
References to studies awaiting assessment
Calabrese 2000 {published data only}
- Calabrese JR. Update on the use of topiramate in bipolar disorder. 153rd Annual Meeting of the American Psychiatric Association; 2000 May 13‐18; Chicago. Vol. No. 44A.
- Calabrese JR. Update on the use of topiramate in bipolar disorder. 155th Annual Meeting of the American Psychiatric Association; 2002 May 18‐23; Philadelphia.
Mirsepassi 2013 {published data only}
- Mirsepassi Z, Mazinani R, Fadai F, Alibeigi N, Nazeri‐Astaneh A. Topiramate add‐on lithium carbonate for treatment of acute mania. Iranian Journal of Psychiatry and Behavioral Sciences 2013;7(2):11‐5. [PMC free article] [PubMed] [Google Scholar]
Yoon 2003 {published data only}
- Yoon BH, Bae SO, Yoon JS, Kim MK, Sea YW. Combination of topiramate in acute mania. 156th Annual Meeting of the American Psychiatric Association; 2003 May 17‐22; San Francisco, CA. NR507.
- Yoon BH, Bae SO, Yoon JS, Lee KL, Choi Y. Combination of topiramate in acute mania. Korean Journal of Psychopharmacology 2002;13(4):246‐53. [Google Scholar]
Additional references
APA 1994
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders (DSM‐IV). 4th Edition. Washington, DC: American Psychiatric Association, 1994. [Google Scholar]
Bourgeois 1998
- Bourgeois BF. New antiepileptic drugs. Archives of Neurology 1998;55(9):1181‐3. [DOI] [PubMed] [Google Scholar]
Cavanagh 2009
- Cavanagh J, Schwannauer M, Power M, Goodwin GM. A novel scale for measuring mixed states in bipolar disorder. Clinical Psychology & Psychotherapy 2009;16:497–509. [DOI: 10.1002/cpp.633] [DOI] [PubMed] [Google Scholar]
DerSimonian 1986
- DerSimonian R, Laird N. Meta‐analysis in clinical trials. Controlled Clinical Trials 1986;7:177‐88. [DOI] [PubMed] [Google Scholar]
Goodwin 2007
- Goodwin RK, Jamison KR. Manic‐Depressive Illness. Bipolar Disorders and Recurrent Depression. 2nd Edition. Oxford University Press, 2007. [Google Scholar]
Hamilton 1980
- Hamilton M. Rating depressive patients. Journal of Clinical Psychiatry 1980;41:21‐4. [PubMed] [Google Scholar]
Higgins 2003
- Higgins JPT, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta‐analyses. BMJ 2003;327:557‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Kushner 2006
- Kushner SF, Khan A, Lane R, Olson WH. Topiramate monotherapy in the managment of acute mania: results of four double‐blind placebo‐controlled trials. Bipolar Disorders 2006;8:15‐27. [DOI] [PubMed] [Google Scholar]
Macritchie 2009
- Macritchie K, Geddes J, Scott J, Haslam DR, Silva de Lima M, Goodwin G. Valproate for acute mood episodes in bipolar disorder. Cochrane Database of Systematic Reviews 2009, Issue 1. [DOI: 10.1002/14651858.CD004052] [DOI] [PubMed] [Google Scholar]
Marcotte 1998
- Marcotte D. Use of topiramate, a new anti‐epileptic as a mood stabiliser. Journal of Affective Disorders 1998;50(2‐3):245‐51. [DOI] [PubMed] [Google Scholar]
McElroy 2000b
- McElroy SL, Suppes T, Keck PE, Frye MA, Denicoff KD, Altshuler LL, et al. Open‐label adjunctive topiramate in the treatment of bipolar disorders. Biological Psychiatry 2000;47(12):1025‐33. [DOI] [PubMed] [Google Scholar]
Meldrum 1996
- Meldrum BS. Update on the mechanism of action of antiepileptic drugs. Epilepsia 1996;37 Suppl 6:S4‐11. [DOI] [PubMed] [Google Scholar]
Merikangas 2011
- Merikangas KR, Jin R, He JP, Kessler RC, Lee S, Sampson NA, et al. Prevalence and correlates of bipolar spectrum disorder in the world mental health survey initiative. Archives of General Psychiatry 2011;68(3):241‐51. [DOI: 10.1001/archgenpsychiatry.2011.12] [DOI] [PMC free article] [PubMed] [Google Scholar]
Montgomery 1979
- Montgomery SA, Asberg M. A new depression scale designed to be sensitive to change. British Journal of Psychiatry 1979;134:382‐9. [DOI] [PubMed] [Google Scholar]
NICE 2006
- National Institute for Health and Care Excellence. Bipolar disorder: the management of bipolar disorder in adults, children and adolescents, in primary and secondary care. National Institute for Health and Care Excellence; 2006 July. NICE Clinical Guideline 38.
Oxman 1992
- Oxman AD, Guyatt GH. A consumer's guide to subgroup analyses. Annals of Internal Medicine 1992;116(1):78‐84. [DOI] [PubMed] [Google Scholar]
Perlis 2006
- Perlis RH, Ostacher MJ, Patel JK, Marangell LB, Zhang H, Wisniewski SR, et al. Predictors of recurrence in bipolar disorder: primary outcomes from the Systematic Treatment Enhancement Program for Bipolar Disorder (STEP‐BD). American Journal of Psychiatry 2006;163(2):217‐24. [DOI] [PubMed] [Google Scholar]
Petty 1995
- Petty F. GABA and mood disorders: a brief review and hypothesis. Journal of Affective Disorders 1995;34:275‐81. [DOI] [PubMed] [Google Scholar]
Phillips 2013
- Phillips ML, Kupfer DJ. Bipolar disorder diagnosis: challenges and future directions. The Lancet 2013;381(9878):1663‐71. [DOI] [PMC free article] [PubMed] [Google Scholar]
Poon 2012
- Poon SH, Sim K, Sum MY, Kuswanto CN, Baldessarini RJ. Evidence‐based options for treatment‐resistant adult bipolar disorder patients. Bipolar Disorders 2012;14:573‐84. [DOI: 10.1111/j.1399-5618.2012.01042.x] [DOI] [PubMed] [Google Scholar]
Power 2004
- Power PS, Sachs GS, Kushner SF, Wang D, Olson W, Capece J, et al. Topiramate in adults with acute bipolar I mania. New Research Abstracts, American Psychiatric Association 2004, 157th Annual Meeting, New York, NY, May 1‐6, 2004 (Abstract NR 365).
Prince 2007
- Prince M, Patel V, Saxena S, Maj M, Maselko J, Phillips MR, et al. No health without mental health. The Lancet 2007;370(9590):859‐77. [DOI] [PubMed] [Google Scholar]
RevMan 2012 [Computer program]
- The Nordic Cochrane Centre, The Cochrane Collaboration. Review Manager (RevMan). Version 5.2. Copenhagen: The Nordic Cochrane Centre, The Cochrane Collaboration, 2012.
Rosa 2011
- Rosa AR, Fountoulakis K, Siamouli M, Gonda X, Vieta E. Is anticonvulsant treatment of mania a class effect? Data from randomized clinical trials. CNS Neuroscience & Therapeutics 2011;17(3):167‐77. [DOI: 10.1111/j.1755-5949.2009.00089.x] [DOI] [PMC free article] [PubMed] [Google Scholar]
Sachs 2000
- Sachs G, Koslow GC, Orsini C, Cosgrove V, Sambur M, Demopulous C, et al. Treatment of refractory bipolar mood disorder: efficacy of topiramate. Acta Neuropsychiatrica 2000;12(3):169. [Google Scholar]
Taylor 2015
- Taylor D, Paton C, Kapur S. The Maudsley Prescribing Guidelines in Psychiatry. 12th Edition. Wiley, 2015. [Google Scholar]
Vasudev 2006
- Vasudev K, Macritchie K, Geddes J, Watson S, Young A. Topiramate for acute affective episodes in bipolar disorder. Cochrane Database of Systematic Reviews 2006, Issue 1. [DOI: 10.1002/14651858.CD003384.pub2] [DOI] [PubMed] [Google Scholar]
Vasudev 2009
- Vasudev A, Macritchie K, Rao SNK, Geddes J, Young AH. Tiagabine in the treatment of acute affective episodes in bipolar disorder. Cochrane Database of Systematic Reviews 2009, Issue 1. [DOI: 10.1002/14651858.CD004694.pub3] [DOI] [Google Scholar]
Vieta 2000
- Vieta E, Gilabert A, Rodriguez A, Garcia‐Castrillon A, Luna MJ, Arrufat F, et al. Topiramate as 'add‐on' therapy in refractory bipolar disorder. Acta Neuropsychiatrica 2000;12(3):173. [Google Scholar]
Vieta 2003
- Vieta E, Sanchez‐Moerno J, Goikolea JM, Torrent C, Benbarre A, Colom F, et al. Adjunctive topiramate in bipolar II disorder. World Journal of Biological Psychiatry 2003;4(4):172‐6. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
WHO 1992
- World Health Organization. The ICD‐10 Classification of Mental and Behavioural Disorders. Clinical descriptions and diagnostic guidelines. Geneva: World Health Organization, 1992. [Google Scholar]
WHO 2004
- World Health Organization. The global burden of disease: 2004 update. Geneva: World Health Organization; published 2008.
Wilding 2004
- Wilding J, Gaal L, Rissanen A, Vercruysse F, Fitchet M. A randomized double‐blind placebo‐controlled study of the long‐term efficacy and safety of topiramate in the treatment of obese subjects. International Journal of Obesity and Related Metabolic Disorders 2004;28(11):1399‐410. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Woods 2004
- Woods TM, Eichner SF, Franks AS. Weight gain mitigation with topiramate in mood disorders. Annals of Pharmacotherapy 2004;38(5):887‐91. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Young 1978
- Young RC, Biggs JT, Ziegler VE, Meyer DA. A rating scale for mania: reliability, validity and sensitivity. British Journal of Psychiatry 1978;133:429‐35. [DOI] [PubMed] [Google Scholar]
Young 2006
- Young AH, Geddes JR, Macritchie K, Rao SN, Watson S, Vasudev A. Tiagabine in the treatment of acute affective episodes in bipolar disorder: efficacy and acceptability. Cochrane Database of Systematic Reviews 2006, Issue 3. [DOI: 10.1002/14651858.CD004694.pub2] [DOI] [PubMed] [Google Scholar]