

Abstract
Background
Gastric cancer is the fifth most common cancer and third leading cause of cancer‐related deaths worldwide. Complete resection of the whole tumor remains the only approach to treat this malignant disease. Since gastric cancer is usually asymptomatic in its early stages, many people are diagnosed at an advanced stage when the tumor is inoperable. In addition, because other conventional treatments (radiotherapy and chemotherapy) have only modest efficacy for those with advanced/metastatic gastric cancer, the prognosis in such cases is poor. Recently, trials have provided some promising results regarding molecular‐targeted therapy, raising the possibility that the development of these agents could be a fruitful approach. However, the benefit of molecular‐targeted therapy for advanced gastric cancer remains inconclusive.
Objectives
To evaluate the efficacy and safety of molecular‐targeted therapy , either alone or in combination with chemotherapy, in people with advanced gastric cancer.
Search methods
We searched the following databases (from inception to December 2015): the Cochrane Central Register of Controlled Trials (CENTRAL), MEDLINE, EMBASE, and CINAHL. In addition, we searched the reference lists of included trials and contacted experts in the field.
Selection criteria
We searched for randomized controlled trials (RCTs) in adults (aged 18 years or older) with histologically‐confirmed advanced adenocarcinoma of the stomach/gastro‐esophageal junction. Trials of participants with esophageal adenocarcinoma were also considered to be eligible. The eligible trials should aim to evaluate the effects of molecular‐targeted agents on participants' prognosis.
Data collection and analysis
Two review authors independently performed selection of eligible trials, assessment of trial quality, and data extraction. We used methods of survival analysis and expressed the intervention effect as a hazard ratio (HR) when pooling time‐to‐event data, and calculated the odds ratio (OR) for dichotomous data and mean differences (MDs) for continuous data, with 95% confidence intervals (CI).
Main results
We included 11 studies randomizing 4014 participants to molecular‐targeted therapy plus conventional chemotherapy or chemotherapy alone. Five were at low risk of bias, and we considered the risk of bias in the other six studies to be high, mainly due to their open‐label design. All identified studies reported data regarding survival. We found low‐quality evidence that molecular‐targeted may have a small effect on mortality (HR 0.92, 95% CI 0.80 to 1.05, 10 studies) compared with conventional chemotherapy alone. Similarly, it may have little effect on progression‐free survival when compared with conventional chemotherapy alone (HR 0.90, 95% CI 0.78 to 1.04, 11 studies; low‐quality evidence). We did not find evidence from subgroup analysis that survival outcomes differed by type of molecular‐targeted agent (EGFR‐ or VEGF‐targeting agents) or tumor type, meaning that we were unable to explain the variation in effect across the studies by the presence or absence of prognostic biomarkers or type of molecular‐targeted agent. From 11 eligible trials, we were able to use data from 3723 participants with measurable tumors. We found low‐quality evidence that molecular‐targeted therapy may increase tumor response (OR 1.24, 95% CI 1.00 to 1.55, low‐quality evidence). Data from one small trial were too limited to determine the effect of treatment on quality of life (very low‐quality evidence). The addition of targeted therapy to chemotherapy probably increases the risk of adverse events (OR 2.23, 95% CI 1.27 to 3.92, 5 trials, 2290 participants, moderate‐quality evidence) and severe adverse event (OR 1.19, 95% CI 1.03 to 1.37, 8 trials, 3800 participants), compared with receiving chemotherapy alone.
Authors' conclusions
There is uncertainty about the effect of adding targeted therapy to chemotherapy on survival outcomes in people with advanced gastric cancer, with very little information on its impact on quality of life. There is more certain evidence of increased risk of adverse events and serious adverse events. The main limitation of the evidence for survival outcomes was inconsistency of effects across the studies, which we could not explain by prespecified subgroups in terms of the type of therapy or tumor type. Ongoing studies in this area are small and unlikely to improve our understanding of the effects of targeted therapy, and larger studies are needed.
Plain language summary
Effect of molecular‐targeted therapy on the progress and survival of people in the late stages of stomach cancer
Review question
Does molecular‐targeted therapy (a type of treatment specifically targeting cancer cells) benefit people with late‐stage stomach cancer?
Background
Due to the lack of clinical symptoms, many stomach cancers are diagnosed at a very late stage (stage III or stage IV), for which surgery cannot be the best option anymore. The effects of chemotherapy and radiotherapy on late‐stage stomach cancer are very limited, leading to a low possibility of survival for people with the disease (fewer than one in five people survive for longer than five years). Recent research suggested that molecular‐targeted therapy may prolong the survival time for people with late‐stage stomach cancer. However, the therapeutic benefit of this treatment is still under debate.
Study characteristics
We searched databases until December 2015 for randomized controlled trials (clinical trials where people are randomly allocated to one of two or more treatment groups) in adults (aged 18 years or over), diagnosed with late‐stage stomach cancer. We found 11 trials (4014 participants) that met our selection requirements and randomized people to receive targeted treatment plus chemotherapy or chemotherapy alone.
Key results
Adding molecular‐targeted treatment to chemotherapy may have a small effect on survival and on stopping further development of the disease, compared with chemotherapy alone, but the evidence is of low quality. The treatment may increase the likelihood that tumors get smaller (low‐quality evidence), but there is insufficient evidence to know how much of a difference it can make to the person's quality of life (very low‐quality evidence). It probably increases the risk of adverse events and serious adverse events (moderate‐quality evidence).
Quality of the evidence
Currently, the evidence is of low quality for survival outcomes, mainly due to the type of study design, and the inconsistencies between the results of individual studies. We therefore suggest that well‐designed clinical trials should be performed, to improve the evidence base.
Summary of findings
Summary of findings for the main comparison. Summary of findings: Molecular‐targeted therapy plus chemotherapy compared to chemotherapy alone for people with advanced gastric cancer.
| Molecular‐targeted therapy plus chemotherapy compared to chemotherapy alone for people with advanced gastric cancer | ||||||
| Patient or population: people with advanced gastric cancer Setting: hospital Intervention: molecular‐targeted therapy plus chemotherapy Comparison: chemotherapy alone | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk with chemotherapy alone | Corresponding risk with molecular‐target therapy plus chemotherapy | |||||
| Overall survival | Study population:12 month mortality rate | HR 0.92 (0.80 to 1.05) | 3843 participants (10 RCTs) |
⊕⊕⊝⊝ LOW 1,2 | Assumed risk calculated based on 12 months overall mortality rates (extracted from corresponding Kaplan‐Meier curves) observed in the control arms of the included trials (53.6%). We could not explain variation in the effect by subgroup analyses examining the type of molecular targeted agent or tumour type. |
|
| 536 per 1000 | 507 per 1000 (459 to 553) | |||||
| Progression‐free survival | Study population: 6 month progression‐free survival rate | HR 0.90 (0.78 to 1.04) | 4014 participants (11 RCTs) |
⊕⊕⊝⊝ LOW 2,3,4 | Assumed risk calculated based on 6‐month progression rate (extracted from corresponding Kaplan‐Meier curves) observed in the control arms of the included trials (55.5%) We could not explain variation in the effect by subgroup analyses examining the type of molecular targeted agent or tumour type. |
|
| 555 per 1000 | 517 per 1000 (432 to 577) | |||||
|
Quality of life (change from baseline in EORTC QOL30 global health status scale) Duration of follow‐up: Median follow‐up differed between treatment groups (28 months with molecular‐targeted therapy and 23 months in control group) |
Higher change scores were obtained for chemotherapy‐alone group (MD 10 ± 33.9 SD) than molecular‐targeted therapy plus chemotherapy group (MD 0.0 ± 28.1 SD), but the results are based on a very small number of participants and the confidence interval is wide | 53 participants (1 RCT) |
⊕⊝⊝⊝ VERY LOW 5,6 | |||
|
Overall response rate Duration of follow‐up: varied considerably between studies (the median follow‐up time ranged from 5.3 months to 28.5 months) |
Study population | OR 1.24 (1.00 to 1.55) | 3723 (11 RCTs) | ⊕⊕⊝⊝ LOW 2,4 | ||
| 365 per 1000 | 417 per 1000 (365 to 472) | |||||
|
Adverse event (any) Duration of follow‐up: varied considerably between studies (the median follow‐up time ranged from 5.3 months to 28.5 months) |
Study population | OR 2.23 (1.27 to 3.92) | 2290 (5 RCTs) | ⊕⊕⊕⊝ MODERATE 7 | ||
| 962 per 1000 | 983 per 1000 (971 to 990) | |||||
|
Severe adverse event (grade ≥ 3 ) Duration of follow‐up: varied considerably between studies (the median follow‐up time ranged from 5.3 months to 28.5 months) |
Study population | OR 1.19 (1.03 to 1.37) | 3800 (8 RCTs) | ⊕⊕⊕⊝ MODERATE8 | ||
| 669 per 1000 | 707 per 1000 (676 to 735) | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; OR: Odds ratio; HR: Hazard ratio. | ||||||
| GRADE Working Group grades of evidence High quality: We are very confident that the true effect lies close to that of the estimate of the effect Moderate quality: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low quality: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low quality: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
1Downgraded one level due to imprecision: the confidence interval obtained under both the primary and sensitivity analysis for risk of bias includes potentially meaningful differences with either intervention.
2Downgraded one level due to inconsistency: The size and direction of effect varied across the studies (significant heterogeneity was detected).
3Removal of studies at high risk of performance bias reduced the statistical heterogeneity and increased the effect size, so we did not downgrade for risk of bias.
4Downgraded one level due to imprecision: the confidence interval around the effect includes both meaningful benefit and little or no effect of molecular‐targeted therapy.
5Downgraded two levels due to imprecision: effect was estimated based on results from 53 participants and the confidence interval includes appreciable harm and little/no effect.
6Downgraded one level due to serious risk of bias: open‐label study was at high risk of performance bias for this outcome.
7Downgraded one level due to risk of bias (selective reporting bias): three included studies did not provide data on this outcome, and three others had no summary data for meta‐analysis.
8Downgraded one level due to risk of bias (performance bias): removing studies at high risk bias due to lack of blinding resulted in a more imprecise effect suggesting that the results may have been sensitive to the exclusion of open‐label studies.
Background
Description of the condition
Gastric cancer is the fifth most common cancer and third leading cause of cancer‐related deaths worldwide (Ferlay 2015). The incidence of gastric cancer has been shown to have declined in many developed countries. However, in recent years the decline appears to be slowing in both men and women. In Sweden, there has been an increased prevalence of atrophic gastritis, a well‐established precancerous lesion of non‐cardia gastric cancer, among young middle‐aged residents (Song 2015a). In addition, based on USA cancer register data, there has been an increase in the incidence of non‐cardia gastric cancer in the white population aged 25 to 39 years (Anderson 2010). These recent trends indicate that the etiology of this malignancy may be changing.
Complete resection of the whole tumor remains the only approach to treat this malignant disease. Evidence shows that additional perioperative chemotherapy or adjuvant chemotherapy increases the chance of survival (40% five‐year survival rate) (Macdonald 2001; Sakuramoto 2007). However, since gastric cancer is usually asymptomatic in its early stages, many people are diagnosed at an advanced stage, when the tumor is inoperable. In addition, because other conventional treatments (radiotherapy and chemotherapy) have only modest efficacy for those with advanced/metastatic gastric cancer, the prognosis in such cases is poor; the five‐year survival rate is less than 10% (Power 2010), and the median survival time in the range of 6 to 11 months (Wagner 2010).
It has been shown that chemotherapy improves quality of life and prolongs survival when compared to best supportive care (BSC) alone (Wagner 2006). Combination chemotherapy, usually comprising fluorouracil (or its oral prodrugs) plus a platinum compound (e.g. cisplatin), with or without the addition of a third drug (typically docetaxel or epirubicin), is currently the standard first‐line regimen for advanced gastric cancer (AGC) (Price 2012). However, the responses are usually partial and limited, with considerable toxicities (Wagner 2010). Also, radiation therapy (RT) is usually reserved only for symptom control in AGC, especially for pain or uncontrolled bleeding. The current limitations in treatment support the need to investigate safer and more effective agents for AGC treatment.
See Appendix 1 for a glossary of topic‐specific terms.
Description of the intervention
With our growing understanding of the underlying molecular basis of carcinogenesis, several targeted agents have been developed, delivering promising outcomes for treating people with lung, colon, breast, or kidney cancer. Nevertheless, compared to other solid tumors which predominantly rely on a particular signal pathway, gastric cancer appears to have a more complicated molecular and genetic carcinogenesis (Wu 2009). Although it might theoretically limit the application of molecular‐targeted therapy in this field, clinical trials have shown favorable efficacy results when adding a targeted agent (trastuzumab) to standard chemotherapy for human epidermal growth factor receptor‐2 (HER‐2)‐positive people with AGC (Bang 2010). Based on this pivotal phase III trial, trastuzumab has been approved as the first targeted agent in the first‐line treatment of people with HER‐2 over‐expressing AGC, both in the European Union (European Medicines Agency 2013) and in the USA (Genentech 2013). More recently, lapatinib plus paclitaxel has also been shown to produce a higher overall response rate compared to paclitaxel alone as the second‐line treatment for HER‐2‐positive AGC, although the improvement in survival time was not statistically significant (11 months versus 8.9 months, P = 0.10) (Satoh 2014). Additional clinical evidence continues to emerge regarding novel agents targeting other signaling pathways. With minor toxicity (e.g. acne‐like rash or diarrhea) (Widakowich 2007), use of molecular‐targeted agents alone or together with standard chemotherapy could be a rational approach to better outcomes for people with AGC.
Briefly, most targeted therapies for AGC focus on vascular endothelial growth factor (VEGF) or epidermal growth factor receptor (EGFR) pathways. Compounds targeting other pathways, such as PI3K/mTOR and HGF/MET, are also under investigation, but mainly in Phase I and Phase II clinical evaluations. The targeted agents under development are listed and summarized in Table 2.
1. Molecular‐targeted agents for advanced gastric cancer.
| Molecular target | Mechanism of action | Targeted agent | Phase of development | |
| EGFR‐targeting agent | Anti‐EGFR mAb | Cetuximab | III | |
| Panitumumab | III | |||
| EGFR TKIs | Erlotinib | II | ||
| Gefitinib | II | |||
| Anti‐HER‐2 mAb | Trastuzumab | III | ||
| HER‐2 and EGFR TKI | Lapatinib | III | ||
| VEGF‐targeting agent | Anti‐VEGF mAb | Bevacizumab | III | |
| Anti‐VEGFR mAb | Ramucirumab | III | ||
| VEGFR TKI | MTI, targeted on VEGFR, PDGFR and Raf | Sunitinib | II | |
| Sorafenib | II | |||
| MTI, targeted on VEGFR, PDGFR and c‐Kit | Cediranib | II | ||
| mainly targeted on VEGFR‐2 | Apatinib | III | ||
| P13K/mTOR‐targeting agent | mTOR inhibitor | Everolimus | III | |
| HGF/MET‐targeting agent | MET TKI | Foretinib | I‐II | |
| Crizotinib | I‐II | |||
| anti‐MET antibody | Onartuzumab | II | ||
| anti‐HGF antibody | Rilotumumab | III | ||
| MMP‐targeting agent | MMP inhibitor | Marimastat | III | |
| FGF‐targeting agent | FGFR inhibitor | AZD 4547 | II | |
| dual inhibitor of FGF and VEGF | Brivanib | II | ||
| HDAC‐targeting agent | HDAC inhibitor | Vorinostat | I‐II | |
| EpCAM‐targeting agent | Trifuctional bispecific antibody | Catumaxomab | II | |
EGFR: epidermal growth factor receptor; FGF: fibroblast growth factor; FGFR: fibroblast growth factor receptor; HDAC: histone deacetylase; HGF: hepatocyte growth factor; mAb: monoclonal antibodies; MMP: matrix metalloproteinase; MTI: multi‐targeted TKI; mTOR: mammalian target of rapamycin; PDGFR: platelet derived growth factor receptor; TKI: tyrosine kinase inhibitor; VEGF: vascular endothelial growth factor; VEGFR: vascular endothelial growth factor receptor
How the intervention might work
EGFR inhibitors
Epidermal growth factor receptor (EGFR) is the cell surface receptor for members of the epidermal growth factor (EGF) family, existing at multiple sites, including skin, gut, and renal tissue. By binding of specific ligands (EGF or transforming growth factor alpha) to its extracellular domain, EGFR can be activated, and thus its intracellular tyrosine kinase domain initiates downstream signals of rat sarcoma (Ras)/rapidly accelerated fibrosarcoma (Raf)/mitogen‐activated protein kinase (MAPK) or the protein kinase B (Akt)/mechanistic target of rapamycin (mTOR) pathway, which eventually lead to DNA synthesis and cell proliferation (Figure 1). The EGFR family consists of four members: HER‐1 (also know as EGFR), HER‐2, HER‐3, and HER‐4, among which the HER‐1 and HER‐2 represent the targets for current drug development for AGC.
1.
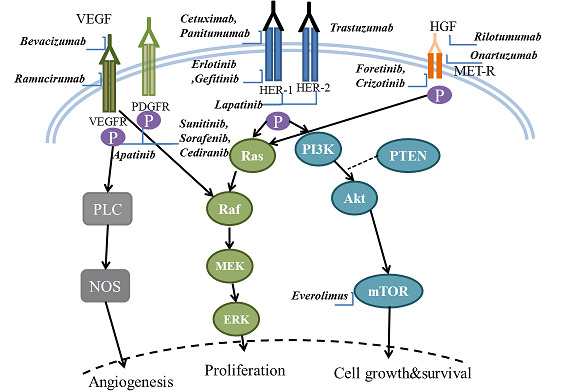
Schematic diagram of key signaling pathways in gastric carcinogenesis and the corresponding molecular targeted agents
Targeting EGFR (HER‐1)
Studies have shown that the over‐expression of this growth factor receptor is present in 27% to 64% of gastric cancers (Ilson 2011; Kim 2008; Lieto 2008), which might also be associated with more aggressive histology and poorer prognosis. The common approach to inhibit the EGFR is either by blocking the extracellular domain of EGFR via monoclonal antibodies (cetuximab/panitumumab), or inhibiting its intracellular tyrosine kinase domain (erlotinib/gefitinib). Although EGFR tyrosine kinase inhibitors (TKIs) have been demonstrated to be effective in lung cancer treatment, poor efficacy was reported for AGC (Draovich 2006). Similarly, a recent trial comparing cetuximab in combination with standard chemotherapy to chemotherapy alone (Lordick 2013) found no significant clinical benefit for the combination treatment.
Targeting HER‐2
HER‐2 over‐expression has been observed in approximately 20% of all gastric cancers (Kim 2011; Lordick 2010b; Yano 2006), with notably higher prevalence in intestinal than in diffuse or mixed gastric cancer (32.2% versus 6.1% or 20.4% respectively) (Bang 2012). Despite emerging data showing poorer outcomes for people with HER‐2‐positive gastric cancer (Begnami 2011; Kim 2011), the prognostic value of HER‐2 over‐expression in gastric cancer remains a controversial issue (Chua 2011; Grabsch 2010). Trastuzumab, a humanized recombinant monoclonal antibody that selectively binds to the extracellular domain of HER‐2, when applied together with standard chemotherapy exhibited a superior outcome for people with HER‐2‐positive AGC compared to chemotherapy alone (Bang 2010). Apart from directly blocking the HER‐2 signaling pathways and its downstream events, this target agent might also indirectly induce antibody‐dependent cellular cytoxicity (Spector 2009). Recently, a dual TKI inhibiting both HER‐2 and EGFR was developed (lapatinib). However, this agent produced disappointing results when applied to people with HER‐2‐negative AGC (Iqbal 2011).
Angiogenesis inhibitors
Vascular endothelial growth factor (VEGF) is a family of proteins produced by cells that stimulate angiogenesis. As ligands interact with VEGF receptors (VEGFRs) located in the cell surface (see Figure 1), VEGFs are crucial promotors to mediate endothelial cell proliferation and new vessel formation (Carmeliet 2003). There are four VEGF members (VEGF‐A, VEGF‐B, VEGF‐C, and VEGF‐D) and three types of VEGFRs (VEGFR‐1, VEGFR‐2, and VEGFR‐3).
For most solid cancers, both tumor growth and metastasis are highly dependent on misregulated angiogenesis (Hanahan 1996). Therefore, the VEGF pathway, as a key regulator of angiogenesis, has become a rational target for the development of therapeutic agents. In gastric cancer, the expression of VEGF and VEGFR was found in approximately 40% of patients (Maeda 1996; Ni 2010). Furthermore, studies revealed that the expression of VEGF was associated with tumor vascularity and metastases, thus indicating a poor prognosis (Lieto 2008; Tanigawa 1996).
Similar to the strategies for EGFR pathways, VEGF‐targeting agents function through neutralizing antibodies to VEGF (bevacizumab), through blocking the extracellular part of its receptor (ramucirumab), or through inhibiting its intracellular tyrosine kinase domain (sunitinib/sorafenib/cediranib/apatinib). Most of the VEGFR TKI agents are actually multi‐targeted TKIs. For example, sorafenib also targets the platelet‐derived growth factor receptor (PDGFR) and RAF pathways. Consequently, sorafenib can theoretically exert antineoplastic action from two different aspects (Wilhelm 2008): firstly, it inhibits tumor proliferation by blocking RAF/mitogen‐activated protein kinase (MEK)/extracellular‐signal‐regulated kinases (ERK) signaling pathways; and secondly, it suppresses angiogenesis by blocking VEGR and PDGFR. However, in a Phase II clinical trial, there was no evidence to indicate its superiority over combination chemotherapy for AGC treatment (Sun 2010).
Other targeted agents
Targeting PI3K‐Akt‐mTOR pathway
The mammalian target of rapamycin (mTOR) is a key protein kinase. After being activated by PI3K through Akt, mTOR could mediate signals responsible for cell growth and proliferation, cellular metabolism, and angiogenesis (Shaw 2006). The activity of mTOR could be positively regulated by many receptors, including EGFRs and VEGFRs, and negatively regulated by some intracellular factors, such as phosphatase and tensin homolog (PTEN) (see Figure 1). Previous research suggests that the PI3K‐Akt‐mTOR pathway is frequently activated in gastric cancer (Murayama 2009; Yu 2009). Notably, relative to upstream receptors with an over‐expression rate of only 20% to 30%, activated Akt was detected in more than 80% of gastric cancer cases (Yu 2009). It might indicate that inhibitors at the PI3K‐Akt level or mTOR level could be more effective than those targeting upstream moleculars. Currently, everolimus is the most investigated agent particularly targeting mTOR.
Targeting HGF/MET
The hepatocyte growth factor receptor (HGF)/MET receptor, together with its ligand hepatocyte growth factor/scatter factor (HGF/SF), mediates the epithelial‐to‐mesenchymal transition (EMT) during embryogenesis, as well as the process of tumor invasion and metastasis. Over‐expression of MET presents in about 50% to 60% of people with gastric cancer (Graziano 2011), and its prognostic value remains unclear. Until now, molecular drugs targeting this part include onartuzumab (anti‐MET antibody), rilotumumab (anti‐HGF antibody), foretinib and crizotinib (MET TKIs).
Targeting MMPs
Matrix metalloproteins (MMPs) are a family of zinc‐dependent endopeptidases that break down the components of the extracellular matrix. The MMPs play an important role in tissue remodeling through association with various physiological and pathological processes, such as morphogenesis, angiogenesis, tissue repair, and metastasis. Aberrant expression of MMPs occurs in several solid tumors, and is therefore considered to be related to the invasive potential of these tumors. Based on promising data from preclinical and early clinical evaluations, two MMP inhibitors (marimastat, prinostat) are now being tested in ongoing Phase III clinical trials.
More potential targeting agents focusing on other cell receptors, such as fibroblasts growth factor receptor (FGFR), or other downstream components such as histone deacetylases (HDAC), are also currently under test.
Why it is important to do this review
The management of AGC has a very limited evolution in the last decade. Despite the development of new chemotherapy regimens and the introduction of novel adjuvant treatments, the prognosis for people with AGC has not substantially improved, with median overall survival remaining less than one year. Recently, trials provided some promising results regarding molecular‐targeted therapy, in particular those targeting HER‐2 receptors, raising the possibility that the development of these agents could be an approach which might produce better outcomes for people with AGC. However, many questions remain, such as:
Are molecular‐targeted agents clinically beneficial for people with AGC?
Which pathway or molecule is the most efficient target for drug development?
-
Which is the most effective way to apply these drugs:
Should they be for all people with AGC or only for those with certain genetic biomarkers?
Should they be used alone or together with other chemotherapy agent?
We therefore feel there is a need for a systematic review to evaluate the effectiveness and safety profile of molecular‐targeted therapy.
Objectives
To evaluate the efficacy and safety of molecular‐targeted therapy , either alone or in combination with chemotherapy, in people with advanced gastric cancer.
Methods
Criteria for considering studies for this review
Types of studies
We included randomized controlled trials (RCTs). We included studies reported as full text, those published as abstract only, and unpublished data.
Types of participants
We included adults (aged 18 years and older) with histologically‐confirmed adenocarcinoma of the stomach or of the gastro‐esophageal junction with locally advanced unresectable (M0) or metastatic (M1) disease. We considered people with esophageal adenocarcinoma, if they had been enrolled in the trial together with those with gastric and gastro‐esophageal junction cancer, to be eligible, since this type of cancer also typically arises adjacent to the stomach (Pohl 2005). We excluded people with the following characteristics:
Previously treated by chemotherapy for metastatic or locally‐advanced unresectable cancer;
With known brain metastasis;
With other malignant disease in the previous five years, apart from basal‐cell cancer of the skin.
Types of interventions
We included trials comparing:
Molecular‐targeted agents (e.g. anti‐EGFR agents, VEGF‐targeting agents) plus conventional chemotherapy versus conventional chemotherapy alone;
Molecular‐targeted agents (e.g. anti‐EGFR agents, VEGF‐targeting agents) versus no treatment.
Types of outcome measures
Primary outcomes
Overall survival (OS): survival until death from all causes;
Progression‐free survival (PFS): time from randomization to either death or disease progression, whichever occurs first. Disease progression is defined according to Response Evaluation Criteria in Solid Tumors (RECIST) (Therasse 2000) as at least a 20% increase in the sum of the longest diameter of target lesions, taking as reference the sum of the longest diameter recorded since the treatment started or the appearance of one or more new lesions.
Secondary outcomes
Overall response: assessed response according to RECIST guidelines (Therasse 2000);
Quality of life, measured by a validated scale (e.g. EORTC QOL30 global health status scale);
Adverse events/side effects: such as anemia/neutropenia, nausea, diarrhea, and skin pigmentation, graded severity with the National Cancer Institute‐Common Terminology Criteria for Adverse Events (NCI‐CTCAE), including the percentage of treatment‐related deaths.
Search methods for identification of studies
Electronic searches
We conducted a literature search to identify all published and unpublished RCTs. The literature search identified potential studies in all languages. We translated the non‐English‐language papers and fully assessed them for potential inclusion in the review as necessary.
We searched the following electronic databases to identify potential studies for inclusion:
Cochrane Central Register of Controlled Trials (CENTRAL) (Issue 12, 2015) (Appendix 2);
MEDLINE (1946 to 4th December 2015) (Appendix 3);
EMBASE (1980 to 4th December 2015) (Appendix 4);
CINAHL (1982 to 4th December 2015) (Appendix 5).
We also conducted a search of ClinicalTrials.gov for planned and ongoing trials.
Searching other resources
We checked reference lists of all primary studies and reviewed articles for additional references. We contacted authors of identified trials and asked them to identify other published and unpublished studies. We also asked experts in the field and manufacturers of relevant drugs to provide details of current clinical trials and any relevant unpublished material.
We handsearched the abstracts from 1995 to 2014 from the American Digestive Disease Week (DDW) published in Gastroenterology, the United European Gastroenterology Week (UEGW) published in Gut, and the proceedings of the American Society of Clinical Oncology (ASCO) and the European Cancer Congress (ESMO‐ECCO).
We searched for errata or retractions from eligible trials on www.ncbi.nlm.nih.gov/pubmed on 4 December 2015.
Data collection and analysis
Selection of studies
Two review authors (HS, JZ) independently screened the titles and abstracts for potential inclusion of all the studies we identified as a result of the search. We coded them as 'retrieve' (eligible or potentially eligible/unclear) or 'do not retrieve'. We then retrieved the full‐text study reports/publications for further assessment. Similarly, two review authors (HS, JZ) independently screened the full text and identified studies for inclusion, and recorded the reasons for exclusion of the ineligible studies. We resolved any disagreement through discussion or, if required, by consulting a third review author (DL). Finally, we identified and excluded duplicates and collated multiple reports of the same study, so that each study rather than each report was the unit of interest in the review. We documented the selection process in sufficient detail to complete a PRISMA flow diagram and Characteristics of included studies and Characteristics of excluded studies tables.
Data extraction and management
We used a standard data collection form for study characteristics and outcome data, which has been piloted on at least one study in the review. One review author (HS) extracted study characteristics from the included studies, which in detail were:
Methods: study design, total duration study and run‐in, number of study centers and location, study setting, withdrawals, date of study
Participants: number (N), mean age, age range, gender, severity of condition, diagnostic criteria, baseline lung function, smoking history, inclusion criteria, exclusion criteria
Interventions: intervention, comparison, concomitant medications, excluded medications
Outcomes: primary and secondary outcomes specified and collected, time points reported
Notes: funding for trial, notable conflicts of interest of trial authors
Two review authors (HS, JZ) independently extracted outcome data from the included studies. We noted in the Characteristics of included studies table whether outcome data were reported in an unusable way. We resolved disagreements by consensus or by involving a third review author (DL). One review author (HS) copied across the data from the data collection form into the Review Manager 5 file. Then we double‐checked that the data were entered correctly by comparing the study reports with how the data were presented in the systematic review. A second review author spot‐checked study characteristics for accuracy against the trial report.
Assessment of risk of bias in included studies
Two review authors (HS, JZ) independently assessed the risks of bias for each study, using the criteria outlined in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). We resolved any disagreement by discussion or by involving a third assessor (DL). We assessed the risk of bias according to the following domains:
Random sequence generation;
Allocation concealment;
Blinding of participants and personnel (for all outcomes);
Blinding of outcome assessment (overall survival, serious adverse events);
Blinding of outcome assessment (progression‐free survival; response, adverse events, quality of life);
Incomplete outcome data (for all outcomes);
Selective outcome reporting;
Other bias.
We graded each potential source of bias as high, low or unclear, and provide a quote from the study report together with a justification for our judgment in the 'Risk of bias' table. We then summarized the 'Risk of bias' judgments across different studies for each of the domains listed. We considered blinding separately for different key outcomes where necessary (e.g. for unblinded outcome assessment, risk of bias for all‐cause mortality may be very different from that for a participant‐reported pain scale). Where information on risk of bias relates to unpublished data or correspondence with a trialist, we noted this in the 'Risk of bias' table.
When considering treatment effects, we took into account the risk of bias for the studies that contribute to that outcome.
Assesment of bias in conducting the systematic review
We conducted the review according to our previously published protocol, and reported any deviations from it in the Differences between protocol and review section of the full review.
Measures of treatment effect
We analyzed dichotomous data as odds ratios (ORs) with 95% confidence intervals (CIs), and continuous data as mean differences (MDs) or standardized mean differences (SMDs) with 95% CIs. We ensured that higher scores for continuous outcomes have the same meaning for the particular outcome, explaining the direction and reporting where the directions were reversed if this was necessary. For time‐to‐event data, we used methods of survival analysis and expressed the intervention effect as a hazard ratio (HR). In all analyses, we calculated the 95% confidence interval (CI). We extracted the HR for each individual trial directly from published data, if available, or alternatively using reported summary statistics or Kaplan‐Meier curves, as suggested in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011; Parmar 1998; Tierney 2007).
We undertook meta‐analyses only where this was meaningful, i.e. if the treatments, participants and the underlying clinical question were similar enough for pooling to make sense.
A common way that trialists indicate when they have skewed data is by reporting medians and interquartile ranges. When we encountered this we noted that the data were skewed and considered the implications of this.
Where multiple trial arms were reported in a single trial, we included only the relevant arms. If two comparisons (e.g. drug A versus placebo and drug B versus placebo) were entered into the same meta‐analysis, we halved the control group to avoid double‐counting.
Unit of analysis issues
We only considered RCTs. We did not find any probable RCTs with non‐standard designs. But if we identify such trials in future updates, we will assess non‐standard designs, such as cluster‐randomized trials, in order to avoid unit‐of‐analysis errors, including:
Recruitment bias;
Baseline imbalance;
Loss of clusters;
Incorrect analysis;
Comparability with individually‐randomized trials.
Dealing with missing data
We contacted investigators or study sponsors in order to verify key study characteristics or to obtain missing numerical outcome data where possible (e.g. when a study was presented as an abstract only). If we could not elicit a reply from the study authors after repeated attempts, we dropped these incomplete data from the analysis, stating this clearly in the Results section and discussing it further under the Potential biases in the review process section of the Discussion.
Assessment of heterogeneity
We conducted tests for heterogeneity using the Chi² test, with significance set at P < 0.1. We used the I² statistic (Higgins 2003) to estimate the total variation across studies due to heterogeneity; we consider I² less than 25% as low‐level, 25% to 50% as moderate‐level, and greater than 50% as high‐level heterogeneity. If we found high levels of heterogeneity (I² > 50%, Higgins 2011) for the primary outcomes, we explored its possible sources using the sensitivity and subgroup analyses described below.
Assessment of reporting biases
We attempted to contact study authors to ask them to provide missing outcome data. Where this was not possible, and the missing data were thought to introduce serious bias, we explored the impact of including such studies in the overall assessment of results by a sensitivity analysis.
We created funnel plot to explore possible publication biases when we were able to pool 10 or more trials.
Data synthesis
We used Review Manager 5 (Review Manager 2014) for pooling data and statistical analysis.For time‐to‐event outcomes (OS, PFS), we combined data using the generic inverse variance (GIV) method, and we presented measurements of treatment effects as HRs and 95% CIs. Since the design of the agents of interest is based on a different mechanism (targeting different pathways), we used a random‐effects model for primary analyses. For subgroup analysis, if we found the studies to be homogeneous in terms of age, diagnostic subtype, intervention type, and intervention duration, we used both the fixed‐effect model and the random‐effects model. We then compared the results from the two different models. In the absence of heterogeneity and significant reporting bias, these two models should yield the same results. In this case, we reported the results from the fixed‐effect model only. Otherwise, if the results were different, indicating significant heterogeneity, we reported the results from the random‐effects model only.
'Summary of findings' table
We created a 'Summary of findings' table using the GRADEpro software (GRADEprofiler 2008). We used the five GRADE considerations (study limitations, consistency of effect, imprecision, indirectness, and publication bias) to assess the quality of a body of evidence as it related to the studies which contribute data to the meta‐analyses for the prespecified outcomes. We used methods and recommendations described in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). We then justified all decisions to down‐ or upgrade the quality of studies using footnotes, and made comments to aid the reader's understanding of the review where necessary. We considered whether there was any additional outcome information that we could not incorporate into meta‐analyses; and if so we noted it in the comments, stating whether it supported or contradicted the information from the meta‐analyses.
Subgroup analysis and investigation of heterogeneity
We planned to carry out the following subgroup analyses:
Participants receiving molecular‐targeted agents for different purposes: adjunctive therapy or monotherapy (molecular‐targeted agents plus conventional chemotherapy versus conventional chemotherapy alone; molecular‐targeted agents versus placebo);
Different types of molecular‐targeted agents: e.g. anti‐EGFR agents, VEGF‐targeting agents;
Participants with and without specifically molecular prognostic markers, such as V‐Ki‐ras2 Kirsten rat sarcoma viral oncogene homolog (K‐Ras) mutation, EGFR gene copy number, and HER‐2 status.
We used the following outcomes in subgroup analyses:
Overall survival (OS);
Progression‐free survival (PFS).
We firstly examined the differences between subgroups by visual inspections of their confidence intervals; non‐overlapping confidence intervals indicate a statistically significant difference in treatment effect between subgroups. We then used the approach of Borenstein 2008 to formally investigate differences between two or more subgroups. To ensure the statistical power, we only conducted subgroup tests for those outcomes with three or more trials contributing data.
Sensitivity analysis
We performed sensitivity analyses defined a priori to assess the robustness of our conclusions. We did this by repeating the analyses in order to explore the influence of the following factors on effect size:
Exclusion of unpublished studies;
Exclusion of lower‐quality studies (those at high or unclear risk of bias relating to randomization, blinding or attrition);
Use of a fixed‐effect model (provided that a random‐effects model was initially used).
Reaching conclusions
We based our conclusions only on findings from the quantitative or narrative synthesis of included studies for this review. We avoided making recommendations for practice and our implications for research give the reader a clear sense of where the focus of any future research in the area should be and what the remaining uncertainties are.
Results
Description of studies
We have summarized the characteristics of the studies in the Characteristics of included studies tables.
Results of the search
The electronic search in December 2015 identified 2791 citations (114 from CENTRAL, 1463 from MEDLINE, 1214 from EMBASE). Handsearching found no relevant abstracts from conference proceedings. Of these 2791 records, we considered 126 (accounting for 78 reports after excluding duplicates) to be highly relevant after checking their abstracts, and we therefore tried to obtain the full texts for detailed assessment. Finally, 45 reports describing 20 different studies met our inclusion criteria. However, ten trials were available only as abstracts, among which seven were ongoing studies (NCT01503372; NCT01662869; NCT02314117; NCT01443065; NCT01123473; NCT01774786; ACTRN12609000109202) with no data reported (ACTRN12609000109202 suspended enrolment because of an unplanned and unfavorable safety review). The other three were completed trials (Hecht 2013; Wahab 2011; Wang 2012). We were unable to access the full data and relevant study information for Wahab 2011 and Wang 2012, having received no response to our attempts to contact the authors. We therefore list these two trials as Studies awaiting classification. For Hecht 2013, we found details about the trial by referring to a presentation at ASCO available on the Internet.
We didn't find any errata or retractions from eligible trials. Figure 2 shows the details of the search results.
2.
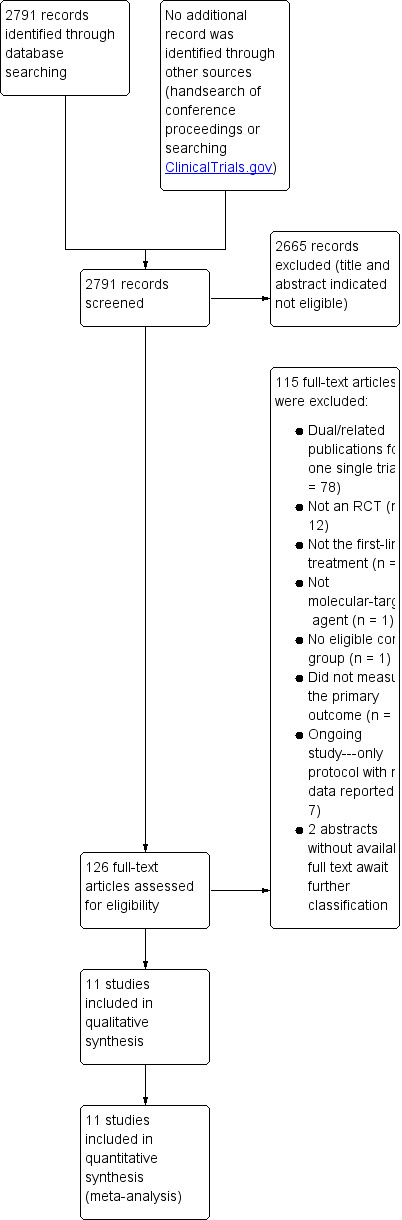
Study Flow diagram for RCTs
Included studies
After evaluation of eligible articles, we include 11 trials, corresponding to 11 individual RCTs (Bang 2010; Eatock 2013; Hecht 2013; Iveson 2014; Koizumi 2013; Lordick 2013; Ohtsu 2011; Rao 2010; Shen 2015; Waddell 2013; Zhang 2014) with 4085 participants. The Characteristics of included studies tables summarize the details of the included studies. Agents under investigations targeted the EGFR (trastuzumab/lapatinib /cetuximab /matuzumab/panitumumab) or the VEGF (TSU‐68/bevacizumab) pathway.
Study design
Six studies had an open‐label design without application of placebo (Bang 2010; Koizumi 2013; Lordick 2013; Rao 2010; Waddell 2013; Zhang 2014), while the other five RCTs used a double‐blinded and parallel‐group design. With the exception of Zhang 2014, all the included trials were multicenter, with seven international trials, and the other three located in Japan (Koizumi 2013), China (Shen 2015), and the UK (Waddell 2013).
Participants
All included trials enrolled participants with histologically‐confirmed advance gastric adenocarcinoma. Most of them allowed the inclusion of adenocarcinomas of gastro‐esophageal junction (except for Koizumi 2013 and Zhang 2014); and three of them also involved adenocarcinomas originating from the esophagus (Hecht 2013) or the distal esophagus (Eatock 2013; Rao 2010). Three studies applied a specific biomarker for participant selection: HER2‐positive was required for participant enrolment in Bang 2010 and Hecht 2013; and all participants were EGFR‐positive in Rao 2010.
Interventions
All included RCTs compared molecular‐targeted agents plus conventional chemotherapy versus conventional chemotherapy alone. No trial applied a molecular‐targeted agent for monotherapy. Six trials assessed the addition of EGFR‐targeting agents: trastuzumab (Bang 2010)/lapatinib (Eatock 2013)/cetuximab (Lordick 2013; Zhang 2014)/matuzumab (Rao 2010)/panitumumab (Waddell 2013) to standard chemotherapy. Three trials focused on VEGF‐targeting agents: TSU‐68 (Koizumi 2013)/bevacizumab (Ohtsu 2011; Shen 2015). The other two trials used experimental drugs targeting the Tie2 receptor and its ligands (angiopoientin‐1 and ‐2) (Eatock 2013), another pathway for tumor angiogenesis, and the HGF/MET pathway (Iveson 2014), respectively.
Eatock 2013 and Iveson 2014 compared two different doses of molecular‐targeted agents plus chemotherapy versus chemotherapy alone, while the other RCTs had only one experimental group and one control group.
Outcome measures
PFS was reported in all included RCTs. Apart from Eatock 2013, the other primary outcome of our review, OS, was reported in 10 of the 11 included RCTs.
For secondary outcomes, all the studies reported the overall response rate. However, since six RCTs (Bang 2010; Eatock 2013; Hecht 2013; Iveson 2014; Ohtsu 2011; Shen 2015) allowed the enrolment of participants with non‐measurable disease, so that this outcome could not be recorded for these people, it was measured in only 3723 participants. Similarly, all studies documented adverse events. However, in three RCTs (Ohtsu 2011; Shen 2015; Waddell 2013), only severe adverse events, classified as severity ≥ grade 3, were reported, with no data for non‐serious adverse events. In addition, three reports (Koizumi 2013; Rao 2010; Zhang 2014) only gave data for each adverse symptom or disease, with no overall percentage of adverse events or overall percentage of any severe adverse events. Quality of life was measured in only one included RCT (Rao 2010).
Due to the inconsistencies of follow‐up duration between different trials (see Included studies), these point estimated results need to be interpreted with caution.
Excluded studies
Please see Characteristics of excluded studies.
Risk of bias in included studies
Figure 3 summarizes the risk of bias in the included studies. Overall, five trials were well‐designed and well‐conducted, and therefore assessed as being at low risk of bias (Eatock 2013; Hecht 2013; Iveson 2014; Ohtsu 2011; Shen 2015), although for three of them (Iveson 2014; Ohtsu 2011; Shen 2015), the influence of the sponsor was apparent during the data analysis/interpretation and manuscript preparation stages. The other six trials were at high risk of bias (Bang 2010; Koizumi 2013; Lordick 2013; Rao 2010; Waddell 2013; Zhang 2014), mainly because there was no placebo comparator (open‐label design). For Zhang 2014, little information was available from the published paper for the risk of bias assessment; despite contacting the study authors we were unable to acquire more details.
3.

Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
Allocation
One included trial (Zhang 2014) did not explicitly state the exact method for 'randomisation'. We therefore considered it to be at unclear risk of selection bias. The other studies all described clearly how the random sequence was generated. For allocation concealment, we considered nine trials to be at low risk: eight allocated participants using a central interactive voice recognition system (Bang 2010; Eatock 2013; Hecht 2013; Iveson 2014; Lordick 2013; Ohtsu 2011; Rao 2010; Shen 2015), and one study involved central allocation via fax (Waddell 2013). Two studies (Koizumi 2013; Zhang 2014) had unclear risk, since they provided no information.
Blinding
We considered five trials with a double‐blind, placebo‐controlled design (Eatock 2013; Hecht 2013; Iveson 2014; Ohtsu 2011; Shen 2015 ) to be at low risk of bias for blinding of participants and researchers, as well as blinding of outcome assessors. Six open‐label trials (Bang 2010; Koizumi 2013; Lordick 2013; Rao 2010; Waddell 2013; Zhang 2014) had no placebo comparator. We judged these studies to have a high risk of performance bias (e.g. through stress‐related mechanisms).
For assessing detection bias, we classified the following outcomes as subjectively ascertained: PFS, quality of life, response and any adverse event. These outcomes could be affected by the outcome assessors' knowledge of treatment received. We considered OS and severe adverse events to be unlikely to be affected by blinding of outcome assessment. We assessed all trials to be at low risk of detection bias for objective outcomes. Besides these five investigator‐masked double‐blind studies, four open‐label RCTs also applied masked review for outcome evaluations to reduce the risk of detection bias (Bang 2010; Koizumi 2013; Lordick 2013; Rao 2010). We therefore considered these trials to have low risk of detection bias for measuring subjective outcomes. We judged Waddell 2013 as 'unclear bias', since although it had no masking for assessors, a central monitoring system was applied to control the quality of outcome measurements. We classified one other study as being at unclear risk of bias, since no relevant information was provided for assessment (Zhang 2014).
Incomplete outcome data
We considered all the included studies to be at low risk of bias, either because the number of participants missing from follow‐up was very low (dropout rates below 5%) (Bang 2010; Eatock 2013; Iveson 2014; Koizumi 2013; Zhang 2014), or the efficacy analysis (analysis for survival time) was done on the intention‐to‐treat population of all participants randomly allocated to treatment (Hecht 2013; Lordick 2013; Ohtsu 2011; Rao 2010; Shen 2015; Waddell 2013). All the studies reported overall response rate, but only among participants with measurable disease.
Selective reporting
We judged all of the included studies to be at low risk of selective outcome reporting. Seven studies (Bang 2010; Eatock 2013; Hecht 2013; Iveson 2014; Ohtsu 2011; Shen 2015; Waddell 2013) had well‐documented study protocols which were consistent with their published full reports. We also classified the other four studies as being at low selective reporting risk (Koizumi 2013; Lordick 2013; Rao 2010; Zhang 2014), since they reported all important outcomes (i.e. OS, PFS, overall response, severe adverse events), although no study protocol was available. However, we found very limited information for quality of life, since only one trial (Rao 2010) evaluated it. All studies included data on severe adverse events. However, summary data were available for only eight of them (Bang 2010; Eatock 2013; Hecht 2013; Iveson 2014; Lordick 2013; Ohtsu 2011; Shen 2015; Waddell 2013). Similarly, we extracted summary data on general adverse events from only five studies (Bang 2010; Eatock 2013; Hecht 2013; Iveson 2014; Lordick 2013).
Other potential sources of bias
Waddell 2013 was identified to be with unclear risk of other potential bias due to its early termination. We considered all other trials to be at low risk of other potential biases. However, all the trials except for Zhang 2014 stated clearly that they were supported by pharmaceutical companies, and the role of the sponsors in some funded studies was critical: in six trials (Bang 2010; Iveson 2014; Lordick 2013; Ohtsu 2011; Rao 2010; Shen 2015), the sponsors were involved in study design, data collection, management and statistical analysis. The other five RCTs confirmed the independence of the conduct and data collection of the study.
Effects of interventions
See: Table 1
We extracted summary data from all 11 included studies. For the trials with multiple intervention groups with different doses of experimental agent but one control group, we summed the number of participants for the primary analysis, irrespective of the dose level they received, to detect the global effect of molecular‐targeted therapy.
Primary outcomes
Effect of molecular‐targeted therapy plus chemotherapy, compared with chemotherapy alone, with or without placebo, on overall survival
With the exception of Eatock 2013, which was a small Phase II trial, all studies, involving 3843 participants, reported an evaluation of overall survival. There was statistically significant heterogeneity between the results of individual trials (I² = 61%, P = 0.005). Therefore, as planned, we used a random‐effects model for pooling the results. The effect of molecular therapy on survival was uncertain due to wide confidence intervals and inconsistency of effect across the studies. The pooled HR was 0.92 (95% CI 0.80 to 1.05) (Analysis 1.1; Figure 4). The quality of the evidence was low, due to performance bias and inconsistency between the results of the included studies (Table 1).
1.1. Analysis.
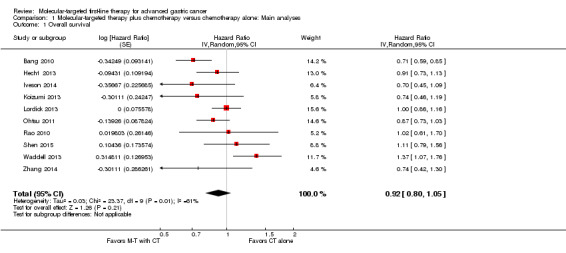
Comparison 1 Molecular‐targeted therapy plus chemotherapy versus chemotherapy alone: Main analyses, Outcome 1 Overall survival.
4.
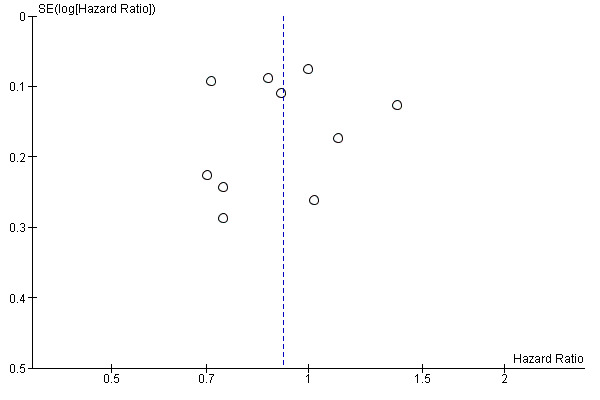
Funnel plot of comparison: 1 Molecular‐targeted therapy plus chemotherapy versus chemotherapy alone: main analyses, outcome: 1.1 Overall survival.
We performed several sensitivity analyses when pooling data from these trials. When we performed a complete‐case analysis but applied a fixed‐effect model, the confidence interval for the main HR (0.92) became narrower (0.85 to 0.99), indicating a statistically significant benefit. In addition, when we restricted the analysis to trials at low risk of bias (Hecht 2013; Iveson 2014; Ohtsu 2011; Shen 2015), we found there was no statistically significant between‐trial heterogeneity (I² = 0%, P = 0.42), and the HR was 0.90 (95% CI 0.79 to 1.01).
Effect of molecular‐targeted therapy plus chemotherapy, compared with chemotherapy alone with or without placebo, on progression‐free survival
All trials provided data regarding progression‐free survival (4014 participants). As with results for overall survival, we found no evidence of significant benefit of additional molecular‐targeted therapy in prolonging progression‐free survival; the main analysis including results from all studies demonstrated a HR of 0.90 (95% CI 0.78 to 1.04) (Analysis 1.2; Figure 5). Also, we detected high heterogeneity between individual trial results (I² = 64%, P = 0.002). The quality of evidence was low due to performance bias and inconsistency between the results of included studies (Table 1).
1.2. Analysis.
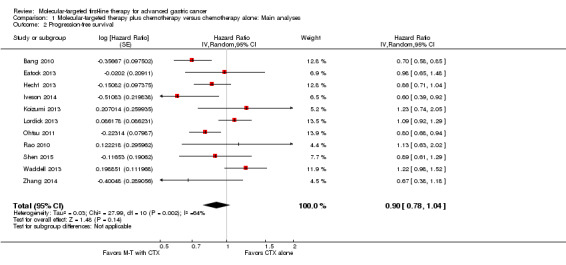
Comparison 1 Molecular‐targeted therapy plus chemotherapy versus chemotherapy alone: Main analyses, Outcome 2 Progression‐free survival.
5.
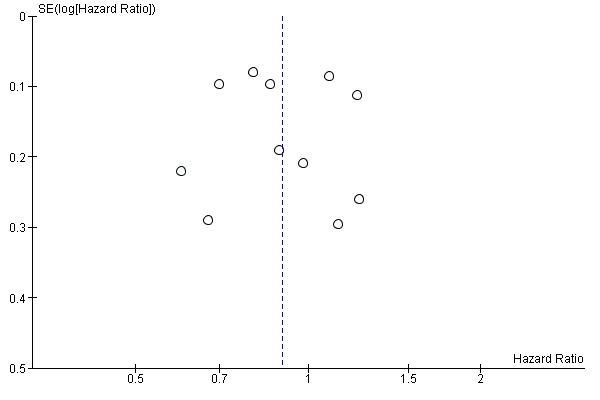
Funnel plot of comparison: 1 Molecular‐targeted therapy plus chemotherapy versus chemotherapy alone: main analyses, outcome: 1.2 Progression‐free survival.
However, results produced by a fixed‐effect model showed a small but significant benefit (HR 0.90, 95% CI 0.83 to 0.97). Restricting the analysis to trials at low risk of bias only (Eatock 2013; Hecht 2013; Iveson 2014; Ohtsu 2011; Shen 2015) indicated a statistically significant benefit of molecular‐targeted treatment (HR 0.82, 95% CI 0.74 to 0.92), without evidence of statistical heterogeneity (I² = 0%, P = 0.51).
Effect of molecular‐targeted therapy plus chemotherapy, compared with chemotherapy alone, with or without placebo, on overall response
All trials provided data regarding overall response. However, since six RCTs (Bang 2010; Eatock 2013; Hecht 2013; Iveson 2014; Ohtsu 2011; Shen 2015) allowed the recruitment of participants with non‐measurable disease, this outcome was restricted to the 3723 participants who had measurable tumors. Overall, 816 (42.1%) of 1936 participants assigned to the molecular‐targeted therapy plus conventional chemotherapy group were subsequently assessed as having either a complete or a partial response, compared with 653 (36.5%) of 1787 participants allocated to the chemotherapy‐only control treatment. There was statistically significant heterogeneity between individual trial results (I² = 52%, P = 0.02). Using a random‐effects model , we observed a statistically significant increase in tumor response rate among participants assigned to the adjuvant molecular‐targeted therapy group (OR 1.24, 95% CI 1.00 to 1.55) (Analysis 1.3; Figure 6). The quality of evidence was low due to performance bias and inconsistency between results of included studies (Table 1).
1.3. Analysis.
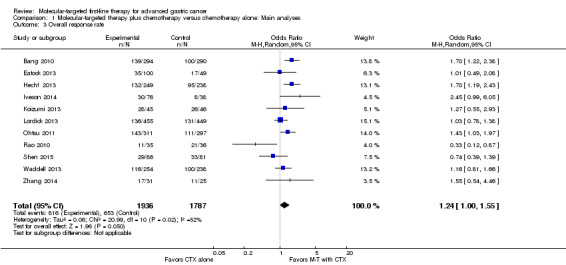
Comparison 1 Molecular‐targeted therapy plus chemotherapy versus chemotherapy alone: Main analyses, Outcome 3 Overall response rate.
6.
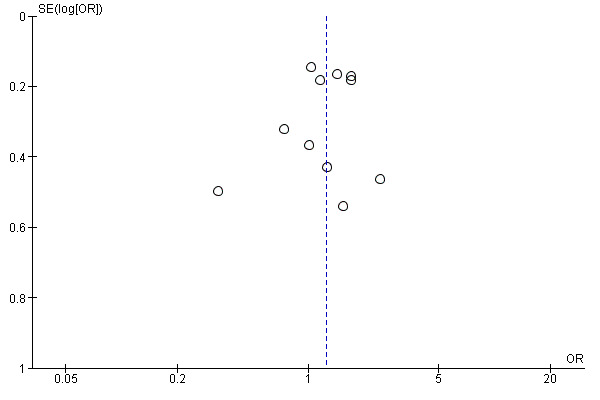
Funnel plot of comparison: 1 Molecular‐targeted therapy plus chemotherapy versus chemotherapy alone: main analyses, outcome: 1.3 Overall response rate.
When we switched to a fixed‐effect model, the OR was 1.28 (95% CI 1.12 to 1.46). Sensitivity analysis with trials at low risk of bias produced similar but more pronounced results (OR 1.41, 95% CI 1.15 to 1.73). Importantly, the duration of follow‐up varied considerably between different studies; the median follow‐up time ranged from 5.3 months (Waddell 2013) to 28.5 months (Rao 2010). It should be noted that this disparity would also influence the observed tumor response rate.
Effect of molecular‐targeted therapy plus chemotherapy, compared with chemotherapy alone, with or without placebo, on quality of life
Only one trial (Rao 2010) reported these data. The absolute number of participants with follow‐up data in the different trial arms was very small, with 21 participants from the experimental groups and 32 from the control group completing the evaluation, using the EORTC QOL30 global health status scale. The chemotherapy‐alone group achieved higher mean scores than the experimental group, both at baseline and at the end of treatment. The score changes indicated there was no relevant difference between these two arms overall, but we downgraded the evidence to very low since the study was at a high risk of bias from the low number of participants and the wide confidence interval around the mean effect (Analysis 1.4; Table 1). The quality of evidence was very low, due to notable performance/detection bias, and imprecision (Table 1).
1.4. Analysis.

Comparison 1 Molecular‐targeted therapy plus chemotherapy versus chemotherapy alone: Main analyses, Outcome 4 Quality of life, measured by EORTC QOL30 global health status scale (score changes between baseline and after treatment).
Effect of molecular‐targeted therapy plus chemotherapy, compared with chemotherapy alone, with or without placebo, on adverse events
Five studies (2290 participants) provided data on the proportion of participants with any adverse event (Bang 2010; Eatock 2013; Hecht 2013; Iveson 2014; Lordick 2013). Overall, there were 1185 (98.3%) out of 1205 participants randomized to the experimental groups who experienced adverse events, compared with 1044 (96.2%) of 1085 participants in the control groups. There was no statistically significant heterogeneity between individual trial results (I² = 0%, P = 0.60). We found evidence (using a random‐effects model) of an increased risk of experiencing any adverse event among participants with molecular‐targeted combination treatment (OR 2.23, 95% CI 1.27 to 3.92) (Analysis 1.5), compared to those with chemotherapy only. Similarly, there was a significant excess risk of serious adverse events with molecular‐targeted therapy, based on data from eight studies (3800 participants): OR 1.19 (95% CI 1.03 to 1.37; I² = 0%) (Analysis 1.6) (Bang 2010; Eatock 2013; Hecht 2013; Iveson 2014; Lordick 2013; Ohtsu 2011; Shen 2015; Waddell 2013).
1.5. Analysis.

Comparison 1 Molecular‐targeted therapy plus chemotherapy versus chemotherapy alone: Main analyses, Outcome 5 Adverse event (any).
1.6. Analysis.
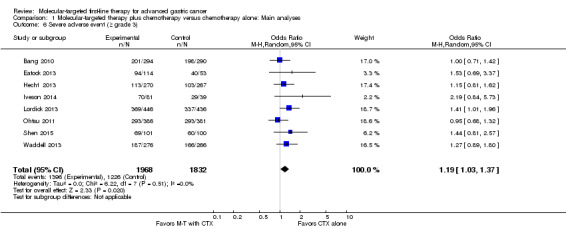
Comparison 1 Molecular‐targeted therapy plus chemotherapy versus chemotherapy alone: Main analyses, Outcome 6 Severe adverse event (≥ grade 3).
As well as performance/detection bias, we considered there to be a risk of possible selective reporting bias, as three studies only provided data for severe adverse events, and another three had no summary data for pooling, prompting us to downgrade the quality of evidence to low (Table 1).
Subgroup analysis
Effect of molecular‐targeted therapy plus chemotherapy, compared with chemotherapy alone, with or without placebo, on overall survival and progression‐free survival, according to the type of molecular‐targeted agents
By pooling results from three RCTs (Koizumi 2013; Ohtsu 2011; Shen 2015), with 1067 participants, we found no evidence that the application of VEGF‐targeting agents could benefit people with AGC, with respect both to overall survival time (HR 0.90, 95% CI 0.76 to 1.06) (Analysis 2.1) and to progression‐free survival (HR 0.87, 95% CI 0.71 to 1.05) (Analysis 2.2). Similarly, we found no survival effect for EGFR‐targeting agents (2655 participants: Bang 2010; Hecht 2013; Lordick 2013; Rao 2010; Waddell 2013; Zhang 2014): for overall survival, the HR is 0.94 (95% CI 0.77 to 1.16) (Analysis 2.1), and for progression‐free survival, the HR is 0.93 (95% CI 0.76 to 1.14) (Analysis 2.2). Due to the presence of high‐level heterogeneity (74% and 75% respectively), we used a random‐effects model for these analyses.
2.1. Analysis.
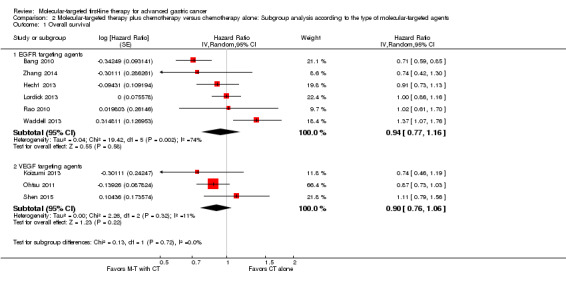
Comparison 2 Molecular‐targeted therapy plus chemotherapy versus chemotherapy alone: Subgroup analysis according to the type of molecular‐targeted agents, Outcome 1 Overall survival.
2.2. Analysis.
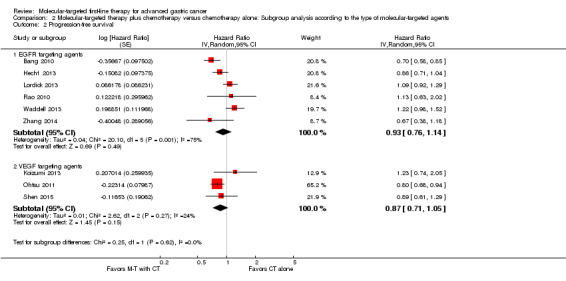
Comparison 2 Molecular‐targeted therapy plus chemotherapy versus chemotherapy alone: Subgroup analysis according to the type of molecular‐targeted agents, Outcome 2 Progression‐free survival.
Tests for subgroup differences for both overall survival and progression‐free survival did not reach statistical significance (test for subgroup differences for overall survival: P = 0.72; I² = 0%, and for progression‐free survival: P = 0.62; I² = 0%).
Effect of molecular‐targeted therapy plus chemotherapy, compared with chemotherapy alone, with or without placebo, on overall survival and progression‐free survival, according to specific molecular prognostic biomarker for participant selection
Three trials used biomarkers for participant selection: Bang 2010 and Hecht 2013 recruited participants only if their tumors demonstrated over‐expression of HER2 protein; and Rao 2010 required an EGFR‐positive tumor when recruiting participants. On the basis of the current data, we found a possible benefit when applying molecular‐targeted agents to participants with HER2‐positive tumors (HR 0.80, 95% CI 0.63 to 1.02 for overall survival (2 trials), and HR 0.78, 95% CI 0.63 to 0.95 for progression‐free survival (2 trials)) ( Analysis 3.1; Analysis 3.2). Among participants without tumor biomarker selection, and with an EGFR‐positive tumor, the possibility of benefiting from adjuvant molecular‐targeted therapy was low (Analysis 3.1; Analysis 3.2).
3.1. Analysis.
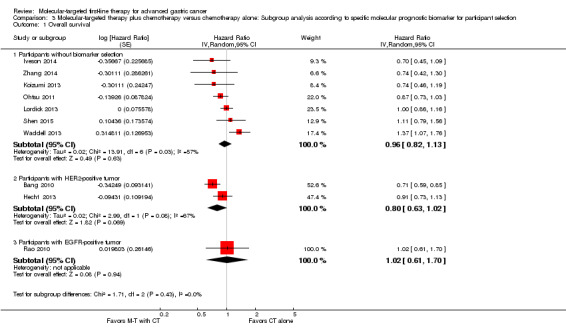
Comparison 3 Molecular‐targeted therapy plus chemotherapy versus chemotherapy alone: Subgroup analysis according to specific molecular prognostic biomarker for participant selection, Outcome 1 Overall survival.
3.2. Analysis.
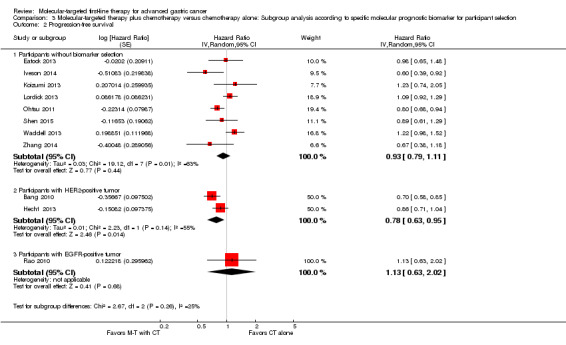
Comparison 3 Molecular‐targeted therapy plus chemotherapy versus chemotherapy alone: Subgroup analysis according to specific molecular prognostic biomarker for participant selection, Outcome 2 Progression‐free survival.
We didn't perform tests for the subgroup differences owing to the limited number of trials involved in the each molecular prognostic biomarker subgroup.
Discussion
Summary of main results
This systematic review and meta‐analysis suggests that there is no clear evidence to support a survival benefit of molecular‐targeted therapy as a first‐line treatment for advance gastric cancer. However, since sensitivity analyses based on a fixed‐effect model and on study quality showed that the addition of molecular‐targeted therapy was significantly associated with improved overall and progression‐free survival, we conclude that the uncertainty of our findings is due to the low quality of the current evidence (inconsistency). In addition, subgroup analyses did not provide evidence that survival outcomes differed by the type of molecular‐targeted agent (EGFR‐ or VEGF‐targeting agents). Although one of the subgroups indicated a possible benefit for participants with HER2‐positive tumors on progression‐free survival, the test for interaction was not significant and this did not translate across to any improvement in survival. It is important to note that there were high levels of statistical heterogeneity within subgroups. This possibly reduced the power to detect significant differences in most of the subgroup analyses that we conducted.
Furthermore, people with AGC with adjuvant molecular‐targeted treatment had an improved overall response rate compared to those receiving chemotherapy only (42.1% versus 36.5%, OR 1.24, 95% CI 1.00 to 1.55, P = 0.05). Also, based on incompletely‐extracted data, this additional treatment was significantly associated with an excess risk of experiencing adverse events (OR 2.23, 95% CI 1.27 to 3.92 for any adverse event; OR 1.19, 95% CI 1.03 to 1.37 for severe adverse events).
Overall completeness and applicability of evidence
To minimize the possible effect of publication bias, we conducted an exhaustive search involving unpublished or ongoing trials, and without any limitation on language. Except for one study without enough information to assess methodological issues (Zhang 2014), the methodological quality of the remaining identified studies can in general be considered as adequate. However, we downgraded the quality of evidence due to wide confidence intervals and inconsistency of effect across the studies.
Data regarding the efficacy of molecular‐targeted therapy for AGC are insufficient, and the included studies only partially addressed the objectives of our review. Firstly, we could not determine the effects of molecular‐targeted therapy on quality of life, since only one small trial provided data for this outcome. Secondly, all included RCTs compared molecular‐targeted agents plus conventional chemotherapy with conventional chemotherapy alone, and so there are no data that enable us to assess the efficacy of a molecular‐targeted agent as a monotherapy.
Quality of the evidence
See Table 1.
The risk of bias in the 11 RCTs varied according to the outcome of interest. The blinding procedures for five double‐blinded RCTs were well‐documented and at low risk of bias (Eatock 2013; Hecht 2013; Iveson 2014; Ohtsu 2011; Shen 2015). However, we noted that for three of them, sponsors were heavily involved in the data analysis/interpretation and manuscript preparation stages (Iveson 2014; Ohtsu 2011; Shen 2015). The effect of this was hard to assess, but the consistent results observed through sensitivity analysis provide some indirect evidence that the involvement of study sponsors did not seriously undermine the findings of our review. The other six trials were at high risk of bias (Bang 2010; Koizumi 2013; Lordick 2013; Rao 2010; Waddell 2013; Zhang 2014), mainly because of their open‐label design.
We could not account for the inconsistency between individual trial results by our predefined subgroups. Although we expected variation in the results across the studies, we found that within subgroups heterogeneity remained high. Sensitivity analyses by risk of bias tended not to change the size or precision of the average effect but it did seem to reduce the amount of between‐study heterogeneity, implying methodological diversity as a possible explanation for the heterogeneity of our results.
We downgraded the quality of evidence for survival outcomes and overall response, due to imprecision indicated by wide confidence intervals, and for quality of life, due to the small sample size of the only study which provided data for this outcome. Selective reporting of adverse events prompted us to downgrade the quality of evidence for this outcome, since minor adverse events were ignored in some reports, while others provided no summary data.
Potential biases in the review process
In order to include as many participants as possible, we decided to conduct combined primary analyses using data from all included studies that applied agents targeting different molecules, irrespective of different participant selection procedures. Between‐study heterogeneity due to clinical diversity was therefore highly probable. We subsequently performed subgroup analyses to accommodate this possible diversity, but they provided little explanation for the high level of heterogeneity detected. The unexplained inconsistency between individual trial results undermines the reliability of our efficacy assessment, and the random‐effects model we used to incorporate the heterogeneity further reduced the accuracy of the estimated effect size.
To prevent bias in the review procedure, we used search strategies guided and developed by the Cochrane Upper Gastrointestinal and Pancreatic Diseases Group. There were no restrictions (e.g. on language, publication type) on the search. Two review authors independently conducted study selection, assessment of the risks of bias, and data collection without blinding. We resolved any disagreements through discussion with a third review author. We dealt with missing information and data by repeated attempts to contact the authors. For studies that had only been published as abstracts, we tried to obtain further details by emailing the authors. Nevertheless, since we did not receive any replies from them, we failed to get enough information to assess the eligibility of two studies (Wahab 2011; Wang 2012). For some outcomes (e.g. adverse events), we extracted only the data available from the study reports, and pooled them for our meta‐analysis. This incomplete study assessment and data pooling could produce bias, which may have influenced the precision and reliability of our results.
In the Differences between protocol and review section, we state that, since some relevant studies involved participants with esophageal adenocarcinoma, we subsequently broadened our criteria of eligible participants. Such post‐protocol change could have some potential impact on our findings.
Agreements and disagreements with other studies or reviews
The benefit of molecular‐targeted therapy for advance gastric cancer in prolonging survival time has been tested in clinical trials during these years. One review (Wagner 2009) found only limited data from Phase I and II studies, with results indicating modest benefits of targeted therapy in the participant population. Another review published four years later (Kim 2013) summarized emerging data from more recent trials (without pooling the data), but also failed to find any conclusive evidence. However, both of these reviews, consistent with our results, emphasized a possibility of survival improvement among selected participants who were identified by molecular predictive and prognostic markers (e.g. HER‐2).
Authors' conclusions
Implications for practice.
There is uncertainty about the effect of adding targeted therapy to chemotherapy on survival outcomes in people with advanced gastric cancer, with very little information available on their quality of life. The main limitation of the evidence for survival outcomes was inconsistent effects across the studies, which we could not explain by prespecified subgroups in terms of the type of therapy or tumor type. There is more certain evidence of an increased risk of adverse events.
Implications for research.
Most of the ongoing studies we found are Phase II clinical trials with a limited number of participants. We would therefore expect that they are unlikely to provide enough evidence for us to further our understanding. In view of the high levels of inconsistency across the studies, further studies would need to explore the relationship between selection of participants and the type of targeted therapy. Randomized controlled trials should maintain blinding of outcome assessors. Study conduct should be independent, i.e. data analysis and results interpretation to be conducted only by statisticians and researchers not employed by sources of commercial sponsorship.
Studies should report adverse events with summary data, and measure quality of life. Focusing recruitment of study populations based on our subgroup analyses would also help to establish whether there are any survival differences between participants with different tumor types.
Acknowledgements
We thank the Cochrane Upper Gastrointestinal and Pancreatic Diseases (UGPD) Group for providing administrative and logistical support for the conduct of this review, and for developing and executing the search strategies. We also thank Eva Tiselius and Olivia Teghararian for helping us screen the updated search results.
Appendices
Appendix 1. Glossary
Adenocarcinoma: is a subtype of cancer that can occur in several parts of the body. It is defined as neoplasia of epithelial tissue that has glandular origin, glandular characteristics, or both.
Adjuvant: is a pharmacological and/or immunological agent that modifies the effect of other agents. Adjuvants may be added to vaccine to modify the immune response by boosting it such as to give a higher amount of antibodies and a longer lasting protection, thus minimizing the amount of injected foreign material.
Angiogenesis: is the physiological process through which new blood vessels form from pre‐existing vessels.
Antineoplastic: refers to actions that prevent, inhibit or halt the development of a neoplasm or tumor.
Asymptomatic: means no symptoms.
Biomarker: short for biological markers, referring to biological measures of a biological state. By definition, a biomarker is "a characteristic that is objectively measured and evaluated as an indicator of normal biological processes, pathogenic processes or pharmacological responses to a therapeutic intervention."
Carcinogenesis: refers to a process by which normal cells are transformed into cancer cells.
Chemotherapy: is a popular method of cancer treatment which uses chemical substances, especially one or more anti‐cancer drugs to kill cancer cells.
Embryogenesis: is the process by which the embryo forms and develops.
Endopeptidase: An enzyme that catalyses the cleavage of peptide bonds within a polypeptide or protein.
Extracellular: is the part outside the cell.
Fibroblasts: is a type of cell that synthesizes the extracellular matrix and collagen, the structural framework for animal tissues, and plays a critical role in wound healing. Fibroblasts are the most common cells of connective tissue in animals.
Hepatocyte growth factor: is a powerful mitogen for hepatocytes and other epithelial tissues. it's secreted by mesenchymal cells and acts as a multi‐functional cytokine on cells of mainly epithelial origin.
Histology: is a branch of biology that focusing on the microscopic anatomy of cells and tissues of plants and animals.
Intracellular: means inside the cell.
Ligand: is a substance (usually a small molecule) that forms a complex with a biomolecule to serve a biological purpose.
Mammalian: refers to something related to a class of animals which are warm‐blooded vertebrates characterized by mammary glands in the female.
Monoclonal antibodies: are monospecific antibodies that are the same because they are made by identical immune cells that are all clones of a unique parent cell, in contrast to polyclonal antibodies which are made from several different immune cells.
Morphogenesis: means the generation of form, and usually in the context of developmental biology where it means the generation of tissue organization and shape in animal and plant embryos
Neutropenia: is a decrease in circulating neutrophils.
Non‐cardia gastric cancer: is the subtype of gastric cancer that originated from all other areas of the stomach, except the top inch where it meets the esophagus.
Pathogenesis: refers to the development of a disease and the chain of events leading to that disease.
Perioperative: pertains to the time before (preoperative), during (intraoperative), and after (postoperative) surgery.
Proliferation: is the growth or production of cells by multiplication of parts.
Receptor: is a protein molecule that receives and responds to a neurotransmitter, or other substance.
Tyrosine kinase: is an enzyme that can transfer a phosphate group from adenosine triphosphate(ATP) to a protein in a cell. It is an important mediator of the signalling cascade.
Appendix 2. CENTRAL search strategy
Via OVID
1. Stomach Neoplasms/
2. ((gastr$ or gut or stomach$) adj2 (carcin$ or cancer$ or neoplas$ or tumour$ or tumor$ or growth$ or adenocarcin$ or malig$)).tw.
3. 1 or 2
4. Antibodies, Monoclonal/
5. Protein Kinase Inhibitors/
6. EGF receptor inhibitor*.tw.
7. (epidermal growth factor receptor adj3 inhibitor*).tw.
8. cetuximab.tw.
9. panitumumab.tw.
10. erlotinib.tw.
11. gefitinib.tw.
12. trastuzumab.tw.
13. lapatinib.tw.
14. exp Angiogenesis Inhibitors/
15. (vascular endothelial growth factor adj2 inhibitor*).tw.
16. VEGF inhibitor*.tw.
17. bevacizumab.tw.
18. ramucirumab.tw.
19. sunitinib.tw.
20. sorafenib.tw.
21. cediranib.tw.
22. apatinib.tw.
23. mTOR inhibitor*.tw.
24. mammalian target of rapamycin inhibitor*.tw.
25. everolimus.tw.
26. foretinib.tw.
27. crizotinib.tw.
28. onartuzumab.tw.
29. rilotumumab.tw.
30. exp Matrix Metalloproteinase Inhibitors/
31. marimastat.tw.
32. prinostat.tw.
33. azd4547.tw.
34. brivanib.tw.
35. Histone Deacetylase Inhibitors/
36. vorinostat.tw.
37. catumaxomab.tw.
38. or/4‐37
39. 3 and 38
Appendix 3. MEDLINE search strategy
Via OVID
1. *Stomach Neoplasms/
2. ((gastr$ or gut or stomach$) adj2 (carcin$ or cancer$ or neoplas$ or tumour$ or tumor$ or growth$ or adenocarcin$ or malig$)).tw.
3. 1 or 2
4. Antibodies, Monoclonal/
5. *Protein Kinase Inhibitors/
6. EGF receptor inhibitor*.tw.
7. (epidermal growth factor receptor adj3 inhibitor*).tw.
8. cetuximab.tw.
9. panitumumab.tw.
10. erlotinib.tw.
11. gefitinib.tw.
12. trastuzumab.tw.
13. lapatinib.tw.
14. exp Angiogenesis Inhibitors/
15. (vascular endothelial growth factor adj2 inhibitor*).tw.
16. VEGF inhibitor*.tw.
17. bevacizumab.tw.
18. ramucirumab.tw.
19. sunitinib.tw.
20. sorafenib.tw.
21. cediranib.tw.
22. apatinib.tw.
23. mTOR inhibitor*.tw.
24. mammalian target of rapamycin inhibitor*.tw.
25. everolimus.tw.
26. foretinib.tw.
27. crizotinib.tw.
28. onartuzumab.tw.
29. rilotumumab.tw.
30. exp Matrix Metalloproteinase Inhibitors/
31. marimastat.tw.
32. prinostat.tw.
33. azd4547.tw.
34. brivanib.tw.
35. *Histone Deacetylase Inhibitors/
36. vorinostat.tw.
37. catumaxomab.tw.
38. or/4‐37
39. 3 and 38
40. randomized controlled trial.pt.
41. controlled clinical trial.pt.
42. randomized.ab.
43. placebo.ab.
44. drug therapy.fs.
45. randomly.ab.
46. trial.ab.
47. groups.ab.
48. or/40‐47
49. exp animals/ not humans.sh.
50. 48 not 49
51. 39 and 50
Appendix 4. EMBASE search strategy
Via OVID
1. stomach cancer/ or stomach tumor/ or cardia carcinoma/ or stomach adenocarcinoma/ or stomach carcinoid/ or stomach carcinoma/
2. ((gastr$ or gut or stomach$) adj2 (carcin$ or cancer$ or neoplas$ or tumour$ or tumor$ or growth$ or adenocarcin$ or malig$)).tw.
3. 1 or 2
4. monoclonal antibody/
5. epidermal growth factor receptor kinase inhibitor/
6. EGF receptor inhibitor*.tw.
7. (epidermal growth factor receptor adj3 inhibitor*).tw.
8. cetuximab/
9. cetuximab.tw.
10. panitumumab/
11. panitumumab.tw.
12. erlotinib/
13. erlotinib.tw.
14. gefitinib/
15. gefitinib.tw.
16. trastuzumab/
17. trastuzumab.tw.
18. lapatinib/
19. lapatinib.tw.
20. exp angiogenesis inhibitor/
21. vasculotropin inhibitor/
22. (vascular endothelial growth factor adj2 inhibitor*).tw.
23. VEGF inhibitor*.tw.
24. bevacizumab/
25. bevacizumab.tw.
26. ramucirumab/
27. ramucirumab.tw.
28. sunitinib/
29. sunitinib.tw.
30. sorafenib/
31. sorafenib.tw.
32. cediranib/
33. cediranib.tw.
34. apatinib.tw.
35. exp "mammalian target of rapamycin inhibitor"/
36. everolimus/
37. everolimus.tw.
38. foretinib/
39. foretinib.tw.
40. crizotinib/
41. crizotinib.tw.
42. onartuzumab/
43. onartuzumab.tw.
44. rilotumumab/
45. rilotumumab.tw.
46. exp matrix metalloproteinase inhibitor/
47. marimastat/
48. marimastat.tw.
49. prinostat.tw.
50. azd 4547/
51. azd4547.tw.
52. brivanib/
53. brivanib.tw.
54. exp histone deacetylase inhibitor/
55. vorinostat/
56. vorinostat.tw.
57. catumaxomab/
58. catumaxomab.tw.
59. or/4‐58
60. 3 and 59
61. random:.tw. or placebo:.mp. or double‐blind:.tw.
62. 60 and 61
Appendix 5. CINAHL search strategy
via EBSCO
| Search ID# | Search Terms |
| S39 | S3 AND S38 |
| S38 | S4 OR S5 OR S6 OR S7 OR S8 OR S9 OR S10 OR S11 OR S12 OR S13 OR S14 OR S15 OR S16 OR S17 OR S18 OR S19 OR S20 OR S21 OR S22 OR S23 OR S24 OR S25 OR S26 OR S27 OR S28 OR S29 OR S30 OR S31 OR S32 OR S33 OR S34 OR S35 OR S36 OR S37 |
| S37 | (MH "Antibodies, Monoclonal+") |
| S36 | catumaxomab |
| S35 | vorinostat |
| S34 | Histone Deacetylase Inhibitor* |
| S33 | brivanib |
| S32 | azd4547 |
| S31 | prinostat |
| S30 | marimastat |
| S29 | Matrix Metalloproteinase Inhibitor* |
| S28 | rilotumumab |
| S27 | onartuzumab |
| S26 | crizotinib |
| S25 | foretinib |
| S24 | everolimus |
| S23 | mammalian target of rapamycin inhibitor* OR mTOR inhibitor* |
| S22 | apatinib |
| S21 | cediranib |
| S20 | sorafenib |
| S19 | sunitinib |
| S18 | ramucirumab |
| S17 | bevacizumab |
| S16 | VEGF inhibitor* |
| S15 | vascular endothelial growth factor N2 inhibitor* |
| S14 | vasculotropin inhibitor* OR angiogenesis inhibitor* |
| S13 | (MH "Angiogenesis Inhibitors+") |
| S12 | lapatinib |
| S11 | trastuzumab |
| S10 | gefitinib |
| S9 | erlotinib |
| S8 | panitumumab |
| S7 | cetuximab |
| S6 | EGF receptor inhibitor* |
| S5 | Protein Kinase N2 Inhibitor* OR (epidermal growth factor receptor kinase) N2 inhibitor* |
| S4 | (MH "Protein Synthesis Inhibitors") |
| S3 | S1 OR S2 |
| S2 | ((gastr* or gut or stomach*) N2 (carcin* or cancer* or neoplas* or tumour* or tumor* or growth* or adenocarcin* or malig*)) |
| S1 | (MH "Stomach Neoplasms") |
Data and analyses
Comparison 1. Molecular‐targeted therapy plus chemotherapy versus chemotherapy alone: Main analyses.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Overall survival | 10 | Hazard Ratio (Random, 95% CI) | 0.92 [0.80, 1.05] | |
| 2 Progression‐free survival | 11 | Hazard Ratio (Random, 95% CI) | 0.90 [0.78, 1.04] | |
| 3 Overall response rate | 11 | 3723 | Odds Ratio (M‐H, Random, 95% CI) | 1.24 [1.00, 1.55] |
| 4 Quality of life, measured by EORTC QOL30 global health status scale (score changes between baseline and after treatment) | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 5 Adverse event (any) | 5 | 2290 | Odds Ratio (M‐H, Random, 95% CI) | 2.23 [1.27, 3.92] |
| 6 Severe adverse event (≥ grade 3) | 8 | 3800 | Odds Ratio (M‐H, Random, 95% CI) | 1.19 [1.03, 1.37] |
Comparison 2. Molecular‐targeted therapy plus chemotherapy versus chemotherapy alone: Subgroup analysis according to the type of molecular‐targeted agents.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Overall survival | 9 | Hazard Ratio (Random, 95% CI) | Subtotals only | |
| 1.1 EGFR targeting agents | 6 | Hazard Ratio (Random, 95% CI) | 0.94 [0.77, 1.16] | |
| 1.2 VEGF targeting agents | 3 | Hazard Ratio (Random, 95% CI) | 0.90 [0.76, 1.06] | |
| 2 Progression‐free survival | 9 | Hazard Ratio (Random, 95% CI) | Subtotals only | |
| 2.1 EGFR targeting agents | 6 | Hazard Ratio (Random, 95% CI) | 0.93 [0.76, 1.14] | |
| 2.2 VEGF targeting agents | 3 | Hazard Ratio (Random, 95% CI) | 0.87 [0.71, 1.05] |
Comparison 3. Molecular‐targeted therapy plus chemotherapy versus chemotherapy alone: Subgroup analysis according to specific molecular prognostic biomarker for participant selection.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Overall survival | 10 | Hazard Ratio (Random, 95% CI) | Subtotals only | |
| 1.1 Participants without biomarker selection | 7 | Hazard Ratio (Random, 95% CI) | 0.96 [0.82, 1.13] | |
| 1.2 Participants with HER2‐positive tumor | 2 | Hazard Ratio (Random, 95% CI) | 0.80 [0.63, 1.02] | |
| 1.3 Participants with EGFR‐positive tumor | 1 | Hazard Ratio (Random, 95% CI) | 1.02 [0.61, 1.70] | |
| 2 Progression‐free survival | 11 | Hazard Ratio (Random, 95% CI) | Subtotals only | |
| 2.1 Participants without biomarker selection | 8 | Hazard Ratio (Random, 95% CI) | 0.93 [0.79, 1.11] | |
| 2.2 Participants with HER2‐positive tumor | 2 | Hazard Ratio (Random, 95% CI) | 0.78 [0.63, 0.95] | |
| 2.3 Participants with EGFR‐positive tumor | 1 | Hazard Ratio (Random, 95% CI) | 1.13 [0.63, 2.02] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Bang 2010.
| Methods | International RCT (122 centers in 24 countries), Phase III | |
| Participants |
Number of participants: 594 (September 2005 to June 2010 ) Number randomized: Experimental group (Trastuzumab in combination with fluoropyrimidine and cisplatin): 298 Control group (fluoropyrimidine and cisplatin): 296 Number evaluated: Experimental group: 294, age (mean ± SD): 59.4 ± 10.8 years Control group: 290, age (mean ± SD): 58.5 ± 11.2 years Diagnosis: Patients with gastric or gastro‐esophageal junction cancer were eligible for inclusion if their tumors showed over‐expression of HER2 protein by immunohistochemistry or gene amplification by fluorescence in situ hybridization Inclusion: eligible patients should: be ≥ 18 years of age; histologically‐confirmed inoperable locally‐advanced, recurrent, or metastatic adenocarcinoma of the stomach or gastro‐esophageal junction; ECOG performance status 0 – 2; adequate organ function; and measurable or non‐measurable disease; tumors were centrally tested for HER2 status with immunohistochemistry (HercepTest, Dako, Denmark) and fluorescence in situ hybridization (FISH; HER2 FISH pharmDx, Dako) |
|
| Interventions |
Experimental group : D1 trastuzumab 8 mg/kg i.v. for cycle 1, and then 6 mg/kg i.v. Q3W until disease progression; D1 ‐ 5 800 mg/m² fluorouracil i.v., Q3W for 6 cycles; D1 80 mg/m² cisplatin i.v., Q3W for 6 cycles; D1 ‐ 15 1000 mg/m² capecitabine by mouth twice daily, every 3 weeks for 6 cycles Control group: D1 ‐ 5 800 mg/m² fluorouracil i.v., every 3 weeks for 6 cycles; D1 80 mg/m² cisplatin i.v., every 3 weeks for 6 cycles; D1 ‐ 15 1000 mg/m² capecitabine by mouth twice daily, every three weeks for 6 cycles |
|
| Outcomes |
Duration of follow‐up (median): 18.6 months (IQR 11 ‐ 25) for experimental group, 17.1 (IQR 9 ‐ 25) for control group OS, FPS, overall response, adverse events |
|
| Notes | Sponsor: Hoffmann‐La Roche The sponsor was involved in the study design, data collection, data analysis, results interpretation, and manuscript preparation Study referred to as "ToGA" |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "The randomisation sequence was created by F Hoff mann‐La Roche" |
| Allocation concealment (selection bias) | Low risk | "Treatment was assigned by use of a randomised block design with block sizes of four patients, via a central interactive voice recognition system (by telephone)" |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | The open‐label design may introduce performance bias (e.g. through stress‐related mechanisms) |
| Blinding of outcome assessment (detection bias) OS, Serious adverse events | Low risk | These outcomes were unlikely to be affected by the blinding status of assessors |
| Blinding of outcome assessment (detection bias) PFS, Response, quality of life & adverse events | Low risk | "Efficacy and safety data were monitored by an independent data monitoring committee." |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 584 randomized participants were included in analysis (98.3%) |
| Selective reporting (reporting bias) | Low risk | Prior protocol was available |
| Other bias | Low risk | No obvious potential source of bias |
Eatock 2013.
| Methods | International RCT (40 centers in 10 countries), Phase II | |
| Participants |
Number of participants: 171 (December 2007 to July 2009) Number randomized: Experimental group A (CX (cisplatin and capecitabine) + AMG 386 10 mg/kg): 56 Experimental group B (CX + intravenous AMG 386 3 mg/kg): 59 Control group (CX + placebo): 56 Number evaluated: Experimental group A: 56, age (median and range): 61 (18 ‐ 80) years Experimental group B:59, age (median and range): 57 (29 ‐ 74) years Control group: 56, age (median and range): 62 (37 ‐ 84) years Diagnosis: Patients with metastatic gastric, gastro‐esophageal junction, or distal esophageal adenocarcinoma Inclusion: eligible patients should: be ≥18 years; have metastatic histologically‐ or cytologically‐confirmed gastric, gastro‐esophageal junction, or distal esophageal adenocarcinoma; measurable or non‐measurable disease per RECIST; ECOG performance status ≤ 1; be able to swallow oral medications; and have adequate hematologic, coagulation, hepatic, cardiac, and renal function |
|
| Interventions |
Experimental group A : CX (cisplatin 80 mg/m² i.v. every 3 weeks; capecitabine 1000 mg/m² by mouth twice a day for 14 days every three weeks) + intravenous AMG 386 10 mg/kg QW Experimental group B : CX (cisplatin 80 mg/m² i.v. every 3 weeks; capecitabine 1000 mg/m² by mouth twice a day for 14 days every three weeks) + intravenous AMG 386 3 mg/kg every week Control group: CX (cisplatin 80 mg/m² i.v. every three weeks; capecitabine 1000 mg/m² by mouth twice a day for 14 days every three weeks) + placebo every week |
|
| Outcomes |
Duration of follow‐up (median): 32 weeks PFS, overall response, adverse events |
|
| Notes | Sponsor: Amgen | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | a computerized interactive voice response system was used for randomization and allocation |
| Allocation concealment (selection bias) | Low risk | a computerized interactive voice response system was used for randomization and allocation |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "Investigators, the study sponsor, and patients were blinded to treatment assignments" |
| Blinding of outcome assessment (detection bias) OS, Serious adverse events | Low risk | These outcomes were unlikely to be affected by the blinding status of assessors |
| Blinding of outcome assessment (detection bias) PFS, Response, quality of life & adverse events | Low risk | "Investigators, the study sponsor, and patients were blinded to treatment assignments" |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 171 participants included in analysis (100%) |
| Selective reporting (reporting bias) | Low risk | Prior protocol was available |
| Other bias | Low risk | No obvious potential source of bias |
Hecht 2013.
| Methods | International RCT (186 centers in 22 countries), Phase III | |
| Participants |
Number of participants: 545 (June 2008 to January 2012) Number randomized: Experimental group (oxaliplatin and capecitabine + lapatinib): 272 Control group (oxaliplatin and capecitabine + placebo): 273 Number evaluated: Experimental group: 249, age (median and range): 61 (19 ‐ 86) years Control group: 238, age (median and range): 59 (27 ‐ 84) years Diagnosis: ErbB2 (HER2)‐positive patients with histologically‐confirmed locally‐advanced unresectable or metastatic adenocarcinoma of the stomach, esophagus or gastro‐esophageal junction Inclusion: eligible patients should: have signed informed consent; have histologically‐confirmed gastric, esophageal, or gastro‐esophageal junction adenocarcinoma; disease that is locally advanced (unresectable), metastatic, or locally recurrent; Measurable or non‐measurable, but radiologically evaluable disease, according to RECIST; ErbB2 (HER2)‐positive; be aged 18+ years; ECOG performance status 0 ‐ 2; adequate organ function, including adequate hematologic, renal and liver function; cardiac ejection fraction within institutional range of normal as measured by echocardiogram; able to swallow and retain oral medications, and/or receive enteral medications via gastrectomy feeding tube; women and men with potential to have children must be willing to practise acceptable methods of birth control during the study; prior gastric surgery is permitted if > 3 weeks prior and recovered; prior chemotherapy for non‐gastric malignancy if > than 5 years; prior neoadjuvant and/or adjuvant chemotherapy for early‐stage gastric cancer if > 6 months since completion; at least 4 weeks since prior radiotherapy; prior biologic, hormonal, or immunologic cancer treatment if > 5 years since treatment |
|
| Interventions |
Experimental group : D1 oxaliplatin 130 mg/m²; D1 ‐ 14 capecitabine 850 mg/m² twice daily; D1 ‐ 21 lapatinib 1250 mg/day Control group: D1 oxaliplatin 130 mg/m²; D1 ‐ 14 capecitabine 850 mg/m² twice daily; D1 ‐ 21 placebo daily |
|
| Outcomes |
Duration of follow‐up (median): not mentioned OS, PFS, overall response, adverse events |
|
| Notes | Sponsored by GlaxoSmithKline (GSK) Initial study design had PFS as the primary endpoint with a sample size of 410 participants. In September 2009, the study was amended to change the primary endpoint to OS and increase sample size to 535 participants Reported in abstract form only |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | The randomization code was created by the GlaxoSmithKline internal randomization system named Randall |
| Allocation concealment (selection bias) | Low risk | A randomization code was blinded to all project members and investigators and locked until unblinding in February 2013 |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blind (participant, caregiver, investigator, outcomes assessor) |
| Blinding of outcome assessment (detection bias) OS, Serious adverse events | Low risk | These outcomes were unlikely to be affected by the blinding status of assessors |
| Blinding of outcome assessment (detection bias) PFS, Response, quality of life & adverse events | Low risk | Double blind (participant, caregiver, investigator, outcomes assessor) |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 487 randomized participants were included in analysis (89.4%); ITT analysis |
| Selective reporting (reporting bias) | Low risk | Prior protocol was available |
| Other bias | Low risk | No obvious potential source of bias |
Iveson 2014.
| Methods | International RCT (43 centers), Phase II | |
| Participants |
Number of participants: 121 (February 2009 to November 2010) Number randomized: Experimental group A (ECX (epirubicin, cisplatin, and capecitabine) + rilotumumab 7.5 mg/kg): 42 Experimental group B (ECX + rilotumumab 15 mg/kg): 40 Control group (ECX + placebo): 39 Number evaluated: Experimental group A: 42, age (median and range): 62 (27 ‐ 78) years Experimental group B(ECX + rilotumumab 15 mg/kg): 40, age (median and range): 59.5 (28 ‐ 76) years Control group: 39, age (median and range): 60 (39 ‐ 79) years Diagnosis: patients with unresectable locally‐advanced or metastatic gastric or esophagogastric junction adenocarcinoma Inclusion: eligible patients should: be > 18 years; have pathologically‐confirmed, unresectable locally‐advanced or metastatic gastric or esophagogastric junction adenocarcinoma (tumors of the distal esophagus within 5 cm of the esophagogastric junction were allowed); measurable and non‐measurable disease; ECOG performance status of 0 or 1; life expectancy of at least 3 months; adequate organ function; hemoglobin concentration of at least 90 g/L; absolute neutrophil count of at least 1.5*10⁹ cells per L; platelet count of at least 100*10⁹ platelets per L (without transfusion < 14 days before enrolment or randomization); creatinine clearance of at least 60 mL/min (calculated or measured); AST and ALT concentrations of ≤ 2·5 × ULN, or AST and ALT of 5 × ULN or less in the presence of liver metastasis; total bilirubin 1·5 × ULN or less; and partial thromboplastin time 1·5 × ULN or less and international normalization ratio ≤ the ULN |
|
| Interventions |
Experimental group A : intravenous infusion of rilotumumab 7.5 mg/kg before administration of ECX + ECX (epirubicin 50 mg/m² administered intravenously on day 1 every 3 weeks; cisplatin 60 mg/m² administered intravenously on day 1 every 3 weeks, and capecitabine 625 mg/m² taken twice a day orally on days 1 – 21 every 3 weeks) Experimental group B : intravenous infusion of rilotumumab 15 mg/kg before administration of ECX + ECX (epirubicin 50 mg/m² administered intravenously on day 1 every 3 weeks; cisplatin 60 mg/m² administered intravenously on day 1 every 3 weeks, and capecitabine 625 mg/m² taken twice a day orally on days 1 – 21 every 3 weeks) Control group: intravenous infusion of placebo before administration of ECX + ECX (epirubicin 50 mg/m² administered intravenously on day 1 every 3 weeks; cisplatin 60 mg/m² administered intravenously on day 1 every 3 weeks, and capecitabine 625 mg/m² taken twice a day orally on days 1 – 21 every 3 weeks) Treatment continued for up to 10 cycles or until disease progression, unacceptable toxicity, or withdrawal of informed consent |
|
| Outcomes |
Duration of follow‐up (median): 21.7 months (IQR 20.5 ‐ 23.5) PFS, OS, overall response, adverse events |
|
| Notes | Sponsor: Amgen The sponsor was involved in the study design, data collection, data analysis, results interpretation, and manuscript preparation |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "The randomisation list was generated using permuted blocks with a block size of six and prepared by an individual independent of the study team" |
| Allocation concealment (selection bias) | Low risk | "Treatment allocation was assigned using an interactive voice response system" |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "Patients, investigators, and the study team were masked to treatment allocation, which was straightforward because rilotumumab is a colourless liquid." |
| Blinding of outcome assessment (detection bias) OS, Serious adverse events | Low risk | These outcomes were unlikely to be affected by the blindness status of assessors |
| Blinding of outcome assessment (detection bias) PFS, Response, quality of life & adverse events | Low risk | "Patients, investigators, and the study team were masked to treatment allocation, which was straightforward because rilotumumab is a colourless liquid." |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 121 randomized participants were included in analysis (100%) |
| Selective reporting (reporting bias) | Low risk | Prior protocol was available |
| Other bias | Low risk | No obvious potential source of bias |
Koizumi 2013.
| Methods | Japanese multicenter RCT (14 centers in Japan), Phase II | |
| Participants |
Number of participants: 93 (December 2008 to February 2012) Number randomized: Experimental group (TSU‐68 + S‐1/CDDP): 46 Control group (S‐1/CDDP): 47 Number evaluated: Experimental group: 45, age (median and range): 62 (30 ‐ 74) years Control group: 46, age (median and range): 63.5 (44 ‐ 76) years Diagnosis: patients with chemotherapy‐naïve unresectable or recurrent AGCs Inclusion: eligible patients should: be > 20 years; with histologically‐ or cytologically‐confirmed adenocarcinoma; unresectable or recurrent gastric cancer; no prior systemic treatment. Recurrent patients were eligible if the last dose of postoperative adjuvant chemotherapy had been received at least 180 days before the start of the study |
|
| Interventions |
Experimental group : TSU‐68: 800 mg m–2 twice a day day 1 – 35 every 5 weeks; S‐1: 40 – 60 mg m–2 twice a day day 1 – 21 every 5 weeks; CDDP: 60 mg m–2 i.v. on day 8 Control group: S‐1: 40 – 60 mg m–2 twice a day day 1 – 21 every 5 weeks; CDDP: 60 mg m–2 i.v. on day 8 The treatments were continued until 1 of the following occurred: progressive disease (PD), unacceptable toxicity, withdrawal of participant consent (regardless of toxicity), or termination of treatment at the discretion of the attending physician |
|
| Outcomes |
Duration of follow‐up (median): not mentioned PFS, OS, overall response, adverse events (reported by different symptoms, no data for any adverse event) |
|
| Notes | This trial was supported by Taiho Pharmaceutical Co., Ltd | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomization was performed according to the minimization method |
| Allocation concealment (selection bias) | Unclear risk | Not mentioned |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | The open‐label design may introduce performance bias (e.g. through stress‐related mechanisms) |
| Blinding of outcome assessment (detection bias) OS, Serious adverse events | Low risk | These outcomes were unlikely to be affected by the blinding status of assessors |
| Blinding of outcome assessment (detection bias) PFS, Response, quality of life & adverse events | Low risk | All measured images were assessed by a central imaging review committee |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 91 randomized participants were included in analysis (97.8%) |
| Selective reporting (reporting bias) | Low risk | No prior protocol was available but all important outcomes were reported |
| Other bias | Low risk | No obvious potential source of bias |
Lordick 2013.
| Methods | International multicenter RCT (164 sites in 25 countries), Phase III | |
| Participants |
Number of participants: 904 (June 2008 to December 2010) Number randomized: Experimental group (capecitabine and cisplatin + cetuximab): 455 Control group (capecitabine and cisplatin): 449 Number evaluated: Experimental group: 455, age (median and range): 60 (23 ‐ 84) years Control group: 449, age (median and range): 59 (18 ‐ 81) years Diagnosis: patients with histologically‐confirmed adenocarcinoma of the stomach or gastro‐esophageal junction with locally‐advanced unresectable (M0) or metastatic (M1) disease Inclusion: eligible patients should: be > 18 years; histologically‐confirmed adenocarcinoma of the stomach or gastro‐esophageal junction with locally‐advanced unresectable (M0) or metastatic (M1) disease; availability of tumor material for EGFR expression assessment; at least 1 radiographically‐documented measurable lesion (≥ 2 cm in at least 1 dimension by conventional techniques or ≥ 1 cm by spiral CT) in a previously non‐irradiated area according to RECIST; ECOG performance status of 0 – 1 with adequate organ function; no previous chemotherapy for metastatic or locally‐advanced unresectable gastric or gastro‐esophageal junction cancer; adjuvant chemotherapy completed at least 1 year before randomization and not more than 300 mg/m² cisplatin administered; no previous treatment with drugs targeting EGFR‐related or VEGFR‐related signalling pathways; and no clinically relevant coronary artery disease, congestive heart failure, cardiomyopathy, history of myocardial infarction in the last 12 months, or high risk of uncontrolled arrhythmia |
|
| Interventions |
Experimental group : oral capecitabine 1000 mg/m² twice daily from the evening of day 1 until the morning of day 15; intravenous cisplatin 80 mg/m² on day 1; once‐weekly cetuximab (400 mg/m² at the first infusion then 250 mg/m² every week) Control group: oral capecitabine 1000 mg/m² twice daily from the evening of day 1 until the morning of day 15; intravenous cisplatin 80 mg/m² on day 1 Treatment was continued until radiographically‐documented tumor progression, unacceptable toxicity, or withdrawal of consent by the participant |
|
| Outcomes |
Duration of follow‐up (median): not mentioned. Final analysis was on March 31, 2012 PFS, OS, overall response, adverse events |
|
| Notes | The sponsor (Merck KGaA) was responsible for data management and statistical analysis, and has been involved in data interpretation and manuscript drafting | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | A stratified, permuted, block randomization procedure (variable block size) was used |
| Allocation concealment (selection bias) | Low risk | Randomization was done centrally with an interactive voice response system |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | The open‐label design may introduce performance bias (e.g. through stress‐related mechanisms) |
| Blinding of outcome assessment (detection bias) OS, Serious adverse events | Low risk | These outcomes were unlikely to be affected by the blinding status of assessors |
| Blinding of outcome assessment (detection bias) PFS, Response, quality of life & adverse events | Low risk | Outcomes were assessed by masked review at an independent review committee |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | ITT analysis was used |
| Selective reporting (reporting bias) | Low risk | No protocol was available but all important outcomes were reported |
| Other bias | Low risk | No obvious potential source of bias |
Ohtsu 2011.
| Methods | International multicenter RCT (93 centers in 17 countries), Phase III | |
| Participants |
Number of participants: 774 (September 2007 to December 2008) Number randomized: Experimental group (capecitabine and cisplatin/FU + bevacizumab): 387 Control group (capecitabine and cisplatin/FU + placebo): 387 Number evaluated: Experimental group: 387, age (median and range): 58 (22 ‐ 81) years Control group: 387, age (median and range): 59 (22 ‐ 82) years Diagnosis: patients with previously untreated, histologically‐confirmed, unresectable locally‐advanced or metastatic adenocarcinoma of the stomach or gastro‐esophageal junction Inclusion: eligible patients should: be > 18 years; with previously untreated, histologically‐confirmed, unresectable locally‐advanced or metastatic adenocarcinoma of the stomach or gastro‐esophageal junction; with ECOG performance status of 0 to 2 and life expectancy of 3 months; measurable and non‐measurable disease; disease had to be evaluable according to RECIST; adjuvant chemotherapy was permitted if completed 6 months before random assignment; surgery or radiotherapy was permitted if completed 28 days before random assignment; adequate bone marrow, hepatic, and renal function (including proteinuria of 1 g/24 hours) |
|
| Interventions |
Experimental group : Bevacizumab 7.5 mg/kg i.v. on day 1 every 3 weeks; cisplatin 80 mg/m² i.v. on day 1 every 3 weeks; oral capecitabine 1000 mg/m² twice daily for 14 days every 3 weeks/FU 800 mg/m²/d i.v. on days 1 ‐ 5 Control group: placebo on day 1 every 3 weeks; cisplatin 80 mg/m² i.v. on day 1 every 3 weeks; oral capecitabine 1000 mg/m² twice daily for 14 days every 3 weeks/FU 800 mg/m²/d i.v. on days 1 ‐ 5 Cisplatin was given for 6 cycles; capecitabine and bevacizumab were administered until disease progression or unacceptable toxicity |
|
| Outcomes |
Duration of follow‐up (median): 11.4 months for experimental group, and 9.4 months for control group PFS, OS, overall response, adverse events (only the incidence for grade ≥ 3 adverse events was reported, not for any adverse events) |
|
| Notes | Sponsor: Genentech, Inc Collaborators: Hoffmann‐La RocheChugai Pharmaceutical The sponsor was involved in the study design, data collection, data analysis, results interpretation, and manuscript preparation Study referred to as "AVAGAST" |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | A stratified, permuted, block randomization procedure was used |
| Allocation concealment (selection bias) | Low risk | Allocation concealment was performed by an interactive voice recognition system supplied by a clinical research organization responsible for appropriate and independent treatment allocation. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blind design |
| Blinding of outcome assessment (detection bias) OS, Serious adverse events | Low risk | These outcomes were unlikely to be affected by the blinding status of assessors |
| Blinding of outcome assessment (detection bias) PFS, Response, quality of life & adverse events | Low risk | Double blind (participant, investigator) |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | ITT analysis has been used |
| Selective reporting (reporting bias) | Low risk | Prior protocol was available. |
| Other bias | Low risk | No obvious potential source of bias |
Rao 2010.
| Methods | Multicenter RCT (22 centres in the UK and Europe), Phase II | |
| Participants |
Number of participants: 72 (Auguest 2005 to November 2006) Number randomised: Experimental group: 36 Control group: 36 Number evaluated: Experimental group: 35, age (median and range): 59 (29 ‐ 79) years Control group: 36, age (median and range): 64 (36 ‐ 76) years Diagnosis: patients with previously untreated, histopathologically‐confirmed metastatic gastric adenocarcinoma or adenocarcinoma of the lower third of the esophagus; Only patients with EGFR‐positive tumors were enrolled into the study Inclusion: eligible patients should: be > 18 years; ECOG performance status 0/1; normal cardiac function (left ventricular ejection fraction within the institutional normal range); a minimum 12‐month interval from completion of any neoadjuvant or adjuvant chemotherapy; a minimum 4‐week interval from completion of radiotherapy; and adequate liver, bone marrow and renal function |
|
| Interventions |
Experimental group : 800 mg matuzumab weekly; epirubicin 50 mg/m², cisplatin 60 mg/m² on day 1 and capecitabine 1250 mg/m² daily in a 21‐day cycle Control group: epirubicin 50 mg/m², cisplatin 60 mg/m² on day 1 and capecitabine 1250 mg/m² daily in a 21‐day cycle Treatment cycles were repeated every 3 weeks for a maximum of 8 cycles of ECX unless there was evidence of disease progression or unacceptable toxicity, death occurred or consent was withdrawn. Matuzumab was continued as a single agent after the 8 cycles of ECX unless there was evidence of disease progression or unacceptable toxicity, death occurred or consent was withdrawn |
|
| Outcomes |
Duration of follow‐up (median): 28.5 months for experimental group, and 23 months for control group OS, PFS, overall response, quality of life, adverse events (reported by different symptoms, no data for any adverse event) |
|
| Notes | Sponsor: Merck KGaA The sponsor involved in the study design, data collection, and data analysis |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Random sequence was generated by a computer programme |
| Allocation concealment (selection bias) | Low risk | Randomization was carried out centrally in a blinded manner by telephone using an interactive voice response system |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | The open‐label design may introduce performance bias (e.g. through stress‐related mechanisms). |
| Blinding of outcome assessment (detection bias) OS, Serious adverse events | Low risk | These outcomes were unlikely to be affected by the blinding status of assessors |
| Blinding of outcome assessment (detection bias) PFS, Response, quality of life & adverse events | Low risk | Blinded radiological review was performed |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 71 participants included in the analysis (98.6%); ITT was used |
| Selective reporting (reporting bias) | Low risk | No prior protocol was available but all important outcomes were reported |
| Other bias | Low risk | No obvious potential source of bias |
Shen 2015.
| Methods | Chinese multicenter RCT (14 hospitals in China), Phase III | |
| Participants |
Number of participants: 202 (March 2009 to July 2010) Number randomised: Experimental group (capecitabine and cisplatin + bevacizumab): 100 Control group (capecitabine and cisplatin + placebo): 102 Number evaluated: Experimental group:100, age (median): 54.2 years. control group: 102, age (median): 55.5 years. Diagnosis: patients with previously untreated, histologically‐confirmed, inoperable, locally‐advanced or recurrent, and/or metastatic adenocarcinoma of the stomach or gastro‐esophageal junction Inclusion: eligible patients should: be > 18 years; with histologically‐confirmed, inoperable, locally advanced or recurrent, and/or metastatic adenocarcinoma of the stomach or gastro‐esophageal junction; with no prior treatment for advanced/metastatic disease; ECOG performance status 0 – 2; adequate organ function; with measurable or non‐measurable but evaluable disease |
|
| Interventions |
Experimental group : Bevacizumab 7.5 mg/kg i.v. on day 1 every 3 weeks; cisplatin 80 mg/m² i.v. on day 1 every 3 weeks; oral capecitabine 1000 mg/m² twice daily for 14 days every 3 weeks Control group: placebo on day 1 every 3 weeks; cisplatin 80 mg/m² i.v. on day 1 every 3 weeks; oral capecitabine 1000 mg/m² twice daily for 14 days every 3 weeks Cisplatin was given for 6 cycles; capecitabine and bevacizumab were administered until disease progression or unacceptable toxicity |
|
| Outcomes |
Duration of follow‐up (median): 10.0 months for experimental group, and 10.5 months for control group OS, PFS, overall response, adverse events (only the incidence for grade ≥ 3 adverse events was reported, not for any adverse events) |
|
| Notes | Sponsor: Hoffmann‐La Roche The sponsor was involved in the study design, data collection, data analysis, and manuscript preparation |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | The dynamic least‐squares minimization randomization method was used |
| Allocation concealment (selection bias) | Low risk | Participants were randomly assigned to 1 of the 2 treatment groups via an interactive voice response system |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blind: "neither patients nor investigators knew which treatment patients were receiving". |
| Blinding of outcome assessment (detection bias) OS, Serious adverse events | Low risk | These outcomes were unlikely to be affected by the blinding status of assessors |
| Blinding of outcome assessment (detection bias) PFS, Response, quality of life & adverse events | Low risk | Double‐blind: "neither patients nor investigators knew which treatment patients were receiving". |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | ITT analysis was used |
| Selective reporting (reporting bias) | Low risk | Prior protocol was available |
| Other bias | Low risk | No obvious potential source of bias |
Waddell 2013.
| Methods | UK multicenter RCT (63 centers in UK), Phase III | |
| Participants |
Number of participants: 553 patients June 2008 to October 2011) Number randomised: Experimental group (epirubicin and oxaliplatin and capecitabine (EOC) + panitumumab): 278 control group (EOC): 275 Number evaluated: Experimental group: 278, age (median and range): 62 (26 ‐ 83) years Control group: 275, age (median and range): 63 (26 ‐ 83) years Diagnosis: patients with histologically‐verified, untreated, metastatic or locally‐advanced inoperable adenocarcinoma or undifferentiated carcinoma of the esophagus, gastro‐esophageal junction, or stomach. Inclusion: eligible patients should: be > 18 years; have measurable disease on CT or MRI; WHO performance status of 0 – 2; adequate cardiac, renal, liver, and bone marrow function |
|
| Interventions |
Experimental group : epirubicin 50 mg/m² i.v. on day 1, every 3 weeks; oxaliplatin 100 mg/m² i.v. on day 1 every 3 weeks; oral capecitabine 1000 mg/m² per day on day 1 – 21 every 3 weeks; and panitumumab 9 mg/kg i.v. on day 1, every 3 weeks Control group: epirubicin 50 mg/m² i.v.on day 1 every 3 weeks; oxaliplatin 130 mg/m² i.v. on day 1 every 3 weeks; and oral capecitabine 1250 mg/m² per day on days 1 – 21 every 3 weeks Participants received a maximum of 8 cycles of treatment |
|
| Outcomes |
Duration of follow‐up (median): 5.3 months (IQR: 2.6 ‐ 9.5) for experimental group, and 4.6 months (IQR: 1.8 ‐ 10.1) for control group OS, PFS, overall response, adverse events (only the incidence for grade ≥ 3 adverse events was reported, not for any adverse events) |
|
| Notes | Sponsor: Royal Marsden NHS Foundation Trust Study closed early due to lack of efficacy |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Randomisation was done independently at the Institute for Cancer Research Clinical Trials and Statistics Unit (ICR‐CTSU) by random permuted blocks (block sizes of six and eight) and stratified by centre region (locations were divided into 11 regions), extent of disease (locally advanced vs metastatic disease), and performance status (0 vs 1 vs 2)" |
| Allocation concealment (selection bias) | Low risk | "Patients were enrolled by trials office staff at the Royal Marsden Hospital, who then faxed confirmation of the allocated treatment group to local site staff" |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | The open‐label design may introduce performance bias (e.g. through stress‐related mechanisms). |
| Blinding of outcome assessment (detection bias) OS, Serious adverse events | Low risk | These outcomes were unlikely to be affected by the blinding status of assessors |
| Blinding of outcome assessment (detection bias) PFS, Response, quality of life & adverse events | Unclear risk | No masking for outcome assessors, but quality of reported data was controlled by trust‐appointed monitoring staff or central monitoring system |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | ITT analysis was used |
| Selective reporting (reporting bias) | Low risk | Prior protocol was available |
| Other bias | Unclear risk | Study closed early due to lack of efficacy |
Zhang 2014.
| Methods | RCT, China | |
| Participants |
Number of participants: 56(August 2010 to September 2012) Number randomised: Experimental group (cetuximab in combination with S‐1 and oxaliplatin): 30 Control group (S‐1 and oxaliplatin): 26 Number evaluated: Experimental group: 30, age(median and range): 56 (26 ‐ 78) years Control group: 26, age(median and range): 56 (26 ‐ 78) years Diagnosis: patients with histologically‐ or cytologically‐proven unresectable gastric cancer or recurrence postoperation Inclusion: All of the participants had at least 1 measurable lesion by the RECIST criteria, were ECOG performance status (PS) 0 ‐ 2 and had anticipated life expectancies > 3 months. Additionally, the participants were required to be chemotherapy naïve or > 6 months past the last adjuvant chemotherapy and to possess favorable bone marrow reservation (hemoglobin ≥ 80 g/L, platelet count ≥ 100 × 109, leukocyte count = 3 to 10 × 109, neutrophil count ≥ 1.5 × 109). Laboratory examination showed adequate liver function (total bilirubin ≤ 1.5 times the upper limit, ALT/AST ≤ 2.5 times the upper limit) and renal function (blood creatinine ≤ 1.5 mg/dL, creatinine clearance ≥ 50 mL/min) |
|
| Interventions |
Experimental group cetuximab in combination with S‐1 and oxaliplatin: once‐weekly cetuximab (400 mg/m² at the first infusion then 250 mg/m² every week) + oxaliplatin (100 mg/m²) i.v. administered on day 1 and S‐1 (80 mg/m²/day) orally twice daily for 14 days. All participants then took 1 week’s rest before the next cycle Control group S‐1 and oxaliplatin: oxaliplatin (100 mg/m²) i.v. administered on day 1 and S‐1 (80 mg/m²/day) orally twice daily for 14 days. All participants then took 1 week’s rest before the next cycle |
|
| Outcomes |
Duration of follow‐up (median):not mentioned. Cut‐off date of final analysis was on 30 September 2012 OS, PFS, overall response, adverse events(reported by different symptoms, no data for any adverse event) |
|
| Notes | The reported median ages for all participants were 49 years in text, but 56 years in corresponding tables | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Unspecified |
| Allocation concealment (selection bias) | Unclear risk | Unspecified |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | The open‐label design may introduce performance bias (e.g. through stress‐related mechanisms). |
| Blinding of outcome assessment (detection bias) OS, Serious adverse events | Low risk | These outcomes were unlikely to be affected by the blinding status of assessors |
| Blinding of outcome assessment (detection bias) PFS, Response, quality of life & adverse events | Unclear risk | Unspecified |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 56 randomized participants were included in analysis (100%) |
| Selective reporting (reporting bias) | Low risk | No prior protocol was available but all important results were reported |
| Other bias | Low risk | No obvious potential source of bias |
ALT: alanine aminotransferase AST: aspartate aminotransferase CT: computerised tomography ECOG: Eastern Cooperative Oncology Group IQR: interquartile range ITT: intention‐to‐treat i.v.: intravenous MRI: magnetic resonance imaging OS: overall survival PFS: progression‐free survival RECIST: Response Evaluation Criteria in Solid Tumors SD: standard deviation ULN: upper limit of normal WHO: World Health Organization
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Liang 2009 | Not first‐line treatment |
| Ohtsu 2013 | Not first‐line treatment |
| Qin 2014 | Not first‐line treatment (focusing on patients who failed second‐line chemotherapy) |
| Richards 2013 | Not first‐line treatment |
| Satoh 2014 | Not first‐line treatment |
| Sun 2013 | A subanalysis of TYTAN trial (second‐line treatment) |
| Takahashi 2014 | Not first‐line treatment |
| Xu 2013 | Not molecular‐targeted agent |
Characteristics of studies awaiting assessment [ordered by study ID]
Wahab 2011.
| Methods | RCT |
| Participants | 41 chemo‐naïve patients with AGC (21 were randomized to experimental group, and 20 to control group) |
| Interventions |
|
| Outcomes | OS, PFS, overall response, adverse events |
| Notes | Not available in full text. We were unable to acquire full details about the trial and its results from the authors |
Wang 2012.
| Methods | RCT |
| Participants | 40 chemo‐naïve patients with advanced or metastatic gastric cancer (20 were randomized to experimental group, and 20 to control group) |
| Interventions |
|
| Outcomes | Overall response, adverse events |
| Notes | Not available in full text. We were unable to acquire full details about the trial and its results from the authors |
Characteristics of ongoing studies [ordered by study ID]
ACTRN12609000109202.
| Trial name or title | A randomised phase II study evaluating weekly docetaxel, cisplatin, fluoropyrimidine (wTCF) plus or minus panitumumab in advanced oesophago‐gastric cancer |
| Methods | RCT |
| Participants | Patients with histological diagnosis of metastatic or locally‐recurrent esophago‐gastric cancer |
| Interventions |
Docetaxel (T): 30 mg/m² calculated to the nearest mg and administered as an i.v. infusion on day 1 and day 8 of each cycle Cisplatin (C):60 mg/m² administered as an i.v. infusion on day 1 of each cycle Peripheral Venous Infusion (PVI) 5‐fluorouracil (5FU) : 160 mg/m² daily, by continuous infusion administered via an indwelling venous line (peripherally inserted central catheter (PICC)) line or infusaport according to local practice or capecitabine: 500 mg/m² oral twice daily for 21 days (42 doses)
Panitumumab: administered i.v. by an infusion pump through a peripheral line or indwelling catheter 9mg/kg on day 1 Treatment described above will continue for a maximum total of 8 cycles ((1 cycle = 3 weeks) x 8 cycles) which equates to 24 weeks |
| Outcomes | RR, OS, PFS |
| Starting date | March 2010 |
| Contact information | attax3@ctc.usyd.edu.au |
| Notes | From April 2010 to November 2011, 77 participants were enrolled from 15 institutions. A safety alert from the REAL3 study (also involving P in OG cancer) prompted an unplanned review of data from ATTAX3 by the IDMC. The IDMC found no evidence of adverse outcomes associated with P, but as it did not appear that P would significantly improve efficacy, they recommended cessation of the study to new enrolment. |
NCT01123473.
| Trial name or title | Effectiveness of first line treatment with lapatinib and ECF/X in histologically proven adenocarcinoma of the stomach or the esophagogastric junction, metastatic or not amenable to curative surgery according to HER2 and EGFR status: a randomized Phase II trial |
| Methods | RCT |
| Participants | Patients with histologically‐confirmed adenocarcinoma of the stomach or the esophago‐gastric junction; metastatic disease OR not amenable to curative surgery; tissue material for HER2 and EGFR assessment must be available; positive HER2 status by IHC OR positive EGFR by either FISH or IHC at time of randomization; no clinical signs of CNS involvement; no prior palliative systemic chemotherapy |
| Interventions |
|
| Outcomes | PFS; response rate; OS; toxicity; concordance of diagnostic tests |
| Starting date | May 13 2010 |
| Contact information | Not available |
| Notes | This study has been terminate since the company withdrew interest |
NCT01443065.
| Trial name or title | MEGA (Met or EGFR Inhibition in Gastroesophageal Adenocarcinoma): FOLFOX alone or in combination with AMG 102 or panitumumab as first‐line treatment in patients with advanced gastroesophageal adenocarcinoma |
| Methods | RCT |
| Participants | Patients with histologically‐proven adenocarcinoma of the stomach, esophagus or gastro‐esophageal junction; locally‐advanced or metastatic disease; measurable disease (RECIST 1.1); no known HER2 over‐expression; no prior palliative chemotherapy |
| Interventions |
|
| Outcomes | PFS rate at 4 months, PFS, OS, time to progression |
| Starting date | January 2011 |
| Contact information | Not available |
| Notes | Phase II. This study is ongoing, but not recruiting participants |
NCT01503372.
| Trial name or title | Pazopanib with 5‐Fluorouracil, leucovorin and oxaliplatin (FLO) as 1st‐line treatment in advanced gastric cancer; a randomized hase‐II‐study of the Arbeitsgemeinschaft Internistische Onkologie |
| Methods | RCT |
| Participants | Patients with histologically‐confirmed adenocarcinoma of the stomach or the gastro‐esophageal junction with either metastatic or locally‐advanced disease, incurable by operation |
| Interventions |
|
| Outcomes | PFS rate at 6 months; PFS rate at 9 and 12 months; median PFS; response rate |
| Starting date | November 2011 |
| Contact information | magenkarzinom@charite.de |
| Notes | The recruitment status of this study is unknown because the information has not been verified recently |
NCT01662869.
| Trial name or title | A study of onartuzumab (MetMAb) in combination with mFOLFOX6 in patients with metastatic HER2‐negative and Met‐positive gastroesophageal cancer (MetGastric) |
| Methods | RCT |
| Participants | Patients with metastatic HER2‐negative and Met‐positive adenocarcinoma of the stomach or gastro‐esophageal junction |
| Interventions |
|
| Outcomes | OS in the Met IHC 2+/3+ patient subgroup; OS in the intention‐to‐treat population; response rate; safety |
| Starting date | November 2012 |
| Contact information | Not available |
| Notes | This study is ongoing, but not recruiting participants |
NCT01774786.
| Trial name or title | A study of perjeta (Pertuzumab) in combination with herceptin (trastuzumab) and chemotherapy in patients with HER2‐positive metastatic gastroesophageal junction or gastric cancer |
| Methods | RCT |
| Participants | Patients with HER2‐positive metastatic adenocarcinoma of the stomach or gastro‐esophageal junction; ≥ 18 years of age; measurable or evaluable non‐measurable disease as assessed by the investigator according to RECIST; ECOG performance status 0 or 1; life expectancy ≥ 3 months |
| Interventions |
|
| Outcomes | OS; PFS; Overall objective response (partial response + complete response) occurring on 2 consecutive occasions ≥ 4 weeks apart; Duration of objective response; Clinical benefit rate (best response of complete response or partial response or stable disease for 6 weeks or longer); safety |
| Starting date | June 2013 |
| Contact information | global.rochegenentechtrials@roche.com |
| Notes | Sponsor: Hoffmann‐La Roche |
NCT02314117.
| Trial name or title | A randomized, double‐blind, placebo‐controlled Phase 3 study of capecitabine and cisplatin with or without ramucirumab as first‐line therapy in patients with metastatic gastric or gastroesophageal junction adenocarcinoma (RAINFALL) |
| Methods | RCT |
| Participants | Patients with a histopathologically‐confirmed diagnosis of metastatic gastric or gastro‐esophageal junction (GEJ) adenocarcinoma |
| Interventions |
|
| Outcomes | The primary endpoint is PFS; OS is the key secondary endpoint |
| Starting date | January 2015 |
| Contact information | Call 1‐877‐CTLILLY (1‐877‐285‐4559) or 1‐317‐615‐4559 Mon ‐ Fri 9 AM ‐ 5 PM Eastern time (UTC/GMT ‐ 5 hours, EST) |
| Notes | This study is ongoing, and now recruiting participants |
Differences between protocol and review
We changed the eligibility criteria of the review to allow the inclusion of studies involving participants with esophageal adenocarcinoma, because this type of cancer typically arises adjacent to the stomach, and has been historically treated with similar chemotherapy to that used for gastric cancer.
We added methods for analyzing time‐to‐event data after publication of the protocol. In preparing the protocol we had anticipated evaluating survival outcomes as a risk ratio, but on reviewing data from the included studies we considered the most appropriate way to analyze survival was to preserve the analysis of survival outcomes as time‐to‐event data.
Contributions of authors
Conceiving the review: HS
Designing the review: HS
Co‐ordinating the review: HS
Designing search strategies: Cochrane UGPD Group
Writing the review: HS, JZ
Providing general advice on the review: DL
Performing previous work that was the foundation of the current study: HS, DL, JZ
Sources of support
Internal sources
Karolinska Institutet, Sweden.
Shandong Provincial Hospital, China.
External sources
No sources of support supplied
Declarations of interest
Huan Song: none known
Jianwei Zhu: none known
DongHao Lu: none known
New
References
References to studies included in this review
Bang 2010 {published data only}
- Bang YJ, Cutsem E, Feyereislova A, Chung HC, Shen L, Sawaki A, et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2‐positive advanced gastric or gastro‐oesophageal junction cancer (ToGA): a phase 3, open‐label, randomised controlled trial. Lancet 2010;376(9742):687‐97. [DOI] [PubMed] [Google Scholar]
- Fukuda T, Shimozuma K. Cost‐effectiveness analysis of trastuzumab to treat HER2‐positive advanced gastric cancer based on the randomised ToGA trial.. British Journal of Cancer 2011;105(9):1273‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye M, Chen H, Shen JF. Costs of trastuzumab in combination with chemotherapy for HER2‐positive advanced gastric or gastroesophageal junction cancer: an economic evaluation in the Chinese context. Clinical Therapeutics 2012;34(2):468‐79. [DOI] [PubMed] [Google Scholar]
Eatock 2013 {published data only}
- Eatock MM, Tebbutt NC, Bampton CL, Strickland AH, Valladares‐Ayerbes M, Swieboda‐Sadlej A, et al. Phase II randomized, double‐blind, placebo‐controlled study of AMG 386 (trebananib) in combination with cisplatin and capecitabine in patients with metastatic gastro‐oesophageal cancer. Annals of Oncology 2013;24(3):710‐8. [DOI] [PubMed] [Google Scholar]
Hecht 2013 {published data only}
- Hecht JR, Bang Y, Qin S, Chung H, Xu J, Park J, et al. Lapatinib in combination with capecitabine plusoxaliplatin in HER2‐positive advanced ormetastatic gastric, esophageal, orgastroesophageal adenocarcinoma: the TRIO‐013/LOGiC trial. Journal of Clinical Oncology 2013;31(Suppl):abstr LBA4001. [Google Scholar]
Iveson 2014 {published data only}
- Doshi S, Gisleskog PO, Zhang Y, Zhu M, Oliner KS, Loh E, et al. Rilotumumab exposure‐response relationship in patients with advanced or metastatic gastric cancer. Clinical Cancer Research Published Online First: February 24, 2015. [DOI: 10.1158/1078-0432] [DOI] [PubMed]
- Iveson T, Donehower RC, Davidenko I, Tjulandin S, Deptala A, Harrison M, et al. Rilotumumab in combination with epirubicin, cisplatin, and capecitabine as first‐line treatment for gastric or oesophagogastric junction adenocarcinoma: an open‐label, dose de‐escalation phase 1b study and a double‐blind, randomised phase 2study. Lancet Oncology 2014;15(9):1007‐18. [DOI] [PubMed] [Google Scholar]
Koizumi 2013 {published data only}
- Koizumi W, Yamaguchi K, Hosaka H, Takinishi Y, Nakayama N, Hara T, et al. Randomised phase II study of S‐1/cisplatin plus TSU‐68 vs S‐1/cisplatin in patients with advanced gastric cancer. British Journal of Cancer 2013;109(8):2079‐86. [DOI] [PMC free article] [PubMed] [Google Scholar]
Lordick 2013 {published data only}
- Lordick F, Kang YK, Chung HC, Salman P, Oh SC, Bodoky G, et al. Capecitabine and cisplatin with or without cetuximab for patients with previously untreated advanced gastric cancer (EXPAND): a randomised, open‐label phase 3 trial. Lancet Oncology 2013;14(6):490‐9. [DOI] [PubMed] [Google Scholar]
Ohtsu 2011 {published data only}
- Ohtsu A, Shah MA, Cutsem E, Rha SY, Sawaki A, Park SRl, et al. Bevacizumab in combination with chemotherapy as first‐line therapy in advanced gastric cancer: a randomized, double‐blind, placebo‐controlled phase III study. Journal of Clinical Oncology 2011;29(30):3968‐76. [DOI] [PubMed] [Google Scholar]
- Shah M, Kang Y, Ohtsu A, Delmar P, Foernzler D, Langer B, et al. Tumor and blood plasma biomarker analyses in the avagast phase III randomized study of first line bevacizumab 1 capecitabine/cisplatin in patients with advanced gastric cancer. Annals of Oncology 2010;21:viii67‐viii68. [Google Scholar]
Rao 2010 {published data only}
- Rao S, Starling N, Cunningham D, Sumpter K, Gilligan D, Ruhstaller T, et al. Matuzumab plus epirubicin, cisplatin and capecitabine (ECX) compared with epirubicin, cisplatin and capecitabine alone as first‐line treatment in patients with advanced oesophago‐gastric cancer: a randomised, multicentre open‐label phase II study. Annals of Oncology 2010;21(11):2213‐9. [DOI] [PubMed] [Google Scholar]
Shen 2015 {published data only}
- Shen L, Li J, Xu J, Pan H, Dai G, Qin S, et al. Bevacizumab plus capecitabine and cisplatin in Chinese patients with inoperable locally advanced or metastatic gastric or gastroesophageal junction cancer: randomized, double‐blind, phase III study (AVATAR study). Gastric Cancer 2015;18(1):168‐76. [DOI] [PMC free article] [PubMed] [Google Scholar]
Waddell 2013 {published data only}
- Waddell T, Chau I, Cunningham D, Gonzalez D, Okines AF, Okines C, et al. Epirubicin, oxaliplatin, and capecitabine with or without panitumumab for patients with previously untreated advanced oesophagogastric cancer (REAL3): a randomised, open‐label phase 3 trial. Lancet Oncology 2013;14(6):481‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Zhang 2014 {published data only}
- Zhang ZD, Kong Y, Yang W, Zhang B, Zhang YL, Ma EM, et al. Clinical evaluation of cetuximab combined with an S‐1 and oxaliplatin regimen for Chinese patients with advanced gastric cancer. World Journal of Surgical Oncology 2014;12(1):115‐21. [DOI] [PMC free article] [PubMed] [Google Scholar]
References to studies excluded from this review
Liang 2009 {published data only}
- Liang J, Liang S, Li G, Liu H, Zhao L, Wang H, et al. Clinical efficacy of rh‐endostatin combined with XELOX for advanced gastric cancer. [Chinese]. Chinese Journal of Clinical Oncology 2009;36(17):976‐9. [Google Scholar]
Ohtsu 2013 {published data only}
- Ohtsu A, Ajani JA, Bai YX, Bang YJ, Chung HC, Pan HM, et al. Everolimus for previously treated advanced gastric cancer: results of the randomized, double‐blind, phase III GRANITE‐1 study. Journal of Clinical Oncology 31;31:3935‐43. [DOI] [PMC free article] [PubMed] [Google Scholar]
Qin 2014 {published data only}
- Qin SK. Phase III study of apatinib in advanced gastric cancer: A randomized, double‐blind, placebo‐controlled trial. Journal of Clinical Oncology 2014;32(5s):suppl; abstr 4003. [Google Scholar]
Richards 2013 {published data only}
- Richards D, Kocs DM, Spira AI, David McCollum A, Diab S, Hecker LI, et al. Results of docetaxel plus oxaliplatin (DOCOX) ± cetuximab in patients with metastatic gastric and/or gastroesophageal junction adenocarcinoma: results of a randomised Phase 2 study. European Journal of Cancer 2013;49(13):2823‐31. [DOI] [PubMed] [Google Scholar]
Satoh 2014 {published data only}
- Satoh T, Doi T, Ohtsu A, Tsuji A, Omuro Y, Mukaiyama A, et al. Lapatinib plus paclitaxel versus paclitaxel alone in the second‐line treatment of HER2‐amplified advanced gastric cancer in Asian populations: TyTAN ‐ A randomized, phase III study. Journal of Clinical Oncology 2014;32(19):2039‐49. [DOI] [PubMed] [Google Scholar]
Sun 2013 {published data only}
- Sun GP, Xu RH, Xu JM, Li J, Wang JW, Qin S, et al. The Chinese subgroup from a randomized phase III study of lapatinib in combination with weekly paclitaxel versus weekly paclitaxel alone as second‐line treatment of HER2‐amplified advanced gastric cancer (AGC) in Asian countries. Journal of Clinical Oncology 2013;suppl:abstr 4109. [Google Scholar]
Takahashi 2014 {published data only}
- Takahashi T, Nishikawa K, Miki A, Noshiro H, Yoshikawa T, Nishida Y, et al. Efficacy and safety result of trastuzumab (T‐mab) and paclitaxel for T‐mab naive patients with HER2‐positive previously treated advanced or recurrent gastric cancer (JFMC45‐1102): Final report. Journal of Clinical Oncology 2014;32(Suppl 3):abstr 79. [Google Scholar]
Xu 2013 {published data only}
- Xu R, Ma N, Wang F, Ma L, Chen R, Chen R, et al. Results of a randomized and controlled clinical trial evaluating the efficacy and safety of combination therapy with Endostar and S‐1 combined with oxaliplatin in advanced gastric cancer. Onco Targets and Therapy 2013;6:925‐9. [DOI: 10.2147/OTT.S46487] [DOI] [PMC free article] [PubMed] [Google Scholar]
References to studies awaiting assessment
Wahab 2011 {published data only}
- Wahab MA, Ezzelarab L, Bendary S. Cetuximab plus capecitabine and oxaloplatin for chemonaive patients with advanced gastric cancer. Annals of Oncology 2011;22:v50. [Google Scholar]
Wang 2012 {published data only}
- Wang JW, Chi Y, Zheng ZX, Qu T, Zhou AP, Yang L, et al. Randomized, single‐centered, phase II clinical trial of nimotuzumab plus cisplatin and S‐1 as first‐line therapy in patients with advanced gastric cancer. Journal of Clinical Oncology 2012;30(Suppl; abstr):e14668. [Google Scholar]
References to ongoing studies
ACTRN12609000109202 {published data only}
- A randomised phase II study evaluating weekly docetaxel, cisplatin, fluoropyrimidine (wTCF) plus or minus panitumumab in advanced oesophago‐gastric cancer. Ongoing study March 2010.
NCT01123473 {published data only}
- Effectiveness of first line treatment with lapatinib and ECF/X in histologically proven adenocarcinoma of the stomach or the esophagogastric junction, metastatic or not amenable to curative surgery according to HER2 and EGFR status: a randomized Phase II trial. Ongoing study May 13 2010.
NCT01443065 {published data only}
- MEGA (Met or EGFR Inhibition in Gastroesophageal Adenocarcinoma): FOLFOX alone or in combination with AMG 102 or panitumumab as first‐line treatment in patients with advanced gastroesophageal adenocarcinoma. Ongoing study January 2011.
NCT01503372 {published data only}
- Pazopanib with 5‐Fluorouracil, leucovorin and oxaliplatin (FLO) as 1st‐line treatment in advanced gastric cancer; a randomized hase‐II‐study of the Arbeitsgemeinschaft Internistische Onkologie. Ongoing study November 2011.
NCT01662869 {published data only}
- A study of onartuzumab (MetMAb) in combination with mFOLFOX6 in patients with metastatic HER2‐negative and Met‐positive gastroesophageal cancer (MetGastric). Ongoing study November 2012.
NCT01774786 {published data only}
- A study of perjeta (Pertuzumab) in combination with herceptin (trastuzumab) and chemotherapy in patients with HER2‐positive metastatic gastroesophageal junction or gastric cancer. Ongoing study June 2013.
NCT02314117 {published data only}
- A randomized, double‐blind, placebo‐controlled Phase 3 study of capecitabine and cisplatin with or without ramucirumab as first‐line therapy in patients with metastatic gastric or gastroesophageal junction adenocarcinoma (RAINFALL). Ongoing study January 2015.
Additional references
Anderson 2010
- Anderson WF, Camargo MC, Fraumeni JF Jr, Correa P, Rosenberg PS, Rabkin CS. Age‐specific trends in incidence of noncardia gastric cancer in US adults. JAMA 2010;303(17):1723‐28. [DOI] [PMC free article] [PubMed] [Google Scholar]
Bang 2012
- Bang YJ. Advances in the management of HER2‐positive advanced gastric and gastroesophageal junction cancer. Journal of Clinical Gastroenterology 2012;46(8):637‐48. [DOI] [PubMed] [Google Scholar]
Begnami 2011
- Begnami MD, Fukuda E, Fregnani JH, Nonogaki S, Montagnini AL, Costa WL Jr, et al. Prognostic implications of altered human epidermal growth factor receptors (HERs) in gastric carcinomas: HER2 and HER3 are predictors of poor outcome. Journal of Clinical Oncology 2011;29(22):3030‐6. [DOI] [PubMed] [Google Scholar]
Borenstein 2008
- Borenstein M, Hedges LV, Higgins JPT, Rothstein HR. Introduction to Meta‐Anaysis (Statistics in Practice). Chichester (UK): John Wiley & Sons, 2008. [Google Scholar]
Carmeliet 2003
- Carmeliet P. Angiogenesis in health and disease. Nature Medicine 2003;9(6):653‐60. [DOI] [PubMed] [Google Scholar]
Chua 2011
- Chua TC, Merrett ND. Clinicopathologic factors associated with HER2‐positive gastric cancer and its impact on survival outcomes‐‐a systematic review. International Journal of Cancer 2012;130(12):2845‐56. [DOI] [PubMed] [Google Scholar]
Draovich 2006
- Dragovich T, McCoy S, Fenoglio‐Preiser CM, Wang J, Benedetti JK, Baker AF, et al. Phase II trial of erlotinib in gastroesophageal junction and gastric adenocarcinomas: SWOG 0127. Journal of Clinical Oncology 2006;24(30):4922‐7. [DOI] [PubMed] [Google Scholar]
European Medicines Agency 2013
- European Medicines Agency. Herceptin 150 mg powder for concentrate for solution for infusion: summary of product characteristics. www.emea.europa.eu/docs/en_GB/document_library/EPAR_‐_Product_Information/human/000278/WC500074922.pdf. (Accessed 6 November 2013).
Ferlay 2015
- Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns inGLOBOCAN 2012. International Journal of Cancer 2015;136(5):E359‐86. [DOI] [PubMed] [Google Scholar]
Genentech 2013
- Genentech Inc. Herceptin (trastuzumab): US prescribing information 2010. www.accessdata.fda.gov/drugsatfda_docs/label/2010/103792s5256lbl.pdf. (Accessed 6 November 2013).
Grabsch 2010
- Grabsch H, Sivakumar S, Gray S, Gabbert HE, Müller W. HER2 expression in gastric cancer: Rare, heterogeneous and of no prognostic value ‐ conclusions from 924 cases of two independent series. Cell Oncology 2010;32(1‐2):57‐65. [DOI] [PMC free article] [PubMed] [Google Scholar]
GRADEprofiler 2008 [Computer program]
- Brozek J, Oxman A, Schünemann H. GRADEprofiler (GRADEpro). Version 3.2. GRADE Working Group, 2008.
Graziano 2011
- Graziano F, Galluccio N, Lorenzini P, Ruzzo A, Canestrari E, D'Emidio S, et al. Genetic activation of the MET pathway and prognosis of patients with high‐risk, radically resected gastric cancer. Journal of Clinical Oncology 2011;29(36):4789‐95. [DOI] [PubMed] [Google Scholar]
Hanahan 1996
- Hanahan D, Folkman J. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell 1996;86(3):353‐64. [DOI] [PubMed] [Google Scholar]
Higgins 2003
- Higgins JPT, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta‐analyses. BMJ 2003;327(7414):557‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Ilson 2011
- Ilson DH, Kelsen D, Shah M, Schwartz G, Levine DA, Boyd J, et al. A phase 2 trial of erlotinib in patients with previously treated squamous cell and adenocarcinoma of the esophagus. Cancer 2011;117(7):1409‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]
Iqbal 2011
- Iqbal S, Goldman B, Fenoglio‐Preiser CM, Lenz HJ, Zhang W, Danenberg KD, et al. Southwest Oncology Group study S0413: a phase II trial of lapatinib (GW572016) as first‐line therapy in patients with advanced or metastatic gastric cancer. Annals of Oncology 2011;22(12):2610‐5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Kim 2008
- Kim MA, Lee HS, Lee HE, Jeon YK, Yang HK, Kim WH. EGFR in gastric carcinomas: prognostic significance of protein overexpression and high gene copy number. Histopathology 2008;52(6):738‐46. [DOI] [PubMed] [Google Scholar]
Kim 2011
- Kim KC, Koh YW, Chang HM, Kim TH, Yook JH, Kim BS, et al. Evaluation of HER2 protein expression in gastric carcinomas: comparative analysis of 1,414 cases of whole‐tissue sections and 595 cases of tissue microarrays. Annals of Surgical Oncology 2011;18(10):2833‐40. [DOI] [PubMed] [Google Scholar]
Kim 2013
- Kim JG. Molecular targeted therapy for advanced gastric cancer. Korean Journal of Internal Medicine 2013;28(2):149‐55. [DOI] [PMC free article] [PubMed] [Google Scholar]
Lieto 2008
- Lieto E, Ferraraccio F, Orditura M, Castellano P, Mura AL, Pinto M, et al. Expression of vascular endothelial growth factor (VEGF) and epidermal growth factor receptor (EGFR) is an independent prognostic indicator of worse outcome in gastric cancer patients. Annals of Surgical Oncology 2008;15(1):69‐79. [DOI] [PubMed] [Google Scholar]
Lordick 2010b
- Lordick F. Trastuzumab: a new treatment option for HER2‐positive metastatic gastric and gastroesophageal junction cancer. Future Oncology 2011;7(2):187‐99. [DOI] [PubMed] [Google Scholar]
Macdonald 2001
- Macdonald JS, Smalley SR, Benedetti J, Hundahl SA, Estes NC, Stemmermann GN, et al. Chemoradiotherapy after surgery compared with surgery alone for adenocarcinoma of the stomach or gastroesophageal junction. New England Journal of Medicine 2001;345(10):725‐30. [DOI] [PubMed] [Google Scholar]
Maeda 1996
- Maeda K, Chung YS, Ogawa Y, Takatsuka S, Kang SM, Ogawa M, et al. Prognostic value of vascular endothelial growth factor expression in gastric carcinoma. Cancer 1996;77(5):858‐63. [DOI] [PubMed] [Google Scholar]
Murayama 2009
- Murayama T, Inokuchi M, Takagi Y, Yamada H, Kojima K, Kumagai J, et al. Relation between outcomes and localisation of p‐mTOR expression in gastric cancer. British Journal of Cancer 2009;100(5):782‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Ni 2010
- Ni XF, Wu CP, Jiang JT. Serum VEGFR‐3 and survival of advanced gastric cancer patients treated with FOLFOX. World Journal of Gastroenterology 2010;16(17):2163‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Parmar 1998
- Parmar MKB, Torri V, Stewart L. Extracting summary statistics to perform meta‐analyses of published literature for survival endpoints. Statistics in Medicine 1998;17(24):2815‐34. [DOI] [PubMed] [Google Scholar]
Pohl 2005
- Pohl H, Welch HG. The role of over diagnosis and reclassification in the marked increase of esophageal adenocarcinoma incidence. Journal of the National Cancer Institute 2005;97(2):142‐6. [DOI] [PubMed] [Google Scholar]
Power 2010
- Power DG, Kelsen DP, Shah MA. Advanced gastric cancer‐‐slow but steady progress. Cancer Treatment Reviews 2010;36(5):384‐92. [DOI] [PubMed] [Google Scholar]
Price 2012
- Price TJ, Shapiro JD, Segelov E, Karapetis CS, Pavlakis N, Cutsem E, et al. Management of advanced gastric cancer. Expert Review of Gastroenterology & Hepatology 2012;6(2):199‐208. [DOI] [PubMed] [Google Scholar]
Review Manager 2014 [Computer program]
- The Nordic Cochrane Centre: The Cochrane Collaboration. Review Manager (RevMan). Version 5.3. Copenhagen: The Nordic Cochrane Centre: The Cochrane Collaboration, 2013.
Sakuramoto 2007
- Sakuramoto S, Sasako M, Yamaguchi T, Kinoshita T, Fujii M, Nashimoto A, et al. Adjuvant chemotherapy for gastric cancer with S‐1, an oral fluoropyrimidine. New England Journal of Medicine 2007;357(18):1810‐20. [DOI] [PubMed] [Google Scholar]
Shaw 2006
- Shaw RJ, Cantley LC. Ras, PI(3)K and mTOR signalling controls tumour cell growth. Nature 2006;441(7092):424‐30. [DOI] [PubMed] [Google Scholar]
Song 2015a
- Song H, Held M, Sandin S, Rautelin H, Eliasson M, Söderberg S, et al. Increase in the prevalence of atrophic gastritis among adults age 35 to 44 years old in Northern Sweden between 1990 and 2009. Clinical Gastroenterology and Hepatology 2015;13(9):1592‐600. [DOI] [PubMed] [Google Scholar]
Spector 2009
- Spector NL, Blackwell KL. Understanding the mechanisms behind trastuzumab therapy for human epidermal growth factor receptor 2‐positive breast cancer. Journal of Clinical Oncology 2009;27(34):5838‐47. [DOI] [PubMed] [Google Scholar]
Sun 2010
- Sun W, Powell M, O'Dwyer PJ, Catalano P, Ansari RH, Benson AB 3rd. Phase II study of sorafenib in combination with docetaxel and cisplatin in the treatment of metastatic or advanced gastric and gastroesophageal junction adenocarcinoma: ECOG 5203. Journal of Clinical Oncology 2010;28(18):2947‐51. [DOI] [PMC free article] [PubMed] [Google Scholar]
Tanigawa 1996
- Tanigawa N, Amaya H, Matsumura M, Shimomatsuya T, Horiuchi T, Muraoka R, et al. Extent of tumor vascularization correlates with prognosis and hematogenous metastasis in gastric carcinomas. Cancer Research 1996;56(11):2671‐6. [PubMed] [Google Scholar]
Therasse 2000
- Therasse P, Arbuck SG, Eisenhauer EA, Wanders J, Kaplan RS, Rubinstein L, et al. New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of Canada, National Cancer Institute of the United States. Journal of the National Cancer Institute 2000;92(3):205‐16. [DOI] [PubMed] [Google Scholar]
Tierney 2007
- Tierney JF, Stewart LA, Ghersi D, Burdett S, Sydes MR. Practical methods for incorporating summary time‐to‐event data into meta‐analysis. Trials 2007;8(16):1‐16. [DOI] [PMC free article] [PubMed] [Google Scholar]
Wagner 2006
- Wagner AD, Grothe W, Haerting J, Kleber G, Grothey A, Fleig WE. Chemotherapy in advanced gastric cancer: a systematic review and meta‐analysis based on aggregate data. Journal of Clinical Oncology 2006;24(18):2903‐9. [DOI] [PubMed] [Google Scholar]
Wagner 2009
- Wagner AD, Moehler M. Development of targeted therapies in advanced gastric cancer: promising exploratory steps in a new era. Current Opinion in Oncology 2009;21(4):381‐5. [DOI] [PubMed] [Google Scholar]
Wagner 2010
- Wagner AD, Unverzagt S, Grothe W, Kleber G, Grothey A, Haerting J, et al. Chemotherapy for advanced gastric cancer. Cochrane Database of Systematic Reviews 2010, Issue 3. [DOI: 10.1002/14651858.CD004064.pub3] [DOI] [PubMed] [Google Scholar]
Widakowich 2007
- Widakowich C, Castro G Jr, Azambuja E, Dinh P, Awada A. Review: side effects of approved molecular targeted therapies in solid cancers. Oncologist 2007;12(12):1443‐55. [DOI] [PubMed] [Google Scholar]
Wilhelm 2008
- Wilhelm SM, Adnane L, Newell P, Villanueva A, Llovet JM, Lynch M. Preclinical overview of sorafenib, a multikinase inhibitor that targets both Raf and VEGF and PDGF receptor tyrosine kinase signaling. Molecular Cancer Therapeutics 2008;7(10):3129‐40. [DOI] [PMC free article] [PubMed] [Google Scholar]
Wu 2009
- Wu K, Nie Y, Guo C, Chen Y, Ding J, Fan D. Molecular basis of therapeutic approaches to gastric cancer. Journal of Gastroenterology and Hepatology 2009;24(1):37‐41. [DOI] [PubMed] [Google Scholar]
Yano 2006
- Yano T, Doi T, Ohtsu A, Boku N, Hashizume K, Nakanishi M, et al. Comparison of HER2 gene amplification assessed by fluorescence in situ hybridization and HER2 protein expression assessed by immunohistochemistry in gastric cancer. Oncology Reports 2006;15(1):65‐71. [PubMed] [Google Scholar]
Yu 2009
- Yu G, Wang J, Chen Y, Wang X, Pan J, Li G, et al. Overexpression of phosphorylated mammalian target of rapamycin predicts lymph node metastasis and prognosis of Chinese patients with gastric cancer. Clinical Cancer Research 2009;15(5):1821‐9. [DOI] [PubMed] [Google Scholar]
References to other published versions of this review
Song 2015b
- Song H, Zhu J, Lu D. Molecular‐targeted therapy for advanced gastric cancer. Cochrane Database of Systematic Reviews 2015, Issue 1. [DOI: 10.1002/14651858.CD011461] [DOI] [PMC free article] [PubMed] [Google Scholar]