

Abstract
Understanding the origin and maintenance of phenotypic variation, particularly across a continuous spatial distribution, represents a key challenge in evolutionary biology. For this, animal venoms represent ideal study systems: they are complex, variable, yet easily quantifiable molecular phenotypes with a clear function. Rattlesnakes display tremendous variation in their venom composition, mostly through strongly dichotomous venom strategies, which may even coexist within a single species. Here, through dense, widespread population-level sampling of the Mojave rattlesnake, Crotalus scutulatus, we show that genomic structural variation at multiple loci underlies extreme geographical variation in venom composition, which is maintained despite extensive gene flow. Unexpectedly, neither diet composition nor neutral population structure explain venom variation. Instead, venom divergence is strongly correlated with environmental conditions. Individual toxin genes correlate with distinct environmental factors, suggesting that different selective pressures can act on individual loci independently of their co-expression patterns or genomic proximity. Our results challenge common assumptions about diet composition as the key selective driver of snake venom evolution and emphasize how the interplay between genomic architecture and local-scale spatial heterogeneity in selective pressures may facilitate the retention of adaptive functional polymorphisms across a continuous space.
Keywords: adaptive trait, phenotypic variation, population structure, structural polymorphism, diet, venom
1. Introduction
The origin and genetic basis of phenotypic variation, and its retention in a population in the face of both random and deterministic forces, are pivotal issues for our understanding of evolutionary adaptations. Functional polymorphisms typically segregate in spatially isolated populations [1,2] and/or discrete ecological conditions [3–5]. In contrast, it is much more challenging to dissect the evolutionary processes involved in adaptive geographical variation across a continuous spatial distribution [6]. As a result, relatively few studies have comprehensively examined the relationship between genomic architecture, the resulting phenotypic variation and the ecological pressures maintaining that variation in continuously distributed organisms [2].
Animal venoms represent exemplar models for investigating the genetic basis of phenotypic variation [7]. Genes encoding venom toxins are uniquely expressed in distinct, specialized glands, and their final product can be easily detected and quantified. This sidesteps the problem of pleiotropy in the genes involved in adaptive polygenic traits, which often obscures the phenotypic effects of individual genetic variants [8,9].
Rattlesnakes (Crotalus) produce highly complex and diverse venoms, with tens to hundreds of individual components. These venoms display a puzzling phenotypic dichotomy, with two largely mutually exclusive strategies: type A venoms are highly lethal and characterized by heterodimeric, presynaptic β-neurotoxic phospholipases A2 (PLA2), whereas type B venoms are less toxic and rich in snake venom metalloproteinases (SVMPs) with haemorrhagic and proteolytic activity [10]. The distribution of these phenotypes across the phylogeny of rattlesnakes is highly irregular: both types occur within most major clades, and even within some individual species (electronic supplementary material, figure S1) [10].
Multiple studies have explored the drivers of intraspecific variation in venom composition and found evidence for the effect of natural selection for the optimization of venom to diet [7,11–13]. Even subtle differences, involving only a few low-expression toxins, appear to have selectively significant consequences [14]. This suggests that the much starker intraspecific variation in species with both venom A and B populations would be likely to have very powerful selective consequences, and thus predicts a strong effect of diet-related factors as drivers of this variation [15–17].
While identifying selective drivers has been a significant research focus, the role of neutral factors, such as past population fragmentation [18] or current gene flow, has received less attention. Evolutionary theory traditionally emphasizes the role of gene flow in either facilitating the transfer of selectively favourable alleles or reducing the potential for local adaptation through genotypic homogenization [19]; nonetheless, the relative importance of gene flow and selection on venom have rarely been compared directly. Although recent studies [20,21] have identified selection and inter-population genetic distances as better predictors of venom composition, those involved subtler differentiation than the A/B dichotomy.
The Mojave rattlesnake (Crotalus scutulatus) represents an ideal system to study the causes and mechanisms underlying variation in this remarkable molecular phenotype. Four highly distinct phylogeographic lineages have been identified across its wide range in the southwestern USA and Mexico [17,22]. Here, we focus on the Mojave-Sonoran clade, ranging from California to southwestern New Mexico, which in itself represents a microcosm of the phenomenon of extreme intraspecific venom variation within a single population [22]: most individuals secrete type A venoms characterized by the neurotoxic Mojave toxin (MTX), whereas snakes from central Arizona secrete type B venoms. Intermediate A+B venoms containing both SVMPs and MTX are found at the contact zones between the two venom types [23,24]. Additional toxins belonging to different gene families, such as other, myotoxin (MYO) and C-type lectins (CTL), also show geographical variation in their expression [16,24]. We therefore used the Mojave-Sonoran clade of C. scutulatus to investigate the causes and mechanisms generating and maintaining polymorphisms across a widespread and continuously distributed species. We performed densely sampled population-level analysis of the genomic basis of venom variation, investigated population structure and diet, and then used in-depth environmental association analysis (EAA) and climate reconstruction to disentangle the dynamics between genotype, phenotype and environment.
2. Material and methods
(a). Approach
Initially, we used in-depth proteomic analysis, genome sequencing and venom gland transcriptomics of two representative field-caught adults of C. scutulatus from venom type A and B areas (figure 1) to identify major toxins, and to design primers to test for the presence of specific toxin genes in additional specimens. We then mapped phenotype onto genotype by comparing proteomic and genomic presence/absence of toxins across a larger sample, and, after establishing a strict linkage, extended this to additional specimens at genomic level only. We then correlated the venom profiles with new, densely sampled population genetic data, geographical variation in diet, and physical, climatic and vegetational parameters to understand the drivers of venom variation.
Figure 1.

Geographical variation in venom and diet of adult C. scutulatus. (a) Distribution of samples for which the major venom types were identified based on toxin genotypes; stars represent the sampling locations of the two representative individuals used for the genome-transcriptome-proteome analyses. (b) Two-ring pie-charts showing the proportion of mammals and reptiles from stomach contents (inner charts) and venom types (outer ring) for each locality.
(b). Draft whole-genome sequencing
For each representative individual we sequenced two genomic libraries on an Illumina HiSeq2500. High-quality reads were assembled de novo using the CLC Genomics Workbench platform v. 6.5, and contigs combined into scaffolds using SSPACE Standard 3.0 [25]. Scaffolds containing putative toxin genes were identified by mapping all toxin transcripts to genome assemblies using GMAP software [26].
(c). Venom-gland transcriptomics
Venom gland cDNA libraries of the two representatives were sequenced on an Illumina HiSeq2500 and high-quality reads assembled de novo using Trinity 2.0.4 [27]. We identified all possible toxin transcripts with blastx searches against the NCBI nonredundant (nr) protein sequences [28], UniProtKB [29] and a custom database containing only toxin protein sequences. Homologous toxin transcripts were identified by reciprocal blast analysis and considered homologous if the coding sequences were 99% identical, with minimum 70% sequence coverage. Absence of toxins due to failure of Trinity to recover venom transcripts was verified by reciprocal mapping of reads against both transcriptomes and investigation of the proteome (see below).
(d). Venom proteomics
To link venom proteins to their corresponding transcripts we analysed the venoms of the two representative snakes by RP-HPLC and obtained molecular masses and peptide sequences [30]. All sequences were blasted against the NCBI non-redundant database and the venom-gland transcriptome assemblies using tblastn adjusted for short sequences. RP-HPLC venom profiles of 50 additional specimens from different geographical areas were then examined to identify the most highly expressed and variable toxins, and to test whether variation in venom composition is caused by genome-level differences (see below).
(e). Toxin genotyping
We selected toxins that were unambiguously scorable as either absent or highly expressed in the proteome, and designed gene-specific primer pairs based on our genomic scaffolds using the Primer-BLAST tool [31]. Amplification specificity was checked against our two transcriptomes and the NCBI nucleotide database. Twelve toxin genes belonging to five families were selected for further investigation (see electronic supplementary material, table S3), in addition to the acidic (MTXa) and basic (MTXb) subunit genes of Mojave toxin [32]. Up to 163 individuals were screened for toxin gene presence, PCR products were checked on 1.5% agarose gels, and a subset were sequenced to verify consistency of primer specificity. Sequences were blasted against the NCBI nucleotide (nt) and whole-genome shotgun contigs (wgs) databases. Pairwise Pearson correlation coefficients were calculated to test for linkage between toxin genes.
Given the absolute link between presence/absence of toxins in the proteome and the corresponding coding genes (see below), we expanded our sampling by genotyping additional individuals without proteomic information (e.g. road-killed specimens) to assess toxin gene distributions.
(f). Venom fingerprinting
Proteomic techniques allow detailed characterization of individual venom components, but do not allow for large-scale, standardized comparisons of overall variation and diversity [30]. To increase our sampling and standardize our phenotype comparisons, we analysed the same 50 venoms (see above) and 48 additional samples by on-chip electrophoresis [30]. All samples were from adult snakes. The binary matrix of protein peak presence/absence was used to calculate Shannon diversity index and pairwise Bray–Curtis dissimilarity matrices for subsequent analyses.
(g). Population genetic analysis
After preliminary analyses, we genotyped 290 specimens at 13 microsatellite loci (electronic supplementary material, table S5) (see electronic supplementary material for details). Population structure was determined using the spatial Bayesian clustering algorithm in TESS 2.3.1 [33]. Partitioning of genetic variation within and across subpopulations as inferred by TESS was examined using analysis of molecular genetic variance (AMOVA) in GenAlex [34]. To test whether spatial genetic patterns and population structure are the results of recent genetic bottlenecks, heterozygosity excess and deficit were tested using the software Bottleneck v1.2.02 [35] and Genepop [36].
Isolation by distance (IBD) was tested between pairs of individuals in GenAlex. A pairwise genetic distance matrix was then estimated based on the proportion of shared alleles (Dps) [37] between localities and used in a Mantel test against Euclidean geographical distances.
(h). Inference of past distributions
To test whether current variation in venom composition could be the result of past range fragmentation due to climatic changes, we performed niche modelling using the program MaxEnt [38]. Georeferenced occurrence localities of the Mojave-Sonoran clade of C. scutulatus were gathered from the VertNet (http://vertnet.org) and Global Biodiversity Information Facility (www.gbif.org) databases. Current climatic data were obtained from the WorldClim 1.4 database (http://www.worldclim.org) at 30 s resolution [39]. To avoid collinearity, highly correlated variables (Pearson's coefficient |r| ≥ 0.8) were pruned based on a pairwise correlation matrix, leaving a total of 13 climatic variables (electronic supplementary material, tables S10 and S11). Past climatic data for the Last Glacial Maximum (LGM) were obtained from simulations with global climate models (GCMs) estimated by community climate system models (CCSM), and data from the Last Interglacial (LIG) were obtained from [40]. All models were run with default regularization and 10 replicates subsampled, using 20% of the points for test and 80% for training each replicate. We generated ecological niche models for the species as well as for each individual toxin gene, and used present-day climate envelopes for inference of past distributions.
(i). Statistical analysis workflow
All statistical analyses were performed in R version 3.4.2 [41] using two approaches. First, we grouped individuals into discrete localities delineated by sampling gaps and valley/mountain ridge systems. Individuals falling between localities were excluded. Although this approach has the drawback of removing samples collected between localities, it can exploit population-based association approaches, such as testing for relationships between venom phenotype and diet composition. We ran Mantel and partial Mantel tests (controlling for geographical distance) in the vegan 2.4-4 package [42] using the following response distance matrices: (i) venom phenotype: mean pairwise Bray–Curtis dissimilarities between localities calculated from on-chip fingerprinting binary matrix; (ii) venom genotype: pairwise Bray–Curtis dissimilarity matrices based on toxin gene frequencies (one per gene).
Second, we used an individual-based approach, including all samples, to allow better detection of association along gradients. For the venom phenotype, we analysed patterns of variation using non-metric multidimensional scaling (NMDS) based on a pairwise Bray–Curtis distance matrix and used the individual scores on the first two axes as response variables in regression models. For the venom genotype, presence or absence of each toxin gene were used as response variables in logistic regression models using the glm (generalized linear model) function with binomial (link="logit") error distribution.
False discovery rates for all p-values of multiple comparison analyses were corrected using the method of Benjamini & Hochberg [43]. One locality (‘Gila’), where we were unable to collect venoms, was only included in the genotype analysis.
(j). Venom variation and current gene flow
Multiple approaches were used to test whether variation in venom composition reflects current patterns of gene flow and neutral genetic structure. First, we used AMOVA in GenAlex to estimate numbers of migrants and compare molecular variance between (i) the three major venom types (i.e. A, B, A+B), and (ii) sampling localities. Second, we ran partial Mantel tests between venom and genetic (Dps) distance matrices based on localities. Finally, we tested for correlations between individual-level venom variation and neutral genetic structure using the admixture proportions estimated by TESS as the explanatory variables.
(k). Venom variation and diet
To test whether geographical variation in venom phenotype and distribution of toxin genes is associated with differences in diet composition, we recorded stomach and gut contents from 463 preserved, geo-referenced specimens from museum collections. All prey items were either mammals or reptiles, except for three amphibians, two arthropods and one bird, which were excluded from further analyses. Altogether, 445 items were identified to class level, 327 to family, 249 to genus, and 192 to species level.
For each taxonomic level we calculated the ‘frequency occurrence’, defined as the number of samples in which a food item occurs expressed as a frequency of the total number of samples with identifiable prey [44], the most commonly used method for diet analysis [45]. For each locality, we used the frequency occurrence to calculate two measures of diet composition: (i) diet niche overlap, ranging from 0 (no overlap) to 1 (complete overlap), describes diet composition similarity between localities and corresponds to the pairwise Bray–Curtis dissimilarity index; (ii) niche width (Shannon diversity index) describes the diet diversity within a locality, with values near 0 indicating a narrow niche and values near 1 a broad niche. Both metrics were calculated with prey identified to class, family, genus and species level. Pairwise distance matrices based on these metrics were used for Mantel tests. Additionally, we tested for correlation between venom diversity and niche width, and between frequencies of individual prey species and toxin genes in order to identify potential key species involved in predator–prey arm races.
(l). Environmental association analysis
To test whether the observed variation in venom phenotype and toxin gene distributions were associated with spatial heterogeneity, and to identify environmental factors potentially contributing to local adaptation and genetic variation, we performed EAA.
In addition to the WorldClim data (see above), we used the high resolution digital elevation model (DEM) raster (http://asterweb.jpl.nasa.gov) to produce additional topographic variables including slope, solar radiation, aspect and topographic position index (TPI) using the Spatial Analyst toolbox in ArcMap 10.3 (ESRI®). Land cover data describing North American ecological areas (level III ‘ecoregions’) were obtained from the US EPA (https://www.epa.gov/eco-research/ecoregions-north-america), and vegetation data from the Gap Analysis Project (https://gapanalysis.usgs.gov/gaplandcover/data/download/).
Patterns of environmental heterogeneity across the study areas were examined using principal component analysis (PCA), and the significance of differences between localities were tested with pairwise t-tests.
For climatic and topographic variables, Euclidean distance matrices were calculated based on the average values within each locality, whereas for categorical variables (ecoregion and vegetation) distance matrices were generated based on the proportion of each factor level within localities. Prior to Mantel test analysis the BIOENV procedure [46] in the vegan package was used to reduce the climatic variables contributing to the final distance matrix. This function calculates Euclidean distances for all possible subsets of scaled climatic variables and finds the maximum Spearman (rank) correlation with the response distance matrix.
In the individual-level analysis, univariate regression models were generated for all variables in order to identify the strength, direction and nature of the relationships between each environmental factor and venom variation/toxin gene presence. We also generated climatic niche models for the individual genes using the WorldClim data, and used MaxEnt to generate predicted distribution maps for each.
(m). Gradient analysis
To investigate local environmental patterns at the interface between the two main venom types, we performed a gradient analysis to test associations between phenotypic or genetic variation and environmental factors along a continuous cline. We identified two suitable venom B – venom A transects, one running west (‘Maricopa’) and the other south (‘Sasabe’) from the core of the venom B area (figure 3b). We intensively sampled these two transects and tested for presence of MTX and SVMP genes. Trends along the transects were analysed for each climatic variable.
Figure 3.

Association between venom phenotypic variation, neutral genetic differentiation and environment. (a) Non-metric multidimensional scaling (NMDS) analysis of venom profiles shows great overall variation. Variation along NMDS1 is strongly correlated with the marked east–west environmental cline across Arizona (electronic supplementary material, table S12 and figures S9 and S10), whereas environmental associations along NMDS2, broadly separating the A–B transition, are weaker because global-scale variation hinders the detection of local-scale patterns. (b–g) Local-scale analysis along two transects (b) reveals sharp clines in various temperature (c–e) and precipitation (f,g) variables (see electronic supplementary material, table S11 for bioclimatic variable description) across the venom A–B transition zone.
3. Results and discussion
(a). Venom variation is due to structural genomic variation
High-throughput genome sequencing of C. scutulatus generated 652865 contigs for the venom type A representative individual and 597176 for the type B, with sequencing coverage of approximately 8× (electronic supplementary material, table S1). RNA-Seq of the venom glands generated 37162 contigs for the venom A and 56627 for the type B (electronic supplementary material, table S2). We identified a total of 96 unique toxin transcripts in the venom A transcriptome and 115 in the venom B. Both venom gland transcriptomes and proteomes showed marked differences, with several toxins highly expressed in either one or the other venom (electronic supplementary material, figures S2 and S3), including SVMPs, PLA2s, serine proteases (SVSPs), C-type lectins (CTLs) and myotoxin (MYO).
Comparison of the proteomic profiles and genotypes of 50 specimens confirmed that the presence or absence of 14 differentially expressed toxins in the proteome was invariably associated with the presence or absence of the corresponding coding genes (electronic supplementary material, figure S4). This was previously documented for MTX, other PLA2s and SVMPs [16,32], and is here confirmed for CTLs and MYO. Based on this strict phenotype–genotype link, we analysed the spatial distribution of toxin genes in a larger sample to identify gene complexes and linkage patterns (figure 2a; electronic supplementary material, table S4). In both main venom types, some genes appeared tightly linked, whereas others varied independently. In the core venom B area there were two main genotypes, both characterized by the presence of SVMPs, PLA2s (gA1, gB1 and gK) and CTL-B7, but differing in the presence of a myotoxin (MyoB). Much greater diversity was observed across the venom A genotypes: all were characterized by the tightly linked neurotoxic MTXa and MTXb, the absence of SVMPs, PLA2gK and gB1, but varied in the occurrence of PLA2gA1, MyoB and CTL-B7, each with unique spatial distribution patterns. While MTXa and MTXb, as well as PLA2gK and gB1, remained linked in all specimens, other linkages between gene complexes were disrupted across the contact zone between venom types, where mixed (A+B) genotypes and multiple different gene combinations occur. Interestingly, the intergrade zones also produced three individuals lacking both neurotoxic MTX and SVMP genes (type O), suggesting that mating between mixed genotypes can not only disrupt adaptive genomic linkages, but even lead to the complete loss of multiple key components. This raises the questions of how these different genomic variants persist in the species, and what determines the distribution of venom phenotypes.
Figure 2.

Toxin genotype and niche modelling. (a) Presence–absence matrix of toxin genes and admixture plot (TESS) with K = 2. (b) Niche models and sample distribution of the Mojave-Sonoran clade of Crotalus scutulatus with individuals represented by proportion of genetic clusters. Grey lines delineate ecoregion boundaries.
(b). Venom variation is not associated with population genetic structure
Our climatic niche modelling suggests a past range fragmentation into western Sonoran (AZW) and eastern Madrean (AZE) refuges (figure 2b). Both TESS and sPCA detected a genetic discontinuity with extensive admixture corresponding to the boundaries between the Sonoran and Madrean ecoregions (figure 2b), reflecting predicted Pleistocene vicariance and consistent with postglacial range expansion. No evidence of recent bottlenecks (electronic supplementary material, table S6) or further subpopulation structuring (electronic supplementary material, figures S6 and S7) was detected. Our results contrast with previous inferences of panmixia within the Mojave-Sonoran clade of C. scutulatus, based on analyses of mtDNA, or RADseq data from much smaller samples [22,47].
Since the two genetic clusters did not predict the distribution of venom types (figure 2a; electronic supplementary material, table S8), we further assessed the relationship between venom composition and genetic structure by grouping the samples geographically into localities (figure 1b) and calculating venom distance matrices and toxin gene frequencies. Overall genetic differentiation was weak, including between venom A and B localities (Fst = 0.003–0.05), with high levels of gene flow (Nm = 8–75). Analysis of genetic variation showed evidence of deviation from Hardy–Weinberg equilibrium (HWE) and heterozygosity deficit in the venom B and adjoining localities, suggesting strong selective regimes (electronic supplementary material, table S7). AMOVA analysis grouping either by venom types or localities confirmed an absence of finer substructure, with most of the variance arising from within individuals (electronic supplementary material, table S8). Partial Mantel tests showed no significant association between venom phenotype variation and neutral genetic distance; similarly, individual toxin gene frequencies were not correlated with gene flow (table 1). While a significant pattern of isolation by distance (IBD) (Mantel r2 = 0.70, p = 0.006), weak genetic structure (Fst = 0.02) and heterozygosity deficit (p = 0.001) are consistent with population expansion following LGM, the complete absence of association between phenotype and neutral genetic differentiation suggests that strong selective forces are driving the distribution of venom types, rather than differentiation in allopatry followed by range expansion.
Table 1.
Environmental association analysis between localities. Heatmap of partial Mantel tests (Spearman R partial correlation coefficients multiplied by 100) between overall venom phenotype or individual toxin gene frequencies against environmental variables, with geographical Euclidean distance matrix as covariate. Isolation by distance (IBD) is the null model. Proportion of shared alleles (Dps) was used as an index for neutral genetic differentiation. Variables selected with the BIOENV procedure to generate climatic and topography distance matrix are reported: BIO1–BIO9 correspond to measures related to temperature and BIO12–BIO19 to precipitation. Values with p < 0.05 are in bold.
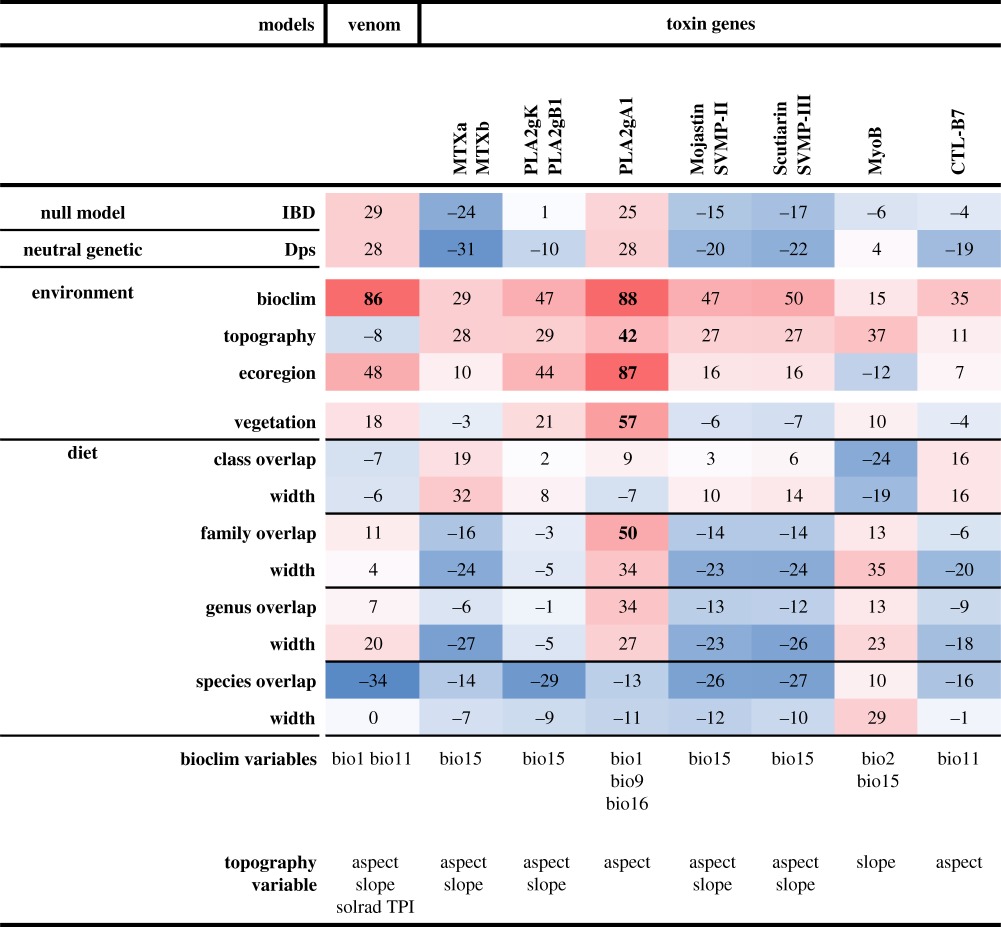 |
(c). Venom composition is not associated with diet spectrum
Because adaptation to diet is generally invoked as the foremost driver of venom evolution [11,14,16,17,47,48], we tested whether the divergent phenotypes are associated with differences in local diet. Our diet data show that C. scutulatus feeds primarily on small mammals, with the rodent families Heteromyidae and Cricetidae alone constituting 78.8% of prey items (figure 1b; electronic supplementary material, figure S8b). Partial Mantel tests found no significant association between overall venom composition and diet spectrum measured as niche overlap or niche width, irrespective of whether the spectrum was resolved to class, family, genus or species level (table 1).
Similarly, we found no significant pairwise relationships between individual toxin gene frequencies and individual prey species; in particular, neither MTX nor SVMPs, the two main players in the venom dichotomy, were linked to any specific prey. We also tested the hypothesis that more complex venoms would allow predation upon a more diverse array of prey [49]. Interestingly, we found the opposite trend: localities with less diverse venoms had broader prey spectra, although this was only weakly significant (electronic supplementary material, figure S8a). None of the frequencies of the individual toxin genes were significantly correlated with either diet composition or niche width, except PLA2gA1, an inhibitor of ADP-induced platelet aggregation [50], which showed a strong association with climate and ecoregion, and a weaker, but significant, correlation with diet composition at the family level (table 1). The functional significance of this is unclear, as this gene is widespread in the genomes of both type A and type B rattlesnakes in general [16]. Whether this association is due to direct selection for diet or a partial correlation between diet and climate or ecoregion is also unclear.
Because the primary function of venom in snakes is prey acquisition [7], adaptation to specific diet as the key selective driver of venom evolution has become the dominant paradigm in the study of snake venom evolution. Since even subtle variation in venom composition can reflect selection for local prey [12,14], we had hypothesized that the stark contrast in toxicity and mode of action (neurotoxic versus haemorrhagic) between A and B venoms in C. scutulatus would have a significant impact on the snakes' foraging biology. Our results thus challenge the widespread assumption of diet composition as the main determinant of the venom dichotomy in this or other rattlesnake species [16,17] and its universality as a selective driver of snake venom evolution in general [7].
(d). Spatial environmental heterogeneity predicts venom variation
Spatial heterogeneity in environmental variables is a key driver of genotypic and phenotypic polymorphism [51]. In the absence of a strong venom–diet association, we performed EAA to understand whether differences in other biotic and/or abiotic factors contribute to geographical variation of venom composition [47,52,53]. Overall venom variation was strongly associated with temperature (table 1), and the longitudinal climatic gradient characterizing the Sonoran desert (electronic supplementary material, figures S9 and S10) was reflected in the differentiation across venom A profiles along the first NMDS axis (figure 3a). In contrast, the second NMDS axis, which broadly separates A and B venoms, showed weaker correlations (electronic supplementary material, table S12). However, across a large, continuous distribution without discrete physical barriers, large-scale analyses may fail to detect the effect of local ecotones and short environmental clines of potential selective importance. We thus analysed local scale climatic trends along two A–B transects and discovered sharp clines for several variables (figure 3b–g). In agreement with this and previous findings [47,53], logistic regression models revealed significant associations of MTX and SVMPs with climatic variables, with venom B areas characterized by larger diurnal thermal fluctuations, milder winters and less seasonal variation in precipitation (electronic supplementary material, table S12).
The other toxin genes, even though highly co-expressed in some phenotypes, showed different correlation patterns, suggesting that different selective forces orchestrate individual loci to create complex, dynamic phenotypes (electronic supplementary material, table S12). Strikingly, genes located a few kb apart, such as some PLA2s [15], also displayed independent associations, demonstrating that divergent selective pressures can differentially affect parts of the same genomic region. Climatic niche modelling of the distribution of individual toxin genes yielded different predictions even for neighbouring genes, and the models proved to be accurate predictors of gene distribution (electronic supplementary material, figure S5), emphasizing the environment–genotype link. This interesting phenomenon deserves further investigation, since genes coding for the same adaptive phenotype are generally brought closer together by means of chromosomal rearrangements such as inversions or supergenes [54].
(e). Genome, environment and the maintenance of geographical variation
The emerging picture of the mechanisms and drivers governing venom variation in C. scutulatus is thus one of adaptive polymorphism with gene flow, with the distribution of toxin genes shaped by directional natural selection for local environmental factors other than diet spectrum or neutral gene flow. Margres et al. [20] recently suggested that gene flow may be more likely to drive venom composition in dietary generalists than in specialists; the lack of association between gene flow and venom composition in the specialist mammal-feeder C. scutulatus is consistent with this, but the lack of association between diet spectrum and venom suggests that other determinants are involved.
The precise nature and mechanism of selection, and especially the association of venom with environmental parameters, remain unclear. It seems to us unlikely that climate by itself exerts strong selection on venom composition. In fact, the generally positive association between type B venoms and higher winter temperatures runs contrary to the hypothesis that SVMPs are needed to assist digestion at lower temperatures [10,55]. However, climatic stability and seasonality may affect other factors, for instance prey community composition and dynamics [52]. These, in turn, could influence snake foraging strategies, and potentially also the exposure of snakes to predation, an understudied source of selection on venom [56]. In widely distributed species occupying diverse environmental conditions, spatial heterogeneity could thus select for local fitness optima, resulting in the maintenance of disparate, locally adaptive gene complexes.
While venom composition does not correlate with diet spectrum, the possibility of more subtle diet-related selection deserves further study: predator–prey arms races, pitting resistance to venom in prey against the snakes' venom, appear to be important drivers of venom evolution in at least some cases [12]. While many desert rodents display resistance to type B venoms [57], there are virtually no corresponding data for type A venoms. Geographical variation in the prevalence of prey resistance to different venom types, perhaps correlated with other environmental variables, could conceivably act as a driver of venom composition in C. scutulatus. This could constitute a fruitful focus for future research. Potential prey-specific toxicity in PLA2gA1, the only diet-associated toxin, may also repay further investigation.
As in previous studies [47,53], we hypothesize that disruptive selection against intermediate A+B phenotypes may ensure spatial segregation, thereby favouring persistence of gene complexes and divergent phenotypes. The role of relatively subtle environmental changes in driving the dramatic differences in venom composition in this species, coupled with selection against intermediate phenotypes, suggests the existence of steep clines in the adaptive fitness landscape, where one phenotype gains a selective advantage over the other across short geographical distances. However, the proximate factors mediating the geographical variation in selection pressures remain to be fully understood.
4. Conclusion
The unique genomic architecture of rattlesnake venom provides an important addition to the catalogue of mechanisms underlying adaptive phenotypic variation, and establishes a promising system for investigating the ecological and evolutionary implications of genomic structural variation in non-model organisms. Together, our results emphasize the importance of combining large-scale genotype, phenotype and ecological data in natural populations to uncover the wide variety of mechanisms and drivers underlying phenotypic variation, and emphasize the need to consider a multitude of factors as potential selective drivers of phenotypic variation.
Supplementary Material
Acknowledgements
We are grateful to the numerous friends and colleagues who helped with fieldwork and sample collection. G. Whiteley, R. A. Harrison and R. Morgan performed venom gland dissection. A. Foote provided insightful comments.
Ethics
Fieldwork carried out under Arizona Game and Fish Department permits SP684786, SP724028 and SP760658 and New Mexico Department of Game and Fish Authorization 3542.
Data accessibility
Raw Illumina sequences have been deposited in the European Nucleotide Archive (ENA) under project accession number PRJEB29193. RNA-seq accession numbers: venom type A: ERS2793705 (right venom gland); ERS2793704 (left venom gland); type B: ERS2793703 (right venom gland). Whole genome sequencing accession numbers: type A: ERS2793891 (300 bp insert) and ERS2793890 (600 bp insert); type B: ERS2793893 (300 bp insert) and ERS2793892 (600 bp insert). Toxin gene sequences are deposited in GenBank with accession numbers: MG948948–MG949116. Sample localities, microsatellite and diet data are available from the Dryad Digital Repository: https://doi.org/10.5061/dryad.1473s5c [58].
Authors' contributions
Conceptualization: W.W., G.Z.; formal analysis: G.Z.; methodology: G.Z., J.J.C., M.J.H.; investigation: all authors; writing—original draft: G.Z.; review and editing: all authors.
Competing interests
We declare we have no competing interests.
Funding
Leverhulme Trust Grant RPG 2013-315 to W.W., Santander Early Career Research Scholarship to G.Z., Ministerio de Economía y Competitividad Grant BFU2013-42833-P to J.J.C.
References
- 1.Mayr E. 1947. Ecological factors in speciation. Evolution 1, 263–288. ( 10.1111/j.1558-5646.1947.tb02723.x) [DOI] [Google Scholar]
- 2.Bosse M, et al. 2017. Recent natural selection causes adaptive evolution of an avian polygenic trait. Science 358, 365–368. ( 10.1126/science.aal3298) [DOI] [PubMed] [Google Scholar]
- 3.Levene H. 1953. Genetic equilibrium when more than one ecological niche is available. Am. Nat. 87, 331–333. ( 10.1086/281792) [DOI] [Google Scholar]
- 4.Hedrick PW. 2006. Genetic polymorphism in heterogeneous environments: the age of genomics. Annu. Rev. Ecol. Evol. Syst. 37, 67–93. ( 10.1146/annurev.ecolsys.37.091305.110132) [DOI] [Google Scholar]
- 5.Chakraborty M, Fry JD. 2016. Evidence that environmental heterogeneity maintains a detoxifying enzyme polymorphism in Drosophila melanogaster. Curr. Biol. 26, 219–223. ( 10.1016/j.cub.2015.11.049) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Foote AD. 2018. Sympatric speciation in the genomic era. Trends Ecol. Evol. 33, 85–95. ( 10.1016/j.tree.2017.11.003) [DOI] [PubMed] [Google Scholar]
- 7.Casewell NR, Wüster W, Vonk FJ, Harrison RA, Fry BG. 2013. Complex cocktails: the evolutionary novelty of venoms. Trends Ecol. Evol. 28, 219–229. ( 10.1016/j.tree.2012.10.020) [DOI] [PubMed] [Google Scholar]
- 8.Stern DL, Orgogozo V. 2008. The loci of evolution: how predictable is genetic evolution? Evolution 62, 2155–2177. ( 10.1111/j.1558-5646.2008.00450.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Seehausen O, et al. 2014. Genomics and the origin of species. Nat. Rev. Genet. 15, 176–192. ( 10.1038/nrg3644) [DOI] [PubMed] [Google Scholar]
- 10.Mackessy SP. 2008. Venom composition in rattlesnakes: trends and biological significance. In Biology of the rattlesnakes (eds Hayes WK, Beaman KR, Cardwell MD, Bush SP), pp. 495–510. Loma Linda, CA: Loma Linda University Press. [Google Scholar]
- 11.Daltry JC, Wüster W, Thorpe RS. 1996. Diet and snake venom evolution. Nature 379, 537–540. ( 10.1038/379537a0) [DOI] [PubMed] [Google Scholar]
- 12.Holding ML, Biardi JE, Gibbs HL. 2016. Coevolution of venom function and venom resistance in a rattlesnake predator and its squirrel prey. Proc. R. Soc. B 283, 20152841 ( 10.1098/rspb.2015.2841) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Smiley-Walters SA, Farrell TM, Gibbs HL. 2017. Evaluating local adaptation of a complex phenotype: reciprocal tests of pigmy rattlesnake venoms on treefrog prey. Oecologia 184, 739–748. ( 10.1007/s00442-017-3882-8) [DOI] [PubMed] [Google Scholar]
- 14.Margres MJ, Wray KP, Hassinger ATB, Ward MJ, McGivern JJ, Lemmon EM, Lemmon AR, Rokyta DR. 2017. Quantity, not quality: rapid adaptation in a polygenic trait proceeded exclusively through expression differentiation. Mol. Biol. Evol. 34, 3099–3110. ( 10.1093/molbev/msx231) [DOI] [PubMed] [Google Scholar]
- 15.Dowell NL, Giorgianni MW, Kassner VA, Selegue JE, Sánchez EE, Carroll SB. 2016. The deep origin and recent loss of venom toxin genes in rattlesnakes. Curr. Biol. 26, 2434–2445. ( 10.1016/j.cub.2016.07.038) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dowell NL, Giorgianni MW, Griffin S, Kassner VA, Selegue JE, Sánchez EE, Carroll SB. 2018. Extremely divergent haplotypes in two toxin gene complexes encode alternative venom types within rattlesnake species. Curr. Biol. 28, 1016–1026. ( 10.1016/j.cub.2018.02.031) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Strickland JL, Mason AJ, Rokyta DR, Parkinson CL. 2018. Phenotypic variation in Mojave rattlesnake (Crotalus scutulatus) venom is driven by four toxin families. Toxins 10, 135 ( 10.3390/toxins10040135) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hewitt G. 2000. The genetic legacy of the Quaternary ice ages. Nature 405, 907–913. ( 10.1038/35016000) [DOI] [PubMed] [Google Scholar]
- 19.Räsänen K, Hendry AP. 2008. Disentangling interactions between adaptive divergence and gene flow when ecology drives diversification. Ecol. Lett. 11, 624–636. ( 10.1111/j.1461-0248.2008.01176.x) [DOI] [PubMed] [Google Scholar]
- 20.Margres MJ, Patton A, Wray KP, Hassinger ATB, Ward MJ, Lemmon EM, Lemmon AR, Rokyta DR. 2018. Tipping the scales: the migration–selection balance leans towards selection in snake venoms. Mol. Biol. Evol. 36, 271–282. ( 10.1093/molbev/msy207) [DOI] [PubMed] [Google Scholar]
- 21.Holding ML, Margres MJ, Rokyta DR, Gibbs HL. 2018. Local prey community composition and genetic distance predict venom divergence among populations of the northern Pacific rattlesnake (Crotalus oreganus). J. Evol. Biol. 31, 1513–1528. ( 10.1111/jeb.13347) [DOI] [PubMed] [Google Scholar]
- 22.Schield DR, et al. 2018. Cryptic genetic diversity, population structure, and gene flow in the Mojave rattlesnake (Crotalus scutulatus), Mol. Phylogenet. Evol. 127, 669–681. ( 10.1016/j.ympev.2018.06.013) [DOI] [PubMed] [Google Scholar]
- 23.Glenn JL, Straight RC, Wolfe MC, Hardy DL. 1983. Geographical variation in Crotalus scutulatus scutulatus (Mojave rattlesnake) venom properties. Toxicon 21, 119–130. ( 10.1016/0041-0101(83)90055-7) [DOI] [PubMed] [Google Scholar]
- 24.Massey DJ, Calvete JJ, Sánchez EE, Sanz L, Richards K, Curtis R, Boesen K. 2012. Venom variability and envenoming severity outcomes of the Crotalus scutulatus scutulatus (Mojave rattlesnake) from Southern Arizona. J. Proteomics 75, 2576–2587. ( 10.1016/j.jprot.2012.02.035) [DOI] [PubMed] [Google Scholar]
- 25.Boetzer M, Henkel CV, Jansen HJ, Butler D, Pirovano W. 2011. Scaffolding pre-assembled contigs using SSPACE. Bioinformatics 27, 578–579. ( 10.1093/bioinformatics/btq683) [DOI] [PubMed] [Google Scholar]
- 26.Wu TD, Watanabe CK. 2005. GMAP: a genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics 21, 1859–1875. ( 10.1093/bioinformatics/bti310) [DOI] [PubMed] [Google Scholar]
- 27.Grabherr MG, et al. 2011. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29, 644–652. ( 10.1038/nbt.1883) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.NCBI Resource Coordinators. 2013. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 41, D8–D20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.The UniProt Consortium. 2014. UniProt: a hub for protein information. Nucleic Acids Res. 43, D204–D212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zancolli G, Sanz J, Calvete JJ, Wüster W. 2017. Venom on-a-chip: a fast and efficient method for comparative venomics. Toxins 9, 179 ( 10.3390/toxins9060179) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ye J, Coulouris G, Zaretskaya I, Cutcutache I, Rozen S, Madsen TL. 2012. Primer-BLAST: a tool to design target-specific primers for polymerase chain reaction. BMC Bioinformatics 13, 134 ( 10.1186/1471-2105-13-134) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zancolli G, et al. 2016. Is hybridization a source of adaptive venom variation in rattlesnakes? A test, using a Crotalus scutulatus x viridis hybrid zone in Southwestern New Mexico. Toxins 8, 188 ( 10.3390/toxins8060188) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen C, Durand E, Forbes F, François O. 2007. Bayesian clustering algorithms ascertaining spatial population structure: a new computer program and a comparison study. Mol. Ecol. Notes 7, 747–756. ( 10.1111/j.1471-8286.2007.01769.x) [DOI] [Google Scholar]
- 34.Peakall R, Smouse PE. 2006. Genalex 6: genetic analysis in Excel. Population genetic software for teaching and research. Mol. Ecol. Notes 6, 288–295. ( 10.1111/j.1471-8286.2005.01155.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Piry S, Luikart G, Cornuet J-M. 1999. BOTTLENECK: a computer program for detecting recent reductions in the effective population size using allele frequency data. J. Hered. 90, 502–503. ( 10.1093/jhered/90.4.502) [DOI] [Google Scholar]
- 36.Raymond M, Rousset F. 1995. GENEPOP (Version 1. 2): population genetics software for exact tests and ecumenicism. J. Hered. 86, 248–249. ( 10.1093/oxfordjournals.jhered.a111573) [DOI] [Google Scholar]
- 37.Bowcock AM, Ruiz-Linares A, Tomfohrde J, Minch E, Kidd JR, Cavalli-Sforza LL. 1994. High resolution of human evolutionary trees with polymorphic microsatellites. Nature 368, 455–457. ( 10.1038/368455a0) [DOI] [PubMed] [Google Scholar]
- 38.Phillips SJ, Anderson RP, Schapire RE. 2006. Maximum entropy modeling of species geographic distributions. Ecol. Modell. 190, 231–259. ( 10.1016/j.ecolmodel.2005.03.026) [DOI] [Google Scholar]
- 39.Hijmans RJ, Cameron SE, Parra JL, Jones PG, Jarvis A. 2005. Very high resolution interpolated climate surfaces for global land areas. Int J Climatol. 25, 1965–1978. ( 10.1002/joc.1276) [DOI] [Google Scholar]
- 40.Otto-Bliesner BL, et al. 2006. Simulating arctic climate warmth and icefield retreat in the last Interglaciation. Science 311, 1751–1753. ( 10.1126/science.1120808) [DOI] [PubMed] [Google Scholar]
- 41.R Development Core Team. 2017. R: a language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; See https://www.R-project.org/. [Google Scholar]
- 42.Oksanen J, et al. 2017. Vegan: Community Ecology Package. R package version 2.4-4. See https://CRAN.R-project.org/package=vegan .
- 43.Benjamini Y, Hochberg Y. 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. B 57, 289–300. [Google Scholar]
- 44.Amundsen PA, Gabler HM, Staldvik FJ. 1996. A new approach to graphical analysis of feeding strategy from stomach contents data—modification of the Costello (1990) method. J. Fish Biol. 48, 607–614. [Google Scholar]
- 45.Davis NE, et al. 2015. Interspecific and geographic variation in the diets of sympatric carnivores: dingoes/wild dogs and red foxes in south-eastern Australia. PLoS ONE 10, e0120975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Clarke KR, Ainsworth M. 1993. A method of linking multivariate community structure to environmental variables. Mar. Ecol. Prog. Ser. 92, 205–219. ( 10.3354/meps092205) [DOI] [Google Scholar]
- 47.Strickland JC, et al. 2018. Evidence for divergent patterns of local selection driving venom variation in Mojave rattlesnakes (Crotalus scutulatus). Sci. Rep. 8, 17622 ( 10.1038/s41598-018-35810-9) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Barlow A, Pook CE, Harrison RA, Wüster W. 2009. Co-evolution of diet and prey-specific venom activity supports the role of selection in snake venom evolution. Proc. R. Soc. B 276, 2443–2449. ( 10.1098/rspb.2009.0048) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Phuong MA, Mahardika GN, Alfaro ME. 2016. Dietary breadth is positively correlated with venom complexity in cone snails. BMC Genomics 17, 401 ( 10.1186/s12864-016-2755-6) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tsai IH, Wang YM, Chen YH, Tu AT. 2003. Geographic variations, cloning, and functional analyses of the venom acidic phospholipases A2 of Crotalus viridis viridis. Arch. Biochem. Biophys. 411, 289–296. ( 10.1016/S0003-9861(02)00747-6) [DOI] [PubMed] [Google Scholar]
- 51.Wang IJ, Bradburd GS. 2014. Isolation by environment. Mol. Ecol. 23, 5649–5662. ( 10.1111/mec.12938) [DOI] [PubMed] [Google Scholar]
- 52.Gren ECK. et al 2017. Geographic variation of venom composition and neurotoxicity in the rattlesnakes Crotalus oreganus and C. helleri: assessing the potential roles of selection and neutral evolutionary processes in shaping venom variation. In Biology of the Rattlesnakes II (eds Dreslik MJ, Hayes WK, Beaupre SJ, Mackessy SP), pp. 228–252. Rodeo, NM: Eco Herpetological Publishing and Distribution, [Google Scholar]
- 53.Zancolli G, et al. 2018. When one phenotype is not enough—divergent evolutionary trajectories govern venom variation in a widespread rattlesnake species. BioRxiv preprint. See . [DOI]
- 54.Thompson MJ, Jiggins CD. 2014. Supergenes and their role in evolution. Heredity 113, 1–8. ( 10.1038/hdy.2014.20) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Thomas RG, Pough FH. 1979. The effect of rattlesnake venom on digestion of prey. Toxicon 17, 221–228. ( 10.1016/0041-0101(79)90211-3) [DOI] [PubMed] [Google Scholar]
- 56.Gangur AN, Seymour JE, Liddell MJ, Wilson D, Smout MJ, Northfield TD. 2017. When is overkill optimal? Tritrophic interactions reveal new insights into venom evolution. Theor. Ecol. 11, 141–149. ( 10.1007/s12080-017-0354-z) [DOI] [Google Scholar]
- 57.Perez JC, Haws WC, Garcia VE, Jennings BM. 1978. Resistance of warm-blooded animals to snake venoms. Toxicon 16, 375–383. ( 10.1016/0041-0101(78)90158-7) [DOI] [PubMed] [Google Scholar]
- 58.Zancolli G, et al. 2014. Data from: When one phenotype is not enough: divergent evolutionary trajectories govern venom variation in a widespread rattlesnake species Dryad Digital Repository. ( 10.5061/dryad.1473s5c) [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Citations
- Zancolli G, et al. 2014. Data from: When one phenotype is not enough: divergent evolutionary trajectories govern venom variation in a widespread rattlesnake species Dryad Digital Repository. ( 10.5061/dryad.1473s5c) [DOI] [PMC free article] [PubMed]
Supplementary Materials
Data Availability Statement
Raw Illumina sequences have been deposited in the European Nucleotide Archive (ENA) under project accession number PRJEB29193. RNA-seq accession numbers: venom type A: ERS2793705 (right venom gland); ERS2793704 (left venom gland); type B: ERS2793703 (right venom gland). Whole genome sequencing accession numbers: type A: ERS2793891 (300 bp insert) and ERS2793890 (600 bp insert); type B: ERS2793893 (300 bp insert) and ERS2793892 (600 bp insert). Toxin gene sequences are deposited in GenBank with accession numbers: MG948948–MG949116. Sample localities, microsatellite and diet data are available from the Dryad Digital Repository: https://doi.org/10.5061/dryad.1473s5c [58].