

Abstract
Background
Sickle cell disease is a group of genetic diseases which is especially prevalent in tropical and subtropical regions; however, forced migration and ongoing population movement have spread it throughout the world, with estimated birth rates reaching 0.49 per 1000 in the Americas, 0.07 per 1000 in Europe, 0.68 per 1000 in South and Southeast Asia, and 10.68 per 1000 in Africa. Life for individuals with sickle cell disease can be affected by repeated acute complications and compounded by progressive organ damage. Studies reveal that when people with chronic illness learn self‐management, their clinical outcomes and quality of life improves; and they show lower dependence on healthcare services. There are, however, no reviews identifying which interventions improve knowledge and little is known about the impact of patient or care‐giver knowledge on clinical and psychosocial outcomes in people with sickle cell disease.
Objectives
1. To determine the effectiveness of patient‐ and caregiver‐centred educational interventions for changing knowledge and understanding of sickle cell disease among patients as well as caregivers of people with the disease.
2. To assess the effectiveness and safety of patient‐ and caregiver‐centred educational interventions and programs for the recognition of signs and symptoms of disease‐related morbidity, adherence to treatment and healthcare utilization in patients with sickle cell disease.
Search methods
The authors searched the Cochrane Cystic Fibrosis and Genetic Disorders Group Haemoglobinopathies Trials Register, compiled from electronic database searches and handsearching of journals and conference abstract books. Additional trials were sought from the reference lists of the trials and reviews identified by the search strategy.
Date of last search: 11 April 2016.
Selection criteria
Randomized and quasi‐randomized controlled trials which evaluate the effectiveness of individual‐ and group‐based interventions for either the patient with sickle cell disease or their caregivers, or both. Eligible interventions will aim to change knowledge, attitudes or skills, improve psychosocial aspects of the disease as well as treatment adherence and healthcare utilization. Trials evaluating the intervention versus no program, comparing two interventions and those which are part of a multi‐faceted intervention to improve a range of sickle cell‐related health outcomes are all eligible for inclusion.
Data collection and analysis
Two review authors independently selected trials based on stated inclusion criteria and thereafter examined each selected report to extract data using a prepared, piloted, data collection form. A third author assisted in reaching consensus if there were any discrepancies. Similarly, risk of bias was assessed by two authors and verified by a third author.
Main results
A total of 12 trials (11 randomized controlled trials and one quasi‐randomized trial) of 563 people with HbSS, HbSC or HbSβthal, aged six to 35 years old, were included in the review; the majority of participants were African‐American. Interventions ranged from a total of one hour to weekly sessions for eight weeks and the post‐intervention assessments ranged from the end of the intervention period to 12 months after completion. The heterogeneity of the included trials, which encompasses setting, inclusion and exclusion criteria, interventional method and time of assessment, ranged from 'not important' to 'moderate to substantial' for different review outcomes. The overall risk of bias was low for selective reporting, unclear for random sequence generation, allocation concealment, blinding of participants and blinding of outcome assessment. Incomplete outcome reporting and blinding of personnel showed mixed bias representations.
Patient knowledge was assessed by four trials (160 participants) with moderate to substantial heterogeneity. There was evidence that educational programs improved patient knowledge, standardised mean difference 0.87 points (95% confidence interval 0.28 to 1.45, moderate quality evidence), which improved further when a trial with high bias was removed in a sensitivity analysis. Caregiver knowledge, reported in a single trial of 20 families, also showed an improvement, standardised mean difference 0.52 points (95% confidence interval 0.03 to 1.00, moderate quality evidence). The effect on patient knowledge was sustained at longer follow‐up periods, whereas the effect on caregiver knowledge was not sustained.
There were two primary outcomes related to the effectiveness of educational programs on the recognition of signs and symptoms of disease‐related morbidity. No comparative data were reported for patients or caregivers (or both) recognising signs and symptoms leading to self‐management. Data from two trials were analysed for the utilization of health services and showed no evidence of an effect, mean difference 0.33 (95% confidence interval ‐0.57 to 1.23, moderate quality evidence).
With regard to the review's secondary outcomes, depression showed a statistically significant decline in intervention groups, standardised mean difference ‐0.66 points (95% confidence interval ‐1.18, to ‐0.14, moderate quality evidence). Adherence to treatment was not assessed in any of the identified trials. No effects of interventions were seen on coping, family relationships or health‐related quality of life of patients.
The quality of evidence was low for positive coping and moderate for child knowledge, healthcare utilization and depression. This suggests that further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimates.
Authors' conclusions
This review identifies important positive effects of educational interventions on improving patient knowledge of sickle cell disease and depression. Effects on patients' knowledge were maintained for longer than for caregivers. The effect on knowledge was significant but small and whether it offers any clinical benefit is uncertain. Significant factors limiting these effects could be trials being under powered as well as attrition rates. Effects were not statistically significant in assessments of secondary outcomes, possibly due to the paucity of the number of trials and patients and caregivers. Trials showed moderate to high heterogeneity which might impact the results. To better study effects on outcomes, further controlled trials are needed with rigorous attention given to improve recruitment and retention and to decrease bias. Predetermined protocols using similar measurements should be used across multiple sites.
Plain language summary
Interventions for patients and caregivers to improve knowledge of sickle cell disease and recognition of its related complications
Review question
We wished to determine if any educational interventions have helped people with sickle cell disease and their caregivers to improve their understanding of the disease, recognize its complications, improve their adherence to treatment, affect how they utilize healthcare and improve other social and psychological problems that they might face.
Background
Sickle cell disease is a lifelong, inherited disorder which can cause a number of complications throughout an individual's life. It may cause a huge burden on both the patient and their family, including frequent visits to healthcare facilities. The illness causes not just physical complications such as painful crises and strokes, but may have many other effects such as depression, poor quality of life, coping issues and poor family relationships. When people with a chronic illness have better understanding about their illness, they manage their illness better and improve their quality of life. We wish to compare effects of different interventions as well as individual interventions to no intervention.
Search date
The evidence is current to 11 April 2016.
Study characteristics
The review included 12 trials (563 people with HbSS, HbSC or HbSβthal aged six to 35 years). Participants were assigned randomly to either educational programs, no program and in some cases to a non‐educational program, e.g. art therapy. Interventions ranged from a total of one hour to weekly sessions for eight weeks and post‐intervention assessments ranged from the end of the intervention period to 12 months after completion.
Key results
Educational programs and other interventions have resulted in improvements in patient knowledge or understanding of sickle cell disease, and a decrease in depression. Effects on patients' knowledge were maintained for longer than for caregivers. The effects are shown to be small but may result from the fact that most studies had very small numbers of participants and there was much variation between studies. The interventions studied showed no effect on patients' utilization of health services, relationships between families, caregiver or patient skills, coping or health‐related quality of life of the patient. No comparative data were reported for patients or caregivers (or both) recognising signs and symptoms leading to self‐management. No trials assessed the adherence to treatment.
Quality of the evidence
Trials varied in the interventions being studied as well as how the different outcomes were measured. The quality of evidence was low for the outcome positive coping and moderate for the outcomes child knowledge, healthcare utilization and depression. This suggests that further research is likely to have an important impact on our confidence in the effect of the treatment. Further research using randomized controlled trials with more people (including different populations) are needed to improve our understanding of which interventions are likely to be useful.
Summary of findings
for the main comparison.
| Psycho‐educational interventions compared with no education for sickle cell disease | ||||||
|
Patient or population: patients with sickle cell disease Settings: all settings Intervention: psycho‐educational intervention Comparison: no education | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No. of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| No Education | Psycho‐educational | |||||
|
SCD knowledge:children Pre‐ and post‐knowledge assessment Duration of follow‐up range ‐ immediately post to 1 year |
The mean knowledge score ranged across control groups from ‐0.90 to 0.71 | The mean knowledge score in the intervention groups was
0.87 higher (0.28 to 1.45) higher |
160 (4 studies) | ⊕⊕⊕⊝ moderate1 | Overall heterogeneity: I² = 67%. Higher score indicates greater knowledge. | |
|
Healthcare utilization clinic visits, emergency room visits and hospital admissions Duration of follow‐up range immediately post ‐ 1 year |
The mean healthcare utilization score ranged across control groups from ‐2.9 to 0.4 | The mean healthcare utilization scores in the intervention groups wereClinic visits 0.90 higher (‐0.95 to 2.76) ER visits 0.10 higher (‐2.35 to 2.55) Hospital admissions 0.16 higher (0.98 to 1.31) |
176 (2 studies) | ⊕⊕⊕⊝ moderate1 | Overall heterogeneity: I² = 0%. Effect is uncertain. | |
|
Positive coping Coping attempts and coping strategies Duration of follow‐up range immediately post ‐ 1 year |
The mean coping score ranged across control groups from 1.0 to 1.7 | The mean coping score in the intervention groups was 0.46 higher (‐0.38 to 1.29) | 208 (5 studies) | ⊕⊕⊝⊝ low1,2 | Higher score indicates better or improved coping. Effect is uncertain. | |
|
Depression standardised depression scores |
The mean depression score ranged across control groups from ‐2.5 to ‐0.7 | The mean depression score in the intervention groups was ‐0.66 lower (‐1.18 to ‐0.14) | 135 (3 studies) | ⊕⊕⊕⊝ moderate1 | overall heterogeneity: I² = 49%. lower scores indicate less depression. | |
| Patient‐reported signs and symptoms/ self‐management | Outcome not reported. | |||||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence High quality: further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: we are very uncertain about the estimate. | ||||||
1Downgraded one level (moderate) as due to unclear risk of bias rated and the lack of blinding and allocation concealment in some studies.
2 Variation in size and direction of the effect.
2.
| Psycho‐educational interventions compared with no education for sickle cell disease | ||||||
|
Patient or population:caregivers of patients with sickle cell disease Settings: all settings Intervention: psycho‐educational intervention Comparison: no education | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No. of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| No Education | Psycho‐educational | |||||
|
SCD knowledge: caregiver Pre‐ and post‐knowledge assessment Duration of follow‐up range ‐ immediately post intervention (6weeks) and 6months (only in intervention group) |
The mean knowledge score in the control group was 0.30 | The mean knowledge score in the intervention groups was
0.52 higher (0.03 to 1.00) higher |
39 (1 study) | ⊕⊕⊝⊝ low1,2 | Higher score indicates greater knowledge. | |
|
Family relationship Pre‐intervention and post‐intervention assessment Home visits and training session for family members Patient‐caregiver relationships assessed pre‐intervention and immediately post‐intervention. |
The mean patient‐caregiver relationship score ranged across control groups from ‐0.74 to ‐0.64 | The mean patient‐caregiver relationship score in the intervention group was 1.22 higher (‐0.24 to 2.68) |
76 (2 studies) | ⊕⊕⊕⊝ moderate1 | Overall heterogeneity : I² = 68%. Effect is uncertain. | |
|
Caregiver‐reported signs and symptoms Lead to self management |
Outcome not reported. | |||||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence High quality: further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: we are very uncertain about the estimate. | ||||||
1Downgraded one level (moderate) as due to unclear risk of bias rated and the lack of blinding and allocation concealment in the studies
2 Downgraded one level due to only a single study available for the patient‐caregiver relationships outcome (imprecision).
Background
Description of the condition
The term sickle cell disease (SCD) covers a group of genetic diseases which are especially prevalent in tropical and subtropical regions that have endemic malaria. Hence, SCD is most common in Africa, which accounts for an estimated 84% of worldwide affected births and has an average birth rate across the continent of 10.68 per 1000 live births (Modell 2008). Historical forced migration and ongoing population movement have spread SCD throughout the world, with estimated birth rates of 0.49 per 1000 in the Americas, and 0.07 per 1000 in Europe, and 0.68 per 1000 in Southeast Asia. Within these continental regions there is a large variability in birth rate. The prevalence of carrier frequency is as equally variable with limited surveys reporting 1% to 38% in Africa, 0% to 29% in the eastern Mediterranean and 17% to 30% in India (Weatherall 2001).
Normal adult haemoglobin AA consists of two alpha and two beta globin chains. Sickle cell disease results from a single nucleotide switch of valine to glutamic acid in the sixth position on the β‐globin chain (Ingram 1959). This may occur in one chain, resulting in the 'carrier' state of HbAS (sickle cell trait) or it may be coupled with another haemoglobinopathy, e.g. the thalassaemias. Substitution of both haemoglobin alleles with the 'sickle' variant results in homozygous SS disease, which is recognised as the most severe form of the group (Pauling 1964; Rees 2010).
Life for people with SCD can be affected by repeated acute complications and compounded by progressive organ damage (Ataga 2000; Di Sabatino 2011; Ebert 2010; Pham 2000; Stuart 2004). Many of these complications can occur in childhood. Stroke is a devastating complication of SCD during childhood or adolescence (or both). In people with homozygous SS disease, it has a reported incidence of 0.75 per 100 patient years in the two‐ to five‐year age group and a recurrence rate of 6.4 events per 100 patient years during a five‐year period in people under 20 years of age (Ohene‐Frempong 1998). Moreover, cognitive impairment has been linked to overt and silent infarcts in children (Kral 2001; Steen 2003). Hyposplenism predisposes to recurrent infections with encapsulated organisms; and although greatly reduced by prophylactic penicillin therapy, infection still remains the leading cause of mortality in children with SCD (Di Sabatino 2011; Leikin 1989). Pulmonary diseases, including acute chest syndrome, are an important cause of morbidity and mortality in both children and adults with SCD (Leikin 1989; Miller 2011; Quinn 2004). Frequent vaso‐occlusive crises are an unwanted trademark of the disease. They can become a major source of healthcare utilization leading to disruptions throughout an individual's lifespan and a reliance on healthcare professionals. As the life expectancy of people with SCD increases, they are more likely to suffer end‐organ damage and present with complications such as renal disease, cardiac problems and retinal damage. The difficulty of adjusting to and coping with these various disease manifestations can often lead to reduced social interaction, anxiety and depression (Anie 2005; Brown 1993). The unpredictable and varied pattern of complications results in uncertainties and a reduced quality of life for people living with SCD.
Description of the intervention
We define the intervention as a patient‐ or caregiver‐centred education program which is based on a specified protocol, manual or curriculum. It is aimed at changing knowledge, attitudes or skills, e.g. improving the knowledge and understanding of SCD as well as the recognition of complications that occur in the disease. We define a caregiver as one who is a non‐healthcare professional and who is in charge of the patient's well‐being for any length of time. This may include, but is not limited to, a parent, other relative, non‐related guardian, school teacher, camp counsellor or childcare provider.
Recent systematic reviews of parenting interventions among healthy children have shown benefits in a range of behavioural and cognitive outcomes (Barlow 2010), with some evidence for cost‐effectiveness (NICE 2007). A review on the effect of maternal education on childhood growth pinpointed two important areas that can be transferred to other parent‐education programs; the need for recognition of symptoms; and knowledge of the solutions (Imdad 2011). The beneficial effects of SCD knowledge have been explored in a small number of observational studies. In‐depth interviews with people with SCD summarized that increasing knowledge and understanding of the illness helped individuals gain better control of their illness and encouraged greater psychosocial skills (e.g. confidence and assertiveness when seeking hospital care) (Maxwell 1999; Tanabe 2010). Those with better knowledge of SCD tend to have more positive health beliefs, such as higher perceived benefits of testing for SCD (Bhatt 2011). Improved parental knowledge of SCD translates into more positive family relations (Hurtig 1994) and is important for adaptive coping (Burlew 1989). Parents with improved knowledge claim better adherence to a child's medical management, are more consistent with their child's school attendance and have better perceptions of their quality of life (QoL) (Kaslow 2000). A recent Cochrane Review highlighted varying effects of patient education and psychological therapies on SCD‐related pain (Anie 2015); there may be some trial overlap between this systematic review and the current review; however, all trials will still be considered in this review.
Given these positive effects, it seems reasonable to hypothesise that educational interventions for the patient or caregiver may be beneficial in equipping patients and caregivers with the necessary skills to manage the physical and psychological demands of the disease.
How the intervention might work
Currently, the brunt of clinical management is placed on the healthcare system, and given the clinical consequences of many SCD complications, this is generally appropriate. However, studies reveal that when people with a chronic illness learn greater self‐management, their clinical outcomes and QoL improve; and they are less dependent on healthcare services (Bodenheimer 2002; Sawyer 2005). The responsibility for the initial recognition of SCD symptoms lies with patients or their caregivers (or both), this may be especially relevant for those caring for younger children without the cognitive or verbal skills to recognise or vocalise their symptoms. Acute splenic sequestration, which occurs in infancy (median age 1.4 years), and dactylitis (inflammation of toes or fingers) (Brousse 2012) are examples of early‐life complications for which symptoms are overt, and so are useful targets for parental educational interventions. Anticipatory and early onset management of painful crises might limit their development to more severe forms as well as resultant complications, such as acute chest syndrome, and could be important areas for patient and caregiver training.
We consider that interventions to improve knowledge may help in many ways. For example, training in the recognition and management of SCD‐related complications could provide patients and caregivers with the skills to identify the symptoms of these complications, and the confidence to determine when referral to the healthcare system is appropriate. Early referral to healthcare providers could lead to improved clinical outcomes, and appropriate decisions on whether referral is necessary could lessen the psychological reliance of the SCD family on the healthcare professional. Although the reduction of adverse clinical outcomes are ultimately a goal of educational interventions, the use of explicit clinical endpoints is unlikely to offer sufficient statistical power in such interventions; because of this we focus on healthcare utilization as a proxy for clinical endpoints.
Why it is important to do this review
Although it is understood that increasing knowledge and understanding improves outcomes in people living with chronic illnesses, there are no reviews identifying which interventions improve knowledge and little is known or thought of in clinical practice about the impact of patient or caregiver knowledge on clinical outcomes in people with SCD. This review will document evidence that can be used to generate interest and to influence practice in patient and caregiver training for improving knowledge as well as improving the clinical outcomes which result from people living with SCD.
Objectives
To determine the effectiveness of patient‐ and caregiver‐centred educational interventions for changing knowledge and understanding of SCD among patients and their caregivers.
To assess the effectiveness and safety of patient‐ and caregiver‐centred educational interventions and programs for the recognition of signs and symptoms of disease‐related morbidity, adherence to treatment and healthcare utilization in patients with SCD.
Methods
Criteria for considering studies for this review
Types of studies
Randomized controlled trials (RCTs) and quasi‐RCTs were eligible for inclusion in this review. Trials where quasi‐randomized methods were used were included when there was sufficient evidence that the treatment and control groups were similar at baseline. Both published or unpublished trials were eligible. Cross‐over trials were not considered a suitable design for knowledge interventions and were not considered in this review.
Types of participants
We included trials in any setting in which the intervention was provided for patients (aged five years and older), parents or adult caregivers of children with SCD of all age groups. The participants were of either sex and confirmed to have one of the SCDs (i.e. SS disease, SC disease, Sβ+ thalassaemia and Sβ0 thalassaemia) by electrophoresis and sickle solubility test, with family studies or DNA tests as appropriate.
Types of interventions
Included trials evaluated the effectiveness of individual‐ and group‐based interventions for either the patient or caregivers. There were no restrictions on who facilitated the intervention, i.e. by a healthcare professional, by a social worker, self‐learning or by any other training body or group; or on the avenue of facilitation, i.e. face‐to‐face, online, written manual or telephone.
The interventions had to have a specified protocol, manual or curriculum aimed at changing knowledge, attitudes or skills covering a range of topics relevant to the identification of SCD‐related illness, including decisions on the use of healthcare services compared to self‐management for any given disease‐related illness. We included interventions to improve psychosocial aspects of the disease in addition to illness. Trials evaluating the intervention versus no program, comparing two interventions as well as those which were a part of a multi‐faceted intervention aimed at improving a range of sickle cell‐related health outcomes were included.
Types of outcome measures
Primary outcomes
Patient and caregiver understanding of SCD and related complications (e.g. the 'Sickle Cell Disease Knowledge Questionnaire' (Armstrong 1993), the 'Illness Perception Questionnaire')
Patient or caregiver (or both) ‐ reported signs and symptoms leading to self‐management
-
Utilization of health services
number of acute hospitalizations (admission to hospital in‐patient services for an acute event)
average length of hospital stay
hospital appointments (scheduled, unscheduled and missed)
primary care appointments (scheduled, unscheduled and missed)
Secondary outcomes
Adherence to treatment
Changes in caregiver or patient skills (e.g. the 'Parenting Clinical Observation Schedule' (Hill 2008))
Changes in caregiver‐patient relationship (e.g. the 'Parent‐Adolescent Relationship Questionnaire' (Robin 1990))
Caregiver or patient stress or anxiety (e.g. the 'Parenting Stress Index' (Abidin 1995), the 'Paediatric Inventory for Parents' (Streisand 2001))
Psychological, social and cognitive functioning of the child (e.g. the 'Social Functioning Questionnaire' (Tyrer 2005), the 'California Verbal Learning Test' (Delis 1988))
Health‐related QoL of patient; generic or disease‐specific or both (e.g. the 'Child Health Questionnaire' (Landgraf 1998); the 'SF‐36'; the 'WHOQOL‐Bref')
Search methods for identification of studies
Electronic searches
Our search identified relevant trials from the Cystic Fibrosis and Genetic Disorders Group's Haemoglobinopathies Trials Register using the terms: sickle cell AND (family interventions OR self help OR education).
The Haemoglobinopathies Trials Register is compiled from electronic searches of the Cochrane Central Register of Controlled Trials (CENTRAL) (updated each new issue of the Cochrane Library) and weekly searches of MEDLINE. Unpublished work is identified by searching the abstract books of five major conferences: the European Haematology Association conference; the American Society of Hematology conference; the British Society for Haematology Annual Scientific Meeting; the Caribbean Public Health Agency Annual Meetings; and the National Sickle Cell Disease Program Annual Meeting. For full details of all searching activities for the register, please see the relevant section of the Cochrane Cystic Fibrosis and Genetic Disorders Group Module.
A search of clinical trial registries was carried out to identify ongoing and completed trials maintained by the European Medicines Agency (www.clinicaltrialsregister.eu/), the US National Institute of Health (http://clinicaltrials.gov/) and the WHO (www.who.int/ictrp/en/). Please refer to the appendices for further details (Appendix 2). Date of searches: 11 April 2016.
Date of the latest search of the Cochrane Cystic Fibrosis and Genetic Disorders Group's Haemoglobinopathies Trials Register: 11 April 2016.
Searching other resources
References of included trials were screened to identify other potentially relevant trials.
Data collection and analysis
Selection of studies
We used Endnote to maintain the compiled list of references obtained from the multiple database searches, without duplicated records. Review authors (KQ, NB) examined titles and abstracts independently to remove obviously irrelevant reports; where there were disagreements a third review author (MA) provided consensus. Two review authors (NB, KQ) retrieved the full text reports of trials that they agreed were potentially relevant and linked together multiple reports of the same trial. Two review authors (MA, KQ) also independently examined the full text of the trials for compliance with the eligibility criteria. One review author (KQ) corresponded with investigators to determine trial eligibility and ascertain additional data from selected trials.
Data extraction and management
Review authors (NB, KQ) independently examined each selected report and extracted data using a data collection form developed using the Cochrane Consumers and Communication Group's Data Extraction template. The data collection form included baseline characteristics of the participants, e.g. age, sex and total number; and information pertaining to the educational program, e.g. the method and duration; and the time points at which the pre‐determined outcomes were monitored. The review authors also extracted characteristics of the population relevant for addressing potential issues in health equity using the PROGRESS‐Plus concept (place of residence; race, ethnicity and culture; occupation; sex; religion; education; socio‐economic status; social capital; age; disability; and sexual orientation) (Welch 2012). The authors extracted data on a single trial from multiple reports of the same trial. The review authors resolved all discrepancies by discussion and review with the third review author (MA).
Authors assessed outcomes at the end of the educational intervention, and at six and 12 months post‐intervention.
Assessment of risk of bias in included studies
Using the relevant Cochrane tool, two authors (DF, KQ) assessed the risk of bias in each trial for:
selection bias – sequence generation and allocation concealment;
performance bias – blinding of participants and personnel;
detection bias – blinding of outcome assessment;
attrition bias ‐ incomplete outcome data;
reporting bias; and
other bias.
Two review authors (NB, KQ) collated judgements and rationale for each bias domain in a tabular form. They based their judgements of 'low', 'high', or 'unclear' risk on the Cochrane criteria (Higgins 2011a). The review authors stratified trials by risk of bias and constructed a summary assessment table, as in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011b).
Measures of treatment effect
The review only included data for continuous outcomes (e.g. knowledge scores and QoL scores), from which the authors calculated the mean change from baseline for each group or the mean post‐intervention values and standard deviation (SD) for each group. They converted all standard errors (SE) to SDs. The authors produced a pooled estimate of treatment effect by calculating the mean difference (MD) and 95% confidence intervals (CIs). Where continuous scores measured the same outcome but in a variety of ways (e.g. different scales to measure knowledge) the authors standardised the change for the outcomes to a uniform scale using the standardised MD (SMD). Where effects were reported on a negative scale in a single trial, the authors converted these to positive scales by the multiplying the mean effect by minus one (‐1). Authors assessed skewness as per chapter 9 of the Cochrane Handbook for Systematic Reviews of Interventions (Altman 1996). As an a priori method, they planned to analyze categorical data such as adherence to treatment using odds ratios (OR), and risk ratios (RR), but they had no such data. The authors will use these methods in future updates of this review, should such data become available.
Unit of analysis issues
Cluster‐randomized trials
Interventions of educational programs included in this review measured outcomes at the level of the participant and did not include cluster‐randomized trials. It is likely that educational interventions may take place in a cluster‐randomized design which would require adjustment for clustering. For future updates, if the authors include such designs, and if they assess that the trial authors did not control appropriately for clustering (e.g. variance‐inflated standard errors, hierarchical linear models) then they will apply the methods outlined in chapter 16 of the Cochrane Handbook for Systematic Reviews of Interventions to inflate the SE (Higgins 2011b). This includes calculating the unadjusted SMD and 95% CIs for the generic inverse variance method, and then inflating the SE of the effect estimate by multiplying by the square root of the variance inflation factor, calculated as: 1 + ((M ‐ 1) multiplied by ICC), where M is the average cluster size. The authors will calculate the SE as the CI divided by 3.92. Where the pooled estimate is the MD, the authors will use the variance inflation factor (VIF) to adjust the SDs in the educational intervention and control groups separately. They will use these calculated SDs in the meta‐analysis, and incorporate in the SE of the MD and the weighting procedures.
Multiple treatment groups
For trials with multiple treatment groups, in one trial the authors combined the (active intervention) psycho‐educational arms for cognitive behavioral therapy (CBT)‐related relaxation technique and art therapy, as these were deemed to have similar mechanism of action on the outcomes addressed in the trial. For the second trial that included two types of controls (attention placebo and a strict control), we included only the strict control in our meta‐analysis as the control group ‐ in keeping with our objectives outlined in protocol to compare intervention to control.
Dealing with missing data
In order to allow an intention‐to‐treat analysis, authors extracted data on the number of participants with each outcome event, as per allocated treated group, irrespective of compliance and whether or not the participant was later thought to be ineligible or otherwise excluded from treatment or follow up. Where data were missing or unclear, one review author (KQ) made contact with the primary investigators for clarification.
Assessment of heterogeneity
The review authors assessed heterogeneity through a visual examination of the combined data presented in the forest plots, and by considering the I² statistic (Higgins 2003). In addition, they used the Chi² values and considered a P value less than 0.10 to be an indication of heterogeneity (Deeks 2011). The I² statistic reflects the likelihood that variation of results across trials are due to heterogeneity rather than by chance, and the review authors interpreted this using the following simple classification:
0% to 40%: might not be important;
30% to 60%: may represent moderate heterogeneity;
50% to 90%: may represent substantial heterogeneity;
75% to 100%: considerable heterogeneity.
Assessment of reporting biases
In order to identify selective outcome reporting, where possible the review authors compared outcomes described in the protocol with those reported in the publication. The review authors requested protocols for all trials from the primary investigators, corresponding author, trial registry portal or relevant organisation. They also compared outcomes listed in the 'Methods' section of the final paper with those presented in the 'Results' section. If the published papers reported negative findings either only partially, or not at all, the review authors contacted the primary investigators for these data.
Data synthesis
Authors performed meta‐analyses according to:
patient outcomes; and
outcomes measured in the caregiver.
The authors analysed data primarily using the fixed‐effect model where they considered trials to have little or no heterogeneity. However, where moderate, substantial or considerable heterogeneity was present (I2 greater than 40%), they used a random‐effects model to analyse data. We assumed statistical significance at an alpha of 5%.
Subgroup analysis and investigation of heterogeneity
Due to the inability to combine a sufficient number of trials (at least 10), we did not investigate any heterogeneity identified using pre‐defined subgroup analyses of potential confounding factors. The predefined factors were:
genotype of the patient (SS and S‐β0 thalassaemia versus SC and S‐β+ thalassaemia);
training method (individual versus group education);
delivery of intervention (indirect use of leaflets, brochures, videos versus direct workshops, face‐to‐face seminars).
Furthermore, there were not sufficient reports in the include trials to conduct subgroup analyses across categories of PROGRESS.
Sensitivity analysis
We investigated the impact of bias on the results examined by comparing meta‐analyses including and excluding trials with concerns of high risk of selection or reporting bias (due to issues relating to randomization, allocation concealment, or masking of interventions from participants or trial personnel).
Summary of findings table
We constructed 'Summary of findings' tables and rated the quality of evidence using GRADE for all reported primary and selected secondary outcomes in patients and caregivers (Guyatt 2011) (Schünemann 2008) (Table 1; Table 2). In addition to the primary outcomes, the authors chose the secondary outcomes of coping and depression for inclusion in the tables as they are clinically meaningful to patients and caregivers with chronic diseases such as SCD. The authors were particularly intrigued with coping and depression as these are also major concerns in the adolescent population living with SCD (Anie 2005). GRADE categorises the quality of the evidence as high, moderate, low, or very low, with RCTs starting out at high or medium quality. Authors may downgrade evidence from RCTs if there is a high risk of bias across trials, if results are inconsistent or imprecise, or in the presence of publication bias.
Results
Description of studies
See 'Characteristics of included studies', 'Characteristics of excluded studies' and 'Characteristics of studies awaiting classification'.
Results of the search
A total of 51 records were retrieved by the electronic searches, 23 of these were full texts and 28 were abstracts. During initial assessment, 20 records (nine full texts and 11 abstracts) were found to be ineligible (Figure 1).
1.
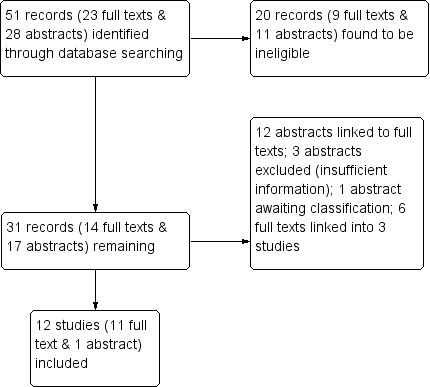
Study flow diagram.
Of the remaining 14 full texts, three of them were linked, leaving 11 full text records to be included in a quantitative synthesis.
Of the remaining 17 abstracts, 12 were also linked to full texts which were already identified; three were subsequently excluded due to failed attempts to retrieve additional information from authors (Carpenter 2007; Robinson 1990; Schatz 2004); one is still awaiting classification (Bhogte 2013); and the remaining abstract (bolstered with unpublished information received from the author) was included in quantitative synthesis (Lemanek 2006).
Included studies
Design
Of the 12 included trials, 11 were RCTs and one was quasi‐randomized (Hazzard 2002). A total of 10 were two‐arm trials and two were three‐arm trials (Broome 2001; Thomas 1999). Interventions ranged from a total of one hour to weekly sessions for eight weeks and the post‐intervention assessments ranged from the end of the intervention period to 12 months after completion.
Sample sizes
The number of people randomized was not stated in all trials; however, the number of people analysed ranged from 24 to 64 participants.
Setting
Where stated, the majority of the trials were conducted in the USA. One trial was conducted in the UK (Thomas 1999). Trials were both single‐ and multi‐centered. They were conducted in both clinic and classroom‐like settings. One trial was conducted in the home (Ketchen 2006).
Participants
A total of 563 people with HbSS, HbSC or HbSβthal were analysed in the 12 trials. The majority of participants were African‐American and the ages ranged from six to 35 years old. The male:female ratio analysed was stated in seven studies and the average was 1:1.2. One trial analysed participants in two different categories – children (aged six to 13 years) and adolescents (aged 14 to 18 years) (Broome 2001). The Hazzard trial recruited a mixed cohort, including children with asthma; however, analyses for children with SCD were performed separately (Hazzard 2002). Caregivers were included in two of the trials (Ketchen 2006; Koontz 2004). The Ketchen report noted that the majority of the caregivers were female (36 female:1 male). The inclusion and exclusion criteria varied between trials and included age limits, length of time in hospital, pain medication use, intelligence quotient and history of cerebrovascular accident (Characteristics of included studies).
Interventions
Nine of the interventions were described as cognitive‐based therapy or psycho‐education interventions (Barakat 2010; Broome 2001; Daniel 2015; Gil 1996;Gil 1997; Kaslow 2000; Ketchen 2006; Schatz 2015; Thomas 1999). The methods utilized in individual trials were varied, including videos, audios, computer programs and written materials. There were also practical demonstrations of relaxation and distraction strategies and other pain management techniques.
Two trials listed education (computer programs and classroom‐like sessions) as the intervention (Hazzard 2002; Koontz 2004).
The final trial used a coaching intervention, where coaches were intentional in their efforts to encourage each child to follow treatment guidelines (Lemanek 2006).
Whereas participants were usually randomized at the beginning of the trial, Broome deviated from this pattern by dividing the intervention into two phases (Broome 2001). Phase 1, which focused on disease education and communication with the healthcare professional, was attended by all participants. Phase 2 was the RCT section of the trial where participants were randomized either to cognitive‐based therapy, art therapy or attention control.
The schedule of the intervention period also differed between trials. The intensity of the interventions ranged from 45 to 60 minutes per week to one hour every two weeks, with the total duration of intervention ranging from a total of one hour to eight weeks. Hazzard differed in that their intervention comprised of four, three‐day educational packages (Hazzard 2002) (Characteristics of included studies). Some interventions also included take‐home materials and booster follow‐up calls.
The times of assessment also varied between trials, ranging from immediately post‐intervention to 12 months post‐intervention. The majority of trials included pre‐assessment values for both the intervention and control groups. Koontz and Lemanek did not present pre‐intervention values for either group (Koontz 2004; Lemanek 2006). Kaslow did not present six‐month post‐intervention assessments of the control group since some of them opted to cross‐over to the intervention (Kaslow 2000).
Outcomes
Two‐thirds of the primary outcomes and five of the six secondary outcomes listed in our protocol were addressed in these trials.
Five trials assessed patient or caregiver (or both) understanding of SCD using varying tools, e.g. the Sickle Cell Knowledge Questionnaire or the "how much do I know about SCD" test (Barakat 2010; Hazzard 2002; Kaslow 2000; Ketchen 2006; Koontz 2004). All five trials reported a post assessment, Kaslow added a follow‐up assessment at six months and Barakat a follow‐up assessment at one year (Barakat 2010; Kaslow 2000).
Four trials reported on utilization of health services, including emergency room visits, clinic visits, calls to healthcare professionals and the number of days spent in hospital (Barakat 2010; Broome 2001; Gil 1996; Lemanek 2006). Thomas mentioned healthcare utilization in their methods but no reports were seen in the results section (Thomas 1999). All four trials reported a post assessment (Barakat 2010; Broome 2001; Gil 1996; Lemanek 2006). Gil also made a three‐month report and Barakat and Broome reported another follow up at one year (Barakat 2010; Broome 2001; Gil 1996).
Ketchen examined the changes in caregiver or patient skills using the health promotion activities scale (Ketchen 2006).
Three trials used several tools, e.g. the Family Adaptability and Cohesion Evaluation Scale, to assess the patient‐caregiver relationship (Barakat 2010; Kaslow 2000; Ketchen 2006 ).
Three trials measured caregiver or patient stress or anxiety using, e.g. General Health Questionnaire–28 (Kaslow 2000; Ketchen 2006; Thomas 1999).
Eight trials assessed the psychological, social and cognitive functioning of the child utilizing scales, e.g. Child Behavior Checklist (Barakat 2010;Broome 2001; Daniel 2015; Gil 1996; Hazzard 2002; Kaslow 2000; Thomas 1999; Schatz 2015). Gil, however, did not provide summary statistics (Gil 1996).
Two trials reported on the health‐related QoL (Daniel 2015; Ketchen 2006) .
The two outcomes not addressed were (1) adherence to medication and (2) patient‐ or caregiver‐reported signs and symptoms leading to self‐management which was mentioned by the 1997 Gil trial but analyses were reported only for the intervention group (Gil 1997).
Excluded studies
See Characteristics of excluded studies.
A total of 20 reports were excluded for various reasons including: not being an RCT (Barakat 2007, Kaslow 1997, Radcliffe 2007); irrelevant participants (Oyeku 2005; Oyeku 2005 b; Oyeku 2010; Yang 2000, Mendolia 1993; Wilkie 2013); irrelevant intervention (Chernoff 2002; Daly 2012; Ireys 2001; King 2006; King 2007; Lemanek 2009;Thomas 2013; Yerys 2003); and irrelevant outcomes (Applegate 2003;Boroffice 1991; Jenerette 2014). A further three were subsequently excluded; two reports appeared to be methods papers without data on relevant outcomes (Carpenter 2007; Robinson 1990) and one was a narrative description in an unpublished abstract (Schatz 2004).
Risk of bias in included studies
The results of the risk of bias assessment are presented graphically (Figure 2; Figure 3). For the 12 trials, the overall risk of bias was low for selective reporting, unclear for random sequence generation, allocation concealment, blinding of outcome assessment and blinding of participants and personnel. Incomplete outcome reporting showed mixed representations.
2.
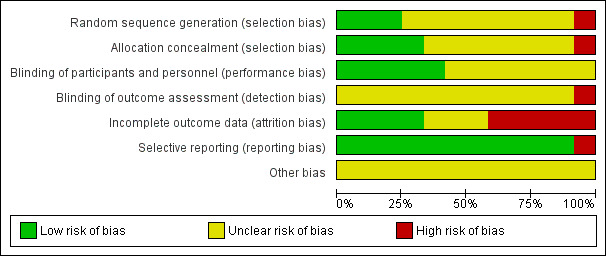
Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
3.
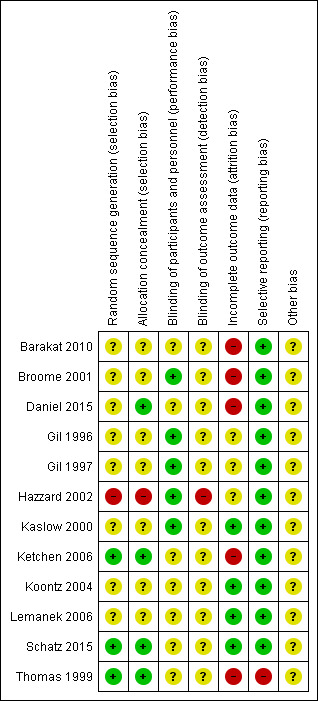
Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
Allocation
Sequence generation
One trial had a high risk of bias for random sequence generation where participants were randomized by months of study where those hospitalized in months one to seven were placed in the "control group" (Hazzard 2002). Three trials had a low risk: in one trial group assignments were generated prior to the start of data collection, envelopes sealed, shuffled and numbers then given to families in consecutive order (Ketchen 2006); in the second trial random allocation was conducted by means of a sequence of labelled cards in sealed envelopes and the sequence was based on a random number table (Thomas 1999); and in the third trial participants were assigned by drawing coloured marbles out of an opaque bag (Schatz 2015). For the remaining eight trials there was insufficient information to permit judgement as being low or high risk and were assessed as being of unclear risk of bias (Barakat 2010; Broome 2001; Daniel 2015; Gil 1996; Gil 1997; Kaslow 2000; Koontz 2004; Lemanek 2006).
Allocation concealment
One trial had a high risk for allocation concealment bias (Hazzard 2002), and four trials had a low risk, where group assignments were through envelopes sealed, shuffled and numbers then given to families in consecutive order (Daniel 2015; Ketchen 2006; Schatz 2015; Thomas 1999). There was insufficient information for the remaining seven trials to permit judgement as being low or high risk for allocation concealment bias (Barakat 2010; Broome 2001; Gil 1996; Gil 1997; Kaslow 2000; Koontz 2004; Lemanek 2006).
Blinding
Blinding of participants and personnel (performance bias)
Considering the nature of the interventions, the authors judged that the outcome was not likely to be influenced by a lack of blinding so five trials were assessed as low risk (Broome 2001;Gil 1996;Gil 1997; Hazzard 2002; Kaslow 2000). There were seven trials where lack of blinding could have introduced bias; however, since the published work did not include any information on blinding, the risk was labelled as unclear (Barakat 2010;Daniel 2015; Ketchen 2006;Koontz 2004;Lemanek 2006;Schatz 2015; Thomas 1999).
Blinding of outcome assessment (detection bias)
The Hazzard trial was assessed as high risk of bias because due to occasional shortages of trial staff, the interventionist assisted the participants during the assessments (Hazzard 2002). All other trials were assessed as being at unclear risk of bias because no information was provided in the publication on blinding to support any other judgement.
Incomplete outcome data
Three trials had low attrition bias where 100% (Lemanek 2006), 96% (Schatz 2015) and 90% (Kaslow 2000) of participants completed the post‐evaluation and were analysed. The Koontz trial had a borderline low attrition rate (85.7%) based on the a priori cut off of 15%. All four of the non‐completers were from the intervention group and were excluded after randomization when it was revealed that they did not meet the inclusion criteria (Koontz 2004). The authors made a judgement to include the study in the low attrition bias category. Five trials had high attrition: the Barakat trial had 35.8% attrition; the Broome trial had up to 30% attrition and did not use intention‐to‐treat analysis; the Ketchen trial had 33% attrition; the Daniel trial had 25% attrition and the Thomas trial had 39% attrition (Barakat 2010; Broome 2001; Daniel 2015; Ketchen 2006; Thomas 1999). The remaining three trials had insufficient information to permit judgement as being low or high risk as we were uncertain of the number of people entering the trial (Gil 1996; Gil 1997; Hazzard 2002).
Selective reporting
All trials included in this review, with one exception, were assessed by the authors to have low risk of reporting bias because all outcomes reported in the objectives and methods were present in the results. However, the Thomas trial did not report one of the three objectives, namely, utilization of health services (Thomas 1999).
Other potential sources of bias
The authors did not find any other potential sources of bias.
Effects of interventions
Primary outcomes
The results of this section are presented sequentially in keeping with our primary and secondary outcomes, with sensitivity analysis where one was conducted.
1. Patient understanding (knowledge of SCD)
Patient knowledge
Four RCTs with 160 patients were included in a random effects meta‐analysis for patient knowledge of SCD with age ranging from six to 18 years (Barakat 2010; Kaslow 2000; Ketchen 2006; Hazzard 2002). Koontz was excluded from the meta‐analysis as pre‐values were not reported and hence change in score could not be calculated (Koontz 2004). However, the study did report a significant improvement in patient knowledge in the intervened group when compared to those in the control group (effect size: 1.83). Education sessions were mainly undertaken in a family setting and lasted a maximum of six weeks. Knowledge was measured using the 'SCD Knowledge Test' which was adapted and renamed the 'How much do I know about SCD' scale for two subsequent studies. Barakat employed a more recently adapted SCD knowledge questionnaire as their measure. Patients who received the psycho‐educational intervention were at 0.87 points higher standardized mean score for knowledge compared to the control or no intervention group (95% CI 0.28 to 1.45) (Analysis 1.1). There was moderate to substantial level of heterogeneity (Chi² = 9.15, df = 3 (P = 0.03); I² = 67%). A sensitivity analysis excluding the Hazzard trial from the meta‐analysis for reasons of high risk of bias (random sequence generation, allocation concealment and blinding of outcome assessors) showed significant positive effects, MD 1.12 (95% CI 0.72 to 1.52) for SCD knowledge in children (Analysis 1.2) (Hazzard 2002) which also removed all heterogeneity from the meta‐analysis. The latter study was designed to test children while hospitalised for complications of their SCD and that is possibly the largest contributor to the heterogeneity introduced by including this study in the meta analysis.
1.1. Analysis.
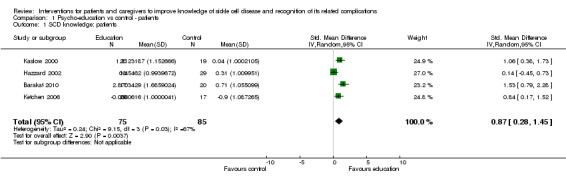
Comparison 1 Psycho‐education vs control ‐ patients, Outcome 1 SCD knowledge: patients.
1.2. Analysis.
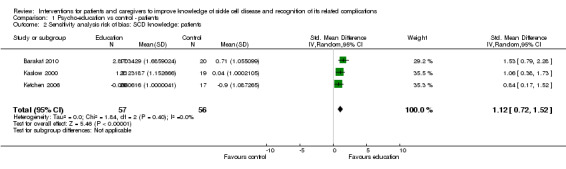
Comparison 1 Psycho‐education vs control ‐ patients, Outcome 2 Sensitivity analysis risk of bias: SCD knowledge: patients.
Interestingly, Barakat and Kaslow both reported on assessments of knowledge at 12 months and six months post intervention, respectively, and have reported sustained effects at those times (Barakat 2010; Kaslow 2000).
2. Patients and caregiver self management
No trials or data were available from any source to address this outcome.
3. Utilization of health services
Two RCTs of 176 patients (130 active treatment: 46 controls) with age ranging from six to 18 years reported on indices of healthcare utilization, i.e. visits to emergency department, primary care or clinic and admission to hospital (Barakat 2010; Broome 2001). Medical chart reviews were conducted to determine healthcare utilization in both trials; and both trials involved aspects of a CBT approach as part of the intervention. Patients receiving the intervention had a higher standardized utilization score, MD 0.33 points (95% CI ‐0.57 to 1.23) (Analysis 1.3) with no heterogeneity (heterogeneity: Chi² = 0.58, df = 3 (P = 0.90); I² = 0%). The Lemanek trial and the 1996 Gil trial were omitted from the fixed‐effect meta‐analysis as they did not report change data (Gil 1996; Lemanek 2006). However, both trials narratively reported that there was no significant effect of intervention on any healthcare utilization outcomes that were measured.
1.3. Analysis.
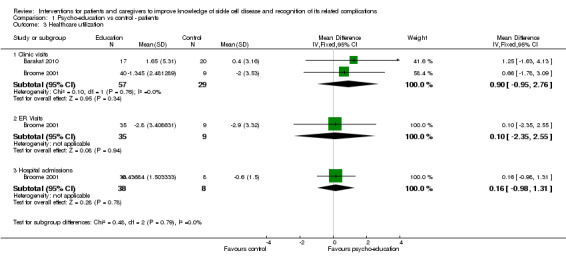
Comparison 1 Psycho‐education vs control ‐ patients, Outcome 3 Healthcare utilization.
Even though formal analyses showed no significant effects, both trials report decreased numbers of clinic and ER visits over the longer follow up after intervention.
Secondary outcomes
1. Adherence to treatment
No trials or data were available from any source to address this outcome.
2. Changes in caregiver or patient skills
One trial reported on the parent's assessment of their child's (age range eight to 18 years) participation in health promotion activities (using an authors' created seven‐item measure which asked questions such as drinking adequate amounts of water, getting sufficient rest and avoiding extremes of temperatures) in 20 children receiving intervention and 17 controls (Ketchen 2006). After six weeks of a computer‐based psycho‐educational intervention, there was no difference among groups or by time, MD 0.54 (95% CI ‐0.23 to 1.31) (Analysis 1.4).
1.4. Analysis.

Comparison 1 Psycho‐education vs control ‐ patients, Outcome 4 Change in caregiver/patient skills.
3. Changes in caregiver‐patient relationship
Family Relationship: child
Two trials assessed family cohesion and adaptability as domains to assess relationships within families in a total of 76 children with SCD (37 children intervened, 39 controls) (Barakat 2010; Kaslow 2000). The 'Family Cohesion Scale' and the 'Family Adaptability and Cohesion Evaluation Scale' (FACES II) scale were used as measures in these studies. Meta‐analysis using random effects revealed an improvement (though not significant) in family relationship assessment in the treated group when compared to controls, MD 4.92 points (95% CI ‐4.22 to 14.06) (Analysis 1.5). There was considerable heterogeneity among the two trials (heterogeneity: Chi² = 6.90, df = 1 (P = 0.009); I² = 86%). The two trials used different intervention modalities, CBT and a family psycho‐educational intervention and this may be contributing to the heterogeneity in this meta‐analysis.
1.5. Analysis.
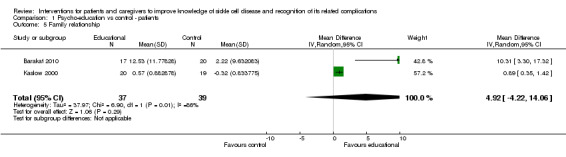
Comparison 1 Psycho‐education vs control ‐ patients, Outcome 5 Family relationship.
4. Caregiver or patient depression or anxiety
Three RCTs with 121 patients (age range seven to 35 years) were included in a random‐effects meta‐analysis for patient depression and anxiety (Kaslow 2000; Ketchen 2006; Thomas 1999). They employed the 'Child Behaviour Checklist', the 'Children's Depression Inventory', and the 'General health Questionnaire‐28' tool respectively as their measures. Psycho‐education sessions were mainly undertaken in a family setting and lasted a maximum of eight weeks. Patients who received the psycho‐educational intervention had a lower depression score compared to the control or no intervention group, SMD 0.66 points (95% CI ‐1.18 to ‐0.14) (Analysis 1.6). There was moderate heterogeneity reported (heterogeneity: Chi² = 1.91, df = 2 (P = 0.39); I² = 49%). The heterogeneity present in the meta‐analysis may be due to the fact that all three studies employed different measurement instruments, and the Thomas study included adults up to age 35 years as well as older children above age 15 years.
1.6. Analysis.
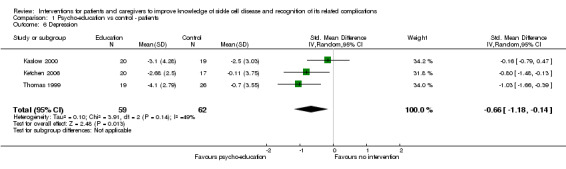
Comparison 1 Psycho‐education vs control ‐ patients, Outcome 6 Depression.
5. Psychological, social and cognitive functioning of the child
Disease self efficacy
One trial reported on patients' confidence in their ability to manage interactions with healthcare providers in 17 patients (age range 12 to 18 years) undergoing a pain management cognitive behaviour intervention and 20 controls (Barakat 2010). The 20‐item 'Disease Self‐Efficacy Scale' was used for the measurement of this outcome and investigators reported a non‐significant difference between groups, MD 0.18 points (95% CI ‐0.11 to 0.47) (Analysis 1.7).
1.7. Analysis.

Comparison 1 Psycho‐education vs control ‐ patients, Outcome 7 Disease self‐efficacy.
Coping
Two separate random‐effects meta‐analyses were performed to assess patient coping skills and attempts. A variety of intervention modalities were employed, with CBT being the most commonly used. Psycho‐education sessions were mainly carried out in hospital or a home setting and lasted a maximum of eight weeks. Three studies used the 'Coping Strategies Questionnaire', whereas other studies employed the child and adolescent versions of the 'Kid‐Cope' measure and the age‐appropriate 'School Age Coping Inventory and Adolescents' (A‐COPE) measures. One meta‐analysis including five RCTs with 208 patients assessed positive coping; 100 patients in intervention group and 108 controls (Barakat 2010; Broome 2001; Hazzard 2002; Schatz 2015; Thomas 1999). Meta‐analysis revealed a non‐significant SMD of 0.46 points (95% CI ‐0.38 to 1.29) (Analysis 1.8). There was significant statistical heterogeneity reported (heterogeneity: Tau² = 0.79; Chi² = 31.93, df = 4 (P = 0.0.0001); I² = 87%). The other meta‐analysis including three trials with 138 patients found lower negative coping attempts, SMD ‐0.82 points (95% CI ‐1.70 to 0.05); however, this was not significant (Analysis 1.9) (Hazzard 2002; Schatz 2015; Thomas 1999). Similar to the analysis of positive coping a considerable level of heterogeneity was observed (heterogeneity: Tau² = 0.50; Chi² = 11.80, df = 2 (P = 0.003); I² = 83%. The diverse types of interventions employed and the measurement tools utilized for the various studies possibly account for much of the heterogeneity in these analyses.
1.8. Analysis.

Comparison 1 Psycho‐education vs control ‐ patients, Outcome 8 Positive coping.
1.9. Analysis.
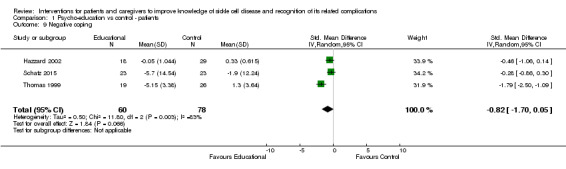
Comparison 1 Psycho‐education vs control ‐ patients, Outcome 9 Negative coping.
Emotional & behavioural difficulties
Kaslow reported using the 'Child Behavioural Checklist' instrument on parent's assessment of the 'Internalizing' and 'Externalizing' emotional and behavioural difficulties faced by their children (age range seven to 16 years; 20 children undergoing six family psycho‐educational sessions as intervention and 19 control children) (Kaslow 2000). Both groups had a reduction in their emotional and behavioural disturbances over time but the MD between groups was not significant, MD 0.47 points (95% CI ‐2.45 to 3.39) (Analysis 1.10).
1.10. Analysis.

Comparison 1 Psycho‐education vs control ‐ patients, Outcome 10 Emotional & behavioural difficulties.
6. Health‐related quality of life of the patient
Two trials reported on the parent's assessment of their child's QoL (using the caregiver version of 'Pediatric Qualiiy of Life Inventory') in 44 children receiving intervention and 55 controls (Daniel 2015; Ketchen 2006). Both trials included a face‐to‐face psycho‐educational intervention ranging from one day (all four sessions lasting between 75 and 90 min done on a single day to reduce burden, followed by three 30‐min booster phone call sessions) to six weeks. A fixed‐effects meta‐analysis showed that parents whose children received the intervention reported a lower score or improved QoL compared to the control group; however, this was not significant (no heterogeneity), MD ‐1.47 points (95% CI ‐5.41 to 2.47) (Analysis 1.11). Additionally, Daniel reported on child assessment of QoL (using the child version of the 'Pediatric Qualiiy of Life Inventory') and the reported improvement in QoL was 0.21 (P value = 0.60) (Daniel 2015).
1.11. Analysis.
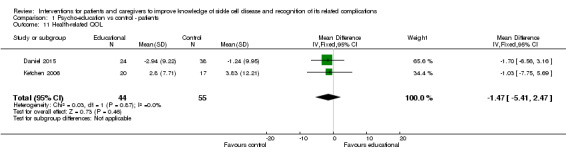
Comparison 1 Psycho‐education vs control ‐ patients, Outcome 11 Health‐related QOL.
Caregiver‐reported outcomes
Caregiver knowledge
A single trial compared 20 families who attended six‐weekly meetings for a one hour psycho‐education session at each time with 19 families who had standard care (Kaslow 2000). They reported a MD in SCD knowledge (measured using the parent version of the 'Sickle Cell Disease Knowledge Test') score of 0.52 points favouring the intervened group (95% CI 0.03 to 1.00) (Analysis 2.1). This improvement in caregiver knowledge was not maintained at six months follow up post intervention.
2.1. Analysis.

Comparison 2 Psycho‐education vs control ‐ caregiver, Outcome 1 SCD knowledge: parent.
Family relationship: parent or caregiver
The Kaslow and Ketchen trials looked at the relationships within families in caregivers/parents of 76 children with SCD (age range seven to 18 years; 37 intervened, 39 controls) (Kaslow 2000; Ketchen 2006). The former utilized the 'Family Adaption and Cohesion Evaluation Scale' (FACES II) scale and latter used the the short form of the 'Interaction Behavior Questionnaire' as measures. A psycho‐educational intervention was used in each trial. A random‐effects meta‐analysis revealed a non‐significant improvement in family relationship assessment in the treatment group compared to controls, MD 1.22 points (95% CI ‐0.24 to 2.68) (Analysis 2.2). Heterogeneity was moderate among the two trials (heterogeneity: Chi² = 3.09, df = 1 (P = 0.08); I² = 68%). The difference in measurement scales employed in the two studies may in part account for this heterogeneity.
2.2. Analysis.

Comparison 2 Psycho‐education vs control ‐ caregiver, Outcome 2 Family relationship.
Discussion
Summary of main results
A comprehensive search of the literature found 12 trials that investigated the effectiveness of interventions for patients and caregivers to improve knowledge of sickle cell disease (SCD) and recognition of its related complications.
Statistically significant improvements were seen in knowledge of SCD in children who received the intervention. The increase in caregivers' knowledge was also significant, but measured in only one small trial. Depression decreased significantly in those who received any intervention.
A meta‐analysis including four trials (160 participants) found that the standardized knowledge score was 0.87 points higher (95% CI 0.28 to 1.45, moderate quality evidence) for patients exposed to psycho‐educational interventions including cognitive behavioral therapy (CBT) and art therapy compared to control patients (Table 1). There was significant heterogeneity (I² = 67%) which was reduced to 0% by the exclusion of one trial with an overall high risk of bias; this also led to the standardized knowledge score increasing to 1.12 points higher after sensitivity analysis. Whether the size of this intervention effect on knowledge is of any clinical benefit is difficult to say. However, the numbers of people recruited were too small to adequately power the studies and the high attrition rates of each study may be part of the reason why the effect sizes were noted to be small. Despite these limitations for each of these studies, there is undoubtedly a trend towards positive effects on knowledge. Better designed studies with more consistently applied interventions are needed to provide more robust results.
Of the three randomized controlled trials (RCTs) (121 participants) assessing the outcome of depression, meta‐analysis showed an improvement in depression scores for those who received a psycho‐educational intervention compared to those who received no educational intervention. This difference was on average ‐0.66 (95% CI ‐1.18 to ‐0.14, moderate quality evidence) (P = 0.01).
Healthcare utilization among those patients receiving a psycho‐educational intervention was higher, though not statistically significant, for: clinic visits, MD 0.90 (95% CI ‐0.95 to 2.76); ER visits, MD 0.10 (95%CI ‐2.35, 2.55); and hospital admissions, MD 0.16 (95%CI ‐0.98 to 1.31) (moderate quality evidence).
Similarly, patient and caregiver interventions made little difference in positive coping attempts and strategies, SMD 0.46 (95%CI ‐0.38 to 1.29) or in negative coping behaviours, SMD ‐0.82 (95% CI ‐1.70 to 0.05) (Table 1).
In the single study that examined the effect on psycho‐educational intervention on caregivers of children with SCD, the knowledge score among caregivers was 0.52 points higher (95% CI 0.03 to 1.00, moderate quality evidence) in the intervention group (Table 2).
What works?
We have found that most trials have employed aspects of CBT to assist in building skills and behaviours to manage pain and other complications associated with SCD. Psycho‐education, involving families especially appears to be effective.
Even though many of the results reported both by the trial authors and within this Cochrane Review appear to be non‐significant, there are many reports of other benefits that have been noted. These include positive behaviours seen in participants, especially those in the intervention groups, such as an increased desire to seek information on SCD, caregivers allowing their children to express greater autonomy, patients seeking healthcare advice in managing problems such as pain and therefore reducing their visits, increasing school attendance and generally increased awareness and engagement regarding their disease.
Overall completeness and applicability of evidence
Most trials were conducted in children and some in young adults, therefore, we are not able to extrapolate results to older people. The wide variety of intervention types and measurement methods for trial outcomes utilized in the various trials, made the present review especially difficult. It is not easy to create similar domains and indices of these measures; however, even the few and varied trials point to some improvements in knowledge and depression, derived from the meta‐analysis of various interventions. The results of this review should encourage healthcare professionals to continue employing these measures. This especially true of the pseudo‐reduction of both patients and their families and CBT approaches, which have been proven to be beneficial in improving knowledge and mental health outcomes in their patients.
Quality of the evidence
Psycho‐educational interventions for patients and caregivers with SCD are often complex and difficult to carry out using conventional methods of clinical randomized trials. Studying their effectiveness therefore requires an understanding for context and the pragmatic approach that may compromise ‘risk of bias’.
The judgement of evidence in this review ranged from low to moderate. The majority of trials were fairly recent with only two trials (Gil 1996; Gil 1997) possibly conducted before the publication of the first Consolidated Standards of Reporting Trials (CONSORT) Statement in 1996 (Begg 1996).
Overall, most risk of bias information from trials was judged as 'unclear', that is, there are potential limitations that are likely to lower confidence in the effect estimate. This resulted in downgrading the quality of evidence by one level for the meta‐analyses (Schünemann 2008a). The trials included in the meta‐analysis for coping were affected by wide confidence intervals, which we assessed as imprecision of the included trials. In addition there were visible variations in both the size and direction of the effect in the same meta‐analysis suggesting inconsistency.
Finally, one trial was primarily found to be at high risk of bias for random sequence generation, allocation concealment and blinding outcome assessors. However, it is not plausible that it would significantly change the direction of the results. A sensitivity analysis based on risk of bias attempting to explain the severe heterogeneity confirmed a stronger effect on improving knowledge when this trial with overall high risk of bias was excluded.
Potential biases in the review process
The strict adherence to Cochrane methodology for conducting systematic review limits bias in the conduct of our review. The review process included at least two authors in critical areas such as selection of potential trials, deciding on inclusion and exclusion of trials, extracting data, risk of bias assessment and conducting analyses. However, it is likely that the review is still affected by potential bias.
Publication bias
Although a comprehensive search was undertaken to include the searching of unpublished work through abstract books of relevant and context‐specific conferences, such as the Caribbean Public Health Agency Annual Meetings and the National Sickle Cell Disease Program Annual Meeting, it is possible that some publications could have been missed. Furthermore, we did not handsearch any relevant journals. The lack of handsearching is a potential limitation but the exhaustive search conducted by the Cochrane Cystic Fibrosis and Genetic Disorders Group's Information Specialist addresses this sufficiently. The search strategy included many key databases and searched websites of relevant organizations.
Bias from missing data
Some trials included in this review were limited by incomplete or unusable data. The review authors made contact with trial authors for additional data with limited success. As a result a few trials were synthesized narratively and not included in the meta‐analyses.
Agreements and disagreements with other studies or reviews
There are few trials that examine the effectiveness of interventions for patients and caregivers to improve knowledge and other outcomes in SCD as well as recognition of its complications and no reviews of reasonable comparability to this one could be found.
Authors' conclusions
Implications for practice.
Our review clearly establishes an effect between patient or caregiver (or both) education and knowledge and understanding of disease, as well as depression associated with disease, even though no clear effects were shown on coping, family relationships, healthcare utilization patterns, patient or caregiver skills in disease management and quality of life.
Education has become an integral part of medical practice and is known to improve self‐management, especially in patients with chronic diseases. This review again highlights the importance of using educational interventions, especially psycho‐education involving both patients and members of their families, as well as training in CBTs, such as distraction and imagery. The effects on depression are important to consider as the presence of depression can result in many adverse physical and other outcomes for the patients.
Implications for research.
The current evidence suggests that patient and caregiver educational programs do offer some benefit to people living with SCD. Additionally using the National Institute of Health management guidelines to develop education outlines for RCT testing can be considered.
In these trials, the majority of participants were persons of African‐Caribbean descent living in the USA, hence it might be worthwhile repeating such trials in different ethnic and cultural groups in varied geographical locations.
Adherence to medication or treatment plans recommended by the healthcare providers was not assessed in these trials. This could be important because if knowledge is gained but not put into practice then clinical outcome would not be seen. Future trials may use a RCT design, where participants are randomly assigned to education or control and measured outcomes are adherence to medications and clinical measures.
Only a limited number of trials have examined whether any of the effects of the interventions are sustained and this is another aspect of work requiring further understanding.
Notes
This review includes details of a previously published protocol (now withdrawn) entitled 'Interventions for caregivers for the recognition of disease‐related complications in children with sickle cell disease' (Quimby 2013).
Acknowledgements
The authors would like to acknowledge the support of the Cystic Fibrosis and Genetic Disorders Group in developing this review.
Appendices
Appendix 1. Glossary of terms
| Term | Meaning |
| Acute chest syndrome | A complication seen in sickle cell disease due to vaso‐occlusion occurring in lungs |
| Allele | An alternative form of a gene at a particular location on the chromosome and responsible for a particular trait |
| HbAS | Sickle cell trait |
| Homozygous | Identical pair of genes for a particular inherited characteristic |
| Hyposplenism | Reduced function of the spleen |
| Incidence | Number of new cases of a disease occurring over a defined period of time |
| Infarct | Tissue death resulting from lack of blood flow to an area |
| Morbidity | A diseased state |
| Mortality | State of death |
| Multifactorial aetiologies | Multiple factors causing a particular complication or presentation |
| Prevalence | Total number of cases with a particular disease measured at a particular time |
| Prophylactic | Any measure taken to prevent disease |
| Vaso‐occlusive crisis | Common complication in people with sickle cell disease due to a blockage of blood flow resulting from sickling in the blood |
Appendix 2. Trials register searches
Search terms considered
Sickle educate education educational psychoeducation psycho‐education psychoeducational psycho‐educational social psychosocial psycho‐social knowledge attitude attitudes self family families parent parents caregiver caregivers promotion promote cognitive behaviour behavioural behaviour behavioural emotion emotions emotional
Strategies
European Medicines Agency (www.clinicaltrialsregister.eu/ctr‐search/search)
Searched 08/04/2016 – 0 results found
+sickle educate education educational psychoeducation psycho‐education psychoeducational psycho‐educational social psychosocial psycho‐social knowledge attitude attitudes self family families parent parents caregiver caregivers promotion promote cognitive behaviour behavioural behaviour behavioural emotion emotions emotional
ClinicalTrials.gov (http://clinicaltrials.gov/)
Searched 08/04/2016 – 45 studies found
Advanced search form:
Study type: interventional studies Condition: sickle Intervention: educate OR psycho‐education OR psycho‐educational OR social OR psychosocial OR psycho‐social OR knowledge OR attitude OR self OR family OR parent OR caregiver OR promote OR cognitive OR behavior OR behaviour OR emotion
WHO International Clinical Trials Registry Platform (http://www.who.int/ictrp/en/)
Searched 11/04/2016 – 12 studies found
Advanced search form:
Condition: Sickle Intervention: educate OR education OR educational OR psychoeducation OR psycho‐education OR psychoeducational OR psycho‐educational OR social OR psychosocial OR psycho‐social OR knowledge OR attitude OR attitudes OR self
Also
Condition: Sickle Intervention: family OR families OR parent OR parents OR caregiver OR caregivers OR promotion OR promote OR cognitive OR behaviour OR behavioural OR behaviour OR behavioural OR emotion OR emotions OR emotional
Data and analyses
Comparison 1. Psycho‐education vs control ‐ patients.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 SCD knowledge: patients | 4 | 160 | Std. Mean Difference (IV, Random, 95% CI) | 0.87 [0.28, 1.45] |
| 2 Sensitivity analysis risk of bias: SCD knowledge: patients | 3 | 113 | Std. Mean Difference (IV, Random, 95% CI) | 1.12 [0.72, 1.52] |
| 3 Healthcare utilization | 2 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 3.1 Clinic visits | 2 | 86 | Mean Difference (IV, Fixed, 95% CI) | 0.90 [‐0.95, 2.76] |
| 3.2 ER Visits | 1 | 44 | Mean Difference (IV, Fixed, 95% CI) | 0.10 [‐2.35, 2.55] |
| 3.3 Hospital admissions | 1 | 46 | Mean Difference (IV, Fixed, 95% CI) | 0.16 [‐0.98, 1.31] |
| 4 Change in caregiver/patient skills | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 5 Family relationship | 2 | 76 | Mean Difference (IV, Random, 95% CI) | 4.92 [‐4.22, 14.06] |
| 6 Depression | 3 | 121 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.66 [‐1.18, ‐0.14] |
| 7 Disease self‐efficacy | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 8 Positive coping | 5 | 208 | Std. Mean Difference (IV, Random, 95% CI) | 0.46 [‐0.38, 1.29] |
| 9 Negative coping | 3 | 138 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.82 [‐1.70, 0.05] |
| 10 Emotional & behavioural difficulties | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 11 Health‐related QOL | 2 | 99 | Mean Difference (IV, Fixed, 95% CI) | ‐1.47 [‐5.41, 2.47] |
Comparison 2. Psycho‐education vs control ‐ caregiver.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 SCD knowledge: parent | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 2 Family relationship | 2 | 76 | Mean Difference (IV, Random, 95% CI) | 1.22 [‐0.24, 2.68] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Barakat 2010.
| Methods | A 2‐group, randomized treatment design was used. Randomization occurred at the end of the baseline assessment. |
|
| Participants | Number of participants: 53 randomized 37 analysed. Inclusion 1. Adolescents between 12 and 18 years old with SCD‐SS or Sß‐thalassemia. 2. Ability to recruit a support person (who could be a relative or a responsible friend) to attend all intervention sessions with them. Exclusion 1. Receiving a medical treatment for SCD to reduce pain (e.g., hydroxyurea or transfusion therapy). 2. Did not speak English; or did not have a caregiver that spoke English. |
|
| Interventions | Four 90‐min sessions: baseline, 2 weeks, 4 weeks, 8 weeks. Family‐based cognitive behavioral pain management intervention for adolescents with SCD education (content developed from existing psycho‐educational programs for youth with SCD) (n = 26). Pain management (training in deep breathing/relaxation, positive coping statements and guided imagery) (n = 27). |
|
| Outcomes | Pre‐, post‐ and at 12 months. 1. Patient and caregiver understanding of SCD and related complications (measured using the 'Sickle Cell Knowledge Questionnaire'). 2. Utilization of health services (measured as counts of clinic visits). 3. Changes in caregiver‐patient relationship (measured as family cohesion). 4. Psychological, social and cognitive functioning of the child (measured as coping attempts and 'Disease Self‐Efficacy' (using Disease self‐efficacy scale)). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Insufficient information to permit judgement of ‘low risk’ or ‘high risk’. |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information to permit judgement of ‘low risk’ or ‘high risk’. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Insufficient information to permit judgement of ‘low risk’ or ‘high risk’. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Insufficient information to permit judgement of ‘low risk’ or ‘high risk’. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 19/53 participants missing at follow up (35.8%). |
| Selective reporting (reporting bias) | Low risk | All outcomes reported in objectives and methods present in results. |
| Other bias | Unclear risk | No other bias anticipated, but insufficient information to permit judgement of ‘low risk’ or ‘high risk’. |
Broome 2001.
| Methods | RCT. Multicentre. Location: USA. Assessment at baseline and 1 year after intervention. |
|
| Participants | Inclusion: male and female African‐American children with SCD divided into 2 age groups: children (5 ‐ 13 years old) and adolescents (14 ‐ 18 years old). Children: 65 (33 boys 32 girls), mean (SD) age 9.2 years (2.1), range 6 ‐ 13.5 years. Adolescents: 32 (11 boys 21 girls), mean (SD) age 15.3 (1.5), range 14 ‐ 18.5 years. |
|
| Interventions | Phase 1 All participants attended self‐care education classes designed to provide them with understanding of their disease and pain associated with it, pain and how to communicate with health professionals about their pain. Schedule: 1 class per week x 2 weeks. Phase 2 Intevention group: CBT for pain: CBT techniques were taught in addition to video tapes, audio tapes and written materials. Control group 1: art therapy: taught ways to express their feelings and concerns about painful events in a supportive sharing environment. Control group 2: attention control groups: engaged in fun activities such as picnics, trips to museum, etc. schedule: 1 class per week x 4 weeks. |
|
| Outcomes | 1. Utilization of health services (measured by number of ER visits / number of clinic visits/ number of hospitalisations). 2. Psychological, social and cognitive functioning of the child (coping) measured by either: a. Schoolagers' coping strategies inventory (SCSI) ‐ 26 item self‐report instrument to measure frequency and effectiveness of coping strategies used by children ages 8 ‐ 12 years; b. Adolescent coping orientation for problem experiences (A‐COPE)‐ 54 item questionnaire with 12 sub‐scales. |
|
| Notes | All participants attendee self‐care education classes designed to provide them with understanding of their disease and pain associated with it, pain and how to communicate with health professionals about their pain. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Insufficient information to permit judgement of ‘low risk’ or ‘high risk’. |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information to permit judgement of ‘low risk’ or ‘high risk’. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Considering the nature of the interventions, the authors judged that the outcome was not likely to be influenced by a lack of blinding. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Insufficient information to permit judgement of ‘low risk’ or ‘high risk’. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Up to 30% attrition and did not use intention‐to‐treat analysis. |
| Selective reporting (reporting bias) | Low risk | All outcomes reported in the objectives and methods were present in the results. |
| Other bias | Unclear risk | No other bias anticipated, but insufficient information to permit judgement of ‘low risk’ or ‘high risk’. |
Daniel 2015.
| Methods | RCT. Multicentre. |
|
| Participants | SCD, exclusion criteria: children with severe developmental delay or children/caregivers with severe psychopathology. | |
| Interventions | Intervention: psycho‐education, 4 sessions, lasting between 75 and 90 min followed by three 30‐min booster phone call sessions. Control: routine care. |
|
| Outcomes | Cognitive functioning and QOL, Pre‐treatment, 6 months post‐treatment. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Stratified by gender in blocks of 10. |
| Allocation concealment (selection bias) | Low risk | Randomization (stratified by gender in blocks of 10) was concealed from the family and the trial team until after completing the baseline assessment when an envelope with randomization status was opened and the family was informed of next steps. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Insufficient information to permit judgement of ‘low risk’ or ‘high risk’. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Insufficient information to permit judgement of ‘low risk’ or ‘high risk’. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 25% not completed. |
| Selective reporting (reporting bias) | Low risk | Both outcomes reported. |
| Other bias | Unclear risk | No other bias anticipated, but insufficient information to permit judgement of ‘low risk’ or ‘high risk’. |
Gil 1996.
| Methods | RCT. Parallel. Single centre. Outpatient clinic. USA. Schedule: 45 min session weekly x 3 weeks, data collected over the course of 3 years. |
|
| Participants | Adults with SCD. 85 approached, 64 randomized and completed the trial; male: 27; female: 37; mean age 33.3 years (SD 9.9). |
|
| Interventions | 1. Cognitive Coping Skills Training Condition (intervention) Patients were trained to used 6 relaxation and distraction strategies where there were laboratory training sessions and home practice tapes. 2. Disease‐Education Control Condition (control) ‐ educated by nurses and health educators in a didactic‐discussion format where educational materials commonly distributed by the SCD centre was used. There were 3 disease‐education sessions focused on hereditary nature of SCD, importance of regular health care maintenance and the incidence of painful episodes. |
|
| Outcomes | 1. Utilization of health services ‐ MANCOVAs used to evaluate measures of SCD pain, and healthcare use (ER visits, hospitalizations, and phone calls or visits to health care providers). 2. Psychological, social and cognitive functioning of the adult patients with SCD (measured using the CSQ‐Coping Attempts Factor). |
|
| Notes | A painful stimulus was applied to determine whether or not there was a change in pain sensitivity to the intervention modalities. All participants were African‐American. Mean (SD) educational level = 13.3 years (1.9) (range 7th grade to graduate work). |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Insufficient information to permit judgement of ‘low risk’ or ‘high risk’. |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information to permit judgement of ‘low risk’ or ‘high risk’. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Considering the nature of the interventions, the authors judged that the outcome was not likely to be influenced by a lack of blinding. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Insufficient information to permit judgement of ‘low risk’ or ‘high risk’. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Insufficient information to permit judgement of ‘low risk’ or ‘high risk’. |
| Selective reporting (reporting bias) | Low risk | All outcomes reported in the objectives and methods were present in the results. |
| Other bias | Unclear risk | No other bias anticipated, but insufficient information to permit judgement of ‘low risk’ or ‘high risk’. |
Gil 1997.
| Methods | RCT. SIngle centre. USA. Schedule: two 45 min sessions 2 weeks apart, baseline at the end of the trial. |
|
| Participants | 49 randomized (25 intervention 24 control). 23 girls, 26 boys, Mean (SD) age 11.9 (3.0) years. (HbSS 67%, HbSC 15%, HbSBthal 18%). Exclusion criteria: CVA. |
|
| Interventions | Cognitive coping skills condition (intervention). 45 min session trained with a variety of coping skills followed by a review session 1 week later. Also had a home practice tapes. Standard care control condition (control). No intervention beyond routine medical care. |
|
| Outcomes | Patient or caregiver (or both) ‐ reported signs and symptoms leading to self‐management (measured using pain on a 10‐point scale). Psychological, social and cognitive functioning of the child (measured using the CSQ‐Negative thinking factor). |
|
| Notes | Number of acute and chronic complications occurring the past year were recorded. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Insufficient information to permit judgement of ‘low risk’ or ‘high risk’. |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information to permit judgement of ‘low risk’ or ‘high risk’. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Considering the nature of the interventions, the authors judged that the outcome was not likely to be influenced by a lack of blinding. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Insufficient information to permit judgement of ‘low risk’ or ‘high risk’. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Insufficient information to permit judgement of ‘low risk’ or ‘high risk’. |
| Selective reporting (reporting bias) | Low risk | All outcomes reported in the objectives and methods were present in the results. |
| Other bias | Unclear risk | No other bias anticipated, but insufficient information to permit judgement of ‘low risk’ or ‘high risk’. |
Hazzard 2002.
| Methods | RCT, single centre, 3‐day curriculum, pre‐ and post‐tests. | |
| Participants | Quasi‐experimental trial. Hospitalized children. 110 children participated in the trial. 60 controls. 47 with SCD (note there was also an asthma group with 63 children). 98% were African‐American. 58% were boys. Mean (SD) age 11.7 (2.71) years. Inclusion criteria: children 8 ‐ 18 years old, hospitalized with SCD (or asthma) for 3 or more days. Exclusion criteria: patients who were hospitalised for less than 3 days, were discharged before completing the post‐test measures, were too ill to complete the trial activities, for whom informed consent could not be obtained who did not complete at least one session of the STARBRIGHT World curriculum if they were in the treatment group were excluded from the final sample. |
|
| Interventions | Intervention: education support via STARBRIGHT World; Four 3‐day curricula were developed according to disease and age (ages 8 – 12 and 13 – 18 years) to guide children’s use of STARBRIGHT World activities. Control: routine care. |
|
| Outcomes | Patient and caregiver understanding of SCD and related complications (measured using the how much do I know about SCD). | |
| Notes | STARBRIGHT World is a private computer network specifically developed for hospitalized children by the STARBRIGHT Foundation. The system includes health‐education information, some from relevant Internet Web sites that are prescreened for appropriateness and accuracy, and some from programs specifically developed by the STARBRIGHT Foundation. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Randomized by months of trial where those hospitalised in months 1 ‐ 7 of the trial were placed in 'control' group. |
| Allocation concealment (selection bias) | High risk | Allocation not concealed (as above). |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Review authors judge that the outcome is not likely to be influenced by lack of blinding. |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | The same staff member who participated in the demonstration / training sessions also sometimes administered the pot‐ test questionnaires to the same child”. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Insufficient information to permit judgement of ‘low risk’ or ‘high risk’ as it was not clear how many persons entered the study. |
| Selective reporting (reporting bias) | Low risk | All outcomes reported in objectives and methods present in results. |
| Other bias | Unclear risk | No other bias anticipated, but insufficient information to permit judgement of ‘low risk’ or ‘high risk’. |
Kaslow 2000.
| Methods | RCT. Single centre. Parallel design. USA. Schedule: 1 hour per week x 6 weeks. |
|
| Participants | 39 participants (24 female, 15 male). Age 7 ‐ 16 years. Mean (SD) age 10 years 2 months (2 years 3 months). HbSS disease = 27. HbSC disease = 11. HbSBthal = 1. All were African‐American. Inclusion criteria (youths were eligible if they met at least one of the following criteria):
Exclusion criteria:
|
|
| Interventions | Experimental condition: weekly meeting of families for family psycho education. Sessions were 1 hour duration. Families paid $10.00/1 hour session for 6 sessions (6 weeks). Families encouraged to invite all family members. Also had access to standard care. Control condition: usual care. Patient and caregiver understanding of SCD and related complications: Changes in caregiver or patient skills; Assessment was done; Pre‐intervention; Immediately post intervention (6 weeks); Follow up (6 months); No follow up assessment done on control group at 6 months because some chose to engage in the experimental intervention. |
|
| Outcomes | Patient and caregiver understanding of SCD and related complications (measured with 'Sickle Cell Disease Knowledge Test' ‐ 10 item disease‐knowledge test). Patient‐caregiver relationship (measured using 'Family Adaptability and Cohesion Evaluation Scale' (FACES II) a 30 item self report instrument). Psychological, social and cognitive functioning of the child (measured by 'Child Behavior Checklist'). Caregiver or patient stress or anxiety (measured using the 'Children's Depression Inventory' (CDI)). Instruments were administered to both the child and caregivers. |
|
| Notes | This was a pilot study. Both children and their parents/legal guardian were educated. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Insufficient information to permit judgement of ‘low risk’ or ‘high risk’. |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information to permit judgement of ‘low risk’ or ‘high risk’. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Review authors judge that the outcome is not likely to be influenced by lack of blinding. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Insufficient information to permit judgement of ‘low risk’ or ‘high risk’. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 35 of 39 completed post test. |
| Selective reporting (reporting bias) | Low risk | All outcomes reported in objectives and methods present in results. |
| Other bias | Unclear risk | No other bias anticipated, but insufficient information to permit judgement of ‘low risk’ or ‘high risk’. |
Ketchen 2006.
| Methods | RCT. Multicenter. USA. Schedule: 1 session per week x 6 weeks. |
|
| Participants | 37 caregiver‐child dyads. Caregivers: 36 females 1 male. Children: 18 females 19 males. Initially 56 African‐American families with at least one child between 8 ‐ 18years. |
|
| Interventions | Intervention: home version of the 'Starbright World' computer program; assessed pre‐intervention and post‐intervention at 2 months. Control: routine care. |
|
| Outcomes | Patient and caregiver understanding of SCD and related complications (measured using the 'how much do I know about sickle cell disease scale' ‐ 2 versions one each for the 8 ‐ 12 age group and 13 ‐ 18 age group). Changes in caregiver or patient skills (measured using the health promotion activities on a Liket‐type scale. Changes in caregiver‐patient relationship (measured using the Interaction behavior questionnaire (IBQ). Caregiver or patient stress or anxiety (measured with the 'Children's Depression inventory' (CDI). Health‐related quality of life of patient; generic or disease specific or both (measured with the caregiver from the 'Pediatric Quality of Life Inventory'). |
|
| Notes | Pilot study. 'Starbright World' is a privately funded web‐based computer network connecting children in over 65 hospitals in the USA and Canada. Scholarships were available to families without computers to get one. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | 90 envelopes containing group assignments generated piror to start of data collection. |
| Allocation concealment (selection bias) | Low risk | Envelopes sealed shuffled numbered and given to families in consecutive order. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Insufficient information to permit judgement of ‘low risk’ or ‘high risk’. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Insufficient information to permit judgement of ‘low risk’ or ‘high risk’. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 34% excluded. |
| Selective reporting (reporting bias) | Low risk | All outcomes reported in objectives and methods present in results. |
| Other bias | Unclear risk | No other bias anticipated, but insufficient information to permit judgement of ‘low risk’ or ‘high risk’. |
Koontz 2004.
| Methods | RCT. Mulitcentre: 22 schools in the USA. |
|
| Participants | 8 ‐ 12 years. 28 randomized, 24 analysed. |
|
| Interventions | Intervention: educational material ‐ package with basic facts, 1 hour with teacher, 1 hour with peers. Control: routine services. |
|
| Outcomes | General knowledge questionnaire: caregiver (teacher and parent) and patient. | |
| Notes | No pre‐values given, only post. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Insufficient information to permit judgement of ‘low risk’ or ‘high risk’. |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information to permit judgement of ‘low risk’ or ‘high risk’. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Insufficient information to permit judgement of ‘low risk’ or ‘high risk’. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Insufficient information to permit judgement of ‘low risk’ or ‘high risk’ |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 24 / 28 (85.7%) participants analysed. |
| Selective reporting (reporting bias) | Low risk | Objective outlined in methods is reported in results. |
| Other bias | Unclear risk | No other bias anticipated, but insufficient information to permit judgement of ‘low risk’ or ‘high risk’. |
Lemanek 2006.
| Methods | RCT. Single centre. |
|
| Participants | 36 children between the ages of 5 ‐ 12 years of age, diagnosed with SCD, and admitted to Columbus Children's Hospital for a VOC. Inclusionary criteria included: enrolment within 24 hours of admission, parental consent and child assent to participate, and no history of neurological impairments. |
|
| Interventions | Intervention group: instructed regarding the inpatient guidelines, supervised to ensure the guidelines were followed correctly, received incentives for following and monitoring the guidelines. Control group: received instruction, but did not receive direct supervision or daily incentives. |
|
| Outcomes | Average length hospital stay. | |
| Notes | Additional unpublished information received from author. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Children were matched by age and randomly assigned to an intervention group or a standard care group. |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information to permit judgement of ‘low risk’ or ‘high risk’. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Insufficient information to permit judgement of ‘low risk’ or ‘high risk’. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Insufficient information to permit judgement of ‘low risk’ or ‘high risk’. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 100% completed post assessments. |
| Selective reporting (reporting bias) | Low risk | Objective outlined in methods is reported in results. |
| Other bias | Unclear risk | No other bias anticipated, but insufficient information to permit judgement of ‘low risk’ or ‘high risk’. |
Schatz 2015.
| Methods | RCT. Multicentrer. USA. |
|
| Participants | Display adherence, 1 major or 3 minor pain episodes in last 6 months, no chronic transfusion, no cognitive limitations. | |
| Interventions | Intervention: CBT. Control: routine care. |
|
| Outcomes | 1 face‐to‐face session, 45 – 60 min; weekly telephone session: 30 – 40 min. Materials on cell phone to take home; follow up 8 weeks. |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Drawing coloured marbles out of opaque bag or computer software using blocks of 10. |
| Allocation concealment (selection bias) | Low risk | Researcher not involved in the trial prepared sequentially numbered opaque sealed envelopes. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Insufficient information to permit judgement of ‘low risk’ or ‘high risk’. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Insufficient information to permit judgement of ‘low risk’ or ‘high risk’. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 4% not completed. |
| Selective reporting (reporting bias) | Low risk | All outcomes reported. |
| Other bias | Unclear risk | No other bias anticipated, but insufficient information to permit judgement of ‘low risk’ or ‘high risk’. |
Thomas 1999.
| Methods | RCT. Multicentre. UK. Parallel design. Schedule: 1 session weekly x 2 months, assessed at 6 and 12 months. |
|
| Participants | 97 randomized. Inclusion criteria:
Exclusion criteria: Are receiving any psychological or psychiatric treatment or medication, or another treatment modality, such as hydroxyurea or a blood transfusion program which could impact on the frequency of painful crises experienced. |
|
| Interventions | CBT: 1‐hour group sessions on a weekly basis over 2 months with a psychologist employing a structured CBT approach based on the INPUT main management program at the St. Thomas' Hospital London. Focused primarily on management of chronic pain. Attention placebo group: weekly hour‐long group sessions designed to provide participant with an opportunity for discussion of the types of problems they encountered in living with SCD and feelings associated with these problems. Control group: received conventional medical treatment. |
|
| Outcomes | Utilization of health services (measured using information concerning the number of accident and emergency visits and hospital admissions for painful crise and duration of hospital stay was collected from each patient at each time point). Data were also collated concerning health service use and associated costs at each interval via a brief questionnaire at each time point. This included the patients’ use of GP, community nurse, social services and charities, and relatives or friends taking time off to look after the management of SCD pain....No results available for this outcome. Caregiver or patient stress or anxiety (measured using the 'General Health Questionnaire–28' (GHQ–28). Psychological, social and cognitive functioning of the child (measured using the 'Coping Strategies Questionnaire, Revised Sickle‐Cell Version for Adults' (CSQR). |
|
| Notes | Other instruments used. 'Pain Self‐Efficacy Questionnaire' (PSEQ). 'Short FormMcGill Pain Questionnaire' (SF–MPQ). 'Beliefs About Pain Control Questionnaire' (BPCQ). |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Random allocation was conducted by means of a sequence of labelled cards contained in sealed numbered envelopes. |
| Allocation concealment (selection bias) | Low risk | The allocation sequence was based on a random number table and was restricted to blocks of 4 balanced to obtain equal numbers in each treatment condition. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Insufficient information to permit judgement of ‘low risk’ or ‘high risk’. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Insufficient information to permit judgement of ‘low risk’ or ‘high risk’. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 44% excluded. |
| Selective reporting (reporting bias) | High risk | 1 of the 3 objectives, namely, utilization of health services was not reported. |
| Other bias | Unclear risk | No other bias anticipated, but insufficient information to permit judgement of ‘low risk’ or ‘high risk’. |
CBT: cognitive behavioural therapy CVA: cerebrovascular accident ER: emergency room RCT: randomized controlled trial SD: standard deviation VOC: vaso‐occlusive crisis
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Applegate 2003 | Irrelevant outcome ‐ participation at appointment. |
| Barakat 2007 | Not a RCT. |
| Boroffice 1991 | Irrelevant outcomes; attitudes not listed in outcomes assessed. |
| Carpenter 2007 | Unsuccessful in contacting authors for necessary information. |
| Chernoff 2002 | Irrelevant intervention; buddy system not considered an educational program. |
| Daly 2012 | Irrelevant intervention ‐ pharmacological. |
| Ireys 2001 | Irrelevant intervention; buddy system not considered an educational program. |
| Jenerette 2014 | Irrelevant outcome ‐ stigma. |
| Kaslow 1997 | Not a RCT. |
| King 2006 | Irrelevant intervention; memory strategy not considered an educational program. |
| King 2007 | Irrelevant intervention, memory strategy not considered an educational program. |
| Lemanek 2009 | Irrelevant intervention; massage therapy not considered an educational program. |
| Mendolia 1993 | Not a RCT. |
| Oyeku 2005 | Irrelevant participant ‐ clinician. |
| Oyeku 2005 b | Irrelevant participant ‐ clinician. |
| Oyeku 2010 | Irrelevant participant ‐ clinician. |
| Radcliffe 2007 | Irrelevant study type, outcome. |
| Robinson 1990 | Unsuccessful in contacting authors for necessary information. |
| Schatz 2004 | Unsuccessful in contacting authors for necessary information. |
| Thomas 2013 | Irrelevant intervention. |
| Wilkie 2013 | Irrelevant participants; SS and AS analysed together. |
| Yang 2000 | Irrelevant participants ‐ SCT. |
| Yerys 2003 | Irrelevant intervention; memory strategy not considered an educational program. |
RCT: randomized controlled trial SCT: sickle cell trait
Characteristics of studies awaiting assessment [ordered by study ID]
Bhogte 2013.
| Methods | Single‐centre RCT. |
| Participants | Adolescents with SCD. |
| Interventions | 30‐min DVD featuring young adults with SCD and an educational website on SCD and transition versus a CD containing motivational music. |
| Outcomes | Questionnaire testing participants on their knowledge of SCD, questionnaire on medical history, the 'Sickle Cell Self‐Efficacy Scale' (SCSES), satisfaction survey. |
| Notes | Abstract only. |
RCT: randomized controlled trial SCD: sickle cell disease
Differences between protocol and review
In our original protocol, the participants were defined as patients (aged 10 years and older) and parents or adult caregivers of children with SCD of all age groups. From our review of the literature, interventions focused on children from as early as six years of age. To be inclusive we extended our age inclusion criteria to include the evidence on this younger age group. We believe it captures important information of the usefulness of interventions for improving knowledge and care in sickle cell disease.
Contributions of authors
Monika Asnani is a co‐lead as well as corresponding author on this review. She was involved in conception and design of the protocol. She contributed to content analysis and writing up of review.
Kim Quimby is a co‐lead author on this review. She was involved in the conception and design of the protocol. She participated in the data management, interpretation of results and writing up of the review.
Damian Francis is a methodologist on this review who participated in the methodological design of the protocol. Damian was responsible for assessing quality of included trials (risk of bias) and data analysis on the review.
Nadia Bennett added methodological skills in data management and content expertise.
Sources of support
Internal sources
None, Other.
External sources
-
National Institute for Health Research, UK.
This systematic review was supported by the National Institute for Health Research, via Cochrane Infrastructure funding to the Cochrane Cystic Fibrosis and Genetic Disorders Group.
Declarations of interest
Monika Asnani: none known.
Kim Quimby: none known.
Nadia Bennett: none known.
Damian Francis: none known.
The first two authors are joint lead authors.
New
References
References to studies included in this review
Barakat 2010 {published data only}
- Barakat LP, Schwartz L, Simon K, Radcliffe J. Coping mediates the association of pain with depression in adolescents with sickle cell disease [abstract]. Proceedings of the 29th Annual Meeting of the National Sickle Cell Disease Program. 2006:187.
- Barakat LP, Schwartz LA, Salamon KS, Radcliffe J. A family‐based randomized controlled trial of pain intervention for adolescents with sickle cell disease. Journal of Pediatric Hematology/Oncology 2010;32(7):540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett KY, Simon K, Barakat LP, Radcliffe J. Factors associated with disease knowledge in adolescence [abstract]. Proceedings of the 29th Annual Meeting of the National Sickle Cell Disease Program. 2006:190.
- Boyd RC, Barakat LP, Thigpen NR, Radcliffe J. The development of a brief psychoeducational intervention for adolescents with sickle cell [abstract]. Proceedings of the 27th Annual Meeting of the National Sickle Cell Disease Program. 2004:84.
- Radcliffe J, Barakat LP, Boyd R, Thigpen N. Sickle cell disease knowledge and self‐efficacy are associated among adolescents and caregivers [abstract]. Proceedings of the 28th Annual Meeting of the National Sickle Cell Disease Program. 2005:119.
- Radcliffe J, Barakat LP, Boyd RC, Thigpen N, Benton TD, Kazak AE. Controlled pain intervention for adolescents with sickle cell disease [abstract]. Proceedings of the 27th Annual Meeting of the National Sickle Sell Disease Program. 2004:57.
- Schwartz L, Radcliffe J, Barakat LP. Pain‐related predictors of parent and family functioning in teens with sickle cell disease [abstract]. Proceedings of the 29th Annual Meeting of the National Sickle Cell Disease Program. 2006:238.
Broome 2001 {published data only}
- Broome ME, Maikler V, Kelber S, Bailey P, Lea G. An intervention to increase coping and reduce health care utilization for school‐age children and adolescents with sickle cell disease. Journal of National Black Nurses' Association: JNBNA 2001;12(2):6‐14. [PubMed] [Google Scholar]
Daniel 2015 {published data only}
- Daniel LC, Li Y, Smith K, Tarazi R, Robinson MR, Patterson CA, et al. Lessons learned from a randomized controlled trial of a family‐based intervention to promote school functioning for school‐age children with sickle cell disease. Journal of Pediatric Psychology 2015;40(10):1085‐94. [CENTRAL: 1102193; CRS: 5500135000001413; PUBMED: 26136404] [DOI] [PMC free article] [PubMed] [Google Scholar]
Gil 1996 {published data only}
- Gil KM, Carson JW, Sedway JA, Porter LS, Schaeffer JJW, Orringer E. Follow‐up of coping skills training in adults with sickle cell disease: analysis of daily pain and coping practice diaries. Health Psychology 2000;19(1):85‐90. [DOI] [PubMed] [Google Scholar]
- Gil KM, Wilson JJ, Abrams MA, Orringer E, Clark WC, Janal MN. Coping skills training in adults with sickle cell disease: Post‐intervention and 3‐month follow‐up results [abstract]. Proceedings of the National Sickle Cell Disease Program Annual Meeting Conference. 1996:8.
- Gil KM, Wilson JJ, Edens JL, Webster DA, Abrams MA, Orringer E, et al. Effects of cognitive coping skills training on coping strategies and experimental pain sensitivity in African American adults with sickle cell disease. Health Psychology 1996;15(1):3. [DOI] [PubMed] [Google Scholar]
Gil 1997 {published data only}
- Gil KM, Anthony KK, Carson JW, Redding‐Lallinger R, Daeschner CW, Ware RE. Daily coping practice predicts treatment effects in children with sickle cell disease. Journal of Pediatric Psychology 2001;26(3):163‐73. [DOI] [PubMed] [Google Scholar]
- Gil KM, Wilson JJ, Edens JL, Workman E, Ready J, Sedway J, et al. Cognitive coping skills training in children with sickle cell disease pain. International Journal of Behavioral Medicine 1997;4(4):364‐77. [DOI] [PubMed] [Google Scholar]
Hazzard 2002 {published data only}
- Hazzard A, Celano M, Collins M. Effects of STARBRIGHT World on knowledge, social support, and coping in hospitalized children with sickle cell disease and asthma. Children's Health Care 2002;31(1):69‐86. [Google Scholar]
Kaslow 2000 {published data only}
- Kaslow NJ, Collins MH, Brown F, Baskin M, Griffith J, Hollins LD, et al. Psychoeducational intervention for pediatric sickle cell disease (SCD) increases disease knowledge [abstract]. Proceedings of the National Sickle Cell Disease Program Annual Meeting Conference. 1997:225.
- Kaslow NJ, Collins MH, Rashid FL, Baskin ML, Griffith JR, Hollins L, et al. The efficacy of a pilot family psychoeducational intervention for pediatric sickle cell disease (SCD). Families, Systems, & Health 2000;18(4):381. [Google Scholar]
Ketchen 2006 {published data only}
- Ketchen B, Hazzard A, Lassiter S, Barber N, Armistead L, Mentz R, et al. STARBRIGHT World: A pilot study of a home‐based sickle cell psychoeducational intervention. Children's Health Care 2006;35(4):321‐38. [Google Scholar]
- Kinlow M, Hazzard A, Adams F, Batts B. Computer based interactive education programs for hospitalized children with asthma and sickle cell disease [abstract]. Respiratory Care. 2001; Vol. 46:1112.
Koontz 2004 {published data only}
- Koontz K, Short AD, Kalinyak K, Noll RB. A randomized, controlled pilot trial of a school intervention for children with sickle cell anemia. Journal of Pediatric Psychology 2004;29(1):7‐17. [DOI] [PubMed] [Google Scholar]
Lemanek 2006 {published and unpublished data}
- Lemanek KL, Ranalli M, Parsons E, DeJacimo L. Effectiveness of auxiliary sickle cell disease management guidelines of children hospitalized with pain [abstract]. Proceedings of the 29th Annual Meeting of the National Sickle Cell Disease Program. 2006:134.
Schatz 2015 {published data only}
- Schatz J, Schlenz AM, McClellan CB, Puffer ES, Hardy S, Pfeiffer M, et al. Changes in coping, pain, and activity after cognitive‐behavioral training: a randomized clinical trial for pediatric sickle cell disease using smartphones. Clinical Journal of Pain 2015;31(6):536‐47. [CENTRAL: 1068102; CRS: 5500133000000033; GR:: F31 HL108582/HL/NHLBI NIH HHS/United States; JID:: 8507389; NLM:: PMC4424076 [Available on 06/01/16]; OID: [Other ID]: NLM: NIHMS639485 [Available on 06/01/16]; PMCID:: PMC4424076; PUBMED: 25503599; R21: HL092336/HL/NHLBI NIH HHS/United States; T32: GM081740/GM/NIGMS NIH HHS/United States] [DOI] [PMC free article] [PubMed] [Google Scholar]
Thomas 1999 {published data only}
- Thomas V. Cognitive behaviour therapy for the management of sickle cell disease pain: an evaluation of a community based intervention [abstract]. Proceedings of the National Sickle Cell Disease Program Annual Meeting Conference. 1999:230.
- Thomas V. Cognitive behavioural therapy for the management of pain in sickle cell disease [abstract]. Proceedings of the National Sickle Cell Disease Program Annual Meeting Conference. 1997:38.
- Thomas V. Cognitive behavioural therapy in pain management for sickle cell disease. International Journal of Palliative Nursing 2000;6(9):434‐42. [DOI] [PubMed] [Google Scholar]
- Thomas V. The longitudinal effects of cognitive behaviour therapy for the management of sick cell disease pain [abstract]. Proceedings of the National Sickle Cell Disease Program Annual Meeting Conference. 2000:15.
- Thomas V, Dixon A, Milligan P, Thomas N. Cognitive‐behaviour therapy for the management of sickle cell disease pain: An evaluation of a community‐based intervention. British Journal of Health Psychology 1999;4(3):209‐29. [Google Scholar]
References to studies excluded from this review
Applegate 2003 {published data only}
- Applegate H, Webb PM, Elkin TD, Neul SKT, Drabman RS. Improving parent participation at pediatric diabetes and sickle cell appointments using a brief intervention. Children's Health Care 2003;32(2):125‐36. [Google Scholar]
Barakat 2007 {published data only}
- Barakat LP, Schwartz LA, Simon K, Radcliffe J. Negative thinking as a coping strategy mediator of pain and internalizing symptoms in adolescents with sickle cell disease. Journal of Behavioral Medicine 2007;30(3):540‐7. [DOI] [PubMed] [Google Scholar]
Boroffice 1991 {published data only}
- Boroffice OB. Self‐help in sickle‐cell disease in Nigeria. Health Promotion International 1991;6(4):241‐6. [Google Scholar]
Carpenter 2007 {published data only}
- Carpenter S, Barakat LP, Radcliffe J. Health education in pediatric sickle cell disease: facilitating increased knowledge by using games [abstract]. Proceedings of the 35th Anniversary Convention of the National Sickle Cell Disease Program. 2007:347.
Chernoff 2002 {published data only}
- Chernoff RG, Ireys HT, DeVet KA, Kim YJ. A randomized controlled trial of a community‐based support program for families of children with chronic illnesses: pediatric outcomes. Archies of Pediatrics and Adolescents Medicine 2002;156(6):533‐9. [DOI] [PubMed] [Google Scholar]
Daly 2012 {published data only}
- Daly B, Kral MC, Brown RT, Elkin D, Madan‐Swain A, Mitchell M, et al. Ameliorating attention problems in children with sickle cell disease: a pilot study of methylphenidate. Journal of Developmental and Behavioral Pediatrics 2012;33(3):244‐51. [Record 14: 6125 ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Ireys 2001 {published data only}
- Ireys HT, Chernoff R, DeVet KA, Kim Y. Maternal outcomes of a randomized controlled Trial of a community‐based support Program for families of children with chronic illnesses. Archies of Pediatrics and Adolescents Medicine 2001;155(7):771‐7. [DOI] [PubMed] [Google Scholar]
Jenerette 2014 {published data only}
- Jenerette CM, Brewer CA, Edwards LJ, Mishel MH, Gil KM. An intervention to decrease stigma in young adults with sickle cell disease. Western Journal of Nursing Research 2014;36(5):599‐619. [CENTRAL: 1068104; CRS: 5500133000000035; GR:: K23NR011061/NR/NINR NIH HHS/United States; JID:: 7905435; PUBMED: 24309381; UL1TR000083/TR/NCATS: NIH HHS/United States] [DOI] [PubMed] [Google Scholar]
Kaslow 1997 {published data only}
- Kaslow NJ, Collins MH, Loundy MR, Brown F, Hollins LD, Eckman J. Empirically validated family interventions for pediatric psychology: sickle cell disease as an exemplar. Journal of Pediatric Psychology 1997;22(2):213‐27. [DOI] [PubMed] [Google Scholar]
King 2006 {published data only}
- King AA, Whit A, Armstrong M, McKinstry R, Noetzel M, DeBaun M. An educational remediation program for children with sickle cell disease and cerebral infarcts is feasible and effective [abstract]. Proceedings of the 29th Annual Meeting of the National Sickle Cell Disease Programme. 2006.
King 2007 {published data only}
- King AA, White DA, McKinstry RC, Noetzel M, Debaun MR. A pilot randomized education rehabilitation trial is feasible in sickle cell and strokes. Neurology 2007;68(23):2008‐11. [CENTRAL: 700281; CRS: 5500050000000263; PUBMED: 17548550] [DOI] [PubMed] [Google Scholar]
Lemanek 2009 {published data only}
- Lemanek KL, Ranalli M, Lukens C. A randomized controlled trial of massage therapy in children with sickle cell disease. Journal of Pediatric Psychology 2009;34(10):1091–6. [DOI] [PubMed] [Google Scholar]
Mendolia 1993 {published data only}
- Mendolia SM, Freed N. Sickle Cell Disease ‐ a comprehensive education for the adult patient [abstract]. Proceedings of the 18th Annual Meeting of the National Sickle Cell Disease Programme. 1993.
Oyeku 2005 {published data only}
- Oyeku SO, Feldman HA, Ryan K, Muret‐Wagstaff S, Neufeld EJ. Clinician knowledge and confidence about newborn screening for sickle cell disease: randomized assessment of educational strategies [abstract]. Proceedings of the 28th Annual Meeting of the National Sickle Cell Disease Program. 2005. [DOI] [PMC free article] [PubMed]
Oyeku 2005 b {published data only}
- Oyeku SO, Feldman HA, Ryan K, Muret‐Wagstaff S, Neufeld EJ. Primary care provider knowledge and confidence about newborn screening for sickle cell disease [abstract]. Proceedings of the Pediatric Academic Societies Annual Meeting. 2005.
Oyeku 2010 {published data only}
- Oyeku SO, Feldman HA, Ryan K, Muret‐Wagstaff S, Neufeld EJ. Primary care clinicians' knowledge and confidence about newborn screening for sickle cell disease: randomized assessment of educational strategies. Journal of the National Medical Association 2010;102(8):676‐82. [DOI] [PMC free article] [PubMed] [Google Scholar]
Radcliffe 2007 {published data only}
- Radcliffe J, Barakat L, Simon K, Schwartz LA, Ohene‐Frempong K. Attitude counts: self‐efficacy, negative thinking and health care costs in adolescent sickle cell disease within the teens taking control study [abstract]. Proceedings of the 35th Anniversary Convention of the National Sickle Cell Disease Program and the Sickle Cell Disease Association of America; 2007 Sep 17‐22; Washington DC, USA . 2007:93, Abstract No: 79. [CRS: 5500125000000683]
Robinson 1990 {published data only}
- Robinson GA, Devane J. Impact of home health care on sickle cell disease patients [abstract]. Proceedings of the National Sickle Cell Disease Program Annual Meeting Conference. 1990:113.
Schatz 2004 {published data only}
- Schatz J, Robinson J, Puffer E. Promoting school success following childhood stroke: standard school advocacy versus a neuropsychologically‐informed team planning approach [abstract]. Proceedings of the 27th Annual Meeting of the National Sickle Cell Disease Program. 2004:180.
Thomas 2013 {published data only}
- Thomas LS, Stephenson N, Swanson M, Jesse DE, Brown S. A pilot study: the effect of healing touch on anxiety, stress, pain, pain medication usage, and physiological measures in hospitalized sickle cell disease adults experiencing a vaso‐occlusive pain episode. Journal of Holistic Nursing 2013;31(4):234‐47. [CENTRAL: 1068103; CRS: 5500133000000034; JID:: 8506709; PUBMED: 23817144] [DOI] [PubMed] [Google Scholar]
Wilkie 2013 {published data only}
- Wilkie DJ, Gallo AM, Yao Y, Molokie RE, Stahl C, Hershberger PE, et al. Reproductive Health CHOICES for young adults with sickle cell disease or trait: randomized controlled trial immediate post test effects. Nursing Research 2013;62(5):352‐61. [DOI] [PMC free article] [PubMed] [Google Scholar]
Yang 2000 {published data only}
- Yang YM, Andrews S, Peterson R, Shah A, Cepeda M. Prenatal sickle cell screening education effect on the follow‐up rates of infants with sickle cell trait. Patient Education and Counselling 2000;39(2‐3):185‐9. [DOI] [PubMed] [Google Scholar]
Yerys 2003 {published data only}
- Yerys BE, White DA, Salorio CF, McKinstry R, Moinuddin A, DeBaun M. Memory strategy training in children with cerebral infarcts related to sickle cell disease. Journal of Pediatic Hematology/Oncology 2003;25(6):495‐8. [DOI] [PubMed] [Google Scholar]
References to studies awaiting assessment
Bhogte 2013 {published data only}
- Bhogte C, Nickel R, Hsu L, Meier E. Pilot intervention to improve sickle cell disease transition with a DVD and website shows education of adolescents should occur in the outpatient clinic [abstract]. Pediatric Blood & Cancer 2013;60:S27‐8. [CENTRAL: 1027515; CRS: 5500050000000264; EMBASE: 71047861] [Google Scholar]
Additional references
Abidin 1995
- Abidin RR. Parenting Stress Index (PSI). Lutz, Florida, USA: Psychological Assessment Resources Inc, 1995. [Google Scholar]
Altman 1996
- Altman DG, Bland JM. Statistics Notes: Detecting skewness from summary information. BMJ 1996;313(7066):1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Anie 2005
- Anie KA. Psychological complications in sickle cell disease. British Journal of Haematology 2005;129(6):723–9. [DOI] [PubMed] [Google Scholar]
Anie 2015
- Anie KA, Green J. Psychological therapies for sickle cell disease and pain. Cochrane Database of Systematic Reviews 2015, Issue 5. [DOI: 10.1002/14651858.CD001916.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
Armstrong 1993
- Armstrong FD, Lemanek KL. Sickle Cell Knowledge Questionnaire. University of Miami School of Medicine 1993.
Ataga 2000
- Ataga KI, Orringer EP. Renal abnormalities in sickle cell disease. American Journal of Hematology 2000;63(4):205–11. [DOI] [PubMed] [Google Scholar]
Barlow 2010
- Barlow J, Smailagic N, Ferriter M, Bennett C, Jones H. Group‐based parent‐training programmes for improving emotional and behavioural adjustment in children from birth to three years old. Cochrane Database of Systematic Reviews 2010, Issue 3. [DOI: 10.1002/14651858.CD003680.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Begg 1996
- Begg C, Cho M, Eastwood S, et al. Improving the quality of reporting of randomized controlled trials: The consort statement. JAMA 1996;276(8):637‐9. [DOI] [PubMed] [Google Scholar]
Bhatt 2011
- Bhatt K, Reid ME, Lewis NA, Asnani MR. Knowledge and health beliefs of Jamaican adolescents with sickle cell disease. Pediatric Blood & Cancer 2011;57(6):1044‐8. [DOI] [PubMed] [Google Scholar]
Bodenheimer 2002
- Bodenheimer T, Wagner EH, Grumbach K. Improving primary care for patients with chronic illness: the chronic care model, Part 2. JAMA 2002;288(15):1909‐14. [DOI] [PubMed] [Google Scholar]
Brousse 2012
- Brousse V, Elie C, Benkerrou M, Odièvre MH, Lesprit E, Bernaudin F, et al. Acute splenic sequestration crisis in sickle cell disease: cohort study of 190 paediatric patients. British Journal of Haematology 2012;156(5):643‐8. [DOI] [PubMed] [Google Scholar]
Brown 1993
- Brown RT, Armstrong FD, Eckman JR. Neurocognitive aspects of pediatric sickle cell disease. Journal of Learning Disabilities 1993;26(1):33–45. [DOI] [PubMed] [Google Scholar]
Burlew 1989
- Burlew AK, Evans R, Oler C. The impact of a child with sickle cell disease on family dynamics. Annals of the New York Academy of Sciences 1989;565:161‐71. [DOI] [PubMed] [Google Scholar]
Deeks 2011
- Deeks J, Higgins J, Altman D. Chapter 9 Analysing data and undertaking meta‐analysis. In: Higgins JPT, Green S editor(s). Cochrane Handbook for Systematic Reviews of Interventions. Version 5.1.0. Available from www.cochrane‐handbook.org. Wiley, 2011. [Google Scholar]
Delis 1988
- Delis DC, Freeland J, Kramer JH, Kaplan E. Integrating clinical assessment with cognitive neuroscience: construct validation of the California Verbal Learning Test. Journal of Consulting and Clinical Psychology 1988;56(1):123‐30. [PUBMED: 3346437] [DOI] [PubMed] [Google Scholar]
Di Sabatino 2011
- Sabatino A, Carsetti R, Corazza GR. Post‐splenectomy and hyposplenic states. Lancet 2011;378(9785):86–97. [DOI] [PubMed] [Google Scholar]
Ebert 2010
- Ebert EC, Nagar M, Hagspiel KD. Gastrointestinal and hepatic complications of sickle cell disease. Clinical Gastroenterology and Hepatology 2010;8(6):483–9. [DOI] [PubMed] [Google Scholar]
Guyatt 2011
- Guyatt GH, Oxman AD, Schunemann HJ, Tugwell P, Knotterus A. GRADE guidelines: a new series of articles in the Journal of Clinical Epidemiology. Journal of Clinical Epidemiology 2011;64(4):380‐2. [DOI] [PubMed] [Google Scholar]
Higgins 2003
- Higgins JP, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta‐analyses. BMJ (Clinical research ed.) 2003;327(7414):557‐60. [PUBMED: 12958120] [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2011a
- Higgins JPT, Altman DG (editors). Chapter 8: Assessing risk of bias in included studies. In: Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Higgins 2011b
- Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org 2011.
Hill 2008
- Hill C, Maskowitz K, Danis B, Wakschlag L. Validation of a clinically sensitive, observational coding system for parental behaviors: The Parenting Clinical Observation Schedule. Parenting: Science and Practice 2008;8(2):153‐85. [DOI] [PMC free article] [PubMed] [Google Scholar]
Hurtig 1994
- Hurtig AL. Relationships in families of children and adolescents with sickle cell disease. Journal of Health & Social Policy 1994;5(3‐4):161‐83. [DOI] [PubMed] [Google Scholar]
Imdad 2011
- Imdad A, Yakoob MY, Bhutta ZA. Impact of maternal education about complementary feeding and provision of complementary foods on child growth in developing countries. BMC Public Health 2011;11 Suppl 3:S25. [DOI] [PMC free article] [PubMed] [Google Scholar]
Ingram 1959
- Ingram VM. Abnormal human haemoglobins. III. The chemical difference between normal and sickle cell haemoglobins. Biochimica et Biophysica Acta 1959;36:402–11. [DOI] [PubMed] [Google Scholar]
Kral 2001
- Kral MC, Brown RT, Hynd GW. Neuropsychological aspects of pediatric sickle cell disease. Neuropsychology Review 2001;11(4):179–96. [DOI] [PubMed] [Google Scholar]
Landgraf 1998
- Landgraf JM, Maunsell E, Speechley KN, Bullinger M, Campbell S, Abetz L, et al. Canadian‐French, German and UK versions of the Child Health Questionnaire: methodology and preliminary item scaling results. Quality of Life Research: An International Journal of Quality of Life Aspects of Treatment, Care and Rehabilitation 1998;7(5):433‐45. [PUBMED: 9691723] [DOI] [PubMed] [Google Scholar]
Leikin 1989
- Leikin SL, Gallagher D, Kinney TR, Sloane D, Klug P, Rida W. Mortality in children and adolescents with sickle cell disease. Cooperative Study of Sickle Cell Disease. Pediatrics 1989;84(3):500‐9. [PubMed] [Google Scholar]
Maxwell 1999
- Maxwell K, Streetly A, Bevan D. Experiences of hospital care and treatment‐seeking behavior for pain from sickle cell disease: qualitative study. Western Journal of Medicine 1999;171(5‐6):306‐13. [PMC free article] [PubMed] [Google Scholar]
Miller 2011
- Miller ST. How I treat acute chest syndrome in children with sickle cell disease. Blood 2011;117(20):5297‐305. [DOI] [PubMed] [Google Scholar]
Modell 2008
- Modell B, Darlison M. Global epidemiology of haemoglobin disorders and derived service indicators. Bulletin of the World Health Organization 2008;86(6):480–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
NICE 2007
- National Institute for Clinical Excellence (NICE), Social Care Institute for Excellence (SCIE). Parent‐training/education programmes in the management of children with conduct disorders. NICE Technology Appraisal Guidance 102. www.nice.org.uk/TA102 (accessed 13 Feb 2014).
Ohene‐Frempong 1998
- Ohene‐Frempong K, Weiner SJ, Sleeper LA, Miller ST, Embury S, Moohr JW, et al. Cerebrovascular accidents in sickle cell disease: Rates and risk factors. Blood 1998;91(1):288–94. [PubMed] [Google Scholar]
Pauling 1964
- Pauling L. Molecular disease and evolution. Bulletin of the New York Academy of Medicine 1964;40(5):334–42. [PMC free article] [PubMed] [Google Scholar]
Pham 2000
- Pham PT, Pham PC, Wilkinson AH, Lew SQ. Renal abnormalities in sickle cell disease. Kidney International 2000;57(1):1–8. [DOI] [PubMed] [Google Scholar]
Quinn 2004
- Quinn CT, Rogers ZR, Buchanan GR. Survival of children with sickle cell disease. Blood 2004;103(11):4023‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Rees 2010
- Rees DC, Williams TN, Gladwin MT. Sickle‐cell disease. Lancet 2010;376(9757):2018–31. [DOI] [PubMed] [Google Scholar]
Robin 1990
- Robin AL, Koepke T, Moye A. Multidimensional assessment of parent‐adolescent relations. Psychological Assessment 1990;2(4):451‐9. [Google Scholar]
Sawyer 2005
- Sawyer SM, Aroni RA. Self‐management in adolescents with chronic illness. What does it mean and how can it be achieved?. Medical Journal of Australia 2005;183(8):405‐9. [DOI] [PubMed] [Google Scholar]
Schünemann 2008
- Schünemann HJ, Oxman AD, Higgins JPT, Vist GE, Glasziou P, Guyatt GH. Chapter 11: Presenting results and ‘Summary of findings’ tables. In: Higgins JPT, Green S editor(s). Cochrane Handbook for Systematic Reviews of Interventions. Chichester: John Wiley & Sons, Ltd, 2008:335‐57. [Google Scholar]
Schünemann 2008a
- Schünemann HJ, Oxman AD, Vist GE, Higgins JPT, Deeks JJ, Glasziou P, et al. Chapter 12: Interpreting results and drawing conclusions. In: Higgins JPT, Green S editor(s). Cochrane Handbook for Systematic Reviews of Interventions. Chichester: John Wiley & Sons, Ltd, 2008:359‐87. [Google Scholar]
Steen 2003
- Steen RG, Miles MA, Helton KJ, Strawn S, Wang W, Xiong X, et al. Cognitive impairment in children with hemoglobin SS sickle cell disease: relationship to MR imaging findings and hematocrit. American Journal of Neuroradiology 2003;24(3):382–9. [PMC free article] [PubMed] [Google Scholar]
Streisand 2001
- Streisand R, Braniecki S, Tercyak KP, Kazak AE. Childhood illness‐related parenting stress: the pediatric inventory for parents. Journal of Pediatric Psychology 2001;26(3):155‐62. [PUBMED: 11259517] [DOI] [PubMed] [Google Scholar]
Stuart 2004
- Stuart MJ, Nagel RL. Sickle‐cell disease. Lancet 2004;364(9442):1343–60. [DOI] [PubMed] [Google Scholar]
Tanabe 2010
- Tanabe P, Porter J, Creary M, Kirkwood E, Miller S, Ahmed‐Williams E, et al. A qualitative analysis of best self‐management practices: sickle cell disease. Journal of the National Medical Association 2010;102(11):1033‐41. [DOI] [PubMed] [Google Scholar]
Tyrer 2005
- Tyrer P, Nur U, Crawford M, Karlsen S, McLean C, Rao B, et al. The Social Functioning Questionnaire: a rapid and robust measure of perceived functioning. International Journal of Social Psychiatry 2005;51(3):265‐75. [PUBMED: 16252794] [PubMed] [Google Scholar]
Weatherall 2001
- Weatherall DJ, Clegg JB. Inherited haemoglobin disorders: an increasing global health problem [Inherited haemoglobin disorders: an increasing global health problem]. Bulletin of the World Health Organization 2001;79(8):704‐12. [S0042‐96862001000800005 [pii]] [PMC free article] [PubMed] [Google Scholar]
Welch 2012
- Welch V, Petticrew M, Tugwell P, Moher D, O'Neill J, Waters E, et al. PRISMA‐Equity 2012 extension: reporting guidelines for systematic reviews with a focus on health equity. PLoS Medicine 2012;9(10):e1001333. [DOI] [PMC free article] [PubMed] [Google Scholar]