

Abstract
Ionic liquids (ILs) have been established as effective promoters for the electrocatalytic upconversion of CO2 to various commodity chemicals. Imidazolium ([Im]+) cathode combinations have been reported to selectively catalyze the 2e−/2H+ reduction of CO2 to CO. Recently our laboratory has reported energy-efficient systems for CO production featuring inexpensive bismuth-based cathode materials and ILs comprised of 1,3-dialkylimidazolium cations. As part of our ongoing efforts to understand the factors that drive CO2 reduction at electrode interfaces, we sought to evaluate the catalytic performance of alternative ILs in combination with previously described Bi cathodes. In this work, we demonstrate that protic ionic liquids (PILs) derived from 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) effectively promote the electrochemical reduction of CO2 to formate (HCOO−) with high selectivity. The use of PILs comprised of the conjugate acid of DBU, [DBU-H]+, efficiently catalyzed the reduction of CO2 to HCOO− (FEFA ≈ 80%) with significant suppression of CO production (FECO ≈ 20%) in either MeCN or MeCN/H2O (95/5) solution. When they were used in combination with [DBU-H]+-based PILs, Bi-based cathodes achieved current densities for CO2 reduction (jtot ≈ 25–45 mA/cm2) that are comparable to or greater than those reported with imidazolium ILs such as [BMIM]PF6. As we demonstrate herein, the selectivity of the 2e− reduction of CO2 toward HCOO− or CO can be dictated through the choice of the IL promoter present in the electrolysis solution, even in cases in which the same electrocatalyst material is studied. These findings highlight the tunability of bismuth/IL systems for the electrochemical reduction of CO2 with high efficiency and rapid kinetics.
Keywords: CO2 reduction, ionic liquid, imidazolium, electrocatalysis, formic acid, carbon monoxide, catalytic plasticity, bismuth
INTRODUCTION
The storage of electrical energy from a source of renewable electricity (e.g., solar) as chemical energy via the reduction of CO2 is an attractive strategy for energy storage.1,2 Advances in CO2 reduction catalysis at heterogeneous surfaces and homogeneous transition-metal complexes highlight the potential for converting CO2 into commodity chemicals, as systems capable of producing useful feedstock molecules such as carbon monoxide (CO), formate (HCOO−), and methanol have been established.3–7 Heterogeneous cathode materials have received significant attention due to their potential for incorporation into electrolytic devices. As such, efficient cathode materials including metallic, composite, and doped carbon electrodes have been developed and widely reported in the literature.8‘9 Much effort is now aimed at improving the energy efficiency and electrocatalytic activity of electrode/catholyte systems, while product selectivity (i.e., Faradaic efficiency (FE)) and high rates (current density (j)) of CO2 reduction at minimal overpotentials (η) are retained.
Thermodynamically, the direct one-electron reduction of CO2 to CO2•− is challenging, due in part to the large reorganization energy associated with the transition from linear CO2 to bent CO2•−.10,11 Coupling the transfer of electrons with proton transfer allows for the formation of more stable intermediates, thereby lowering the potential required for CO2 activation by more than 1 V. Additionally, the incorporation of ionic liquids (ILs) as cocatalysts, solvents, and/or electrolytes has been shown to enhance CO2 electrocatalysis at metal electrodes.12–15 For example, our laboratory has demonstrated that in the presence of 1,3-dialkyl-substituted imidazolium-based ILs (e.g., 1-butyl-3-methylimidazolium, [BMIM]+), affordable and readily prepared Bi-based cathodes can drive the electrocatalytic reduction of CO2 to CO with metrics similar to those obtained using precious-metal catalysts based upon Ag and Au.16 The high rate of and selectivity for CO evolution using Bi-based cathodes, in the presence of imidazolium ([Im]+) ILs, has motivated further investigation into the role(s) of ILs in the electrochemical activation of CO2. A combination of in-depth electrochemical17,18 and spectroscopic13,19 techniques suggests that interaction of [Im]+ with certain cathode materials engenders energy-efficient CO2 reduction, independent of anion effects.
In aprotic nonaqueous solvents, such as MeCN, the acidity of the proton at the 2-position of [Im]+ heterocycles (Figure 1) has been calculated to be as high as pKa ≈ 32.20 As a result, [Im]+ cations such as [BMIM]+ are very weak proton donors. It is to be expected that the intermolecular forces that engender favorable CO2 electrocatalysis are limited by the low Bronsted and Lewis acidities of [Im]+-based electrolytes.21,22 Through combined inspiration from the widely studied roles of IL–electrode interactions in heterogeneous CO2 electrocatalysis,12–15 and the established ability of exogenous acids to boost homogeneous catalysis,23–28 we sought to evaluate the performance of Bi-modified electrodes in the presence of protic ionic liquids (PILs) that are more acidic than prototypical [Im]+-based ILs.
Figure 1.
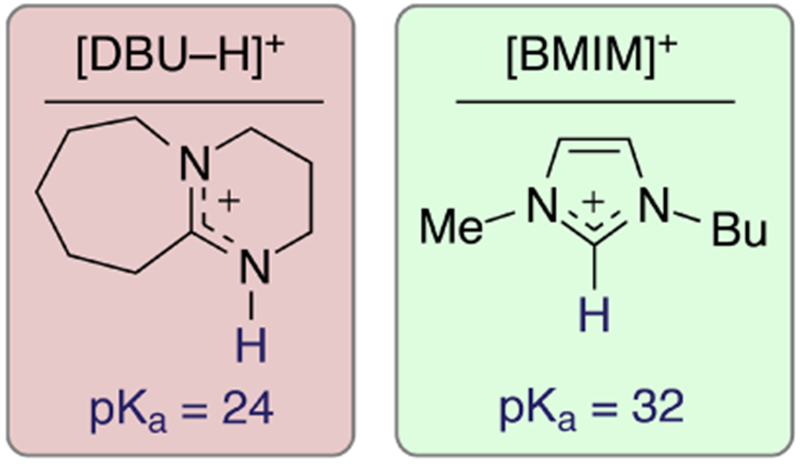
Structures of the IL cations studied in this work. The most acidic proton of each cation is shown in blue, and pKa values reflect the proton donor ability of each in MeCN.
As a subset of ILs, PILs feature an available proton on the positively charged atom(s) that make up the IL cation.29,30 Although imidazolium-based ILs such as [BMIM]+ can serve as very weak proton donors, they are formally classified as aprotic ILs, since the most acidic protons on this cation do not reside on the cationic nitrogen atoms that make up the [Im]+ heterocycle. In contrast, the N-heterocycle 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) is an organosuperbase, which can readily be protonated to form its corresponding conjugate acid ([DBU-H]+), the structure of which is shown in Figure 1. The most acidic proton of [DBU-H]+ (pKa ≈ 24.3 in MeCN;31 pKa ≈ 13.4 in H2O32–34) is bound to the amindinium nitrogen of this cation, which readily forms PILs with weakly coordinating anions. Moreover, PILs based upon [DBU-H]+ have been employed in CO2 capture studies,35–40 as well as other synthetic applications.41–45 Additionally, DBU has been utilized as a cocatalyst for the hydrogenation of CO2 to HCOO− (requiring elevated temperatures and excess H2);46–49 however to the best of our knowledge no studies have been reported that utilize DBU or [DBU-H]+ as a promoter or electrolyte for electrochemical CO2 reduction.
Studying CO2 electroreduction in the presence of a [DBU-H]+-based PIL provides an opportunity to probe how variations in the proton availability and overall structure of nonaqueous electrolytes affect the outcome and kinetics of CO2 reduction at Bi-based cathodes. In this work, we sought to determine how varying the identity and structure of the IL promoter used in conjunction with Bi-film cathodes affects the course of CO2 reduction at the Bi surface. In particular, we were interested to see whether variation of the IL in solution would lead to the promotion of alternative CO2 activation pathways other than the 2e− reduction of CO2 to CO, which is exclusively observed using Bi cathodes and [IM]+ additives. Herein we demonstrate that the IL additive not only tunes the activity of Bi-based cathodes but also directly affects the distribution of CO2 reduction products that can be generated via the two 2e− CO2 reductions embodied by eqs 1 and 2. In particular, these studies demonstrate that the pathway by which CO2 is activated at Bi cathodes in the presence of [BMIM]+ is distinct from that which is operative when [DBU-H]+ is used in place of the [Im]+-based IL and establishes that Bi cathodes demonstrate a high degree of catalytic plasticity as platforms for CO2 reduction.
| (1) |
| (2) |
RESULTS AND DISCUSSION
With the advantageous properties of [Im]+ ILs in mind, PILs comprised of [DBU-H]+ were attractive candidates for study due to the structural and functional similarities of each (Figure 1). Previous studies have shown that interaction of [BMIM]+ with CO2 and negatively polarized Bi cathodes gives rise to efficient and rapid CO production.16–18 Similar to the case for [BMIM]+, [DBU-H]+ is a monocation that exhibits charge delocalization through the carbon atom bridging its amidinium N–C═N structure. The alicyclic six- and seven-membered rings of DBU also provide an organic cation of size similar to that of the [BMIM]+ cation. Moreover, the increased acidity of [DBU-H]+ (pKa(MeCN) ≈ 24.3),31 in comparison to [BMIM]+ (pKa ≈ 32),20 should therefore promote an environment of significantly higher proton activity for CO2 reduction at the Bi cathode. On the basis of the Nernst equation, this decrease in pKa will thermodynamically allow for the production of reduced CO2 species at potentials ~450 mV more positive (see the Supporting Information for full analysis of E° values associated with CO2 reduction in the presence of [BMIM]+ or [DBU-H]+ in MeCN-based electrolytes).
To investigate the viability of [DBU-H]+-promoted CO2 electrocatalysis, we initially prepared and evaluated PILs comprised of [DBU-H]+. For direct comparison to our previously reported bismuth–carbon monoxide evolving catalyst (Bi-CMEC) system,16 which utilizes Bi-modified cathodes and millimolar concentrations of [Im]+ ILs (e.g., [BMIM]+) in pure MeCN, the protic salt [DBU-H]PF6 was synthesized on an ~20 g scale in high yield via treatment of DBU with 1 equiv of [NH4]PF6 in H2O, which led to precipitation of a white solid that was isolated and characterized as [DBU-H]PF6 via 1H and 13C NMR spectroscopy (see the Supporting Information). We note that we chose to use the hexafluorophosphate salt of [DBU-H]+, since this anion is stable under the electrolysis conditions described in this work and since [Im]PF6-based ionic liquids have been some of the most thoroughly studied with Bi cathodes.16–18
In order to determine whether [DBU-H]+ promoted activation of CO2 at heterogeneous supports, linear sweep voltammograms (LSVs) were recorded using a Bi-modified glassy-carbon electrode (GCE) in an electrolyte solution containing 0.1 M TBAPF6 under an atmosphere of CO2. The electrochemical response of 250 mM solutions of either [BMIM]PF6 or [DBU-H]PF6 in CO2-saturated MeCN containing 0.1 M TBAPF6 were compared using identically prepared Bi-based cathodes. LSVs recorded in the presence of [BMIM]+ and [DBU-H]+ are represented by the solid orange and blue traces, respectively, in Figure 2a and are both consistent with electrocatalytic processes.
Figure 2.
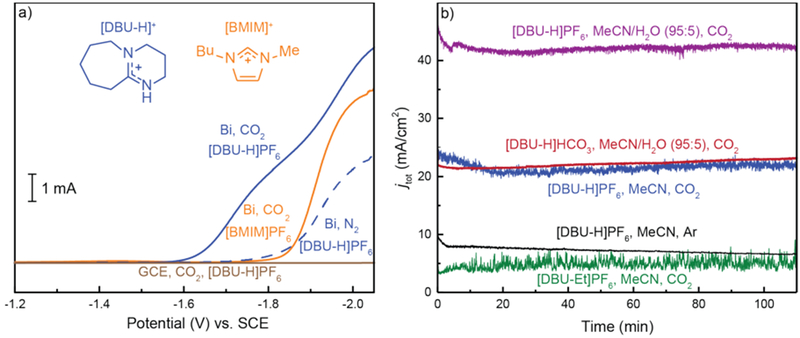
(a) Linear sweep voltammograms (LSVs) recorded for Bi-based and bare GCEs in MeCN containing 250 mM IL and 0.1 M TBAPF6 under an atmosphere of Ar, N2, or CO2. (b) Total current density profiles recorded for Bi-based cathodes in either MeCN or MeCN/H2O (95/5) containing 250 mM IL and 0.1 M TBAPF6 at Eappl = −1.80 V vs SCE.
The large current response observed in the presence of [DBU-H]+ (Figure 2a, blue trace) features a cathodic shift in onset potential of ~300 mV, relative to that observed when [BMIM]+ is present in the catholyte. The less negative onset potential observed in the presence of [DBU-H]+ likely reflects the increased acidity of this PIL relative to the [Im]+ based IL (vide supra). Most notably, the LSV experiment recorded in the presence of [DBU-H]+ shows significant current density over the potential range spanning −1.6 to −1.8 V vs SCE, which is a potential window in which [BMIM]+-based catholytes do not show any appreciable rise in current (Figure 2a, orange trace). Identical experiments performed using Bi cathodes and [DBU-H]+ in the absence of CO2 do not show any current response at potentials less negative than −1.8 V (Figure 2a, dashed blue trace). The lack of significant current densities under a nitrogen atmosphere suggests that the large catalytic wave observed in the presence of [DBU-H]+ and CO2 (Figure 2a, blue trace) cannot be attributed to reduction of protonated DBU at the electrode surface but rather corresponds to activation of CO2 at the Bi cathode. Homogeneous redox processes mediated by [DBU-H]+ can also be ruled out, as no cathodic features were observed for LSVs recorded using a bare GCE in place of a Bi cathode, in the presence of 250 mM [DBU-H]+ (Figure 2a, brown trace). As previously established for Bi/[Im]+ electrocatalysis, these results demonstrate that both the Bi-based cathode and IL promoter are intimately involved in the reduction of CO2.
Controlled-potential electrolysis (CPE) experiments were conducted to establish that the electrochemical responses of Figure 2a correspond to the conversion of CO2 to reduced carbon products. Electrolysis experiments were performed using Bi-modified GCEs with MeCN catholytes containing 0.1 M TBAPF6 and 250 mM [DBU-H]PF6. Upon initiating the CPE at −1.80 V vs SCE, gaseous products were identified by periodic analysis of the reaction headspace by gas chromatography (GC). Consistent with our previously reported Bi/[Im]+ CO2 electroreduction systems,16–18 CO was identified as the major gaseous product during electrolysis, with coproduction of a very small amount of H2 (FEH2 ≈ 2%). Surprisingly, quantification of the CO produced showed a diminished FECO of ~20%, which is in stark contrast to the highly selective evolution of CO (FECO ≈ 85%) observed when imidazolium-based ILs are used in conjunction with Bi-based cathodes (Table 1).16–18 Analysis of the catholyte solution by 1H NMR spectroscopy showed that the vast majority of charge passed during the CPE experiment was directed toward the production of formate (HCOO−) with selectivity of FEFA = 77%. Electrolysis experiments carried out at E = −1.80 V vs SCE showed no decrease in performance over the course of 2 h, demonstrating that the Bi/[DBU-H]+ system is robust and operates with impressive kinetics, as evidenced by the average overall current density of jtot = 27 ± 3 mA/cm2 (Figure 2b, blue trace; Table 1). Identical CPE experiments carried out using a bare GCE in place of the Bi cathode showed virtually no current response (Table 1 and Figure S1), again demonstrating that the Bi-based electrode is critical to the HCOO− production we observe when [DBU-H]+ is added to the catholyte in place of the traditionally employed [BMIM]+ promoter. Similarly, in the absence of the [DBU-H]+ promoter the activity (jtot) for HCOO− production is significantly curtailed, further indicating that, under the conditions described here, both the Bi cathode and [DBU-H]+ are critical to the observed electrocatalysis.
Table 1.
Faradaic Efficiencies (FE) and Current Densities (j) for Electrocatalytic Reduction of CO2 in MeCN or MeCN/H2O (95/5) Containing 0.1 M TBAPF6 and 250 mM of either [BMIM]PF6 or [DBU-H]PF6 Ionic Liquid (IL) at Eappl = −1.80 V vs SCE
| cathode (solv) | IL | FEFA (%) | FECO (%) | FEH2 (%) | jtot (mA/cm2) | jFA (mA/cm2) | jCO (mA/cm2) |
|---|---|---|---|---|---|---|---|
| Bi (MeCN) | [DBU-H]PF6 | 77 ± 5 | 21 ± 1 | 2 ± 1 | 27 ± 3 | 21 ± 4 | 6 ± 1 |
| Bi (MeCN/H2O) | [DBU-H]PF6 | 75 ± 4 | 14 ± 1 | 7 ± 3 | 43 ± 5 | 32 ± 3 | 6 ± 1 |
| Bi (MeCN/H2O) | [DBU-H]HCO3 | 73 ± 5 | 14 ± 2 | 12 ± 3 | 27 ± 2 | 20 ± 3 | 4 ± 1 |
| Bi (MeCN/H2O) | no IL | 70 ± 6 | 10 ± 1 | 14 ± 2 | 4 ± 2 | 3 ± 1 | <0.5 |
| Bi (MeCN) | [DBU-Et]PF6 | 8 ± 1 | 14 ± 2 | 5 ± 1 | 0.4 ± 0.1 | ||
| Bi (MeCN)a | [BMIM]PF6 | 10 ± 2 | 84 ± 3 | <0.5 | 15 ± 2 | 1.5 ± 0.5 | 13 ± 2 |
| GCE (MeCN) | [DBU-H]PF6 | <0.05 | |||||
| Bi (MeCN)b | [DBU-H]PF6 | 48 ± 3 | 6 ± 2 |
Trials performed at Eappl = −1.95 V vs SCE;
Trials performed in the absence of CO2, under a saturated atmosphere of Ar. Note that electrolyses for the Bi/[DBU-H]PF6 and Bi/[BMIM]PF6 systems are both carried out at potentials that are ~200 mV more negative than the onset potentials for electrocatalysis under each set of conditions, as shown by the LSV traces in Figure 2a.
The ability of [DBU-H]+ to donate protons at the electrode/electrolyte interface is critical in establishing the Eeq value for HCOO− production in the MeCN catholyte employed in the above experiments. Under the electrolysis conditions described above, we have determined the equilibrium potential for reduction of CO2 to HCOO− and HCOOH to be Eeq = −0.99, −1.14 V vs SCE, respectively, on the basis of tabulated and calculated thermodynamic values (see the Supporting Information for a full description of Eeq determination). The overpotential for [DBU-H]+-promoted CO2 reduction therefore is ~660–800 mV and to the best of our knowledge represents the first simple heterogeneous electrochemical platform capable of converting CO2 to HCOOH/HCOO− in an organic catholyte with high selectivities and fast kinetics (jFA > 20 mA/cm2).
To further establish that the proton activity of [DBU-H]+ is important to the electrocatalytic responses shown in Figure 2, the ability of an aprotic DBU-based cation to promote CO2 activation at a Bi-modified GCE was probed. Alkylation of DBU with ethyl bromide50 followed by salt metathesis with [NH4]PF6 afforded the ethyl-appended IL [DBU-Et]PF6, in good yield. Since [DBU-Et]+ lacks the relatively acidic amidinium proton of [DBU-H]+, [DBU-Et]PF6 may be considered an aprotic IL, as it cannot readily provide protons to facilitate CO2 reduction at the Bi/electrolyte interface. Satisfyingly, electrochemical experiments carried out in CO2-saturated solutions of MeCN containing 0.1 M TBAPF6 and 250 mM [DBU-Et]PF6 do not show evidence of CO2 electroreduction, reflecting the inability of [DBU-Et]+ to engage in proton transfer. For example, comparison of LSVs recorded with a Bi-modified GCE in the presence of [DBU-H]+ and [DBU-Et]+ shows that only the protonated IL gives rise to significant current response at potentials below −1.95 V (Figure S2). Similarly, CPE experiments carried out in the presence of [DBU-Et]+ give rise to low current densities and no HCOO− production (Figure 2b, green trace, and Table 1).
Although significant quantities of nonvolatile CO2 reduction products have not been observed for CPE experiments employing Bi-modified electrodes in the presence of [Im]+-based ILs,16–18 examples of HCOO− production at Bi cathodes in aqueous electrolytes have been reported.51,52 These studies demonstrated that bulk and nanostructured bismuth electrodes can produce HCOO− in CO2-saturated aqueous HCO3− solution upon application of the absolute overpotential η ≈ 0.9 V with much slower kinetics in comparison to the Bi/[DBU-H]+ systems highlighted above. Despite the limited precedence showing that bismuth cathodes can drive electrochemical HCOO− production in CO2-saturated aqueous HCO3− solutions, extension of our Bi/[DBU-H]+ catalysis to 0.5 M NaHCO3 (pH 7.2) did not produce HCOO− upon CPE. Quite the contrary, over the course of a 2 h CPE (Eappl = −1.45 V vs SCE) quantitative hydrogen production (FEH2 ≈ 100%) was observed with an average partial current density of jH2 = 9.2 ± 1.7 mA/cm2 (Figure S3).
Although the Bi/[DBU-H]+ system proved to be ineffective for CO2 activation in 0.5 M aqueous NaHCO3, further investigation into the scope of Bi/[DBU-H]+-promoted CO2 electrocatalysis was evaluated by examining the response of this catalyst system in CO2-saturated MeCN/H2O (95/5). CPE experiments in which [DBU-H]PF6 (250 mM) was directly added to CO2-saturated MeCN/H2O (95/5) catholyte containing 0.1 M TBAPF6 resulted in an exceptionally rapid rate of CO2 reduction (jtot > 40 mA/cm2). Under these conditions formate production still accounted for ~75% of the charge passed during CPE (Figure 2b, purple trace, and Table 1). To the best of our knowledge, these current densities exceed those of any previously reported catalysts for HCOO− production, owing to the increased solubility of CO2 in the MeCN-based catholyte and the high activity of the Bi/[DBU-H]+ system. Repetition of the above CPE experiments with a bismuth-modified GCE in CO2-saturated MeCN/H2O (95/5) in the absence of the [DBU-H]+ promoter also led to HCOO− production (FEFA ≈ 70%); however, the rate of CO2 reduction was found to be 1 order of magnitude slower in comparison to that obtained for the Bi/[DBU-H]+ system (jtot < 5 mA/cm2 in the absence of [DBU-H]+; Table 1). As such, these experiments clearly highlight the extent to which the [DBU-H]+-based promoter enhances the rate of HCOO− production in MeCN-based catholytes.
By taking advantage of established reactivity of DBU with CO2 in the presence of H2O,53–55 [DBU-H]+-containing catholyte solutions can be generated in situ, which eliminates the need for preparation of ex situ derived [DBU-H]PF6. Solutions of DBU (250 mM) in MeCN/H2O (95/5) were sparged with CO2 to generate [DBU-H]HCO3 in situ. Voltammetry conducted for these solutions with Bi-modified GCE cathodes under either CO2 or N2-saturated MeCN/H2O (95/5), with 0.1 M TBAPF6 supporting electrolyte, produced the polarization curves shown in Figure 3. The LSV recorded in CO2-saturated MeCN/H2O (95/5) containing 250 mM [DBU-H]HCO3 depicts the onset of a large cathodic wave at E ≈ −1.45 V vs SCE (Figure 3, red trace). A similar current response was not observed for Bi cathodes in the absence of CO2, as N2-saturated MeCN/H2O (95/5) did not show a prominent reduction wave under analogous conditions (Figure 3, blue trace), or upon use of a GCE under CO2-saturated conditions (Figure 3, black trace).
Figure 3.
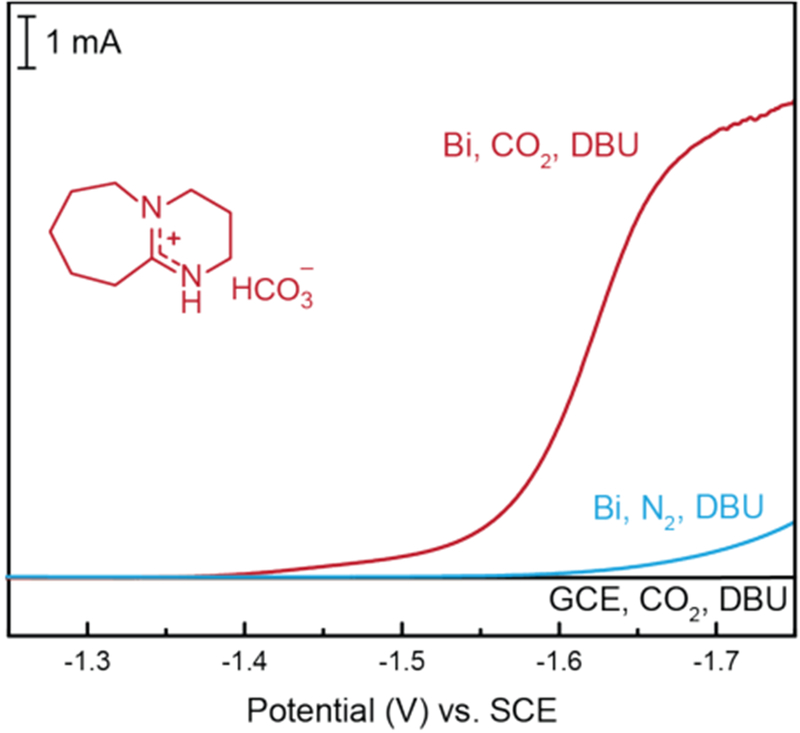
LSVs recorded for Bi-modified and bare GCEs in MeCN/H2O (95/5) containing 0.1 M TBAPF6 and 250 mM DBU under an atmosphere of CO2 (red and black traces) or N2 (blue trace).
The ability of in situ generated [DBU-H]HCO3 IL to promote the electrocatalytic conversion of CO2 to HCOO− at the Bi-modified GCE was confirmed through CPE experiments. CPEs analogous to those described above for catholytes containing ex situ derived [DBU-H]PF6 were performed using equivalent concentrations of [DBU-H]HCO3 prepared in CO2-saturated MeCN/H2O (95/5). The ability of [DBU-HCO3 to promote CO2 reduction at E = −1.80 V in MeCN/H2O (95/5) was confirmed, as equivalent HCOO− (FEFA ≈ 75%) and CO (FECO < 20%) production levels and current densities (jtot ≈ 25 mA/cm2) were observed over the course of2 h CPE experiments (Figure 2b, red trace, and Table 1), demonstrating that in situ generated [DBU-H]+ electrolytes are equally as efficient at promoting CO2 reduction at Bi cathodes as are those prepared exogenously. While the selectivities for formate production are nearly identical for the Bi/[DBU-H]PF6 and Bi/[DBU-H]HCO3 systems, the current density for CO2 reduction in the former is ~15 mA/cm2 higher than that observed when in situ generated [DBU-H]HCO3 is used as the IL promoter. This difference in CO2 reduction activity may reflect the different concentrations of [DBU-H]+ that are present in the catholyte between the two experiments.
Electrocatalytic conversion of CO2 to reduced carbon products offers a way to store renewable sources of electric current when it is coupled with the 4e−/4H+ oxidation of H2O. A two-compartment cell was utilized to demonstrate that the Bi/[DBU-H]+ system can promote HCOO− production coupled with water oxidation. The anode compartment was comprised of either a platinum gauze or Co-OEC56 based electrode submerged in aqueous phosphate buffer (pH 7.4), while the cathode compartment contained a CO2-saturated solution of MeCN/H2O (95/5) supporting 0.1 M TBAPF6 and 250 mM [DBU-H]+. Both compartments were juxtaposed via a Nafion (NRE-212) membrane.
LSV and CPE analyses carried out for this split cell electrolyzer showed the same kinetics and selectivity for HCOO− production encountered for the CPE experiments described in the preceding paragraph (j ≈ 35 mA/cm2; FEFA ≈ 75%), demonstrating that the Bi/[DBU-H]+ system is amenable to energy-storing catalysis that couples CO2 reduction to water oxidation. Longer CPE experiments showed that this mixed solvent electrolyzer configuration is robust, as it operated with these metrics for at least 8 h (Figure S4) with over 200000 cumulative surface turnovers of HCOO− produced per hour of electrolysis. This activity corresponds to a TOF of HCOO− of ~0.5 mmol/cm2 of active catalyst each hour. While these kinetics are already impressive, we expect that improved mass transport using a flow cell or other advanced cell design will enable additional improvements in the kinetics of HCOO− production using the Bi/[DBU-H]+ system.
CONCLUSIONS AND FUTURE DIRECTIONS
The efficient, selective, and rapid electrocatalytic conversion of CO2 to fuels and other value-added compounds are of key importance toward the development of sustainable carbon cycles. While precious metals have been established as excellent catalysts for CO2 conversion, the cost and scarcity of these materials has limited their use on the scale required for commercial chemical/fuel production. Accordingly, recent efforts have been devoted to development of new electrocatalysts for CO2 reduction that are based on less expensive and more abundant materials.
In prior work, we have demonstrated that inexpensive thin film Bi cathodes16–18 and related Bi-based materials57 can promote the 2e−/2H+ reduction of CO2 to CO with fast kinetics (jCO ≈ 5–20 mA/cm2) and high selectivity (FECO ≈ 85%) from MeCN-based catholytes that contain millimolar concentrations of 1,3-dialkylimidazolium based ILs such as [BMIM]PF6. The impressive efficacy with which the Bi/[Im]+ system catalyzes CO evolution is believed to be driven, at least in part, by the manner in which the imidazolium cation interacts with CO2 and the Bi surface at the cathode/electrolyte interface. Moreover, this model suggests that changing the structure and/or electronic nature of the IL cation in the CO2 electrolysis solution might influence the outcome of CO2 activation at the Bi cathode surface.
In this study, we have characterized the ability of thin film Bi cathodes to activate CO2 in MeCN-based electrolyte solutions containing ILs derived from the inexpensive organic base 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU). Protonation of DBU allows for generation and isolation of a [DBU-H]+-containing IL, which contains a protic amidinium moiety. Electrolysis of CO2-saturated MeCN containing 250 mM [DBU-H]+ with a Bi-modified GCE results in a significantly different CO2 reduction catalysis in comparison to that observed in the presence of [BMIM]+. Instead of driving the 2e−/2H+ reduction of CO2 to CO, CPE with the Bi/[DBU-H]+ system promotes the reduction of CO2 via an orthogonal 2e− pathway, to yield predominantly HCOO−, with selectivities as high as FEFA ≈ 75% at jtot ≈ 20–45 mA/cm2. Notably, the selectivity for production of CO is significantly suppressed in the presence of [DBU-H]PF6 (FECO ≈ 20%) in comparison to analogous CPE experiments in which 250 mM [BMIM]PF6 is present in the electrolyte solution (FECO ≈ 85%). The ability to rapidly and selectively generate HCOO− with the Bi/[DBU-H]+ system is an important advance, since both formate and formic acid are high-volume commodity chemicals and are useful feedstocks for the synthesis of multicarbon products and for fuel cell applications.58–60
In addition to promoting the production of HCOO− by Bi cathodes in CO2-saturated MeCN, [DBU-H]+ also greatly enhances formate production in electrolyte solutions containing a significant fraction of water. Electrolysis of CO2-saturated solutions of MeCN/H2O (95/5) containing 250 mM [DBU-H]PF6 at E = −1.80 V resulted in the production of HCOO− with FEFA ≈ 75% and a minor amount of CO (FECO ≈ 15%). Most notably, the kinetics of CO2 reduction under these conditions are exceptionally fast (jtot ≈ 45 mA/cm2), with the vast majority of this current being directed to HCOO− generation. Moreover, CO2 electrolysis in MeCN/H2O (95/5) based catholytes allows for the in situ generation of [DBU-H]HCO3. CPE of CO2-saturated solutions of MeCN/H2O (95/5) containing 250 mM of in situ generated [DBU-H]+results in a catalysis (FEFA ≈ 75%; FECO < 20%) similar to that observed when exogenous [DBU-H]PF6 is added to the electrolyte. In addition to simplifying the generation of [DBU-H]+ based electrolytes, the MeCN/H2O catholyte solution is also readily juxtaposed with an aqueous anolyte, which allows CO2 reduction at the Bi/[DBU-H]+ cathode to be coupled with anodic water oxidation. This electrolysis configuration is robust and allows for CO2 to be converted to HCOO− with a TOF of ~0.5 mmol/cm2 (jFA ≈ 25 mA/cm2) of active catalyst surface area and produces more than 1.5 million cumulative surface turnovers over the course of an 8 h CPE experiment.
While the ability to electrochemically produce formate from CO2 with the impressive selectivities and kinetics that are highlighted above is notable, the ability to switch the distribution of 2e− CO2 reduction products as a function of electrolyte composition using a single inexpensive heterogeneous electrocatalyst (i.e., Bi) is an intriguing finding in and of itself. The observation that Bi cathodes rapidly and selectively drive the electrocatalytic reduction of CO2 to CO in the presence of [Im]+-based ILs, as opposed to HCOO− in the presence of [DBU-H]+, highlights the versatility of Bi/IL catalyst systems for electrochemical CO2 reduction. As such, this work demonstrates that Bi/IL CO2 reduction platforms exhibits a high degree of catalytic plasticity, as the mode and efficiency of CO2 activation that is promoted at the Bi/IL interface largely depends on the identity of the IL cation dissolved in the electrolyte solution. These studies strongly suggest that the electronics of the IL cation and the manner in which these species interact with CO2 and the Bi cathode are critical in determining the mechanistic pathway by which CO2 reduction takes place. In particular, understanding how different electrolyte additives, including ILs, alter the structure and roughness of heterogeneous electrocatalysts at the electrode/electrolyte interface61 will likely be important to understanding how the catalytic plasticity of Bi and other materials for CO2 reduction arises. Additionally, efforts to understand how electrolyte additives organize on polarized electrode surfaces and stabilize critical intermediates62 en route to distinct CO2 reduction products is also a point worthy of major consideration. Accordingly, further studies dedicated to understanding the role(s) of ILs and other electrolyte additives in directing the selectivity of CO2 electroreduction at heterogeneous catalyst platforms are ongoing in our laboratory.
Supplementary Material
ACKNOWLEDGMENTS
Electrochemical studies carried out by J.L.D. were supported by the Fluid Interface Reactions, Structures & Transport (FIRST), an Energy Frontier Research Center funded by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences. A.A. was supported through a Camille and Henry Dreyfus postdoctoral fellowship in Environmental Chemistry. We also thank the Alfred P. Sloan Foundation for support of this work.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscatal.7b03433.
Full descriptions of experimental methods and electrochemistry data (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Lewis NS; Nocera DG Proc. Natl. Acad. Sci. U. S. A 2006, 103, 15729–15735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Olah GA; Prakash GKS; Goeppert AJ Am. Chem. Soc 2011, 133, 12881–12898. [DOI] [PubMed] [Google Scholar]
- (3).Aresta M; Dibenedetto A; Angelini A Chem. Rev 2014, 114, 1709–1742. [DOI] [PubMed] [Google Scholar]
- (4).Kang P; Chen Z; Brookhart M; Meyer TJ Top. Catal 2015, 58, 30–45. [Google Scholar]
- (5).Appel AM; Bercaw JE; Bocarsly AB; Dobbek H; DuBois DL; Dupuis M; Ferry JG; Fujita E; Hille R; Kenis PJA; Kerfeld CA; Morris RH; Peden CHF; Portis AR; Ragsdale SW; Rauchfuss TB; Reek JNH; Seefeldt LC; Thauer RK; Waldrop GL Chem. Rev 2013, 113, 6621–6658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Costentin C; Robert M; Savéant J-M Chem. Soc. Rev 2013, 42, 2423–2436. [DOI] [PubMed] [Google Scholar]
- (7).Rosenthal J Prog. Inorg. Chem 2014, 59, 299–338. [Google Scholar]
- (8).Hori Y In Modern Aspects of Electrochemistry; Vayenas CG, White RE, Gamboa-Aldeco ME, Eds.; Springer: New York, 2008; Vol. 42, p 89. [Google Scholar]
- (9).Lu Q; Rosen J; Jiao F ChemCatChem 2015, 7, 38–47. [Google Scholar]
- (10).Gutsev GL; Bartlett RJ; Compton RN J. Chem. Phys 1998, 108, 6756–6762. [Google Scholar]
- (11).Compton RN; Reinhardt PW; Cooper RN J. Chem. Phys 1975, 63, 3821–3827. [Google Scholar]
- (12).Rosen BA; Salehi-Khojin A; Thorson MR; Zhu W; Whipple DT; Kenis PJA; Masel RI Science 2011, 334, 643–644. [DOI] [PubMed] [Google Scholar]
- (13).Rosen BA; Haan JL; Mukherjee P; Braunschweig B; Zhu W; Salehi-Khojin A; Dlott DD; Masel RI J. Phys. Chem. C 2012, 116, 15307–15312. [Google Scholar]
- (14).Rosen BA; Zhu W; Kaul G; Salehi-Khojin A; Masel RI J. Electrochem. Soc 2013, 160, H138–H141. [Google Scholar]
- (15).Oh Y; Hu X Chem. Commun 2015, 51, 13698–13701. [DOI] [PubMed] [Google Scholar]
- (16).DiMeglio JL; Rosenthal JJ Am. Chem. Soc 2013, 135, 8798–8801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Medina-Ramos J; DiMeglio JL; Rosenthal JJ Am. Chem. Soc 2014, 136, 8361–8367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Medina-Ramos J; Pupillo RC; Keane TP; DiMeglio JL; Rosenthal JJ Am. Chem. Soc 2015, 137, 5021–5027. [DOI] [PubMed] [Google Scholar]
- (19).Santos VO; Leite IR; Brolo AG; Rubim JC J. RamanSpectrosc 2016, 47, 674. [Google Scholar]
- (20).Magill AM; Cavell KJ; Yates BF J. Am. Chem. Soc 2004, 126, 8717–8724. [DOI] [PubMed] [Google Scholar]
- (21).Gazitúa M; Fuentealba P; Contreras R; Ormazábal-Toledo R J. Phys. Chem. B 2015, 119, 13160–13166. [DOI] [PubMed] [Google Scholar]
- (22).Matsubara Y; Grills DC; Kuwahara Y ACS Catal 2015, 5, 6440–6452. [Google Scholar]
- (23).Ishida H; Tanaka H; Tanaka K; Tanaka TJ Chem. Soc., Chem. Commun 1987, 131–132. [Google Scholar]
- (24).Rail MD; Berben LA J. Am. Chem. Soc 2011, 133, 18577–18579. [DOI] [PubMed] [Google Scholar]
- (25).Smieja JM; Benson EE; Kumar B; Grice KA; Seu CS; Miller AJM; Mayer JM; Kubiak CP Proc. Natl. Acad. Sci. U. S. A 2012, 109, 15646–15650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Costentin C; Drouet S; Robert M; Savéant J-M Science 2012, 338, 90–94. [DOI] [PubMed] [Google Scholar]
- (27).Sampson MD; Nguyen AD; Grice KA; Moore CE; Rheingold AL; Kubiak CP J. Am. Chem. Soc 2014, 136, 5460–5471. [DOI] [PubMed] [Google Scholar]
- (28).Therrien JA; Wolf MO; Patrick BO Inorg. Chem 2014, 53, 12962–12972. [DOI] [PubMed] [Google Scholar]
- (29).Greaves TL; Drummond CJ Chem. Rev 2008, 108, 206–237. [DOI] [PubMed] [Google Scholar]
- (30).Greaves TL; Drummond CJ Chem. Rev 2015, 115, 11379–11448. [DOI] [PubMed] [Google Scholar]
- (31).Kaljurand I; Kütt A; Sooväli L; Rodima T; Mäemets V; Leito I; Koppel IA J. Org. Chem 2005, 70, 1019–1028. [DOI] [PubMed] [Google Scholar]
- (32).Miran MS; Kinoshita H; Yasuda T; Susan MA; Watanabe M Phys. Chem. Chem. Phys 2012, 14, 5178–5186. [DOI] [PubMed] [Google Scholar]
- (33).Yang Z-Z; He L-N; Miao C-X; Chanfreau S Adv. Synth. Catal 2010, 352, 2233–2240. [Google Scholar]
- (34).Nowicki J; Muszyński M; Mikkola J-P RSC Adv 2016, 6, 9194–9208. [Google Scholar]
- (35).Heldebrant DJ; Yonker CR; Jessop PG; Phan L Chem. - Eur. J 2009, 15, 7619–7627. [DOI] [PubMed] [Google Scholar]
- (36).Wang C; Luo H; Jiang D.-e.; Li H; Dai S Angew. Chem., Int. Ed 2010, 49, 5978–5981. [DOI] [PubMed] [Google Scholar]
- (37).Wang C; Mahurin SM; Luo H; Baker GA; Li H; Dai S Green Chem 2010, 12, 870–874. [Google Scholar]
- (38).Wang C; Luo H; Luo X; Li H; Dai S Green Chem 2010,12, 2019–2023. [Google Scholar]
- (39).Brennecke JF; Gurkan BE J. Phys. Chem. Lett 2010, 1, 3459–3464. [Google Scholar]
- (40).Carrera GVSM; Jordão N; Branco LC; Nunes da Ponte M Faraday Discuss 2015, 183, 429–444. [DOI] [PubMed] [Google Scholar]
- (41).Yang Z-Z; Li Y-N; Wei Y-Y; He L-N Green Chem 2011, 13, 2351–2353. [Google Scholar]
- (42).Das Neves Gomes C; Jacquet O; Villiers C; Thuéry P; Ephritikhine M; Cantat T Angew. Chem., Int. Ed 2012, 51, 187–190. [DOI] [PubMed] [Google Scholar]
- (43).Yu B; Zhang H; Zhao Y; Chen S; Xu J; Hao L; Liu Z ACS Catal 2013, 3, 2076–2082. [Google Scholar]
- (44).Zhao Y; Yu B; Yang Z; Zhang H; Hao L; Gao X; Liu Z Angew. Chem., Int. Ed 2014, 53, 5922–5925. [DOI] [PubMed] [Google Scholar]
- (45).Hu J; Ma J; Zhu Q; Zhang Z; Wu C; Han B Angew. Chem., Int. Ed 2015, 54, 5399–5403. [DOI] [PubMed] [Google Scholar]
- (46).Li Y-N; He L-N; Liu A-H; Lang X-D; Yang Z-Z; Yu B; Luan C-R Green Chem 2013, 15, 2825–2829. [Google Scholar]
- (47).Yadav M; Linehan JC; Karkamkar AJ; van der Eide E; Heldebrant DJ Inorg. Chem 2014, 53, 9849–9854. [DOI] [PubMed] [Google Scholar]
- (48).Li Y-N; He L-N; Lang X-D; Liu X-F; Zhang S RSC Adv 2014, 4, 49995–50002. [Google Scholar]
- (49).Reller C; Pöge M; Lißner A; Mertens FORL Environ. Sci. Technol 2014, 48, 14799–14804. [DOI] [PubMed] [Google Scholar]
- (50).Lethesh KC; Shah SN; Abdul Mutalib MI J. Chem. Eng. Data 2014, 59, 1788–1795. [Google Scholar]
- (51).Komatsu S; Yanagihara T; Hiraga Y; Tanaka M; Kunugi A Denki Kagaku 1995, 63, 217–224. [Google Scholar]
- (52).Zhang H; Ma Y; Quan F; Huang J; Jia F; Zhang L Electrochem. Commun 2014, 46, 63–66. [Google Scholar]
- (53).Pérez ER; Santos RHA; Gambardella MTP; de Macedo LGM; Rodrigues-Filho UP; Launay J-C; Franco DW J. Org. Chem 2004, 69, 8005–8011. [DOI] [PubMed] [Google Scholar]
- (54).Heldebrant DJ; Jessop PG; Thomas CA; Eckert CA; Liotta CL J. Org. Chem 2005, 70, 5335–5338. [DOI] [PubMed] [Google Scholar]
- (55).Jessop PG; Heldebrant DJ; Li X; Eckert CA; Liotta CL Nature 2005, 436, 1102. [DOI] [PubMed] [Google Scholar]
- (56).Kanan MW; Nocera DG Science 2008, 321, 1072–1075. [DOI] [PubMed] [Google Scholar]
- (57).Zhang Z; Chi M; Veith GM; Zhang P; Lutterman DA; Rosenthal J; Overbury SH; Dai S; Zhu H ACS Catal 2016, 6, 6255–6264. [Google Scholar]
- (58).Ha S; Larsen R; Masel RI J. Power Sources 2005, 144, 28–34. [Google Scholar]
- (59).Enthaler S; von Langermann J; Schmidt T Energy Environ. Sci 2010, 3, 1207–1217. [Google Scholar]
- (60).Grasemann M; Laurenczy G Energy Environ. Sci 2012, 5, 8171–8181. [Google Scholar]
- (61).Medina-Ramos J; Lee SS; Fister TT; Hubaud AA; Sacci RL; Mullins DR; DiMeglio JL; Pupillo RC; Velardo SM; Lutterman DA; Rosenthal J; Fenter P ACS Catal 2017, 7, 7285–7295. [Google Scholar]
- (62).Lau GPS; Schreier M; Vasilyev D; Scopelliti R; Grätzel M; Dyson PJ J. Am. Chem. Soc 2016, 138, 7820–7823. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.