

Abstract
Objective:
Erythropoietin (EPO), a glycoprotein cytokine essential to hematopoiesis, has neuroprotective effects in rodent models of Alzheimer’s disease (AD). However, high therapeutic doses or invasive routes of administration of EPO are required to achieve effective brain concentrations due to low blood-brain barrier (BBB) penetrability, and high EPO doses result in hematopoietic side effects. These obstacles can be overcome by engineering a BBB-penetrable analog of EPO, which is rapidly cleared from the blood, by fusing EPO to a chimeric monoclonal antibody targeting the transferrin receptor (cTfRMAb), which acts as a molecular Trojan horse to ferry the EPO into the brain via the transvascular route. In the current study, we investigated the effects of the BBB-penetrable analog of EPO on AD pathology in a double transgenic mouse model of AD.
Methods:
5.5 month old male APPswe PSEN1dE9 (APP/PS1) transgenic mice were treated with saline (n=10), or the BBB-penetrable EPO (n=10) 3 days/week intraperitoneally for 8 weeks, compared to same-aged C57BL/6J wild-type mice treated with saline (n=8) with identical regiment. At 9 weeks following treatment initiation, exploration and spatial memory were assessed with the open-field and Y-maze test, mice were sacrificed, and brains were evaluated for Aβ peptide load, synaptic loss, BBB disruption, microglial activation and microhemorrhages.
Results:
APP/PS1 mice treated with the BBB-penetrable cTfRMAb-EPO fusion protein had significantly lower cortical and hippocampal Aβ peptide number (p<0.05) and immune-positive area (p<0.05), a decrease in hippocampal synaptic loss (p<0.05) and cortical microglial activation (p<0.001), and improved spatial memory (p<0.05) compared with APP/PS1 saline controls. BBB-penetrating EPO was not associated with microhemorrhage development.
Conclusions:
The cTfRMAb-EPO fusion protein offers therapeutic benefits by targeting multiple targets of AD pathogenesis and progression (Aβ load, synaptic loss, microglial activation) and improving spatial memory in the APP/PS1 mouse model of AD.
Keywords: blood-brain barrier, erythropoietin, Alzheimer’s disease, transferrin receptor, monoclonal antibody, transcytosis
Graphical Abstract
Introduction
Erythropoietin (EPO), a 30.4 kDa glycoprotein, is a potential treatment for Alzheimer’s disease (AD) since it targets a spectrum of processes involved in AD pathology and progression. Experimental work shows that EPO targets pathways involved in the pathogenesis of AD (amyloid beta (Aβ) toxicity; tau phosphorylation),1–3 and also exhibits robust neuroprotective and neurotrophic effects.4 In vitro, EPO is protective against Aβ induced cell death.2, 5–6 In vivo, EPO attenuates cognitive deficits,7–10 endothelial degeneration,8 Aβ burden,9 mitochondrial dysfunction and apoptosis,7 and promotes neurogenesis.11 In addition, EPO expression is reduced in AD brain,12 whereas EPO receptor (EPOR) expression is increased in rodent models of AD12 and AD patients.13 Notably, neuroprotective effects of EPO are more pronounced with higher EPOR expression.14 Overall, EPO has the potential to prevent new brain insult (by targeting Aβ and tau), and reverse and repair brain damage resulting from prodromal AD pathways (by promoting neuronal survival and neurogenesis).
One major obstacle to the development of EPO for AD is poor blood-brain barrier (BBB) penetration. Only 0.05–0.1% of systemically administered EPO crosses the intact BBB, and this low brain distribution of EPO is confined to the blood volume of brain.15–16 As a result, EPO must be administered intracranially.17–18 EPO does enter the CSF following systemic administration,19 however, CSF entry is not a measure of EPO entry into the brain parenchyma across the BBB.20
Recent studies show the protective effects of systemically adminstered EPO in mouse models of AD.7–8 However, clinical applicability of systemic EPO for neural diseases is limited by the hematopoietic side effects associated with high EPO doses, which become especially relevant while treating chronic diseases like AD that require long term treatment.21–22 Non-hematopoietic variants of EPO (e.g. carbamylated EPO, neuro-EPO) have been developed as alternatives and recent studies show protective effects of these variants in mouse models of AD.9, 23 However, these EPO variants are also large molecules and the BBB permeability of these molecules has not been confirmed. To overcome the BBB, neuro-EPO was administered intranasally in a mouse model of AD.23 It should, however, be noted that the intranasal route relies on drug diffusion for drug transport from olfactory CSF to brain entry sites,24 and whether this approach can be translated to humans, considering the much greater diffusion distances in the brain in humans compared with rodents, still needs further investigation. Hence, an analog of EPO, that is both BBB-penetrable following systemic administration and has minimal hematopoietic effects, is needed for chronic diseases like AD.
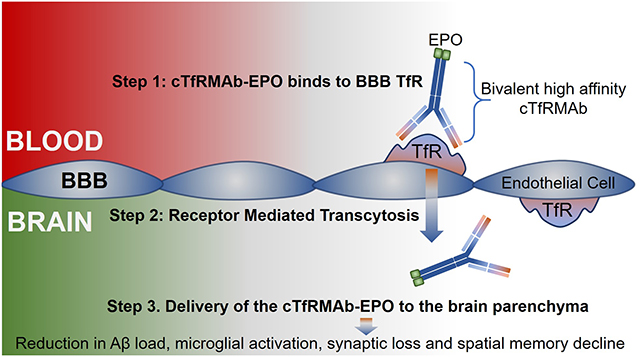
The transferrin receptor (TfR) is expressed on the brain capillary endothelium,25 and the expression of the BBB TfR is unaltered in patients with AD.26 The BBB TfR acts as a receptor mediated transcytosis (RMT) system,27 and among the 2 isoforms of the TfR (TfR1 and TfR2), the isoform expressed at the BBB is TfR1.28 TfR1 is highly expressed at the BBB and ligands to TfR1 can thus be used to deliver drugs into the brain, non- invasively via the transvascular route across the BBB.29–30 The TfR1 is saturated by transferrin, the natural ligand for TfR1, under normal condition, and an alternate drug delivery vector is a monoclonal antibody (MAb) against the TfR1 that binds to a site distinct from the transferrin binding site. One such MAb derived from the rat 8D3 MAb is a rat/mouse chimeric antibody which is approximately 85% mouse amino acid sequence and is specific for the mouse TfR1.29–30 To facilitate the transvascular brain delivery of EPO using this RMT approach, a fusion protein of EPO and the chimeric MAb against the mouse TfR1 (referred to as cTfRMAb) has been engineered.31 The fusion protein is designated as cTfRMAb-EPO and offers several advantages. First, the cTfRMAb domain of the fusion protein binds to the BBB TfR in a bivalent format with high affinity and undergoes RMT, thus acting as a molecular Trojan horse (MTH) to ferry the EPO into the brain via the transvascular route.31 The transvascular route across the BBB results in widespread drug delivery throughout the brain owing to the dense network of the cerebral vasculature compared with the intranasal route.32 The bifunctional cTfRMAb-EPO fusion protein also binds mouse EPOR with high affinity (ED50 = 0.33 ± 0.04 nM).31 The ED50 for EPO binding to EPOR is reported to be 0.14 nM.33 Second, the peripheral TfR allows for rapid TfR mediated peripheral clearance of the cTfRMAb-EPO fusion protein from blood resulting in 10-fold lower plasma area under the curve (AUC) as compared to EPO.31 The hematopoietic action of EPO is proportional to the plasma AUC, and previous work has shown that chronic dosing with the cTfRMAb-EPO fusion protein, equivalent to very high EPO doses of systemic EPO, results in only a 10% increase in hematocrit in a mouse model of neural disease.34
The aim of the current study was to investigate the effect of this BBB-penetrating analog of EPO in a transgenic mouse model of AD. In the current study, 5.5 month old male APPswe, PSEN1dE9 (APP/PS1) transgenic mice were treated with either saline or 3mg/kg cTfRMAb-EPO fusion protein 3 days per week. The fusion protein was administered via intraperitoneal injections to enable long-term treatment of the AD mice. Nine-weeks after treatment initiation, spatial memory and exploration were assessed using the modified Y-maze and open field tests. After 10-weeks of treatment initiation, mice were sacrificed and mouse brains were examined for Aβ load, neuroinflammation, synaptic loss, BBB disruption and cerebral microhemorrhage (CMH) development.
Experimental Section
Fusion protein.
The cTfRMAb-EPO fusion protein was produced in stably transfected Chinese hamster ovary (CHO) cells and purified by protein G affinity chromatography as described previously31 and formulated in 98 mM glycine, 148 mM NaCl, 28 mM Tris, 0.01% Polysorbate 80, pH=5.5, sterile filtered and stored at either 4°C or −80°C till use.
Mouse treatment.
All animal procedures were approved by the University of California, Irvine Institutional Animal Care and Use Committee (IACUC) and were carried out in compliance with University Laboratory Animal Resources regulations. Mice were provided constant access to food and water, and maintained on a 12-h light/12-h dark cycle. Male heterozygous APP/PS1 mutant mice (strain B6C3-Tg APPswe, PSEN1dE9, 85Dbo/Mmjax, stock 004462, Jackson Laboratories, Bar Harbor, ME) were 5.5 months old at the start of the study and were injected intraperitoneally (i.p.) three days a week on Monday, Wednesday and Friday, for eight weeks either with saline (n=10) or the cTfRMAb-EPO fusion protein (3 mg/kg per dose) (n=10). A separate series of age-matched wild type (WT) C57BL/6J littermate controls were treated with equivalent volume of saline i.p. using the same dosing regimen as described above. Based on amino acid sequence, the EPO domain comprises 20% of the cTfRMAb-EPO fusion protein.31
Mice were observed daily, weighed weekly and carefully monitored after each injection for signs of an immune reaction (prostrate, unresponsive or scruffy appearance). Ten-weeks after treatment initiation, mice were anesthetized with a lethal dose of Nembutal (150 mg/kg, i.p.), cardiac perfusions were performed using ice cold phosphate buffer saline (PBS; 0.01 M Na2HPO4, 0.15 M NaCl, KCl, KPO4, pH 7.0) for 5 min to clear the cerebral vasculature, and the brains were harvested.
Cryosectioning.
The brains were processed as described previously.35 Briefly, the right cerebral hemisphere was immersion-fixed with 4% paraformaldehyde in PBS for 24h followed by serial incubation for 24h each in 10%, 20% and 30% sucrose solution at 4°C, frozen, mounted in Tissue-Tek OCT compound (Fisher Scientific, Hampton, NH) and sliced into 20μm-thick sagittal sections at −20°C using a cryostat (Micron Instruments, San Marcos, CA). Three sections per mouse, approximately 600 μm apart, were used for histochemistry as described below.
Amyloid beta (Aβ) fluorescence microscopy.
Thioflavin-S (Thio-S) staining to stain mature Aβ plaques and 6E10 MAb immunofluorescence to stain Aβ peptide was performed as described previously.35–36 Briefly, for Thio-S staining, tissue sections subbed onto glass slides were washed sequentially with 70% and 80% ethanol for 1 min each followed by incubation in 1% Thio-S (Sigma Aldrich, St. Louis, MO) solution in 80% ethanol for 15 min. This was followed by sequential washing with 70% and 80% ethanol for 1 min each. For Aβ immunofluorescence, free floating sections were washed in PBS for 5 min and treated with 70% formic acid for 10 min at room temperature (RT). Sections were blocked with 0.5% bovine serum albumin (BSA) in PBS containing 0.3% TritonX-100 for 1h at RT, and incubated with 1μg/mL of Alexa Fluor 488-conjugated 6E10 MAb (BioLegend; San Diego, CA) overnight at 4°C. All the sections (6E10 and Thio-S) received a final wash with distilled water for 5 min and were cover slipped using Vectamount aqueous mounting media (Vector Laboratories, Burlingame, CA) and sealed with nail polish. Slides were stored at 4°C until imaging. Fluorescent staining was examined using a Leica SP5 Confocal Microscope. For each brain section, two regions in the cortex and the entire hippocampus were scanned serially through a thickness of 5 μm (z-stack depth) and imaged at a 10x magnification. Digitized images were analyzed using the NIH Image J software (version 1.46r) and the following parameters were determined by an observer blinded to the experimental groups: (a) number of positive stains/mm2 of tissue analyzed; (b) stain positive area expressed as a percentage of analyzed area; and (c) average stain size (μm2).
Neuroinflammation, BBB disruption and CMH assessment.
Iba1 (ionized calcium binding adaptor molecule 1; macrophage/microglial marker) and brain parenchymal IgG (marker of BBB disruption) immunohistochemistry were performed as described previously.35, 37 For IgG immunohistochemistry, brain sections were incubated in 0.5% hydrogen peroxide in 0.1 M PBS containing 0.3% Triton-X100 (PBST) for 30 min at room temperature to block endogenous peroxidase activity. After washing with PBST, sections were incubated for 30 min with PBST containing 2% BSA to block nonspecific protein binding and then incubated overnight at 4°C with a rabbit anti-mouse IgG antibody (1:200 dilution; JacksonImmunoResearch, West Grove, PA). After washing with PBST, sections were incubated at RT for 1h with biotinylated anti-rabbit IgG (1:500 dilution; JacksonImmunoResearch, West Grove, PA), followed by 1h incubation at RT with ABC complex according to manufacturer instructions (Vector Laboratories, Burlingame, CA). Sections were developed with 3,3’-diaminobenzidine (DAB) (Vector Laboratories, Burlingame, CA).
For Iba-1 immunohistochemistry, brain sections were incubated in 3% hydrogen peroxide in 0.1 M PBS for 30 min at RT to block endogenous peroxidase activity. After washing with PBS, sections were incubated for 60 min with PBS containing 0.5% BSA and 0.3% Triton-X100 to block non-specific protein binding and incubated overnight at 4°C with a biotin-conjugated rabbit anti-Iba1 (1:200 dilution, Wako Chemicals USA, Richmond, VA). Sections were then incubated for 1h at RT with ABC complex according to manufacturer instructions (Vector Laboratories, Burlingame, CA) and developed using the ImmPACT AEC Peroxidase (HRP) Substrate kit as per the vendors instructions (Vector Labs, Burlingame, CA). After washing, sections were mounted on slides using the Vectamount aqueous mounting media (Vector labs, Burlingame, CA), coverslipped, and sealed with clear nail polish. Two cortical and one hippocampal images per brain section were acquired at 10x magnification using a light microscope (Motic, British Columbia, Canada) and the total immunopositive area was quantified using NIH Image J software by an observer blinded to the experimental groups. Immunopositive area was expressed as % of total analyzed area.
To determine if chronic treatment with the cTfRMAb-EPO fusion protein results in CMH development, brain sections were stained with hematoxylin and eosin (H&E) and Prussian blue (PB) to detect CMH, as described previously.35, 37 For H&E staining, briefly, brain sections subbed onto glass slides were washed in distilled water for 5 min, followed by dipping in Mayer’s hematoxylin (Fisher Scientific, Hampton, NH) for 10 min. After consecutive rinses in tap water, Scott’s tap water and tap water, slides were dipped in Eosin Y (Sigma Aldrich, St. Louis, MO) for 10 min. Sections were washed sequentially in 95% and 100% ethanol and coverslipped with Permount (Fisher Scientific, Hampton, NH) after air drying. PB staining was performed as described previously.38 Briefly, sections were stained using 5% potassium hexacyanoferrate trihydrate (Sigma, St. Louis, MO) and 5% hydrochloric acid (Sigma, St. Louis, MO) for 30 min, rinsed in water and counterstained with Nuclear Fast Red (Sigma, St. Louis, MO), dehydrated, and covered slipped. PB staining was scored semi-quantitatively in 3 sections/mouse as follows: 0 (no microhemorrhage or reactivity detected in the entire brain section); 1 (≤2 microhemorrhages/section), 2 (3 to 10 microhemorrhages/section), or 3 (≥11 microhemorrhages/section); and the average across each mouse was determined.36 The entire brain section was examined to detect CMH and representative images were captured at a 40X magnification using a light microscope (Motic, British Columbia, Canada).
Synaptophysin immunofluorescence:
To assess synaptic loss, brain sections were washed in PBS, followed by incubation in PBS containing 1% BSA and 0.3% Triton-X100 for 60 min at RT to block non-specific binding. Sections were then incubated with anti-synaptophysin H-8 antibody (1:100 dilution, Santa Cruz Biotechnology, Dallas, TX) in PBS containing 1% BSA and 0.3%Triton-X 100 overnight at 4°C followed by incubation with a mouse IgG kappa binding protein (m-IgGκ BP) conjugated to CruzFluor™ 488 (CFL 488) (Santa Cruz Biotechnology, Dallas, TX) in the blocking solution for 60 min at RT. After washing in PBS, sections were incubated in DRAQ5 (BioLegend, San Diego, CA) for 5 min, washed in PBS and mounted onto glass slides using the Vectamount aqueous mounting media (Vector labs, Burlingame, CA), coverslipped, and sealed with clear nail polish. Brain sections incubated without the primary antibody served as controls. Fluorescent staining was examined using the Leica SP5 Confocal Microscope. For each brain section, the entire hippocampus was imaged at a 10x magnification. Mean fluorescent intensities (MFI) in the dentate gyrus (DG), CA3 and entire hippocampus of each image was determined using Adobe Photoshop by 2 observers blinded to the experimental group, and normalized to WT values.
Behavior analysis:
Six and nine-weeks after treatment initiation, behavior analysis was performed. For all the behavior analysis, animals were placed in the behavior suite at least 30 min before testing for acclimatization.
Modified Y-maze:
Spatial memory was assessed using the modified Y-maze using an apparatus that consisted of three-arms each measuring 42cm long × 15cm high × 8 cm wide. Modified Y-maze relies on the innate tendency of mice to explore areas that have not been explored before.39 The modified Y-maze protocol consisted of a training and a testing phase. During the training phase, one arm of the Y-maze was blocked using a removable door, and the animal was placed in the start arm and was allowed to explore two arms for 8 minutes before being placed back into its original cage. Thirty-minutes later, during the testing phase, the animal was placed back into the start arm, this time the door blocking the third (novel) arm was removed so that the animal has access to all the arms of the Y-maze. This testing phase lasted for 8 minutes after which the animal was placed back into its original cage. % entries into the novel arm was estimated, and the apparatus was cleaned with 70% ethanol between animals to avoid odor cues.
Open Field Test:
Locomotion and exploration was assessed in the current study by performing the open-field test using a white open box (72 cm × 72cm with 36 cm walls). Animals were placed in the center of the arena and movements were tracked for 5 min to measure (a) total distance travelled and (b) mean speed.
Nesting:
Animals were singly housed and one pressed cotton square (Nestlets) was placed in each cage 1 h before the dark phase for assessment of nesting behavior seven-weeks after treatment initiation. Mice were left to nest overnight. The next morning, each nest was assigned a score from 1–5 using the following criteria as described previously40: a score of 1 was defined as almost no shredding and no nest, with majority of Nestlet (>90%) still intact, a score of 2 was defined as some shredding, but still no nest (50–90% of the Nestlet remains intact), a score of 3 was assigned when majority of the Nestlet was torn (<50% remains intact); however the nest was spread around the full area of the cage and there was no noticeable nest, a score of 4 was defined as an almost fully shredded Nestlet (>90% has been shredded), with a noticeable, but flat nest. A noticeable nest was defined as >90% of the shreds within one quarter of the cage floor area and a flat nest was defined as one with walls higher than the height of the animal (a mouse curled up on its side) for less than 50% of nest circumference, and a score of 5 was defined as an almost fully shredded (>90%) Nestlet and a perfect nest, with walls higher than mouse body height for >50% of nest circumference.
Statistical Analysis.
Data were represented as mean ± SD and all statistical analysis was performed using GraphPad Prism 5 (GraphPad Software Inc., La Jolla, CA), unless otherwise stated. One-way ANOVA with Bonferroni’s post hoc test for equal variances or Games-Howell for unequal variances (Minitab, State College, PA) were used to compare more than two groups. Y-maze data was compared using the one- sample t test with a hypothesized mean = 33%. Two-way ANOVA with repeated measures was used for the open-field data. A two-tailed p-value of <0.05 was considered statistically significant.
Results
One APP/PS1 saline treated mouse died 7 weeks after treatment initiation and was excluded from the study; all other mice survived the duration of the study. No signs of an immune reaction were observed throughout the study. The average weights of the mice at the beginning of the study were: 35±1 g, 33±2 g and 32±2 g in the WT-saline, APP/PS1-saline and APP/PS1-cTfRMAb-EPO groups, respectively. The average weights of the mice at the end of the study were: 40±2 g, 38±2 g and 35±1 g in the WT- saline, APP/PS1-saline and APP/PS1-cTfRMAb-EPO groups, respectively.
Decreased Aβ load in mice treated with the cTfRMAb-EPO fusion protein:
6E10 MAb-positive Aβ peptide load in WT-saline, APP/PS1-saline and APP/PS1-cTfRMAb- EPO 8 month old mice is shown in figure 1. As expected, WT mice treated with saline did not have any 6E10-positive Aβ-peptide. There was a significant reduction in cortical 6E10-positive Aβ peptide area (30% reduction, p<0.05) and number (34% reduction, p<0.01) in the APP/PS1 mice treated with the cTfRMAb-EPO fusion protein compared with saline treated APP/PS1 mice following 8 weeks of treatment (Fig 1B–C). Similarly, cTfRMAb-EPO fusion protein treated APP/PS1 mice had significantly lower hippocampal 6E10-positive Aβ peptide area (42% reduction, p<0.05) and number (32% reduction, p<0.05) compared with the saline treated APP/PS1 mice (Fig 1E–F). Average size of the 6E10-positive Aβ peptide did not differ between the saline and cTfRMAb- EPO treated APP/PS1 mice (Fig 1D and G).
Figure 1: Effect of the cTfRMAb-EPO fusion protein on Aβ peptide load in the APP/PS1 mice with 6E10 immuno-staining.

Representative images of 6E10 MAb- positive Aβ peptide in the cortex and hippocampus of saline- and cTfRMAb-EPO-treated APP/PS1 and saline treated WT mice (A). Significant reduction in cortical and hippocampal 6E10 MAb-positive Aβ peptide area (B, E) and number (C, F) in the cTfRMAb-EPO treated APP/PS1 mice compared with APP/PS1 treated saline controls. No difference in the 6E10 MAb-positive Aβ peptide size between saline and cTfRMAb- EPO treated APP/PS1 mice (D, G). Scale bar = 100μm. Data are presented as mean ± SD of 8–10 mice per group. One-way ANOVA with Bonferroni’s correction or Kruskal Wallis with Dunn’s correction. *p<0.05, **p<0.01.
Thio-S positive Aβ plaque load in the WT-saline, APP/PS1-saline and APP/PS1- cTfRMAb-EPO treated mice is shown in figure 2. No Aβ plaque deposition was observed in the WT mice. Cortical thio-S positive Aβ plaque area and number were significantly lower in the APP/PS1 mice treated with the cTfRMAb-EPO fusion protein compared with saline treated APP/PS1 mice (Fig 2 A, B and C), however, no significant difference was observed in the average size of thio-S positive cortical Aβ plaque. No significant difference was observed in the hippocampal thio-S positive Aβ plaque area, number or size between the saline and cTfRMAb-EPO treated APP/PS1 mice (data not shown).
Figure 2: Effect of the cTfRMAb-EPO fusion protein on mature Aβ plaque load in the APP/PS1 mice with thio-S staining.

Representative images of thio-S positive mature Aβ plaque in the cortex of saline- and cTfRMAb-EPO-treated APP/PS1 and saline treated WT mice (A). Significant reduction in cortical thio-S positive mature Aβ plaque area (B) and number (C) in the cTfRMAb-EPO treated APP/PS1 mice compared with saline treated APP/PS1 mice. Cortical thio-S positive mature Aβ plaque size did not differ between saline and cTfRMAb-EPO treated APP/PS1 mice (D). Scale bar = 100μm. Data are presented as mean ± SD of 8–10 mice per group. One-way ANOVA with Bonferroni’s correction for equal variance or Games-Howell for unequal variance, or Kruskal Wallis with Dunn’s correction. *p<0.05, **p<0.01.
Decreased microglial activation and no CMH development in mice treated with the cTfRMAb-EPO fusion protein:
There was a significant increase in the Iba-1 positive immunoreactive area (marker of microglial activation) in the APP/PS1-saline mice compared with the WT-saline mice both in the cortex (p<0.0001) and the hippocampus (p<0.05) (Fig 3 A–C). In the APP/PS1 mice, chronic treatment with the cTfRMAb-EPO fusion protein resulted in a 51% reduction (p<0.001) in the cortical Iba-1 positive immunoreactive area (Fig 3A–B). Hippocampal Iba-1 positive immunoreactive area was 37% reduced in the APP/PS1 mice treated with the cTfRMAb-EPO fusion protein compared with APP/PS1-saline mice, but this reduction did not reach statistical significance (Fig 3A and C). No significant difference in the brain parenchymal IgG- positive immunoreactive (marker of BBB disruption) was observed between any experimental groups in the current study (Fig 4A–C). Further, 8 month old APP/PS1 mice did not develop spontaneous acute H&E-positive CMH and chronic treatment with the cTfRMAb-EPO fusion protein did not result in acute CMH (data not shown). Though there was a trend towards increased PB-positive subacute CMH in the APP/PS1-saline mice, no significant difference was observed in the PB-positive sub-acute CMH between any experimental groups, and the average score was 0.8 ± 0.8 in WT-saline, 1.3 ± 0.7 in APP/PS1-saline, and 0.7 ± 0.6 in APP/PS1-cTfRMAb-EPO treated mice (Fig 5).
Figure 3: Effect of the cTfRMAb-EPO fusion protein on Iba-1, a marker of microglial activation, in the APP/PS1 mice.

Representative images of cortical and hippocampal Iba-1 immunostaining in the saline treated WT, saline treated APP/PS1 and cTfRMAb-EPO treated APP/PS1 mice (A). WT mice had significantly lower cortical and hippocampal Iba-1 positive immunoreactive area compared with saline treated APP/PS1 mice (B-C). Iba-1 positive immunostaining was found concentrated around plaque like structures in the APP/PS1 mice (A; black arrow heads). Significant reduction in the cortical Iba-1 positive immunoreactive area in the cTfRMAb-EPO treated APP/PS1 mice compared with saline treated APP/PS1 mice (B). A trend towards reduction in the hippocampal Iba-1 positive immunoreactive area in the cTfRMAb-EPO treated APP/PS1 mice compared with saline treated APP/PS1 mice (C). Data are presented as mean ± SD of 5–8 mice per group. One-way ANOVA with Bonferroni’s correction. *p<0.05, ***p<0.001, ****p<0.0001.
Figure 4: Effect of the cTfRMAb-EPO fusion protein on brain IgG, marker of blood- brain barrier disruption, in the APP/PS1 mice.

Representative images of cortical and hippocampal brain parenchymal IgG immunostaining in the saline treated APP/PS1, cTfRMAb-EPO treated APP/PS1 and saline treated WT mice (A). IgG positive immunostaining was found concentrated around plaque like structures in the APP/PS1 mice (A; black arrow heads). No significant difference in cortical or hippocampal brain IgG between any experimental groups (B-C). Data are presented as mean ± SD of 8–10 mice per group. One-way ANOVA with Bonferroni’s correction for equal variance or Games-Howell for unequal variance, or Kruskal Wallis with Dunn’s correction.
Figure 5: Effect of the cTfRMAb-EPO fusion protein on cerebral microhemorrhage development.
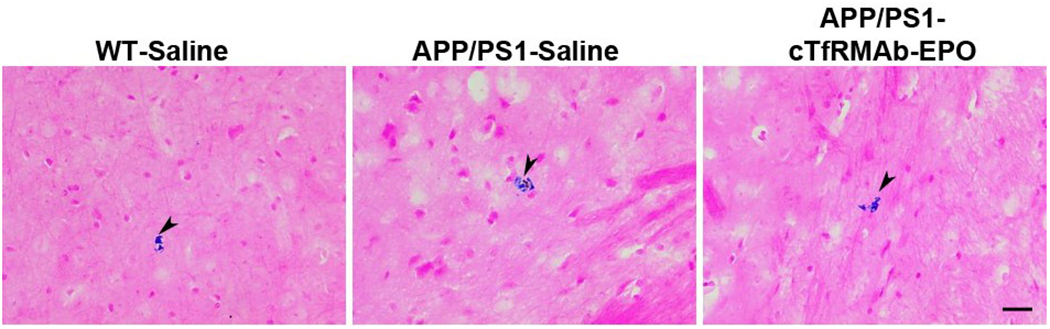
Prussian blue stained (indicated by arrowheads) representative images showing no significant increase in CMH development with chronic cTfRMAb-EPO treatment. Scale bar = 25μm.
cTfRMAb-EPO fusion protein treatment rescues synaptic loss:
Synaptic loss was assessed by measuring synaptophysin positive MFI (synaptic marker). There was a significant reduction in synaptophysin positive MFI in the APP/PS1-saline mice compared with the WT-saline mice in the DG (p<0.05) region of the hippocampus (Fig 6A and B) and the entire hippocampus (p<0.05) (Fig 6A and 6D). In the CA3 region of the hippocampus there was a trend towards reduction in the synaptophysin positive MFI in the APP/PS1 mice compared with the WT-saline mice (Fig 6A and C). The synaptophysin positive MFI was significantly higher in the APP/PS1 mice treated with the cTfRMAb-EPO fusion protein compared with APP/PS1 mice treated with saline in the DG (p<0.05), CA3 (p<0.05) and the entire hippocampus (p<0.05) (Fig 6A–D). No significant difference was found in the hippocampal synaptophysin positive MFI between the WT-saline and APP/PS1-cTfRMAb-EPO treated mice (Fig 6A–D). Omitting the synaptophysin primary antibody resulted in no synaptophysin positive immunofluorescence (Fig 6A).
Figure 6. Effect of the cTfRMAb-EPO fusion protein on synaptophysin, a marker of synaptic loss, in the APP/PS1 mice.

Representative images of hippocampal synaptophysin positive immunostaining (green) in the saline treated WT, saline treated APP/PS1 and cTfRMAb-EPO treated APP/PS1 mice. Primary antibody omitted section represents the control. Blue stain represents DRAQ5 positive nuclei (A). Significantly higher synaptophysin positive MFI in the DG (B), CA3 (C) and total (D) hippocampal region in the cTfRMAb-EPO treated APP/S1 mice compared with the saline treated APP/PS1 mice. Data are presented as mean ± SD of 8–10 mice per group. One-way ANOVA with Bonferroni’s correction for equal variance or Games-Howell for unequal variance, or Kruskal Wallis with Dunn’s correction. *p<0.05.
Improvement in spatial memory in mice treated with the cTfRMAb-EPO fusion protein:
Trajectories of the mice during the modified Y-maze are shown in Fig 7A. Six- weeks after treatment initiation, only the WT-saline mice showed a preference for the novel arm in the modified Y-maze (p<0.05 compared with a hypothesized value of 33%), however, 9 weeks following treatment initiation, both the WT-saline (p<0.01) and APP/PS1-cTfRMAb-EPO mice (p<0.05) showed an increased preference for the novel arm (Fig 7B). APP/PS1 mice treated with saline did not show an increased preference for the novel arm 6 or 9 weeks following treatment initiation.
Figure 7. Effect of the cTfRMAb-EPO fusion protein on spatial memory and exploration in the APP/PS1 mice.

Representative trajectories of saline treated WT, saline- and cTfRMAb-EPO-treated APP/PS1 mice during the training and testing phase of the Y-maze (A). Six-weeks after treatment initiation, saline treated WT mice showed a preference for the novel arm however, nine-weeks after treatment initiation, both the saline treated WT and cTfRMAb-EPO treated APP/PS1 mice showed a preference for the novel arm (B). Representative trajectories of saline treated WT, saline- and cTfRMAb-EPO-treated APP/PS1 mice during the open-field test (C). Six-weeks after treatment initiation, cTfRMAb-EPO treated APP/PS1 mice had significantly lower distance travelled (D) and mean speed (E) compared with saline treated WT mice. At 9 weeks, no differences in distance travelled or mean speed were observed between any experimental groups (D-E). Data are presented as mean ± SD of 8–10 mice per group. For Y-maze, one-sample t test with a hypothesized value of 33% (indicated by the dotted line). Two-way repeated measures ANOVA for the open-field analysis with Bonferroni’s correction. *p<0.05, **p<0.01.
Trajectories of the mice during the open-field test to assess exploration and locomotion are shown in figure 7C. Six-weeks after treatment initiation, there was a significant reduction (p<0.05) in the mean speed and distance traveled in the APP/PS1- cTfRMAb-EPO mice compared with WT-saline mice, however, no significant difference was observed between the APP/PS1-saline and APP/PS1-cTfRMAb-EPO mice. Further, no significant difference in mean speed or distance traveled was observed between any experimental group following 9 weeks of treatment initiation (Fig 7D and7E). No significant difference was observed in nesting scores between any experimental groups (data not shown).
Discussion
The results of the current study show that a BBB-penetrating analog of EPO, the cTfRMAb-EPO fusion protein, reduces Aβ burden, microglial activation and synaptic loss, and improves spatial memory in the APP/PS1 mouse model of AD. Further, chronic treatment with the cTfRMAb-EPO fusion protein does not result in CMH development.
EPO is a potential treatment for AD, however, is a large molecule with limited permeability across either the mouse15 or primate BBB.16 Recent studies show protective effects of systemic EPO through multiple mechanisms in mouse models of AD, and the dose of EPO in these studies ranged from 75μg/kg-2000μg/kg per week, which is higher than the doses at which the cTfRMAb-EPO fusion protein is therapeutic in mouse models of chronic neural diseases (dose range between 60–180μg/kg per week).8–9, 34, 41 This is expected since the cTfRMAb-EPO fusion protein is a BBB- penetrable analog of EPO that rapidly enters the brain parenchyma following systemic administration via TfR mediated transcytosis at the BBB.16, 31 Following systemic administration, the brain volume of distribution of the cTfRMAb-EPO fusion protein is > 50-fold higher than the brain volume of distribution of an IgG that does not cross the BBB, and ~ 70% of the brain cTfRMAb-EPO is found in the postvascular compartment using the capillary depletion method, confirming transcytosis of the fusion protein into the brain parenchyma across the BBB.31 Using capillary depletion, the brain volume of distribution of EPO was found to be comparable to brain blood volume.16
Our previous work shows that the peak brain concentration of a cTfRMAb-based fusion protein is ~ 1.25 μg/g, and the brain concentration remains at 0.7 μg/g at 24h following a single 3mg/kg IP dose in mice.42 Since the cTfRMAb-EPO is 20% EPO, the brain concentration of EPO is predicted to be between 140–250 ng/g (~4–8 nM EPO) between 6–24h following a single 3mg/kg IP dose administration.42 Brain uptake measurements for EPO, on the other hand, have primarily been limited to CSF measurements, and the CSF concentration in rodents injected with a single comparable IP dose of EPO and EPO-TAT (to increase brain penetrability) peaks at ~ 1 pM and 2.5 pM respectively.43 The concentration of EPO in the CSF of AD patients is reported to be about 2 pM.44 With respect to neuroprotection, a 0.3 nM EPO concentration protects against neurotoxicity.43 These reported CSF and therapeutic concentrations are thus lower than the predicted concentration of EPO delivered to the brain with a 3mg/kg IP dose of the cTfRMAb-EPO fusion protein. Based on our previous work,42 since the elimination half-life of the cTfRMAb-based fusion protein following a single 3mg/kg IP dose is ~ 7 h, negligible plasma accumulation of the fusion protein is expected over the course of the study with a dosing frequency of three days a week (Monday, Wednesday and Friday) used in the current study.
In the present proof-of-concept study, the dose of the cTfRMAb-EPO fusion protein is 9mg/kg per week (3mg/kg three days a week). This is equivalent to an EPO dose of 1800μg/kg per week since the fusion protein is 20% EPO based on molecular weight.31 It should be noted that the cTfRMAb-EPO fusion protein has a 10-fold lower plasma AUC compared with EPO due to rapid peripheral clearance by peripheral TfR.31 As a result, the equivalent dose of EPO administered in the current study is 180μg/kg per week. Notably, the cTfRMAb-EPO fusion protein is also neuroprotective at a 3 fold lower dose (equivalent to an EPO dose of 60μg/kg per week) in a mouse model of neural disease.34
One of the major hallmark pathology of AD is the extracellular deposition of Aβ peptide and its aggregation into plaques. EPO exerts anti-Aβ effects and EPO mediated Aβ lowering is shown to be associated with reduced expression of brain endothelial receptor for advanced glycation end products (RAGE) that is involved in the transport of Aβ into the brain.8, 45 In the current study, APP/PS1 mice treated with the cTfRMAb- EPO fusion protein had significantly reduced cortical and hippocampal Aβ peptide (assessed by 6E10-immunostaining) and cortical mature Aβ plaque (assessed by thio-S staining) load. These findings are consistent with recent studies in which peripheral administration of EPO resulted in significant reduction of cortical8–9 and hippocampal9 Aβ load in mouse models of AD. We, however, did not observe a significant reduction in hippocampal thio-S positive mature Aβ plaque in the current study and the low hippocampal thio-S positive mature Aβ plaque load seen in the 8 month old APP/PS1 in the current study mice may account for this inconsistency. The APP/PS1 mice used in the current study begin to develop Aβ deposits by 6 months and deposition increases up to 12 months of age with Aβ deposition in the cortex preceding deposition in the hippocampus.46–47 Future studies in older mice with increased Aβ plaque deposits at the time of sacrifice are needed to determine the hippocampal Aβ plaque lowering effects of the cTfRMAb-EPO fusion protein.
Progressive and gradual decline in cognitive function often correlates with the neuropathological finding of synaptic loss in AD brains.48 In the current study, chronic treatment with the cTfRMAb-EPO fusion protein rescued spatial memory assessed by the modified Y-maze. This rescue of spatial memory was accompanied by a rescue in hippocampal synaptic loss in the mice treated with the cTfRMAb-EPO fusion protein. These results are in agreement with other studies showing beneficial effects of EPO on memory function and decreased neuronal or synaptic loss.8,41,49 Though we saw a small but significant reduction in locomotion of APP/PS1 mice treated with the cTfRMAb-EPO fusion protein 6 weeks after treatment initiation, no such reduction in locomotion was observed 9 weeks after treatment initiation compared with WT mice. The reason for this modest decline in locomotion is not clear and more studies will be needed to determine the significance of this finding. Notably, no significant difference in locomotion was observed between the saline- and cTfRMAb-EPO-treated APP/PS1 mice throughout the study, and the cTfRMAb-EPO fusion protein treated mice appeared healthy (did not appear prostrate, unresponsive or scruffy).
Besides decline in cognitive function, AD is also associated with deterioration of daily life activities and overall well-being.50 We evaluated the effect of the cTfRMAb- EPO on nest building in the present study which is equivalent to activities of daily living in humans and is used to evaluate overall well-being in mice.51 However, we did not observe a significant difference in the nest building scores between the WT and APP/PS1 mice in the current study and no meaningful conclusion could be drawn about the effect of the cTfRMAb-EPO on nest building abilities. This result is consistent with a recent report in which no difference was observed between the genotypes (WT and APP/PS1) in nest building activity.52
Microglia, the innate immune cells of the brain, are often found concentrated around Aβ plaques in the AD brain.53 Reactive microglia around Aβ plaque can mediate plaque phagocytosis and clearance, and also form a protective barrier around plaques to prevent the accretion of new Aβ peptides into existing plaques.53 On the other hand, there is abundant evidence showing that microglia contribute to synaptic loss, promote neuroinflammation and AD pathology.23 The role of microglia around plaques is thus debated and studies show both a protective and detrimental role of microglia in AD.53 Consistent with other studies, we saw a significant increase in microglial activation (assessed by Iba-1 immunohistochemistry) in the APP/PS1 mice compared with WT mice.54 Treatment with the cTfRMAb-EPO fusion protein caused a significant reduction in cortical microglial activation and a similar trend was observed in the hippocampus. This result is in agreement with the reported attenuation of microglial activation by EPO.55
Brain microvascular endothelial damage contributes to neuronal cell death and cognitive decline is AD56 and studies show that EPO is involved in the regulation of brain vascular permeability.57–58 In the current study, though there was an upward trend in the brain IgG positive immunoreactive area (marker of permeability across the brain vascular endothelium) in the 8 month old saline treated APP/PS1 mice, no statistical difference was observed between any experimental groups. CMH are often accompanied with Aβ modifying agents, and loss of cerebral vascular integrity is associated with CMH development.59–60 To determine if Aβ-lowering effects of the cTfRMAb-EPO fusion protein result in CMH development, H&E and PB staining were used to identify acute and sub-acute CMH as described by us previously.37 No significant changes in cerebral vascular permeability in our study corroborates our finding of no significant differences in CMH development between any experimental groups in the current study.
In summary, chronic treatment with the BBB-penetrating EPO, the cTfRMAb-EPO fusion protein, attenuated Aβ load, microglial activation and synaptic loss which was accompanied with an improvement in spatial memory in the APP/PS1 mouse model of AD. These results highlight the protective effects of a BBB-penetrable analog of EPO for AD. The chimeric cTfRMAb used herein is specific for the mouse TfR1 and a recent study shows robust brain uptake and protective effects of humanized TfRMAb in the primate brain.61 Specie specific TfRMAbs may thus be used in future work to translate these results in other species including humans.
Acknowledgements
We would like to thank Kelley Kilday, Austin Ing, Joshua Yang and Dr. Jiahong Sun for their assistance. The authors also thank Dr. Eric Ka-Wai Hui, Dr. Jeff Zhiqiang Lu, Winnie Tai and Phuong Tram of ArmaGen, Inc., for production of the cTfRMAb-EPO fusion protein. Funding for this study was provided by the Joseph H. Stahlberg foundation and NIA R21 AG055949 (RKS).
Footnotes
Conflict of Interest
William Pardridge is a consultant to and Ruben Boado is an employee of ArmaGen, Inc.
References:
- 1.Chong ZZ; Li F; Maiese K, Erythropoietin requires NF-kappaB and its nuclear translocation to prevent early and late apoptotic neuronal injury during beta- amyloid toxicity. Curr Neurovasc Res 2005, 2 (5), 387–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhi-Kun S; Hong-Qi Y; Zhi-Quan W; Jing P; Zhen H; Sheng-Di C, Erythropoietin prevents PC12 cells from beta-amyloid-induced apoptosis via PI3KAkt pathway. Transl Neurodegener 2012, 1 (1), 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sun ZK; Yang HQ; Pan J; Zhen H; Wang ZQ; Chen SD; Ding JQ, Protective effects of erythropoietin on tau phosphorylation induced by beta-amyloid. J Neurosci Res 2008, 86 (13), 3018–27. [DOI] [PubMed] [Google Scholar]
- 4.Brines M; Cerami A, Emerging biological roles for erythropoietin in the nervous system. Nat Rev Neurosci 2005, 6 (6), 484–94. [DOI] [PubMed] [Google Scholar]
- 5.Li G; Ma R; Huang C; Tang Q; Fu Q; Liu H; Hu B; Xiang J, Protective effect of erythropoietin on beta-amyloid-induced PC12 cell death through antioxidant mechanisms. Neurosci Lett 2008, 442 (2), 143–7. [DOI] [PubMed] [Google Scholar]
- 6.Ma R; Hu J; Huang C; Wang M; Xiang J; Li G, JAK2/STAT5/Bcl-xL signalling is essential for erythropoietin-mediated protection against apoptosis induced in PC12 cells by the amyloid beta-peptide Abeta25–35. Br J Pharmacol 2014, 171 (13), 3234–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li YP; Yang GJ; Jin L; Yang HM; Chen J; Chai GS; Wang L, Erythropoietin attenuates Alzheimer-like memory impairments and pathological changes induced by amyloid beta42 in mice. Brain Res 2015, 1618, 159–67. [DOI] [PubMed] [Google Scholar]
- 8.Lee ST; Chu K; Park JE; Jung KH; Jeon D; Lim JY; Lee SK; Kim M; Roh JK, Erythropoietin improves memory function with reducing endothelial dysfunction and amyloid-beta burden in Alzheimer’s disease models. J Neurochem 2012, 120 (1), 115–24. [DOI] [PubMed] [Google Scholar]
- 9.Armand-Ugon M; Aso E; Moreno J; Riera-Codina M; Sanchez A; Vegas E; Ferrer I, Memory Improvement in the AbetaPP/PS1 Mouse Model of Familial Alzheimer’s Disease Induced by Carbamylated-Erythropoietin is Accompanied by Modulation of Synaptic Genes. J Alzheimers Dis 2015, 45 (2), 407–21. [DOI] [PubMed] [Google Scholar]
- 10.Samy DM; Ismail CA; Nassra RA; Zeitoun TM; Nomair AM, Downstream modulation of extrinsic apoptotic pathway in streptozotocin-induced Alzheimer’s dementia in rats: Erythropoietin versus curcumin. Eur J Pharmacol 2016, 770, 52–60. [DOI] [PubMed] [Google Scholar]
- 11.Arabpoor Z; Hamidi G; Rashidi B; Shabrang M; Alaei H; Sharifi MR; Salami M; Dolatabadi HR; Reisi P, Erythropoietin improves neuronal proliferation in dentate gyrus of hippocampal formation in an animal model of Alzheimer’s disease. Adv Biomed Res 2012, 1, 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lourhmati A; Buniatian GH; Paul C; Verleysdonk S; Buecheler R; Buadze M; Proksch B; Schwab M; Gleiter CH; Danielyan L, Age-dependent astroglial vulnerability to hypoxia and glutamate: the role for erythropoietin. PLoS One 2013, 8 (10), e77182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Assaraf MI; Diaz Z; Liberman A; Miller WH Jr.; Arvanitakis Z; Li Y; Bennett DA; Schipper HM, Brain erythropoietin receptor expression in Alzheimer disease and mild cognitive impairment. J Neuropathol Exp Neurol 2007, 66 (5), 389–98. [DOI] [PubMed] [Google Scholar]
- 14.Sanchez PE; Fares RP; Risso JJ; Bonnet C; Bouvard S; Le-Cavorsin M; Georges B; Moulin C; Belmeguenai A; Bodennec J; Morales A; Pequignot JM; Baulieu EE; Levine RA; Bezin L, Optimal neuroprotection by erythropoietin requires elevated expression of its receptor in neurons. Proc Natl Acad Sci U S A 2009, 106 (24), 9848–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Banks WA; Jumbe NL; Farrell CL; Niehoff ML; Heatherington AC, Passage of erythropoietic agents across the blood-brain barrier: a comparison of human and murine erythropoietin and the analog darbepoetin alfa. Eur J Pharmacol 2004, 505 (1–3), 93–101. [DOI] [PubMed] [Google Scholar]
- 16.Boado RJ; Hui EK; Lu JZ; Pardridge WM, Drug targeting of erythropoietin across the primate blood-brain barrier with an IgG molecular Trojan horse. J Pharmacol Exp Ther 2010, 333 (3), 961–9. [DOI] [PubMed] [Google Scholar]
- 17.Bernaudin M; Marti HH; Roussel S; Divoux D; Nouvelot A; MacKenzie ET; Petit E, A potential role for erythropoietin in focal permanent cerebral ischemia in mice. J Cereb Blood Flow Metab 1999, 19 (6), 643–51. [DOI] [PubMed] [Google Scholar]
- 18.Genc S; Kuralay F; Genc K; Akhisaroglu M; Fadiloglu S; Yorukoglu K; Fadiloglu M; Gure A, Erythropoietin exerts neuroprotection in 1-methyl-4-phenyl- 1,2,3,6-tetrahydropyridine-treated C57/BL mice via increasing nitric oxide production. Neurosci Lett 2001, 298 (2), 139–41. [DOI] [PubMed] [Google Scholar]
- 19.Juul SE; McPherson RJ; Farrell FX; Jolliffe L; Ness DJ; Gleason CA, Erytropoietin concentrations in cerebrospinal fluid of nonhuman primates and fetal sheep following high-dose recombinant erythropoietin. Biol Neonate 2004, 85 (2), 138–44. [DOI] [PubMed] [Google Scholar]
- 20.Pardridge WM, Drug transport in brain via the cerebrospinal fluid. Fluids Barriers CNS 2011, 8 (1), 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wolf RF; Peng J; Friese P; Gilmore LS; Burstein SA; Dale GL, Erythropoietin administration increases production and reactivity of platelets in dogs. Thromb Haemost 1997, 78 (6), 1505–9. [PubMed] [Google Scholar]
- 22.McDonald TP; Clift RE; Cottrell MB, Large, chronic doses of erythropoietin cause thrombocytopenia in mice. Blood 1992, 80 (2), 352–8. [PubMed] [Google Scholar]
- 23.Rodriguez Cruz Y; Strehaiano M; Rodriguez Obaya T; Garcia Rodriguez JC; Maurice T, An Intranasal Formulation of Erythropoietin (Neuro-EPO) Prevents Memory Deficits and Amyloid Toxicity in the APPSwe Transgenic Mouse Model of Alzheimer’s Disease. J Alzheimers Dis 2017, 55 (1), 231–248. [DOI] [PubMed] [Google Scholar]
- 24.Lochhead JJ; Thorne RG, Intranasal delivery of biologics to the central nervous system. Adv Drug Deliv Rev 2012, 64 (7), 614–28. [DOI] [PubMed] [Google Scholar]
- 25.Jefferies WA; Brandon MR; Hunt SV; Williams AF; Gatter KC; Mason DY, Transferrin receptor on endothelium of brain capillaries. Nature 1984, 312 (5990), 162–3. [DOI] [PubMed] [Google Scholar]
- 26.Kalaria RN; Sromek SM; Grahovac I; Harik SI, Transferrin receptors of rat and human brain and cerebral microvessels and their status in Alzheimer’s disease. Brain Res 1992, 585 (1–2), 87–93. [DOI] [PubMed] [Google Scholar]
- 27.Fishman JB; Rubin JB; Handrahan JV; Connor JR; Fine RE, Receptor-mediated transcytosis of transferrin across the blood-brain barrier. J Neurosci Res 1987, 18 (2), 299–304. [DOI] [PubMed] [Google Scholar]
- 28.Li JY; Boado RJ; Pardridge WM, Blood-brain barrier genomics. J Cereb Blood Flow Metab 2001, 21 (1), 61–8. [DOI] [PubMed] [Google Scholar]
- 29.Pardridge WM, Blood-brain barrier drug delivery of IgG fusion proteins with a transferrin receptor monoclonal antibody. Expert Opin Drug Deliv 2015, 12 (2), 207–22. [DOI] [PubMed] [Google Scholar]
- 30.Boado RJ; Zhang Y; Wang Y; Pardridge WM, Engineering and expression of a chimeric transferrin receptor monoclonal antibody for blood-brain barrier delivery in the mouse. Biotechnol Bioeng 2009, 102 (4), 1251–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhou QH; Boado RJ; Lu JZ; Hui EK; Pardridge WM, Re-engineering erythropoietin as an IgG fusion protein that penetrates the blood-brain barrier in the mouse. Mol Pharm 2010, 7 (6), 2148–55. [DOI] [PubMed] [Google Scholar]
- 32.Pardridge WM, The blood-brain barrier: bottleneck in brain drug development. NeuroRx 2005, 2 (1), 3–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Elliott S; Egrie J; Browne J; Lorenzini T; Busse L; Rogers N; Ponting I, Control of rHuEPO biological activity: the role of carbohydrate. Exp Hematol 2004, 32 (12), 1146–55. [DOI] [PubMed] [Google Scholar]
- 34.Zhou QH; Hui EK; Lu JZ; Boado RJ; Pardridge WM, Brain penetrating IgG-erythropoietin fusion protein is neuroprotective following intravenous treatment in Parkinson’s disease in the mouse. Brain Res 2011, 1382, 315–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chang R; Knox J; Chang J; Derbedrossian A; Vasilevko V; Cribbs D; Boado RJ; Pardridge WM; Sumbria RK, Blood-Brain Barrier Penetrating Biologic TNF-alpha Inhibitor for Alzheimer’s Disease. Mol Pharm 2017, 14 (7), 2340–2349. [DOI] [PubMed] [Google Scholar]
- 36.Sumbria RK; Hui EK; Lu JZ; Boado RJ; Pardridge WM, Disaggregation of amyloid plaque in brain of Alzheimer’s disease transgenic mice with daily subcutaneous administration of a tetravalent bispecific antibody that targets the transferrin receptor and the Abeta amyloid peptide. Mol Pharm 2013, 10 (9), 3507–13. [DOI] [PubMed] [Google Scholar]
- 37.Sumbria RK; Grigoryan MM; Vasilevko V; Krasieva TB; Scadeng M; Dvornikova AK; Paganini-Hill A; Kim R; Cribbs DH; Fisher MJ, A murine model of inflammation-induced cerebral microbleeds. J Neuroinflammation 2016, 13 (1), 218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sumbria RK; Grigoryan MM; Vasilevko V; Paganini-Hill A; Kilday K; Kim R; Cribbs DH; Fisher MJ, Aging exacerbates development of cerebral microbleeds in a mouse model. J Neuroinflammation 2018, 15 (1), 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rial D; Castro AA; Machado N; Garcao P; Goncalves FQ; Silva HB; Tome AR; Kofalvi A; Corti O; Raisman-Vozari R; Cunha RA; Prediger RD, Behavioral phenotyping of Parkin-deficient mice: looking for early preclinical features of Parkinson’s disease. PLoS One 2014, 9 (12), e114216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Deacon RM, Assessing nest building in mice. Nat Protoc 2006, 1 (3), 1117–9. [DOI] [PubMed] [Google Scholar]
- 41.Maurice T; Mustafa MH; Desrumaux C; Keller E; Naert G; de la CG-BM; Rodriguez Cruz Y; Garcia Rodriguez JC, Intranasal formulation of erythropoietin (EPO) showed potent protective activity against amyloid toxicity in the Abeta(2)(5)(−)(3)(5) non-transgenic mouse model of Alzheimer’s disease. J Psychopharmacol 2013, 27 (11), 1044–57. [DOI] [PubMed] [Google Scholar]
- 42.Sumbria RK; Zhou QH; Hui EK; Lu JZ; Boado RJ; Pardridge WM, Pharmacokinetics and brain uptake of an IgG-TNF decoy receptor fusion protein following intravenous, intraperitoneal, and subcutaneous administration in mice. Mol Pharm 2013, 10 (4), 1425–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang F; Xing J; Liou AK; Wang S; Gan Y; Luo Y; Ji X; Stetler RA; Chen J; Cao G, Enhanced Delivery of Erythropoietin Across the Blood-Brain Barrier for Neuroprotection against Ischemic Neuronal Injury. Transl Stroke Res 2010, 1 (2), 113–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brettschneider J; Widl K; Ehrenreich H; Riepe M; Tumani H, Erythropoietin in the cerebrospinal fluid in neurodegenerative diseases. Neurosci Lett 2006, 404 (3), 347–51. [DOI] [PubMed] [Google Scholar]
- 45.Deane R; Du Yan S; Submamaryan RK; LaRue B; Jovanovic S; Hogg E; Welch D; Manness L; Lin C; Yu J; Zhu H; Ghiso J; Frangione B; Stern A; Schmidt AM; Armstrong DL; Arnold B; Liliensiek B; Nawroth P; Hofman F; Kindy M; Stern D; Zlokovic B, RAGE mediates amyloid-beta peptide transport across the blood-brain barrier and accumulation in brain. Nat Med 2003, 9 (7), 907–13. [DOI] [PubMed] [Google Scholar]
- 46.Jankowsky JL; Fadale DJ; Anderson J; Xu GM; Gonzales V; Jenkins NA; Copeland NG; Lee MK; Younkin LH; Wagner SL; Younkin SG; Borchelt DR, Mutant presenilins specifically elevate the levels of the 42 residue beta- amyloid peptide in vivo: evidence for augmentation of a 42-specific gamma secretase. Hum Mol Genet 2004, 13 (2), 159–70. [DOI] [PubMed] [Google Scholar]
- 47.Garcia-Alloza M; Robbins EM; Zhang-Nunes SX; Purcell SM; Betensky RA; Raju S; Prada C; Greenberg SM; Bacskai BJ; Frosch MP, Characterization of amyloid deposition in the APPswe/PS1dE9 mouse model of Alzheimer disease. Neurobiol Dis 2006, 24 (3), 516–24. [DOI] [PubMed] [Google Scholar]
- 48.Scheff SW; Price DA; Schmitt FA; Mufson EJ, Hippocampal synaptic loss in early Alzheimer’s disease and mild cognitive impairment. Neurobiol Aging 2006, 27 (10), 1372–84. [DOI] [PubMed] [Google Scholar]
- 49.Leconte C; Bihel E; Lepelletier FX; Bouet V; Saulnier R; Petit E; Boulouard M; Bernaudin M; Schumann-Bard P, Comparison of the effects of erythropoietin and its carbamylated derivative on behaviour and hippocampal neurogenesis in mice. Neuropharmacology 2011, 60 (2–3), 354–64. [DOI] [PubMed] [Google Scholar]
- 50.Lawton MP, Quality of life in Alzheimer disease. Alzheimer Dis Assoc Disord 1994, 8 Suppl 3, 138–50. [PubMed] [Google Scholar]
- 51.Jirkof P, Burrowing and nest building behavior as indicators of well-being in mice. J Neurosci Methods 2014, 234, 139–46. [DOI] [PubMed] [Google Scholar]
- 52.Janus C; Flores AY; Xu G; Borchelt DR, Behavioral abnormalities in APPSwe/PS1dE9 mouse model of AD-like pathology: comparative analysis across multiple behavioral domains. Neurobiol Aging 2015, 36 (9), 2519–32. [DOI] [PubMed] [Google Scholar]
- 53.Hansen DV; Hanson JE; Sheng M, Microglia in Alzheimer’s disease. J Cell Biol 2018, 217 (2), 459–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Guo HB; Cheng YF; Wu JG; Wang CM; Wang HT; Zhang C; Qiu ZK; Xu JP, Donepezil improves learning and memory deficits in APP/PS1 mice by inhibition of microglial activation. Neuroscience 2015, 290, 530–42. [DOI] [PubMed] [Google Scholar]
- 55.Tamura T; Aoyama M; Ukai S; Kakita H; Sobue K; Asai K, Neuroprotective erythropoietin attenuates microglial activation, including morphological changes, phagocytosis, and cytokine production. Brain Res 2017, 1662, 65–74. [DOI] [PubMed] [Google Scholar]
- 56.van de Haar HJ; Burgmans S; Jansen JF; van Osch MJ; van Buchem MA; Muller M; Hofman PA; Verhey FR; Backes WH, Blood-Brain Barrier Leakage in Patients with Early Alzheimer Disease. Radiology 2016, 281 (2), 527–535. [DOI] [PubMed] [Google Scholar]
- 57.Martinez-Estrada OM; Rodriguez-Millan E; Gonzalez-De Vicente E; Reina M; Vilaro S; Fabre M, Erythropoietin protects the in vitro blood-brain barrier against VEGF-induced permeability. Eur J Neurosci 2003, 18 (9), 2538–44. [DOI] [PubMed] [Google Scholar]
- 58.Uzum G; Sarper Diler A; Bahcekapili N; Ziya Ziylan Y, Erythropoietin prevents the increase in blood-brain barrier permeability during pentylentetrazol induced seizures. Life Sci 2006, 78 (22), 2571–6. [DOI] [PubMed] [Google Scholar]
- 59.Wilcock DM; Colton CA, Immunotherapy, vascular pathology, and microhemorrhages in transgenic mice. CNS Neurol Disord Drug Targets 2009, 8 (1), 50–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ungvari Z; Tarantini S; Kirkpatrick AC; Csiszar A; Prodan CI, Cerebral microhemorrhages: mechanisms, consequences, and prevention. Am J Physiol Heart Circ Physiol 2017, 312 (6), H1128–H1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yu YJ; Atwal JK; Zhang Y; Tong RK; Wildsmith KR; Tan C; Bien-Ly N; Hersom M; Maloney JA; Meilandt WJ; Bumbaca D; Gadkar K; Hoyte K; Luk W; Lu Y; Ernst JA; Scearce-Levie K; Couch JA; Dennis MS; Watts RJ, Therapeutic bispecific antibodies cross the blood-brain barrier in nonhuman primates. Sci Transl Med 2014, 6 (261), 261ra154. [DOI] [PubMed] [Google Scholar]