

Abstract
Background
Alpha‐1 antitrypsin deficiency is an inherited disorder that can cause chronic obstructive pulmonary disease (COPD). People who smoke are more seriously affected and have a greater risk of dying from the disease. Therefore, the primary treatment is to help people give up smoking. There are now also preparations available that contain alpha‐1 antitrypsin, but it is uncertain what their clinical effect is.
Objectives
To review the benefits and harms of augmentation therapy with intravenous alpha‐1 antitrypsin in patients with alpha‐1 antitrypsin deficiency and lung disease.
Search methods
We searched the Cochrane Central Register of Controlled Trials (CENTRAL), PubMed and ClinicalTrials.gov to 25 March 2016.
Selection criteria
We included randomised trials of augmentation therapy with alpha‐1 antitrypsin compared with placebo or no treatment.
Data collection and analysis
The two review authors independently selected trials, extracted outcome data and assessed the risk of bias.
Main results
We included three trials (283 participants in the analyses) that ran for two to three years. All participants were ex‐ or never‐smokers and had genetic variants that carried a high risk of developing COPD. Only one trial reported mortality data (one person of 93 died in the treatment group and three of 87 died in the placebo group). There was no information on harms in the oldest trial. Another trial reported serious adverse events in 10 participants in the treatment group and 18 participants in the placebo group. In the most recent trial, serious adverse events occurred in 28 participants in each group. None of the trials reported mean number of lung infections or hospital admissions. In the two trials that reported exacerbations, there were more exacerbations in the treatment group than in the placebo group, but the results of both trials included the possibility of no difference. Quality of life was similar in the two groups. Forced expiratory volume in one second (FEV1) deteriorated more in participants in the treatment group than in the placebo group but the confidence interval (CI) included no difference (standardised mean difference ‐0.19, 95% CI ‐0.42 to 0.05; P = 0.12). For carbon monoxide diffusion, the difference was ‐0.11 mmol/minute/kPa (95% CI ‐0.35 to 0.12; P = 0.34). Lung density measured by computer tomography (CT) scan deteriorated significantly less in the treatment group than in the placebo group (mean difference (MD) 0.86 g/L, 95% CI 0.31 to 1.42; P = 0.002). Several secondary outcomes were unreported in the largest and most recent trial whose authors had numerous financial conflicts of interest.
Authors' conclusions
This review update added one new study and 143 new participants, but the conclusions remain unchanged. Due to sparse data, we could not arrive at a conclusion about the impact of augmentation therapy on mortality, exacerbations, lung infections, hospital admission and quality of life, and there was uncertainty about possible harms. Therefore, it is our opinion that augmentation therapy with alpha‐1 antitrypsin cannot be recommended.
Plain language summary
Intravenous alpha‐1 antitrypsin augmentation therapy for treating patients with alpha‐1 antitrypsin deficiency and lung disease
Background
Alpha‐1 antitrypsin deficiency is an inherited disorder that can cause lung disease (chronic obstructive pulmonary disease or COPD, which is a chronic lung condition that prevents the air supply from getting to the lungs). It affects about 1 in 1600 to 1 in 5000 people. Patients with lung disease suffer from shortness of breath, reduced ability to exercise and wheezing. People who smoke are more seriously affected and have a greater risk of dying from the disease.
Study characteristics
We reviewed the benefits and harms of treating patients who have the form of the disease that affects the lungs with alpha‐1 antitrypsin extracted from blood donations. We found three randomised clinical trials (283 participants in the analyses) comparing treatment with alpha‐1 antitrypsin with placebo (a pretend treatment) for two to three years. All participants were ex‐smokers or had never smoked but had the genetic problem that carried a high risk of developing lung problems. The evidence is current to March 2016.
Key results
Only one trial reported deaths (one of 93 participants died taking the medicine and three of 87 died taking placebo). There was no information on harms in the oldest trial. In another trial, serious adverse events occurred in 10 participants in the medicine group and 18 participants in the placebo group. In the most recent trial, serious adverse events occurred in 28 participants in each group.
None of the trials reported on the number of lung infections or hospital admissions. There were more exacerbations (acute worsening in lung function) in the medicine group than in the placebo group, whereas quality of life was similar in the two groups.
All trials measured lung function using forced expiratory volume in one second (how much air a person can breathe out during a forced breath) and carbon monoxide diffusion (a medical test that measures how much gas travels from the lungs to the blood). Lung function was slightly worse in participants taking the medicine but the differences were not significant. Lung function deteriorated significantly less when measured by a special type of X‐ray called a computer tomography (CT) scan. Several secondary outcomes were unreported in the largest and most recent trial whose authors had numerous financial conflicts of interest.
Quality of the evidence
Due to a lack of information, we cannot be sure whether this treatment works or not. Therefore, it is our opinion that treatment with alpha‐1 antitrypsin augmentation cannot be recommended.
Background
Description of the condition
Alpha‐1 antitrypsin deficiency is an inherited disorder that can cause lung or liver disease (Genetics Home Reference 2007). The prevalence of the genotype associated with severe alpha‐1 antitrypsin deficiency is about 1 in 1600 to 1 in 5000 newborns (O'Brien 1978; Sveger 1978). Alpha‐1 antitrypsin helps to regulate protease activity. Proteases are enzymes, and enzymes need to be carefully regulated, otherwise they can attack and damage normal tissues.
Cigarette smokers often develop chronic obstructive pulmonary disease (COPD). A major constituent of the lung pathology in people with COPD is pulmonary emphysema, which is characterised by loss of lung tissue and enlarged alveolar spaces. Smokers with hereditary alpha‐1 antitrypsin deficiency have a particularly high risk of developing pulmonary emphysema, for example, almost all smokers with the Z phenotype (PI*ZZ, i.e. who are homozygotic for the deficiency), will develop emphysema in early adult life and their life expectancy is reduced (Evald 1990; Hutchison 1988).
The major cause of morbidity and death in severe alpha‐1 antitrypsin deficiency is COPD with pulmonary emphysema (Larsson 1978), and liver disease is the second most common complication (Sharp 1971). The emphysema is mainly located in the lower lobes of the lung, whereas smokers with normal phenotype have predominantly upper lobe disease.
The first symptoms of lung disease rarely develop before the age of 30 years in patients with alpha‐1 antitrypsin deficiency, and includes shortness of breath following mild activity, reduced ability to exercise and wheezing (Silverman 2009). About 10% to 15% of people with alpha‐1 antitrypsin deficiency have liver damage. In rare cases, alpha‐1 antitrypsin deficiency also causes a skin condition known as panniculitis, which is characterised by hardened skin with painful lumps or patches (Genetics Home Reference 2007).
Description of the intervention
Preparations of alpha‐1 antitrypsin are made from normal human plasma from blood donors. The drug is generally infused at a dose of 60 mg/kg intravenously every week and is available in some countries for replacement therapy in patients with symptomatic emphysema although a clinical effect has not been documented in randomised controlled trials (RCTs).
How the intervention might work
The mechanism behind the lung damage is believed to be well understood. Alpha‐1 antitrypsin inhibits protein degrading enzymes and protects the pulmonary tissue against the destructive activity of elastase (Silverman 2009; Sveger 1976). Elastase is released by neutrophils when they penetrate into the alveolar wall by chemotaxis induced by cigarette smoke. Therefore, replacement therapy with alpha‐1 antitrypsin might be beneficial.
Why it is important to do this review
It is important to know whether treatment with alpha‐1 antitrypsin is effective for lung disease and might postpone death or lung transplantation. This is an update of a review originally published in 2010 (Gøtzsche 2010).
Objectives
To review the benefits and harms of augmentation therapy with intravenous alpha‐1 antitrypsin in patients with alpha‐1 antitrypsin deficiency and lung disease.
Methods
Criteria for considering studies for this review
Types of studies
Randomised clinical trials (RCTs) in any language, published or unpublished.
Types of participants
Patients with alpha‐1 antitrypsin deficiency, with or without a formal diagnosis of COPD. We did not include trials in newborns, as there is a separate Cochrane review on this (Shah 2001).
Types of interventions
Experimental intervention: augmentation therapy with alpha‐1 antitrypsin.
Control intervention: placebo or no intervention.
Types of outcome measures
Primary outcomes
Mortality.
Harms of the intervention.
Secondary outcomes
Number of exacerbations as defined in the trial report.
Number of lung infections.
Number of hospital admissions.
Quality of life.
Carbon monoxide diffusion.
Forced expiratory volume in one second (FEV1).
Lung density measured by computed tomography (CT) scan.
If studies collected data at several time points, we used the data at end of treatment.
Search methods for identification of studies
Electronic searches
We searched the Cochrane Central Register of Controlled Trials (CENTRAL) (Appendix 1), PubMed (limited to randomised trials, the PubMed clinical queries function) (Appendix 2), and ClinicalTrials.gov (Appendix 3), with no restrictions for publication year. Date of last searches was 25 March 2016.
Searching other resources
We accepted letters, abstracts and unpublished trials in an attempt to reduce the impact of selective reporting of trials and outcomes.
Data collection and analysis
For each step below, we resolved disagreements by discussion.
Selection of studies
Two review authors (PG, HKJ) independently selected the trials to be included in the review.
Data extraction and management
Two review authors (PG, HKJ) independently extracted outcome data; one review author (PG) extracted descriptive data, and one review author (HKJ) checked them.
Assessment of risk of bias in included studies
Two review authors (PG, HKJ) independently assessed the risk of bias. In particular, we recorded generation of the randomisation sequence, concealment of treatment allocation, any blinding and exclusions of participants from the analysis.
Measures of treatment effect
We sought data on all randomised participants, that is including participants that the investigators might have excluded because of poor compliance, ineligibility or loss to follow‐up (intention‐to‐treat analysis).
For dichotomous data, we used the risk ratio (RR). For continuous outcomes, we preferred end of treatment values when available rather than change from baseline values, as baseline recordings are not always available in clinical trials, and as investigators are inclined to show baseline differences and adjust for them when this procedure favours the experimental treatment (Gøtzsche 2006a). For continuous data and for mean numbers of exacerbations, infections and hospital admissions per participant, we used the mean difference (MD) or standardised mean difference (SMD), as appropriate, but abstained from doing a meta‐analysis if the distribution of the data was non‐Gaussian. For time‐to‐event data, we preferred to use the hazard ratio (HR), but accepted the RR if that was the only statistic available. We present data with 95% confidence intervals (CI).
Unit of analysis issues
There were no such issues, as the unit of analysis was the participant in all trials.
Dealing with missing data
When trial reports provided insufficient information of potential significance for the results, we contacted the corresponding study author.
Assessment of heterogeneity
We assessed heterogeneity statistically and also used the I2 statistic (0% to 100%) as a guide to its magnitude (Higgins 2003).
Assessment of reporting biases
We attempted to assess selective reporting of outcomes within trials, and publication bias related to non‐publication of whole trials. If there are enough trials in future updates of this review (more than 10), we will look for funnel plot asymmetry.
Data synthesis
We used a fixed‐effect model for meta‐analysis (which we believe should more appropriately be called a weighted mean, as there is no such thing as a 'fixed‐effect') unless there was heterogeneity (P < 0.10) or other good reasons for using a random‐effects model (e.g. if the interventions were of a very different nature).
Subgroup analysis and investigation of heterogeneity
We explored the reasons for any heterogeneity (e.g. by comparing the characteristics of participants, interventions and outcomes in the included trials). We planned no subgroup analyses.
Sensitivity analysis
If possible, we plan in future to perform a sensitivity analysis where only trials with low risk of bias for allocation concealment and blinding are included (Wood 2008).
Results
Description of studies
Results of the search
We retrieved 46 records in CENTRAL and 27 records on PubMed. We identified three placebo‐controlled RCTs that were eligible for the review and found a fourth ongoing trial on ClinicalTrials.gov that planned to include 339 participants (NCT01983241).
Included studies
This review update added one new study and 143 new participants. All three trials included in this review had recruited patients with genetic variants that carried a high risk of developing COPD (Silverman 2009). One study was publicly funded (Dirksen 1999); the manufacturers of alpha‐1 antitrypsin funded the two other studies. Talecris Biotherapeutics, Inc. financed the EXACTLE trial (EXAcerbations and Computed Tomography scan as Lung End‐points), registered in ClinicalTrials.gov as NCT00263887, which involved co‐authors from the company (Dirksen 2009). CSL Behring financed the RAPID trial (Randomized, placebo‐controlled trial in Alpha‐1 Proteinase Inhibitor Deficiency), registered in ClinicalTrials.gov as NCT00261833, which involved co‐authors from the company (Chapman 2015).
The trials enrolled 330 ex‐ or never‐smokers who had an FEV1 of 30% to 80% of the predicted normal value. The participants were treated for at least three years with four‐weekly infusions of alpha‐1 antitrypsin 250 mg/kg (brand name not stated) or albumin 625 mg/kg as placebo (Dirksen 1999); for two years (with an optional six‐month extension) with weekly infusions of alpha‐1 antitrypsin 60 mg/kg (Prolastin) or 2% albumin as placebo (Dirksen 2009); or for two years (with an optional open‐label treatment with drug for a further two years in some of the included countries) with weekly infusions of alpha‐1 antitrypsin 60 mg/kg (Zemaira) or "a lyophilised placebo" (Chapman 2015).
The primary effect measure was FEV1 (Dirksen 1999) or lung density measured by CT scans (Chapman 2015). The third trial described FEV1 as the 'gold standard' and lung density as an exploratory outcome (Dirksen 2009), but in a meta‐analysis of Dirksen's two studies (Stockley 2010), lung density had changed status, as it was now described as a primary outcome in the second trial.
Excluded studies
We excluded no RCTs of alpha‐1 antitrypsin in patients with alpha‐1 antitrypsin deficiency and lung disease that had an untreated control group (see Figure 1).
1.
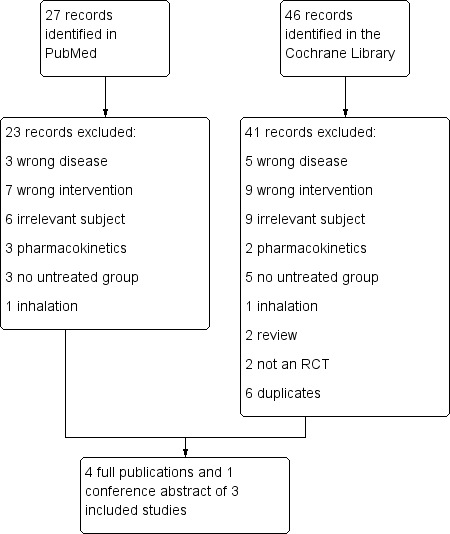
Study flow diagram.
Risk of bias in included studies
See details in the Characteristics of included studies table. Figure 2 shows a summary of all trials. There were some serious limitations in the trials (e.g. some outcome data were unreported).
2.

Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
Allocation
The randomisation method in one trial was minimisation (Dirksen 1999). The procedure was not described, and it was not possible to judge whether it had led to comparable groups, as participant characteristics at baseline were shown for the two included countries and not for the two randomised groups. Another table showed that the groups were comparable at baseline for lung function measurements and CT scan values (Dirksen 1999). Another trial had adequate sequence generation whereas it was not clear whether there was allocation concealment (Dirksen 2009). There were more males in the treatment group than in the placebo group, but this could be a chance finding, as the two groups were comparable for other baseline characteristics (Dirksen 2009). The remaining trial randomised participants in blocks of four and did not describe the blinding procedures clearly; thus, it is uncertain whether allocation concealment was adequate (Chapman 2015).
Blinding
The trials were described as double‐blind and placebo controlled, but one trial did not describe the blinding procedure and it was not clear whether the attempted blinding was effective (Dirksen 1999). It was unclear whether another trial was adequately blinded (Chapman 2015), whereas the remaining trial was effectively blinded (Dirksen 2009).
Incomplete outcome data
In one trial, there were no outcome data for two of the 58 participants who dropped out because they resumed smoking, and it was not described to which groups they were randomised (Dirksen 1999). Another trial described 82 enrolled participants but only 77 were randomised (Dirksen 2009). Three of the 77 participants withdrew from the treatment group and seven from the placebo group; data from the CT scans were included from 71 participants, but change from baseline was only available for 67 participants after two years, and for 34 participants after 2.5 years. Therefore, we used CT scan data after two years. In the remaining trial, it was not clear how many participants contributed to the various analyses (Chapman 2015). One outcome table simply gave the number of randomised participants but a flow chart suggested that data were missing for 10 participants in the treatment group and 20 participants in the placebo group.
Selective reporting
We found no signs of selective reporting for the oldest trial (Dirksen 1999), apart from the fact that the table of baseline values did not give data for the two randomised groups, but from the two countries that were included in the trial.
The trial registration for another trial noted that mortality would be recorded, but this was not reported (Dirksen 2009). Furthermore, the trial report only addressed CT scan measurements, exacerbations and quality of life (Dirksen 2009). For lung function measurements, the report stated that "Values for FEV1, DLCO [carbon monoxide diffusing capacity] and KCO [carbon monoxide transfer coefficient] decreased slightly in both treatment groups during the study but, since these measures were less sensitive than CT, no significant differences were found between the groups (see online supplement for details)". Although this was not selective reporting, we find it curious not to give data on the FEV1 finding in the main report because FEV1 was the accepted method (described as the 'gold standard' in the report) (Dirksen 2009), whereas the CT scan measurements were described as 'exploratory', both when the trial was registered and in the trial report.
The largest and most recent trial did not report several secondary outcomes (see Characteristics of included studies table) (Chapman 2015). We wrote to the corresponding author and an author from the company (Zhenling) and asked for these data. We also requested the trial protocol and the clinical study report. We received no reply from these two people but did make contact with the company through its website. The company spokesperson informed us that we should contact the authors. However, we did not get access to the protocol or replies to any of our questions.
Other potential sources of bias
There was no information about possible conflicts of interest in the original report of the oldest trial (Dirksen 1999). In the final version of another trial, the published paper contained a link to "Statement of Interest" (Dirksen 2009). However, due to a misprint in the journal, the published link did not work; the statement of interest was available on the European Respiratory Society website (erj.ersjournals.com/misc/statements33.dtl#D). According to other publications, it seems that the first author of both trial reports, Dirksen, may have financial conflicts of interest in relation to companies that produce, sell or research alpha‐1 antitrypsin (Alpha‐1 Foundation 2008; CLS Behring 2008; Dirksen 2009; Seersholm 2007; Stockley 2010). Dirksen was listed as a co‐author on the protocol for our Cochrane review but stepped down from that role when he saw the draft for the full review.
The acknowledgments in Dirksen's 2009 trial report mentioned that "Editorial assistance was provided by M. Kenig at PAREXEL and was supported by Talecris Biotherapeutics, Inc.". We have reported previously that such descriptions may conceal that the data analysis and the writing of the manuscript was performed by a commercial company, and that, as a result of this, the investigators may not have had much influence on the manuscript (Gøtzsche 2006b). Another indication of possible commercial influence is the fact that the trade name was preferred over the generic name in the trial report.
The most recent trial was also industry supported, and the academic authors had numerous financial conflicts of interest. The sponsor collected the data and three employees of the company participated in data analysis and writing of the report (Chapman 2015). For this trial, we found remarkably little variation in measures of uncertainty. For example, the width of the CI for annual number of exacerbations was exactly the same for the two groups, 0.38, and the standard deviation for change in patient reported symptoms (St. George's Respiratory Questionnaire (SGRQ) scores) was also exactly the same 16.5 (calculated by us based on the reported standard errors of the mean). This does not necessarily mean that the statistical analyses were biased but it does raise questions about the statistical procedures used or the reporting of the trial.
Effects of interventions
As end‐of‐treatment data were generally not available, we preferably used changes from baseline. We did not detect heterogeneity in any of the analyses (I2 = 0).
Primary outcomes
Mortality
One trial reported mortality data; one patient of 93 in the treatment group and three patients of 87 in the placebo group died (Chapman 2015). One trial recorded, but did not report, mortality (Dirksen 2009).
Harms of the intervention
The oldest trial did not report harms (Dirksen 1999). In another trial, reporting of serious adverse events was inconsistent (Dirksen 2009). This trial reported one or more serious adverse events to have occurred in 10 participants in the treatment group and in 18 participants in the placebo group according to the published trial report, but according to the trial register, these numbers were nine in the treatment group versus 15 in the placebo group (Dirksen 2009). In the remaining trial, 28 participants in each group experienced serious adverse events (Chapman 2015). As most of these events were unlikely to have any relation to the treatment, we did not find it worthwhile to meta‐analyse these data. For example, there were cases of malaria, psoriasis and fracture among the nine serious adverse events recorded in one trial in the treatment group (Dirksen 2009).
Secondary outcomes
Number of exacerbations
The annual exacerbation rate could not be meta‐analysed, as the distribution of the values was highly skewed. In one trial, for example, the mean annual exacerbation rates were 2.55 (SD 2.14) in the treatment group and 2.19 (SD 1.33) in the placebo group (Dirksen 2009). In both trials that reported number of exacerbations, there were more exacerbations in the treatment group than in the placebo group, but the results of both trials included the possibility of no difference between groups. In the most recent trial, the numbers were 1.70 (95% CI 1.51 to 1.89) in the treatment group and 1.42 (1.23 to 1.61) in the placebo group; the RR from a negative binomial regression model in which country and treatment were fixed effects and where adjustment was made for treatment duration was 1.26 (95% CI 0.92 to 1.74) (Chapman 2015).
Number of lung infections
We found no data on number of lung infections.
Number of hospital admissions
We found no data on number of hospital admissions.
Quality of life
Two trials reported quality of life using the SGRQ. One trial reported that SGRQ deteriorated by 1.5 in the treatment group and 2.4 in the placebo group (P = 0.70), which are very small, and clinically irrelevant changes from a mean score at baseline of 44 on a scale that goes up to 100 (Dirksen 2009). We abstained from meta‐analysing these data, as they were contradictory in one trial (Chapman 2015). Data for participant‐reported outcomes were listed on ClinicalTrials.gov as changes from baseline, which were ‐1.19 in the treatment group and ‐0.09 in the placebo group (Chapman 2015), but in the published trial report in The Lancet, these changes were listed as ‐1.4 in the treatment group and 2.0 in the placebo group.
Forced expiratory volume in one second
On average, FEV1 deteriorated more in the treatment group than in the placebo group, but there was no significant between‐group difference (SMD ‐0.19, 95% CI ‐0.42 to 0.05; P = 0.12) (Analysis 1.1).
1.1. Analysis.
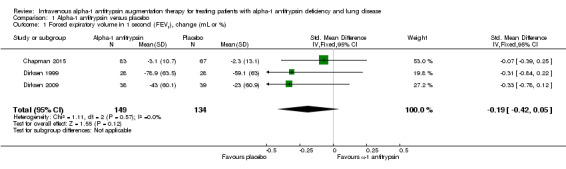
Comparison 1 Alpha‐1 antitrypsin versus placebo, Outcome 1 Forced expiratory volume in 1 second (FEV1), change (mL or %).
Carbon monoxide diffusion
On average, carbon monoxide diffusion deteriorated more in the treatment group than in the placebo group, but there was no significant between‐group difference (SMD ‐0.11, 95% CI ‐0.35 to 0.12; P = 0.34) (Analysis 1.2).
1.2. Analysis.
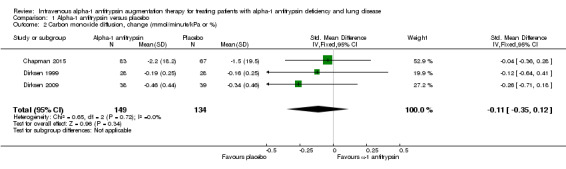
Comparison 1 Alpha‐1 antitrypsin versus placebo, Outcome 2 Carbon monoxide diffusion, change (mmol/minute/kPa or %).
Lung density measured by computer tomography scan
One of the trials measured lung density in four different ways in an exploratory manner (Dirksen 2009). Therefore, we used the mean of the four estimates, but it would make virtually no difference if we had chosen any of them, as the results were very similar. When we combined data from all three studies, lung density deteriorated significantly less in the treatment group than in the placebo group, (MD 0.86 g/L, 95% CI 0.31 to 1.42; P = 0.002).
Discussion
Summary of main results
The three trials were small and there were no differences in clinically relevant outcomes. For the surrogates, there was some evidence of a beneficial effect of alpha‐1 antitrypsin. The lung function decline measured as FEV1 and carbon monoxide diffusion was not slowed down with the drug, but the CT scans of lung density found that treatment might decrease the loss of lung tissue, and this difference was statistically significant. The newest trial was adequately powered for this outcome and also found an effect, but the CI in the meta‐analysis was wide, and in all three trials, the CT scans showed considerable lung density loss, consistent with emphysema progression, in both the treatment and placebo groups. This does not suggest that alpha‐1 antitrypsin augmentation has a major effect on the progression of the disease.
In the oldest trial, the drug was not given weekly but every four weeks. This could potentially have affected the results but we saw no such dosing effect in the analyses, and protective levels seem to have been obtained for at least three out of the four weeks (Stockley 2010).
Overall completeness and applicability of evidence
The trials did not elucidate the harms well. In clinical use, serious reactions occurred in 1% of the patients in the form of dyspnoea, deterioration of serious heart failure and serious allergic reactions (Chen 2007). One report on 747 patients mentioned 720 reactions in 174 patients, 72% of which were moderate and 9% were serious (Heresi 2008).
Quality of the evidence
The studies we reviewed were not powered to assess mortality but the crucial question for this very expensive treatment, which can amount up to EUR70,000 annually for each patient (Chen 2007), or far more, $150,000, in USA (Silverman 2009), is whether it decreases mortality. As only four deaths were reported in the trials, we don't know whether this is the case, and more generally, we had concerns about risk of bias how the included studies were conducted (see Figure 2).
According to the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011), primary outcomes should be essential for decision‐making and should usually have an emphasis on patient‐important outcomes. This is why we decided that FEV1 and lung density measured by CT scan should be secondary outcomes in the protocol that we published for our review, in much the same way as one would consider temperature and thorax X‐ray secondary outcomes in a review of an antibiotic for pneumonia. There have been many attempts in healthcare at validating surrogate outcomes and at substituting them for clinically relevant outcomes in trials, but these approaches are unlikely to be successful (Gøtzsche 1996), and diabetes is a good example. Both tolbutamide and rosiglitazone lower blood glucose but they also increase cardiovascular mortality (Chalmers 1990; Nissen 2007).
Potential biases in the review process
We are not aware of any potential biases in the review process.
Agreements and disagreements with other studies or reviews
One Canadian health technology assessment report concluded that there was no evidence showing health improvement in patients receiving augmentation therapy with alpha‐1 antitrypsin (Chen 2007). This report reviewed only results from the oldest trial (Dirksen 1999). One 2009 meta‐analysis found a positive effect of alpha‐1 antitrypsin on FEV1 but it was unreliable, as it included historically controlled before‐and‐after studies (Chapman 2009). An individual patient data meta‐analysis of the first two trials was published in 2010 (Stockley 2010). Talecris Biotherapeutics, Inc. sponsored the meta‐analysis and "technical editorial assistance was provided". It reported on surrogate outcomes only, and we preferred to use the original published trial data for our meta‐analyses.
The authors of a 2009 review had substantial conflicts of interest related to companies selling alpha‐1 antitrypsin (Silverman 2009). They advised that augmentation therapy should be considered in patients with alpha‐1 antitrypsin deficiency "although compelling evidence of benefit is lacking from randomized trials". They furthermore noted that the guidelines of the American Thoracic Society and the European Respiratory Society recommend augmentation therapy for patients with airflow obstruction related to alpha‐1 antitrypsin deficiency. In 2012, a clinical practice guideline appeared from the Canadian Thoracic Society (Marciniuk 2012). The authors of this guideline had numerous financial conflicts of interest in relation to manufacturers of the drug and their literature review for the guideline did not adhere to common standards for systematic reviews (e.g. there were no search strategies). The authors also included observational studies. Their recommendation was, "We suggest A1AT [alpha‐1 antitrypsin] augmentation therapy may be considered in nonsmoking or exsmoking patients with COPD (FEV1 25% to 80% predicted) attributable to emphysema and documented A1AT (level ≤11 μmol/L), who are receiving optimal pharmacological and nonpharmacological therapies (including comprehensive case management and pulmonary rehabilitation) because of benefits in CT scan lung density (Grade of recommendation: 2B) and mortality (Grade of recommendation: 2C)".
In our opinion, these recommendations are not supported by the evidence presented in this review. The drug has not shown any clinical benefit, it has important adverse effects and it is extremely costly.
Authors' conclusions
Implications for practice.
This review update added one new study and 143 new participants, but the conclusions remained unchanged. Due to sparse data, we could not arrive at a conclusion about the impact of augmentation therapy on mortality, exacerbations, lung infections, hospital admission and quality of life, and there was uncertainty about possible harms. Therefore, it is our opinion that augmentation therapy with alpha‐1 antitrypsin cannot be recommended.
Implications for research.
Further studies with surrogate markers cannot be recommended, if the aim is to elucidate whether or not augmentation therapy with alpha‐1 antitrypsin has a relevant clinical effect. Studies should be large enough to detect a possible effect on clinically relevant outcomes including mortality.
Feedback
Comment on use of Cochrane methodology for an uncommon disease, 27 October 2010
Summary
The review was recently discussed by the Medical and Scientific Advisory Committee (MASAC) of the US‐based Alpha‐1 Foundation. There was concern as to whether the methodology was appropriate for an uncommon disease. Prof. Jamie Stoller detailed these issues.
Principle issues
The recent Cochrane review regarding augmentation therapy for alpha‐1 antitrypsin (AAT) leverages the significant reputation of Cochrane reports as a methodological standard.
The possibility of shortcomings of the methodology, perhaps especially as it applies to rare diseases where the preponderance of concordant data come from observational cohort studies rather than from randomised controlled trials (RCTs), frames an important issue for the Cochrane methods.
Several questions about the recent Cochrane review regarding augmentation therapy frame the specific concerns of applying the Cochrane methodology to a rare disease, where the number of available RCTs may be especially small and the number of participants in these RCTs is small, especially compared with the much larger number of participants in available observational cohort studies.
Possible issues
Can the data from the two RCTs be pooled in view of the fact that neither the drugs used for the two studies nor the dosing regimens compared (monthly vs. weekly infusion, for which there are known pharmacokinetic and pharmacodynamic differences [Hubbard, Barker, Piitulainen]) were the same?
Can the data be pooled when one of the studies (EXACTLE [EXAcerbations and Computed Tomography scan as Lung End‐points]) was hypothesis‐generating regarding which CT [computer tomography] densitometric algorithm was best to detect differences (and only one of the four proposed methods achieved statistical significance)?
Is there concern about pooling the results of two RCTs when the patient populations in the two studies are not independent (i.e., some patient participated in both studies)?
Is there concern that the recommendation in the report about non‐efficacy is countermanded by the included forest plot, which, in fact, showed a significant effect of augmentation therapy on loss of lung density (even though this forest plot may violate poolability criteria?)
Is there concern about pooling the results of the two studies when there is a concern about differential drop‐out rates from the compared groups (i.e., larger drop‐out rates among placebo recipients)?
Is there concern that the Cochrane methodology of pooling only data from RCTs may pose problems under conditions (like rare diseases such as alpha‐1 antitrypsin deficiency), where the preponderance of data come from several concordant observational studies and the number of RCTs and the number of participants in these studies is a small fraction of participants in the observational data sets (Krumholz et al. NEJM)?
Conflict of interest statements
Prof. David Lomas, Cambridge Institute for Medical Research (CIMR); Deputy Director, University Chair, University of Cambridge, UK
I have worked on the basic mechanisms of antitrypsin deficiency for almost 20 years. I have served on the grants committee of the Alpha‐1 Foundation (US) since it was formed and took over as Chair last year. The Chair also sits on the Medical and Scientific Advisory Committee. I receive no personal remuneration for my work on either committee but have been a PI [principle investigator] and co‐PI on grants from the Foundation. In addition I serve on the European eALTA grants committee that awards two post‐doctoral fellowships per year funded by Talecris. I receive an honorarium for my work on this committee and two of my post‐doctoral fellows have won the award. I have in the past spoken on the basic mechanisms of antitrypsin deficiency at symposia arranged by Bayer and Talecris for which I have received honoraria. Finally I do not know either of the authors nor am I familiar with their work.
Prof. Jamie Stoller, M.D., M.S. Jean Wall Bennett Professor of Medicine; Chair, Education Institute, Cleveland Clinic, USA
Advisory Committee/Consultant for: Talecris Biotherapeutics, Inc.; Boehringer‐Ingelheim; Kamada; AsthmaTx; COPD Foundation; Alpha‐1 Foundation.
Lectures supported: Talecris; Baxter; CSL‐Behring; Grifols.
Reply
Some of the questions David Lomas raised have already been addressed in our review.
Scientific principles are universal. They do not change according to the condition being studied, or depend on whether few or many patients are suffering from a given disease. The shortcomings of using observational studies when deciding whether a treatment has a beneficial effect are therefore also universal. We concluded that further studies with surrogate markers cannot be recommended, if the aim is to elucidate whether or not augmentation therapy with alpha‐1 antitrypsin has a relevant clinical effect. Studies should be large enough to detect a possible effect on mortality, and alpha‐1 antitrypsin deficiency isn't so rare that this cannot be done.
The other point raised is whether it is appropriate to pool the two randomised trials that we identified. Yes, it is appropriate and the effects in the two trials were very similar for the three outcomes we meta‐analysed. Furthermore, it would not have made any changes to our conclusions, if we had not pooled the two trials.
David Lomas says that some patients participated in both trials, but he does not document that this is the case. We have not found any information in support of this postulate. Furthermore, even if some patients did participate in both studies, it would not change our findings or conclusions.
Finally, David Lomas notes that CT density was measured in four different ways in one of the trials, and that only one of the results "achieved statistical significance". This observation is irrelevant. We furthermore explain in our review that since lung density was measured in four different ways in an exploratory fashion, we used the average of the four estimates. It would make virtually no difference, if we had chosen any one of them, as the results were very similar.
Contributors
Comment made (on behalf of the Medical and Scientific Advisory Committee (MASAC) of the US‐based Alpha‐1 Foundation) by Prof. David Lomas and Prof. Jamie Stoller.
Response from Prof. Peter C Gøtzsche and Dr Helle Krogh Johansen.
Feedback Editor: Prof Felix Ratjen.
Managing Editor: Miss Tracey Remmington.
What's new
| Date | Event | Description |
|---|---|---|
| 25 March 2016 | New citation required but conclusions have not changed | New trial added. |
| 25 March 2016 | New search has been performed | New literature search run |
History
Protocol first published: Issue 2, 2009 Review first published: Issue 7, 2010
| Date | Event | Description |
|---|---|---|
| 15 February 2011 | Feedback has been incorporated | Amendment made to previous feedback [Entry error by Managing Editor]. Feedback and response on the subject of 'Comment on use of Cochrane methodology for an uncommon disease' has now been incorporated. |
Acknowledgements
We thank Prof. Asger Dirksen for comments on our protocol for this review.
Rebecca Normansell was the Editor for this review and commented critically on the review.
This project was supported by the UK National Institute for Health Research (NIHR), via Cochrane Infrastructure funding to the Cochrane Airways Group. The evidence‐based views and opinions expressed therein are those of the authors and do not necessarily reflect those of the Systematic Reviews Programme, NIHR, National Health Service (NHS) or the Department of Health.
Appendices
Appendix 1. CENTRAL search strategy (all years)
| Search terms |
| #1 MeSH descriptor: [alpha 1‐Antitrypsin] explode all trees #2 antitrypsin OR "proteinase inhibitor" OR Prolastin OR Aralast OR Zemaira OR Trypsone #3 #1 or #2 #4 lung or pulmonary #5 deficiency #6 random* #7 #4 and #5 and #6 #8 #3 and #7 |
Appendix 2. PubMed search strategy (all years)
| Search terms |
| #1 antitrypsin OR "proteinase inhibitor" OR Prolastin OR Aralast OR Zemaira OR Trypsone #2 lung or pulmonary #3 deficiency #4 #1 and #2 and #3 limited to randomised trials (the PubMed clinical queries function) |
Appendix 3. Clinicaltrials.gov search strategy (all years)
| Search terms |
| antitrypsin AND placebo |
Data and analyses
Comparison 1. Alpha‐1 antitrypsin versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Forced expiratory volume in 1 second (FEV1), change (mL or %) | 3 | 283 | Std. Mean Difference (IV, Fixed, 95% CI) | ‐0.19 [‐0.42, 0.05] |
| 2 Carbon monoxide diffusion, change (mmol/minute/kPa or %) | 3 | 283 | Std. Mean Difference (IV, Fixed, 95% CI) | ‐0.11 [‐0.35, 0.12] |
| 3 Computer tomography (CT) lung density, change (g/L) | 3 | 273 | Mean Difference (IV, Fixed, 95% CI) | 0.86 [0.31, 1.42] |
1.3. Analysis.
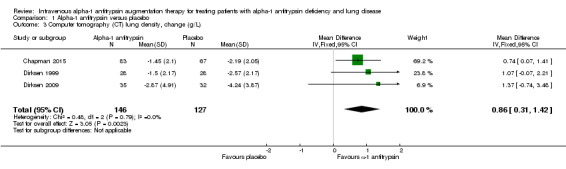
Comparison 1 Alpha‐1 antitrypsin versus placebo, Outcome 3 Computer tomography (CT) lung density, change (g/L).
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Chapman 2015.
| Methods | Double‐blind, placebo‐controlled | |
| Participants | 180 ex‐smokers from Australia, Canada, Czech Republic, Denmark, Estonia, Finland, Germany, Ireland, Poland, Romania, Russia, Sweden, The Netherlands and the US with alpha‐1 antitrypsin deficiency of the ZZ phenotype (168 participants) and moderate emphysema (FEV1 between 35% and 70% of the predicted normal value) | |
| Interventions | Treated for 2 years Treatment: weekly infusions of alpha‐1 antitrypsin 60 mg/kg Placebo: weekly infusions of a lyophilised preparation |
|
| Outcomes | Primary: lung density measured by CT scan Secondary: number of exacerbations, exacerbation duration and severity, FEV1, diffusion capacity, baseline and achieved A1PI concentrations (functional and antigenic assays), incremental shuttle walk test results, health status established with the SGRQ |
|
| Notes | Trial sponsored by the manufacturer, CSL Behring. There were no data reported in the published paper for several of the secondary outcomes | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computerised pseudo random number generator |
| Allocation concealment (selection bias) | Unclear risk | Masked study treatments were supplied to each site in blocks of 4 containing sequential participant numbers. It is not clear whether this procedure could prevent selection bias: "To achieve treatment concealment, A1PI and placebo were packaged identically as lyophilised preparations and individual packages were identified only by patient number. Study drug material was suspended in sterile water for injection and placed in an intravenous bag that was covered with an opaque sleeve by a designated study nurse or pharmacist who did not interact with the patients. Clinical trial associates monitored compliance with the masking procedure throughout the trial". There was no information about the actual compliance with the procedures |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | See 'Allocation concealment (selection bias)' above |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Data on surrogate outcomes were available for 187 of the 190 randomised participants. Not clear whether data were missing for the other outcomes |
| Selective reporting (reporting bias) | High risk | Data on several secondary outcomes were entirely missing, and there were no explanations for this |
| Other bias | High risk | The trial was industry supported, and the academic authors had numerous financial conflicts of interest. The sponsor collected the data and 3 employees of the company participated in data analysis and writing of the report |
Dirksen 1999.
| Methods | Double‐blind, placebo‐controlled | |
| Participants | 58 ex‐smokers from Denmark and The Netherlands with alpha‐1 antitrypsin deficiency of PI*ZZ phenotype and moderate emphysema (FEV1 between 30% and 80% of predicted) | |
| Interventions | Treated for at least 3 years Treatment: 4‐weekly infusions of alpha‐1 antitrypsin 250 mg/kg Placebo: 4‐weekly infusions of albumin 625 mg/kg |
|
| Outcomes | Primary: FEV1 Secondary: carbon monoxide diffusion, participant‐administered serial spirometry (PASS) at home, FVC, VC, lung density with CT scan |
|
| Notes | Trial supported by The Danish State Serum Institute, Laboratoire Français du Fractionnement et des Biotechnologies, The National Danish Research Council for Public Health, The Danish Lung Foundation and The Netherlands Asthma Foundation | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "Patients were stratified by age, level of FEV1, and nationality and randomized by the minimization method". Randomisation procedure not described |
| Allocation concealment (selection bias) | Unclear risk | No details given |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Described as double‐blind and placebo controlled. No information on method |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Data and group assignment not available for 2 participants who dropped out |
| Selective reporting (reporting bias) | Low risk | The table of baseline values did not give data for the 2 randomised groups, but from the 2 countries that were included in the trial |
| Other bias | High risk | No information about possible conflicts of interest, but according to other publications, the first author had financial conflicts of interest |
Dirksen 2009.
| Methods | Double‐blind, placebo‐controlled | |
| Participants | 82 ex‐ or never‐smokers from Copenhagen (Denmark), Malmö (Sweden) and Birmingham (UK) with severe alpha‐1 antitrypsin deficiency (serum concentration < 11 μM) | |
| Interventions | Treated for 2 years (with an optional 6 months' extension) Treatment: weekly infusions of alpha‐1 antitrypsin 60 mg/kg Placebo: weekly infusions of 2% albumin |
|
| Outcomes | Primary: lung density measured by CT scan Secondary: FEV1, carbon monoxide diffusion, frequency of exacerbations, health status (SGRQ) |
|
| Notes | Trial sponsored by the manufacturer, Talecris Biotherapeutics, Inc. 1 of the authors had received a research grant from the Alpha‐1 Foundation | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Participants were randomised in blocks of 4 for each city; block size was not disclosed to the study sites. Computer‐generated random code was used to produce randomisation envelopes |
| Allocation concealment (selection bias) | Unclear risk | Randomisation envelopes were issued to the unblinded pharmacist or designee at each study centre and were kept confidential. Randomisation envelopes were sent to the pharmacist with the study medication. Clinical site pharmacy personnel who prepared the study medication were not blinded. Unclear whether the envelopes were opaque and sealed |
| Blinding (performance bias and detection bias) All outcomes | Low risk | All participants received same total volume per kilogram bodyweight of study medication with no visible difference in external aspect between drugs, as variation in colour by lot was masked by using opaque sleeves. Throughout the course of the trial, individual treatment assignments were unknown to the clinicians; the monitors; the CT scan facility; and the sponsor's data management, clinical and biostatistical teams |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | 82 participants enrolled, 77 randomised, 10 of whom later withdrew; change from baseline for the CT scans only available for 67 participants after 2 years |
| Selective reporting (reporting bias) | High risk | Mortality was recorded but not reported. Values for FEV1, DLCO and KCO were not available in the trial report, only on the journal's website |
| Other bias | High risk | Paper stated, "This study was sponsored by Talecris Biotherapeutics, Inc (Research Triangle Park, NC 27709, USA) and was conducted between November 2003 and January 2007. Two of the authors of the manuscript (MW and CD) are employees of Talecris and participated in the design of the study, in the collection, analysis and interpretation of data (CD was the statistician for the study), in the writing of the manuscript and in the decision to submit the manuscript for publication. The article‐processing charge would be sponsored by Talecris Biotherapeutics, Inc. Editorial assistance was provided by M. Kenig at PAREXEL and was supported by Talecris Biotherapeutics, Inc." |
A1PI: alpha‐1 proteinase inhibitor; CT: computed tomography; DLCO: carbon monoxide diffusing capacity; KCO: carbon monoxide transfer coefficient; FEV1: forced expiratory volume at one second; FVC: forced vital capacity; SGRQ: St. George's Respiratory Questionnaire; VC: vital capacity.
Characteristics of ongoing studies [ordered by study ID]
NCT01983241.
| Trial name or title | SPARTA |
| Methods | Placebo‐controlled |
| Participants | 339 participants |
| Interventions | Alpha‐1‐proteinase inhibitor, 60 or 120 mg/kg per week, or placebo |
| Outcomes | Primary: computer tomography lung scans |
| Starting date | 2013, projected finish date 2021 |
| Contact information | |
| Notes |
Differences between protocol and review
We had planned to include head‐to‐head trials where both groups had received alpha‐1 antitrypsin (e.g. in different doses or regimens); however, we abstained from doing this, as such trials have little relevance while it has not been shown that augmentation therapy with alpha‐1 antitrypsin has any clinical value compared with placebo or no treatment.
Prof. Dirksen was listed as a co‐author on the protocol but stepped down from that role when he saw the draft for the full review.
Forced expiratory volume in one second (FEV1) was a secondary outcome in our protocol. However, according to a request from the Editor in the Cochrane Cystic Fibrosis & Genetic Disorders Review Group, where our review was first published in 2010, FEV1 changed status from being a secondary outcome to being a primary outcome. We disagreed with the request that was enforced upon us and have, therefore, now again made FEV1 a secondary outcome, in accordance with our published protocol and its status as a surrogate outcome.
In the first version of this review, we also searched in the Cochrane Cystic Fibrosis & Genetic Disorders Review Group's Inborn Errors of Metabolism Trials Register. Our review is now published in the Cochrane Airways Group and we did not find it necessary to search this register.
Contributions of authors
PCG wrote the first draft of the protocol and of the review and did the statistical analyses.
HKJ participated in data extraction and provided comments.
Sources of support
Internal sources
The Nordic Cochrane Centre, Denmark.
External sources
No sources of support supplied
Declarations of interest
We have no conflicts of interest.
New search for studies and content updated (no change to conclusions)
References
References to studies included in this review
Chapman 2015 {published data only}
- Chapman KR, Burdon JG, Piitulainen E, Sandhaus RA, Seersholm N, Stocks JM, et al. Intravenous augmentation treatment and lung density in severe α1 antitrypsin deficiency (RAPID): a randomised, double‐blind, placebo‐controlled trial. Lancet 2015;386:360‐8. [DOI] [PubMed] [Google Scholar]
- Chapman KR, Burdon JGW, Piitulainen E, Sandhaus RA, Seersholm N, Stocks JM. Alpha‐1 antitrypsin (A1AT) preserves lung density In homozygous Alpha‐1 antitrypsin deficiency (A1ATD): a randomized, placebo‐controlled trial. American Journal of Respiratory and Critical Care Medicine 2013;187:A6069. [Google Scholar]
- NCT00261833. Zemaira in subjects with emphysema due to alpha1‐proteinase inhibitor deficiency. clinicaltrials.gov/ct2/show/NCT00261833 (accessed 7 August 2015).
Dirksen 1999 {published data only}
- Dirksen A, Dijkman JH, Madsen F, Stoel B, Hutchison DC, Ulrik CS, et al. A randomized clinical trial of alpha(1)‐antitrypsin augmentation therapy. American Journal of Respiratory and Critical Care Medicine 1999;160(5 Pt 1):1468‐72. [DOI] [PubMed] [Google Scholar]
Dirksen 2009 {published data only}
- Dirksen A, Piitulainen E, Parr DG, Deng C, Wencker M, Shaker SB, et al. Exploring the role of CT densitometry: a randomised study of augmentation therapy in alpha‐1 antitrypsin deficiency. European Respiratory Journal 2009;33(6):1345‐53. [DOI: 10.1183/09031936.00159408] [DOI] [PubMed] [Google Scholar]
- NCT00263887. Alpha‐1‐antitrypsin (AAT) to treat emphysema in AAT‐deficient patients (EXACTLE) (trial registration). clinicaltrials.gov/ct2/show/NCT00263887?term=antitrypsin&rank=7 (accessed 5 January 2010).
- Parr DG, Dirksen A, Piitulainen E, Deng C, Wencker M, Stockley RA. Exploring the optimum approach to the use of CT densitometry in a randomised placebo‐controlled study of augmentation therapy in alpha 1‐antitrypsin deficiency. Respiratory Research 2009;10:75. [DOI] [PMC free article] [PubMed] [Google Scholar]
References to ongoing studies
NCT01983241 {published data only}
- Efficacy and safety of alpha1‐proteinase inhibitor (human), modified process (alpha‐1 MP) in subjects with pulmonary emphysema due to alpha1 antitrypsin deficiency (AATD) (SPARTA). clinicaltrials.gov/ct2/show/NCT01983241 (accessed 16 June 2016).
Additional references
Alpha‐1 Foundation 2008
- Alpha‐1 Foundation Homepage. www.alphaone.org/ (accessed 9 May 2008).
Chalmers 1990
- Chalmers TC, Frank CS, Reitman D. Minimizing the three stages of publication bias. JAMA 1990;263:1392‐5. [PubMed] [Google Scholar]
Chapman 2009
- Chapman KR, Stockley RA, Dawkins C, Wilkes MM, Navickis RJ. Augmentation therapy for alfa‐1 antitrypsin deficiency: a meta‐analysis. COPD: Journal of Chronic Obstructive Pulmonary Disease 2009;6(3):177‐84. [DOI] [PubMed] [Google Scholar]
Chen 2007
- Chen S, Farahati F, Marciniuk D, Mayers I, Boudreau R, Keating T. Human α1‐proteinase inhibitor for patients with α1‐antitrypsin deficiency [Technology report no 74]. Ottawa: Canadian Agency for Drugs and Technologies in Health 2007.
CLS Behring 2008
- CLS Behring 2008. www.cslbehring.com/s1/cs/enco/1154398192290/content/1154398189443/content.htm (accessed 9 May 2008).
Evald 1990
- Evald T, Dirksen A, Keittelmann S, Viskum K, Kok‐Jensen A. Decline in pulmonary function in patients with alpha‐1‐antitrypsin deficiency. Lung 1990;168(Suppl):579‐85. [DOI] [PubMed] [Google Scholar]
Genetics Home Reference 2007
- Genetics Home Reference. Alpha‐1 antitrypsin deficiency, 2007. ghr.nlm.nih.gov/condition=alpha1antitrypsindeficiency (accessed 16 November 2008).
Gøtzsche 1996
- Gøtzsche PC, Liberati A, Luca P, Torri V. Beware of surrogate outcome measures. International Journal of Technology Assessment in Health Care 1996;12:238‐46. [DOI] [PubMed] [Google Scholar]
Gøtzsche 2006a
- Gøtzsche PC. Believability of relative risks and odds ratios in abstracts: cross‐sectional study. BMJ 2006;333(7561):231‐4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Gøtzsche 2006b
- Gøtzsche PC, Hróbjartsson A, Johansen HK, Haahr MT, Altman DG, Chan A‐W. Constraints on publication rights in industry‐initiated clinical trials. Journal of the American Medical Association 2006;295:1645‐6. [DOI] [PubMed] [Google Scholar]
Heresi 2008
- Heresi GA, Stoller JK. Augmentation therapy in a‐1 antitrypsin deficiency. Expert Opinion on Biological Therapy 2008;8:515‐26. [DOI] [PubMed] [Google Scholar]
Higgins 2003
- Higgins JPT, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta‐analyses. BMJ 2003;327(7414):557‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Hutchison 1988
- Hutchison DCS. Natural history of alpha1‐protease inhibitor deficiency. American Journal of Medicine 1988;84(Suppl 6A):3‐12. [DOI] [PubMed] [Google Scholar]
Larsson 1978
- Larsson C. Natural history and life expectancy in severe alpha1‐antitrypsin deficiency, Pi Z. Acta Medica Scandinavica 1978;204(5):345‐51. [DOI] [PubMed] [Google Scholar]
Marciniuk 2012
- Marciniuk DD, Hernandez P, Balter M, Bourbeau J, Chapman KR, Ford GT, et al. Alpha‐1 antitrypsin deficiency targeted testing and augmentation therapy: a Canadian Thoracic Society clinical practice guideline. Canadian Respiratory Journal 2012;19:109‐16. [DOI] [PMC free article] [PubMed] [Google Scholar]
Nissen 2007
- Nissen SE, Wolski K. Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. New England Journal of Medicine 2007;356:2457‐71. [DOI] [PubMed] [Google Scholar]
O'Brien 1978
- O'Brien ML, Buist NR, Murphey WH. Neonatal screening for alpha1‐antitrypsin deficiency. Journal of Pediatrics 1978;92(6):1006‐10. [DOI] [PubMed] [Google Scholar]
Seersholm 2007
- Seersholm N, Dirksen A, Hansen NCG, Harving H og Dansk Lungemedicinsk Selskab. Retningslinjer for erstatningsbehandling til patienter med alfa‐1‐antitrypsinmangel, 2007 [Guidelines for replacement therapy in patients with alpha‐1 antitrypsin deficiency]. www.lungemedicin.dk/guidelines/alfa1erstat.pdf (accessed 09 May 2008).
Shah 2001
- Shah P, Ohlsson A. Alpha‐1 proteinase inhibitor (a1PI) for preventing chronic lung disease in preterm infants. Cochrane Database of Systematic Reviews 2001, Issue 3. [DOI: 10.1002/14651858.CD002775] [DOI] [PMC free article] [PubMed] [Google Scholar]
Sharp 1971
- Sharp HL. Alpha‐one‐antitrypsin deficiency. Hospital Practice 1971;6:83‐96. [Google Scholar]
Silverman 2009
- Silverman EK, Sandhaus RA. Alpha1‐antitrypsin deficiency. New England Journal of Medicine 2009;360:2749‐57. [DOI] [PubMed] [Google Scholar]
Stockley 2010
- Stockley RA, Parr DG, Piitulainen E, Stolk J, Stoel BC, Dirksen A. Therapeutic efficacy of α‐1 antitrypsin augmentation therapy on the loss of lung tissue: an integrated analysis of 2 randomised clinical trials using computed tomography densitometry. Respiratory Research 2010;11:136. [DOI] [PMC free article] [PubMed] [Google Scholar]
Sveger 1976
- Sveger T. Liver disease in alpha1‐antitrypsin deficiency detected by screening of 200,000 infants. New England Journal of Medicine 1976;294(24):1316‐21. [DOI] [PubMed] [Google Scholar]
Sveger 1978
- Sveger T. Alpha 1‐antitrypsin deficiency in early childhood. Pediatrics 1978;62(1):22‐5. [PubMed] [Google Scholar]
Wood 2008
- Wood L, Egger M, Gluud LL, Schulz KF, Jüni P, Altman DG, et al. Empirical evidence of bias in treatment effect estimates in controlled trials with different interventions and outcomes: meta‐epidemiological study. BMJ 2008;336(7644):601‐5. [DOI] [PMC free article] [PubMed] [Google Scholar]
References to other published versions of this review
Gøtzsche 2010
- Gøtzsche PC, Johansen HK. Intravenous alpha‐1 antitrypsin augmentation therapy for treating patients with alpha‐1 antitrypsin deficiency and lung disease. Cochrane Database of Systematic Reviews 2010, Issue 7. [DOI: 10.1002/14651858.CD007851.pub2] [DOI] [PubMed] [Google Scholar]