

1. Introduction
The enzyme dihydroorotate dehydrogenase (DHODH) is essential for the de novo production of pyrimidines starting with the generation of uridine monophosphate (UMP), and small molecule inhibitors of DHODH are highly effective in vitro and in vivo in pre-clinical models of malignancy. DHODH is an unlikely cancer target, as it is ubiquitously expressed and is not known to be mutated or overexpressed in cancer. However, malignant cells seem to be more metabolically-dependent on de novo pyrimidine production, forming the potential basis of a therapeutic window. The use of DHODH inhibitors in solid tumor malignancies has been clinically disappointing. However, the use of DHODH inhibitors in the treatment of acute myeloid leukemia (AML) has been more recently shown to have both a cytotoxic and a pro-differentiation effect, making AML an attractive new disease indication. As of 2018, trials of DHODH inhibitors are underway, and will provide better insight into their potential utility in patients with hematologic malignancies.
Historically, the use of small molecule inhibitors of DNA and RNA synthesis is common in cancer chemotherapy, and we have decades of experience with effective molecules such as 5-Fluorouracil, cytarabine, and methotrexate. Given the essential role of uridine monophosphate (UMP) in DNA and RNA synthesis, it is no surprise that multiple inhibitors of de novo pyrimidine synthesis have been identified as hits during in vitro cancer cell line screening efforts through the 1970s and 1980s. Indeed, inhibitors at every step of pyrimidine synthesis have been studied in clinical trials; N-(phosphonacetyl)-L-aspartate (PALA) as an inhibitor of the aspartate-transcarbamylase function of CAD, brequinar sodium as an inhibitor of DHODH, and pyrazofurin as an inhibitor of the orotate-phosphoribosyltransferase function of UMPS (Figure 1).
Figure 1. de novo pyrimidine synthesis feeds myriad downstream metabolic pathways.
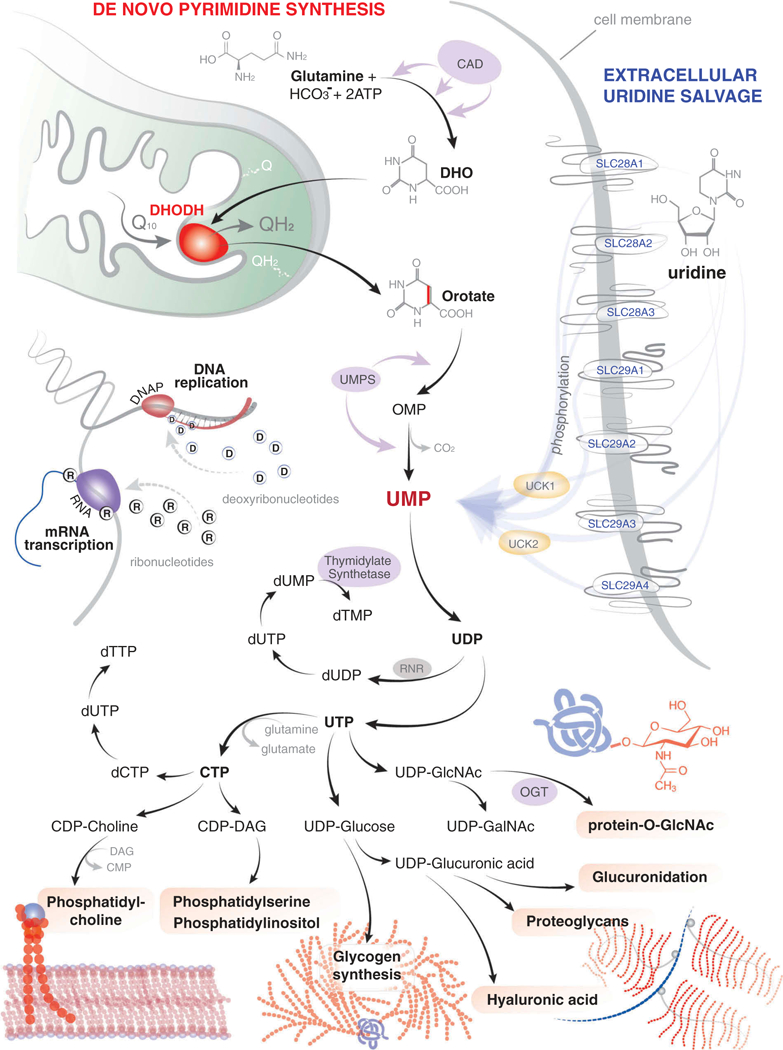
de novo pyrimidine synthesis begins with the conversion of glutamine to dihydroorotate (DHO) through the action of the trifunctional enzyme CAD, which acts as a carbamoyl-phosphate synthetase 2, aspartate transcarbamylase, and dihydroorotase. DHODH resides in the inner mitochondrial membrane and catalyzes the ubiquinone-dependent fourth step in the production of orotate. UMPS (uridine monophosphate synthetase) encodes the bifunctional enzyme to catalyze the final two steps; first as an orotate phosphoribosyltransferase which generates orotidine-5’-monophosphate (OMP) and then as an OMP decarboxylase which converts OMP to UMP. Extracellular uridine can be salvaged by the SLC28A and SLC29A family of membrane transporters. Once intracellular, uridine is then phosphorylated to UMP by the action of UCK (uridine-cytidine kinase 1 or 2).UMP is the initial building block in the production of ribonucleotides and deoxyribonucleotides and is therefore critical for the synthesis of RNA and DNA. However, UMP also feeds the production of phosphatidylcholine, phospatidylserine, phosphatidylinositol, glycogen, hyaluronic acid, proteoglycans, glucuronidation, and the post-translational modification of proteins by O-linked N-acetylglucosamine (GlcNAc).
1.1. What is DHODH?
Dihydroorotate dehydrogenase (DHODH) is a ubiquitous enzyme located within the inner membrane of the mitochondria. Using the cofactor ubiquinone, DHODH catalyzes the fourth step of de novo pyrimidine synthesis: the conversion of dihydroorotate to orotate. DHODH is the only enzyme capable of performing this conversion and it is therefore essential for the cell’s ability to produce uridine monophosphate (Figure 1).
UMP is the first building block in the production of pyrimidine ribonucleosides and deoxyribonuclesides for RNA and DNA synthesis, respectively. Inhibitors of DHODH function by blocking this proximal step leading to the rapid depletion of UMP, UDP, and UTP. Beyond RNA and DNA synthesis, UDP acts as a shuttle for myriad other metabolic pathways in the form of UDP-glucose, UDP-galactose, UDP-glucuronic acid, and UDP-N-acetylglucosamine (UDP-GlcNAc) (Figure 1).
Cells are capable of scavenging uridine from the extracellular environment via nucleoside transporters [1]. However, while the concentrations of extracellular uridine are as high as 10 μM, this is insufficient to sustain a dividing cell, and therefore the lack of DHODH activity is not compatible with life. Indeed, there are no DHODH-deficient animals. Even in the very rare Miller syndrome in humans, these severely affected individuals have hypomorphic alleles rather that complete loss of DHODH activity [2].
Of interest, leukemia cell lines (mouse and human) can be rendered DHODH-deficient by CRISPR/Cas9 gene editing; these knockout lines are dependent on supraphysiologic concentrations (~ 100 μM) of extracellular uridine (D.B. Sykes, unpublished data). However, the observation that these DHODH-null cells are viable suggests that the electron-transport function of DHODH is dispensable and is not the mechanistic target of the anti-leukemia effect of small molecule DHODH inhibitors.
1.2. DHODH inhibitors: initial clinical trial experience
The DHODH inhibitor brequinar (NSC 368,390, DuP-785) demonstrated potent in vitro and in vivo anti-tumor activity across multiple tumor models [3]. Brequinar was quickly advanced to phase I clinical trials in patients with advanced solid tumor malignancies [4–6]. While brequinar was generally well-tolerated, and demonstrated occasional durable responses, the clinical experience was disappointing across more than a dozen trials encompassing more than 500 patients [7–11].
1.3. Renewed interest in DHODH
Further clinical development of brequinar was halted as the patent life neared an end. However, the DHODH inhibitor leflunomide was FDA-approved in 1998 as an immune-suppressive medication for the treatment of rheumatoid arthritis. In the ensuing years, investigators suggested that leflunomide may have utility in the treatment of leukemia [12], of multiple myeloma [13], and of melanoma [14]. Indeed, these findings have led to small trials of leflunomide in myeloma (ClinicalTrials.gov Identifier: NCT02509052) and melanoma (NCT01611675). The potential utility of leflunomide as a chemotherapeutic agent has been limited by its relative low-potency and very long half-life.
1.4. DHODH as a target in acute myeloid leukemia (AML)
The DHODH inhibitor brequinar reappeared following a high-throughput phenotypic screen to identify small molecules that could overcome differentiation arrest in AML [15]. In this screen of more than 330,000 compounds, 11 of the 12 hits were ultimately found to be DHODH inhibitors, speaking to the importance of this pathway. When starved of pyrimidines, the acute myeloid leukemic blasts demonstrated both cell death and differentiation as shown by morphology, cell sur-face-marker expression, and gene expression. Furthermore, the surviving cells were functionally more differentiated, as demonstrated by the loss of leukemia-initiating cell activity [15]. Perhaps it is not surprisingly that prolonged pyrimidine starvation results in a cell-fate decision where the leukemic blast chooses differentiation over self-renewal. These findings were confirmed by other investigators in another model of AML using a different inhibitor of DHODH [16].
With the promise that AML cells may be particularly sensitive to pyrimidine starvation, there has been renewed interest in the clinical development of DHODH inhibitors. The phase I trials of BAY2402234 (Bayer, NCT03404726) and ASLAN003 (Aslan Pharmaceuticals, NCT03451084) are currently underway with other companies (Agios, PTC Therapeutics) likely to be entering the clinical arena (Table 1). While early trials are likely to focus on the population of relapsed and refractory patients with AML, it seems likely that trials in myelodysplastic syndrome (MDS) and other hematologic malignancies will soon follow.
Table 1.
The landscape of DHODH inhibitors in 2018.
| Inhibitor | Company | Stage of development (2018) |
Disease indication | Notes |
|---|---|---|---|---|
| AG-636 | Agios | Pre-clinical | AML and DLBCL (diffuse large B-cell lymphoma) | 1 |
| ASLAN003 | Aslan | Dose optimization clinical trial |
AML | 2 |
| BAY2402234 | Bayer | Phase I clinical trial | Myeloid malignancies | 3 |
| Brequinar sodium | DuPont | Clinical development on hold |
Previously tested in > 500 patients across multiple solid tumor indications |
|
| IMU-838 | Immunic therapeutics | Phase II clinical trial | Inflammatory bowel disease (ulcerative colitis and Crohn’s disease) | 4 |
| Leflunomide (Arava) | Sanofi | FDA approved | Rheumatoid arthritis | |
| Leflunomide (Arava) | Sanofi | Phase I/II clinical trial | Multiple myeloma | 5 |
| PP-001 | Panoptes Pharma | Pre-clinical | Autoimmune eye disease (uveitis) | 6 |
| PTC299 | PTC Therapeutics | Pre-clinical | Hematologic malignancies | 7 |
| Teriflunomide (Aubagio) | Sanofi Genzyme | FDA approved | Multiple sclerosis | |
1.5. DHODH in other malignancies
The dependence on de novo pyrimidine synthesis does not appear to be limited to AML. Since our publication in 2016, studies have suggested that DHODH may be a relevant target in the treatment of triple-negative breast cancer [17], PTEN-mutant tumors [18], and KRAS-driven tumors [19]. Presumably, the drivers of these diverse malignancies must all converge on a similar pathway of metabolic reprogramming to drive their dependence on pyrimidine synthesis and sensitivity to DHODH-inhibition.The metabolic underpinnings of this sensitivity are not known.
2. Expert opinion
DHODH is not a typical cancer target. DHODH is expressed in every cell, in every tissue, in every organ. The protein is not mutated in malignant cells, it is the not the target of recurrent chromosomal translocation, and it is not clearly overexpressed in malignant cells. For these reasons, the basis of the therapeutic window is not obvious. However, studies of brequinar, leflunomide, and teriflunomide suggest that DHODH inhibitors can be both effective and well-tolerated.
Inhibitors of DHODH lead to the rapid depletion of intracellular UMP and downstream metabolites. A cell’s capacity to salvage uridine is limited, and no cell can tolerate complete DHODH-inhibition and pyrimidine starvation indefinitely. I propose that leukemic cells, and possibly malignant cells along many other lineages, have a lower tolerance for pyrimidine starvation when compared to their non-malignant counterparts. Whether this is a result of a larger pyrimidine pool at baseline, or of a higher rate of flux through the pathway of de novo pyrimidine synthesis, is not clear. Regardless, the therapeutic window appears to result from the observation that malignant cells are at the very least more dependent on DHODH, and that they are thus more prone to differentiation (in systems where this can be assessed) and to cytotoxicity when faced with prolonged periods of pyrimidine starvation.
I would like to highlight the possibility that pyrimidine starvation secondary to DHODH inhibition results in death-from-starvation as compared to the death-from-cumulative-damage that one might expect from other cytotoxic chemotherapeutic agents (Figure 2). Cumulative damage in normal tissues (e.g. hematopoietic, gastrointestinal) results in dose-limiting clinical side-effects. Indeed, in the short-term, many chemotherapies are given at the maximum tolerated dose (MTD) followed by long periods of recovery. In the long-term, cumulative damage results in secondary malignancies, a devastating consequence in the patient who survives one cancer only to develop a treatment-related secondary malignancy.
Figure 2. The action of DHODH-inhibitor therapy as compared to cytotoxic chemotherapy.
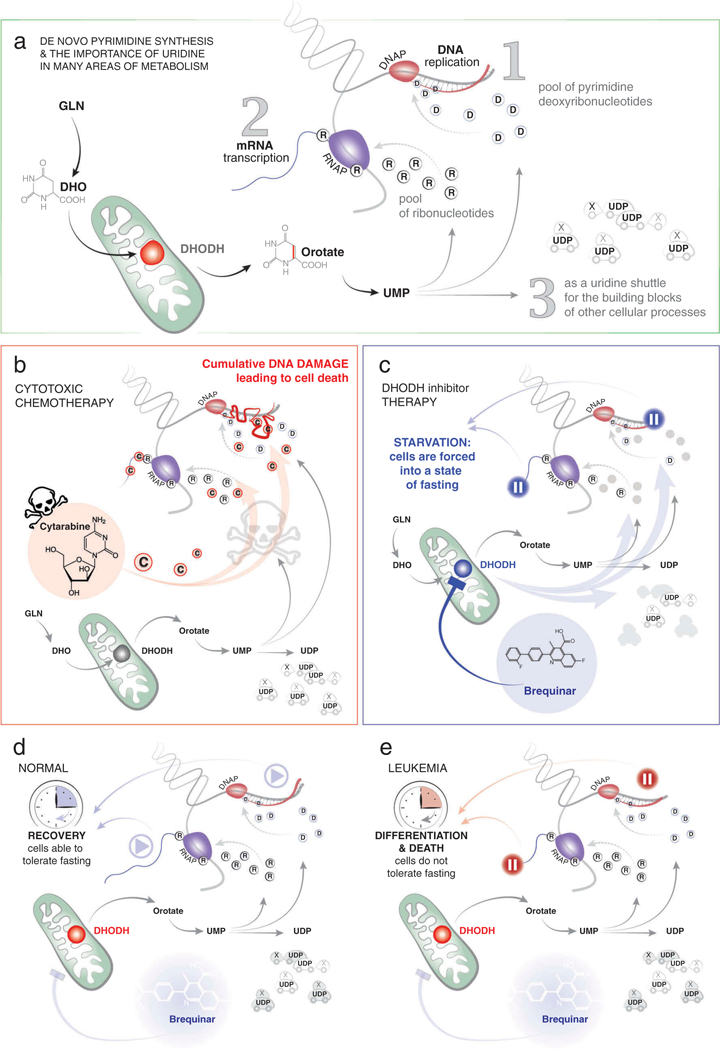
(A) The action of DHODH is essential for the formation of ribonucleotides, deoxyribonucleotides, and essential building blocks for many other metabolic pathways as outlined in Figure 1.(B) Many types of traditional cytotoxic chemotherapy, here exemplified by cytarabine, lead to inhibition of RNA synthesis, DNA synthesis, and cumulative DNA damage in both normal and leukemic cells. (C) Treatment with a DHODH inhibitor (e.g. brequinar) leads to a rapid depletion of intracellular UMP stores, placing cells into a state of starvation, and forcing cells to pause in RNA and DNA synthesis. (D) Normal cells can tolerate a limited period of starvation and resume nucleic acid synthesis and other cellular processes when the inhibition of DHODH is relieved. (E) Leukemic blasts do not tolerate the same period of starvation and undergo differentiation and/or death.
An interesting possibility for DHODH inhibitors is that MTD-level dosing may not be necessary to derive clinical benefit. It may be possible to achieve the therapeutically-desired effect of pyrimidine starvation with doses lower than those that are frankly cytotoxic and lead to cumulative damage. It remains to be seen whether normal tissues will better tolerate starvation without the short-term or long-term side-effects of traditional chemotherapy.
If this rational-dosing hypothesis holds true, it may allow for extended courses of treatment, similar to the 6 months of differentiation therapy (in the form of all-trans retinoic acid and arsenic trioxide) provided to our patients with acute promyelo-cytic leukemia. Theoretically, this long period of drug exposure would systematically eradicate that small pool of minimal residual disease, or leukemia-initiating cells, that commonly result in relapse following traditional induction chemotherapy.
As multiple companies move forward with new clinical trials (Table 1) we will soon have the opportunity to evaluate the potential of DHODH inhibition in the treatment of patients with hematologic malignancies. My sense is that the various inhibitors will not suffer from insufficient potency, but that dose and schedule will be critical in balancing efficacy and toxicity.
With the renewed interest in DHODH as a drug target, now with hematologic malignancies as a primary indication, I hope that we have identified an effective and well-tolerated treatment for our patients. If successful, these trials will spawn interest in widening the scope of indications to other malignancies with the capacity to differentiate or with a special dependence on de novo pyrimidine synthesis.
Acknowledgments
A very sincere thank you to Dr. Olga Kharchenko for her wonderful illustrations and to Dr. David Scadden for his mentorship and careful review of the text.
Funding
This paper was not funded.
Footnotes
Declaration of interest
D.B. Sykes is a co-founder of and scientific advisor for Clear Creek Bio. The author has no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Notes
IMU-838 derived from Vidofludimus calcium salt originally from 4SC AG (https://www.immunic-therapeutics.com/imu-838/).
References
Papers of special note have been highlighted as either of interest (•) or of considerable interest (••) to readers.
- 1.Young JD, Yao SYM, Baldwin JM, et al. The human concentrative and equilibrative nucleoside transporter families, SLC28 and SLC29. Mol Aspects Med. 2013;34:529–547.0 [DOI] [PubMed] [Google Scholar]
- 2.Ng SB, Buckingham KJ, Lee C, et al. Exome sequencing identifies the cause of a mendelian disorder. Nat Genet. 2010;42:30–35.• A description of the genetic basis of the Miller syndrome, an extremely rare inherited disorder resulting from decreased activity of the enzyme dihydroorotate dehydrogenase (DHODH).
- 3.Dexter DL, Hesson DP, Ardecky RJ, et al. Activity of a novel 4- quinolinecarboxylic acid, NSC 368390 [6-fluoro-2-(2”-fluoro-1,1- “biphenyl-4-yl)-3-methyl-4-quinolinecarb oxylic acid sodium salt], against experimental tumors. Cancer Res. 1985;45:5563–5568.•• The first description of brequinar sodium; Dr. David Hesson is the chemist who synthesized and patented brequinar.
- 4.Arteaga CL, Brown TD, Kuhn JG, et al. Phase I clinical and pharmacokinetic trial of Brequinar sodium (DuP 785; NSC 368390). Cancer Res. 1989;49:4648–4653. [PubMed] [Google Scholar]
- 5.Bork E, Vest S, Hansen HH. A phase I clinical and pharmacokinetic study of Brequinar sodium, DUP 785 (NSC 368390), using a weekly and a biweekly schedule. Eur J Cancer Clin Oncol. 1989;25:1403–1411. [DOI] [PubMed] [Google Scholar]
- 6.Schwartsmann G, van der Vijgh WJ, van Hennik MB, et al. Pharmacokinetics of Brequinar sodium (NSC 368390) in patients with solid tumors during a phase I study. Eur J Cancer Clin Oncol. 1989;25:1675–1681. [DOI] [PubMed] [Google Scholar]
- 7.Peters GJ, Schwartsmann G, Nadal JC, et al. In vivo inhibition of the pyrimidine de novo enzyme dihydroorotic acid dehydrogenase by brequinar sodium (DUP-785; NSC 368390) in mice and patients. Cancer Res. 1990;50:4644–4649.•• Dr. Frits Peters is a world leader in the field of purines and pyrimidines. This paper rigorously examines the relationship between DHODH enzyme activity and in vivo plasma uridine concentration.
- 8.Urba S, Doroshow J, Cripps C, et al. Multicenter phase II trial of brequinar sodium in patients with advanced squamous-cell carcinoma of the head and neck. Cancer Chemother Pharmacol. 1992;31:167–169. [DOI] [PubMed] [Google Scholar]
- 9.Moore M, Maroun J, Robert F, et al. Multicenter phase II study of brequinar sodium in patients with advanced gastrointestinal cancer. Invest New Drugs. 1993;11:61–65. [DOI] [PubMed] [Google Scholar]
- 10.Natale R, Wheeler R, Moore M, et al. Multicenter phase II trial of brequinar sodium in patients with advanced melanoma. Ann Oncol. 1992;3:659–660. [DOI] [PubMed] [Google Scholar]
- 11.Maroun J, Ruckdeschel J, Natale R, et al. Multicenter phase II study of brequinar sodium in patients with advanced lung cancer. Cancer Chemother Pharmacol. 1993;32:64–66. [DOI] [PubMed] [Google Scholar]
- 12.Huang M, Wang Y, Collins M, et al. A77 1726 induces differentiation of human myeloid leukemia K562 cells by depletion of intracellular CTP pools. Mol Pharmacol. 2002;62:463–472. [DOI] [PubMed] [Google Scholar]
- 13.Baumann P, Mandl-Weber S, Volkl A, et al. Dihydroorotate dehydrogenase inhibitor A771726 (leflunomide) induces apoptosis and diminishes proliferation of multiple myeloma cells. Mol Cancer Ther. 2009;8:366–375. [DOI] [PubMed] [Google Scholar]
- 14.White RM, Cech J, Ratanasirintrawoot S, et al. DHODH modulates transcriptional elongation in the neural crest and melanoma. Nature. 2011;471:518–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sykes DB, Kfoury YS, Mercier FE, et al. Inhibition of dihydroorotate dehydrogenase overcomes differentiation blockade in acute myeloid leukemia. Cell. 2016;167:171–186.e15.•• In David Scadden’s laboratory, we established a model system that allowed for a high-throughput phenotypic screen to identify compounds that triggered differentiation in acute myeloid leukemia. The top hits from this screen were all inhibitors of DHODH.
- 16.Sainas S, Pippione AC, Lupino E, et al. Targeting myeloid differentiation using potent 2-hydroxypyrazolo[1,5-a]pyridine scaffold-based human dihydroorotate dehydrogenase (hDHODH) inhibitors. J Med Chem. 2018. acs.jmedchem.8b00373. D0I: 10.1021/acs.jmedchem.8b00373. [DOI] [PubMed] [Google Scholar]
- 17.Brown KK, Spinelli JB, Asara JM, et al. Adaptive reprogramming of de novo pyrimidine synthesis is a metabolic vulnerability in triplenegative breast cancer. Cancer Discov. 2017;7:391–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mathur D, Stratikopoulos E, Ozturk S, et al. PTEN regulates glutamine flux to pyrimidine synthesis and sensitivity to dihy-droorotate dehydrogenase inhibition. Cancer Discov. 2017;7: 380–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Koundinya M, Sudhalter J, Courjaud A, et al. Dependence on the pyrimidine biosynthetic enzyme DHODH is a synthetic lethal vulnerability in mutant KRAS-driven cancers. Cell Chem Biol. 2018;25:705–717.e11.•• References 17–19 suggest that malignancies of certain tissue- types (e.g. triple-negative breast cancer) or carrying certain mutational profiles (e.g. loss of PTEN or activated KRAS) may be particularly sensitive to pyrimidine starvation due to inhibition of DHODH.