

Abstract
The preferred conformations of peptides and proteins are dependent on local interactions that bias the conformational ensemble. The n→π* interaction between consecutive carbonyls promotes compact conformations, including α-helix and polyproline II helix. In order to further understand the n→π* interaction and to develop methods to promote defined conformational preferences through acyl N-capping motifs, a series of peptides was synthesized in which the electronic and steric properties of the acyl group were modified. Using NMR spectroscopy, van’t Hoff analysis of enthalpies, X-ray crystallography, and computational investigations, we observe that more electron-rich donor carbonyls (pivaloyl, iso-butyryl, propionyl) promote stronger n→π* interactions and more compact conformations, compared to acetyl or to less electron-rich donor carbonyls (methoxyacetyl, fluoroacetyl, formyl). X-ray crystallography indicates a strong, electronically tunable preference for the α-helix conformation, as observed directly on the ϕ and ψ torsion angles. Electron-donating acyl groups promote the α-helical conformation, even in the absence of the hydrogen bonding that stabilizes the α-helix. In contrast, electron-withdrawing acyl groups exhibited more extended conformations. More sterically demanding groups can promote trans amide bonds independent of the electronic effect on n→π* interactions. Chloroacetyl groups additionally promote n→π* interactions via the interaction of the chlorine lone pair with the proximal carbonyl π*. These data provide additional support for an important role of n→π* interactions in the conformational ensemble of disordered or unfolded proteins. Moreover, this work suggests that readily incorporated acyl N-capping motifs that modulate n→π* interactions may be employed rationally to promote conformational biases in peptides, with potential applications in molecular design and medicinal chemistry.
Keywords: amino acids, conformation analysis, hyperconjugation, peptides, stereoelectronic effects
Graphical Abstract
How is the α-helix stabilized in the absence of hydrogen bonds? The strength of n→π* interactions was systematically modulated via the identity of the peptide acyl N-cap, with strong donors promoting α-helix and polyproline II helix conformations, but weak donors resulting in more extended conformations. These results suggest the broader consideration of specific capping motifs for conformational control of peptides.
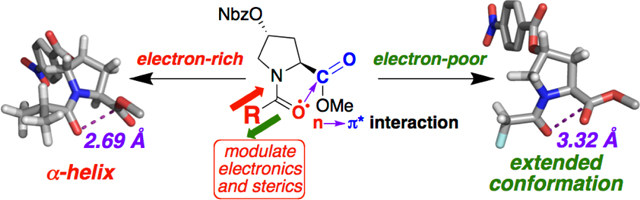
The structure of peptides and proteins is dependent on the sum of a host of non-covalent interactions that are individually weak, but that collectively cause biomolecules to adopt a limited number of conformations, in the process solving the Levinthal paradox.[1] The n→π* interaction is a favorable interaction which is manifested locally in proteins between the lone pair (n) of one carbonyl and the π* molecular orbital of the subsequent carbonyl (Figure 1).[2] The n→π* interaction has been identified as an important determinant of the backbone conformation of proteins, particularly in the α-helix and polyproline II helix (PPII) conformations.[3] Importantly, the n→π* interaction provides a significant basis for biases in the protein main chain conformation. Thus, the n→π* interaction might especially impact structure in the disordered or unfolded states of proteins,[3b, 4] due to the reduced long-range contacts and hydrogen bonding in disordered proteins.[5]
Figure 1.
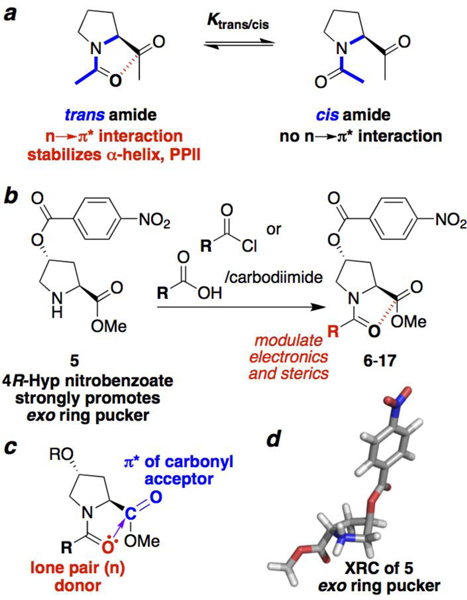
Proline cis-trans isomerism to probe n→π* interactions. (a) Proline trans and cis amide bonds (blue) are in slow exchange on the NMR timescale. Only the trans amide bond may be stabilized by an n→π* interaction. Thus, molecular properties that promote an n→π* interaction lead to a larger Ktrans/cis. (b) Synthesis of peptides with different acyl N-caps from the common intermediate 5. (c) Overlap of the donor (red) carbonyl Oi lone pair (n) with the acceptor carbonyl Ci+1=Oi+1 (blue) π* molecular orbital leads to electron delocalization. The extent of orbital overlap is associated with the Oi…Ci+1 distance (purple), with n→π* interactions exhibiting distances significantly below the 3.22 Å sum of the van der Waals radii of O and C. (d) Crystal structure of 5, which exhibits an exo ring pucker due to the strong stereoelectronic effect of the nitrobenzoate ester.
The strength of the n→π* interaction is impacted by the identities of both the donor and acceptor carbonyls.[3a, 6] For example, thioamides function better than amides as n→π* electron donors due to the greater nucleophilicity of sulfur and the higher energies of the lone pair orbitals of sulfur compared to oxygen.[7] Similarly, the strength of the n→π* interaction may be modulated by changing the electronic properties of the acceptor carbonyl: esters are better acceptors than amides, with the acceptor strength further enhanced by the incorporation of electron-withdrawing groups on the ester, which render the carbonyl more electrophilic and lower the energy of the π* orbital.[8]
We sought to further investigate the nature of the n→π* interaction and the ability to electronically tune peptide structure via the systematic modulation of the electronic properties of the donor carbonyl. The incorporation of electron-donating acyl capping groups on the N-terminus should strengthen an n→π* interaction, and thus promote α-helix and polyproline II helix conformations. In contrast, the introduction of electron-withdrawing substituents would be expected to weaken the n→π* interaction and yield relatively more extended conformations. The N-terminus of peptides is readily modified as the last step of solid-phase peptide synthesis prior to cleavage from resin/side chain deprotection. As such, this approach to control the strength of the n→π* interaction via the identity of the acyl N-capping[9] group could be used to change the conformational preferences of peptides in a general, predictable, and highly accessible manner.
In order to investigate the effects of modification of N-terminal electronics on peptide conformation, we synthesized a series of peptides based on the 4-nitrobenzoate ester of 4R-hydroxyproline (Hyp(4-NO2-Bz)) (Figure 1). This proline derivative strongly promotes an exo ring pucker[10] and readily crystallizes due to the nitrobenzoate group.[11] Peptides (Figure 2) were synthesized with pivaloyl, iso-butyryl, and propionyl N-caps, which are more electron-donating than the acetyl group typically employed at the N-terminus of peptides. In addition, a series of N-caps was incorporated with more electron-deficient substituents than acetyl, including methoxyacetyl, fluoroacetyl, and formyl groups, as well as dichloro-, trichloro-, difluoro-, and trifluoro- acetyl groups.
Figure 2.
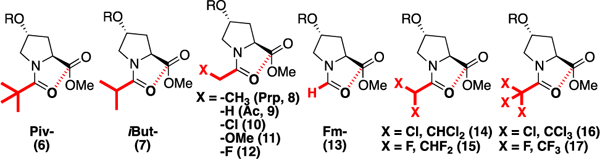
Modulation of the electronic and steric properties of acyl N-capping motifs (N-caps). R = 4-NO2-benzoate ester.
All peptides were analyzed by NMR spectroscopy, to quantify how the identity of the acyl N-cap changes the preference for a trans amide bond, which provides one measure of the strength of the n→π* interaction, with a stronger n→π* interaction resulting in a higher population of trans amide bond (larger Ktrans/cis) (Table 1), if all other factors are equal. For monosubstituted acetyl groups, other than chlorine substitution, an electron-donating substituent increased the population of trans amide bond, while electron-withdrawing substituents increased the population of cis amide bond, relative to acetyl (for N-caps C(O)-CH2-X, X = -CH3 > -H > -OMe > -F).
Table 1.
Effects of acyl N-cap substitution on the thermodynamic properties of peptides in CDCl3. – = not determined. Errors are in the Supporting Information.
| X-(C=O)- Hyp(Nbz)-OMe X= |
Ktrans/cis 298 K |
Ktrans/cis 263 K |
ΔG298 K,
kcal mol−1 |
ΔH, kcal mol−1 |
ΔS, cal mol−1 K−1 |
|---|---|---|---|---|---|
| C(CH3)3 | >20 | — | — | — | — |
| CH(CH3)2 | 7.6 | 10.8 | −1.20 | −1.88 | −2.4 |
| CH2CH3 | 5.7 | 8.1 | −1.03 | −1.62 | −2.0 |
| CH3 | 4.4 | 6.2 | −0.88 | −1.50 | −2.1 |
| CH2CI | 6.0 | 8.0 | −1.06 | −1.39 | −1.2 |
| CH2OCH3 | 3.2 | 4.0 | −0.69 | −0.87 | −0.5 |
| CH2F | 2.6 | 2.9 | −0.57 | −0.54 | −0.1 |
| H | 1.8 | 2.0 | −0.35 | −0.67 | −1.1 |
| CHCI2 | 11.1 | — | −1.43 | — | — |
| CHF2 | 5.1 | — | −0.96 | — | — |
| CCI3 | >20 | — | — | — | — |
| CF3 | 7.8 | — | −1.22 | — | — |
The most strongly trans-promoting N-cap was the pivaloyl group, which exhibited no evidence of cis amide bond by NMR. Proline cis-trans isomerism importantly also depends on steric effects, with larger groups destabilizing the cis amide conformation. The pivaloyl group thus was effective in promoting trans amide via both electronic and steric effects.[3h] Similarly, the most cis-promoting N-cap was the formyl group, which is both less electron-donating than an acetyl group and is smaller, with both effects leading to a reduced[3a] preference for a trans over a cis amide bond.
The effect of steric versus electronic effects in impacting cis-trans isomerism was most observable in the halogen series, with dihalogenation and trihalogenation increasing the population of trans amide bond, despite the expectation of weaker n→π* interactions with increasing F or Cl substitution. Notably, fluorine α-substitution on acyl groups has been previously identified to have effects on conformation that are dependent on the F-C-C-O torsion angle, via the balance of attractive and repulsive interactions between the fluorine and the carbonyl.[6, 12]
In order to further understand how electronic factors impact the n→π* interaction, a subset of peptides was examined by temperature-dependent NMR, to determine the effect of substitution on proline cis-trans isomerism enthalpies (ΔHtrans/cis) via van’t Hoff analysis (Figure 3, Table 1). These data indicate that more electron-donating substituents on the donor carbonyl increased the enthalpic preference for a trans amide bond. In contrast, electron-withdrawing substituents or formyl substitution on the donor carbonyl significantly reduced this preference (CH(CH3)2 > CH2CH3 > CH3 ~ CH2Cl >> CH2OMe > H > CH2F). Again, chloromethyl substitution exhibited anomalous behavior if only electronic effects are considered.
Figure 3.
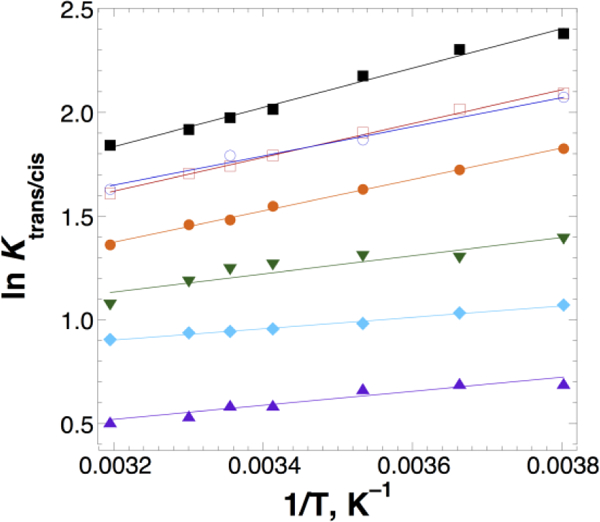
van’t Hoff analysis of the temperature dependence of Ktrans/cis for the peptides R–C(O)–Hyp(4-NO2-Bz)-OMe. From top to bottom, R = –CH(CH3)2 (black squares),–CH2CH3 (open red squares), –CH2Cl (open blue circles), –CH3 (orange circles), –CH2OCH3 (green inverted triangles), –CH2F (cyan diamonds), –H (purple triangles).
X-ray crystal structures were solved for nearly all derivatives (Figure 4, Table 2), with most structures exhibiting two molecules in each asymmetric section of their unit cell, providing the ability to examine electronic effects on structure in detail. The pivaloyl derivative adopted a polyproline II helix conformation, with a very close 2.68 Å Oi…Ci+1 intercarbonyl distance, indicating a particularly favorable n→π* interaction for the most electron-rich donor carbonyl studied. Notably, the iso-butyryl, propionyl, chloroacetyl, acetyl, and methoxyacetyl derivatives all adopted an α-helical conformation in the crystalline form, indicating the ability of the n→π* interaction to promote the α-helix independent of hydrogen bonding. The iso-butyryl N-cap exhibited quite close Oi…Ci+1 intercarbonyl distances (2.69, 2.74 Å) as well as surprisingly compact values of ϕ (–45˚, –47˚), which are consistent with particularly favorable n→π* interactions. Among -CH2-X N-caps, the propionyl (2.83, 2.88 Å) and chloroacetyl (2.85, 2.88 Å) groups exhibited the closest intercarbonyl distances, indicating the most favorable n→π* interactions, consistent with the observations that these derivatives had the largest enthalpies and free energies favoring trans amide bonds. In contrast, the methoxyacetyl N-cap exhibited longer n→π* interaction distances (2.89, 3.05 Å), consistent with its less favorable enthalpy and a lower Ktrans/cis.
Figure 4.
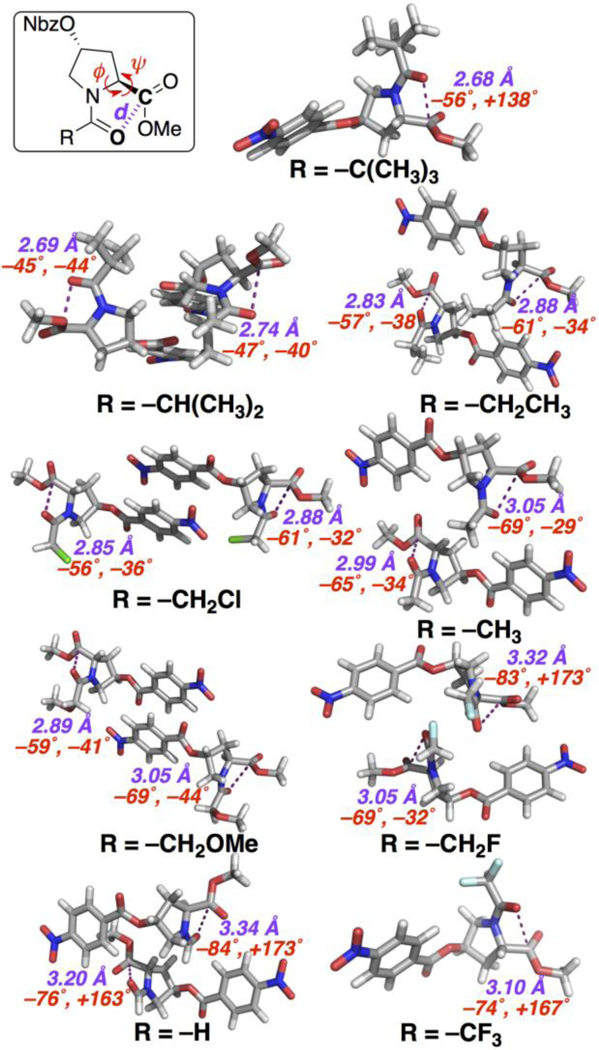
X-ray crystal structures of derivatives, with the Oi…Ci+1 intercarbonyl distance d and the peptide main chain (ϕ,ψ) torsion angles in each molecule indicated. Nbz = 4-NO2-benzoate ester.
Table 2.
| X-(C=O)- Hyp(Nbz)-OMe X= |
X-ray crystallography | calculations. Flp | |||||
|---|---|---|---|---|---|---|---|
| molecule 1 | molecule 2 | α-helix | PPII | ||||
| ϕ, Ψ ° | d, Å | ϕ, Ψ ° | d, Å | d, Å | NBO Ec | d, Å | |
| C(CH3)3 | −56, +138 | 2.681 | — | — | 2.723 | 2.27d | 2.726 |
| CH(CH3)2 | —45, —44 | 2.687 | −47,−40 | 2.738 | 2.785 | 1.97 | 2.781 |
| CH2CH3 | −57,−38 | 2.828 | −61,−34 | 2.883 | 2.807 | 1.87 | 2.816 |
| CH3 | −65,−34 | 2.991 | −69, −29 | 3.049 | 2.812 | 1.85 | 2.816 |
| CH2CI | −56, −36 | 2.847 | −61,−32 | 2.879 | 2.837 | 1.68 | 2.834 |
| CH2OCH3 | −59, −41 | 2.893 | −69, −44 | 3.046 | 2.841 | 1.70 | 2.844 |
| CH2F | −69,−32 | 3.053 | −83, −173 | 3.320 | 2.838 | 1.71 | 2.837 |
| H | −76, +163 | 3.197 | −84, +173 | 3.336 | 2.961 | 1.11 | 2.963 |
| CHF2 | −56, −36 | 2.858 | −63,−30 | 2.921 | 2.861 | 1.40 | 2.864 |
| CF3 | −74, +167 | 3.099 | — | — | 2.854 | 1.41 | 2.859 |
Additional details are in the Supporting Information. d = distance between the donor O (Oi) and the acceptor carbon (Ci+1).
Calculations on X-C(O)-Flp-OMe at the MP2 level of theory and 6–311++G(2d,2p) basis set in implicit water, on peptides with an exo ring pucker and the indicated general conformation. ϕ and ψ torsion angles are tabulated in the Supporting Information, as are calculations using the M06–2X DFT method and similar calculations on X-C(O)-Pro-OMe.
NBO energies for orbital overlap between the Oi p-like orbital and the C=Oi+1 π* orbital in kcal mol–1.
An interaction energy of 0.66 kcal mol–1 was in addition calculated between the Oi s-like orbital and the C=Oi+1 π* orbital.
The peptide with the fluoroacetyl N-cap exhibited two structures: one was in an α-helical conformation, though with a longer (3.05 Å) intercarbonyl distance and a more extended value of ϕ (–69˚, compared to –57˚ for propionyl); the other structure exhibited an extended conformation ((ϕ,ψ) = –83˚, –173˚). The trifluoroacetyl and formyl derivatives also exhibited longer intercarbonyl distances and extended conformations, consistent with weak n→π* interactions in these more electron-poor derivatives. The observation of extended conformations is noteworthy given the inherent preference of the 4R-nitrobenzoate ester of Hyp for an exo ring pucker, and thus for more compact conformations. While crystal packing obviously impacts the specific details of the structures observed, collectively these data are consistent with a substantial ability of donor carbonyl electronic properties to modulate the strength of the n→π* interaction, and thus the preference for a compact versus extended backbone conformation.
In order to obtain further insights into the effect of N-cap identity on the n→π* interaction, all derivatives were examined computationally, using DFT (M06–2X) and MP2 methods.[13] Structures were examined as X-Pro-OMe and X-Flp-OMe peptides, with an exo ring pucker. Initial models were developed using crystallographically observed structures and then subjected to geometry optimization. 4R-Fluoroproline (Flp) and 4R-hydroxyproline nitrobenzoate have similar effects on conformation.[10] Therefore, Flp was used in modeling, due to its much lower computational cost, which allowed the use of larger basis sets and analysis using the MP2 method, which better addresses electron correlation than DFT methods. Peptides were analyzed with an exo ring pucker in both the α-helix and PPII conformations, in order to specifically examine effects on both secondary structures stabilized by n→π* interactions.
Computational results corroborated observations by NMR spectroscopy and X-ray crystallography (Table 2, Figure 5). Using the distance between the donor carbonyl O (Oi) and the acceptor carbonyl C (Ci+1) as a reporter of n→π* interaction strength, the pivaloyl group exhibited the closest Oi…Ci+1 distances, while the formyl group exhibited the longest Oi…Ci+1 distances, with the distances Piv > iBut > CH2CH3 > CH3 > CH2Cl ~ CH2OMe ~ CH2F > CHF2 ~ CF3 > H. In addition, a stronger donor resulted in a more compact geometry in a given conformation (e.g. PPII conformation, by DFT: propionyl (ϕ,ψ) = –58˚, +143˚, versus formyl (ϕ,ψ) = –64˚, +151˚).
Figure 5.
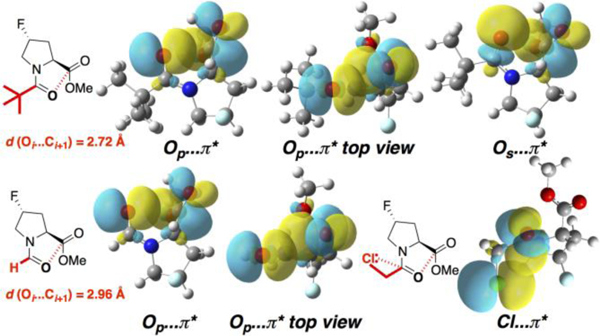
NBO analysis of n→π* interactions in the limiting cases of pivaloyl and formyl N-caps. The extent of orbital overlap between the p-like Oi orbital and the C=Oi+1 π* orbital is significantly greater with the geometry observed in the pivaloyl N-cap than that in the formyl N-cap. The former also exhibits overlap between the s-like Oi and π* orbitals. Bottom right: the chloroacetyl N-cap exhibits an n→π* interaction between the chlorine lone pair and the adjacent carbonyl, resulting in serial n→π* interactions. Calculations were conducted at the MP2 level with the 6–311++G(2d,2p) basis set in implicit water.
The computational results also provided insights into the basis for the higher-than-expected preference of the chloroacetyl group for a trans amide bond and a close n→π* interaction. This derivative exhibited a Cl-C-C-O torsion angle of ~ –90˚ both crystallographically and computationally. A torsion angle scan confirmed that this conformation was preferred.[14] NBO analysis[15] revealed that this conformation allows for favorable orbital overlap between one chlorine lone pair (n) and the donor carbonyl π* orbital, as an n→π* interaction with an NBO interaction energy of 2.5 kcal mol–1. These results suggest that the chloroacetyl group enhances the carbonyl/carbonyl n→π* interaction via two serial n→π* interactions, whereby one n→π* interaction (here, Cl…C=O) makes that acceptor carbonyl a better donor for a subsequent n→π* interaction. These serial effects have been proposed to stabilize both the α-helix and polyproline helix conformations in proteins.[3b, 3d, 3e, 4]
We have described the application of peptide acyl N-capping motifs to modulate peptide structure, via the tunable control of the strength of n→π* interactions. We have observed crystallographically that more electron-donating N-cap motifs promote closer n→π* interactions and more compact conformations, including α-helix and polyproline II helix, whereas more electron-withdrawing N-cap motifs relatively promote more extended conformations. Notably, electronic modulation of the n→π* donor carbonyl was observed to directly impact the ϕ and ψ main chain torsion angles, as expected based on prior, more indirect analyses that employed readout of the ω torsion angle. Collectively, these results provide further support for the importance of n→π* interactions in protein structure, with particular relevance in understanding disordered states of proteins. The unfolded states of proteins remain not well understood, yet are central to understanding protein folding and recognition. In addition, these results should have significant applications in peptide design and in medicinal chemistry, whereby the identity of the acyl N-capping motif could be used to predictably promote defined conformations, for optimized structure, function, and molecular recognition.
Supplementary Material
Acknowledgements
We thank NSF (CHE-1412978) for support. Instrumentation support was provided by NIH (GM110758) and NSF (CHE-1229234).
References
- [1].Levinthal C, J. Chim. Phys. Physico-Chimie Biol. 1968, 65, 44–45. [Google Scholar]
- [2].a Bretscher LE, Jenkins CL, Taylor KM, DeRider ML, Raines RT, J. Am. Chem. Soc. 2001, 123, 777–778; [DOI] [PubMed] [Google Scholar]; b Newberry RW, Raines RT, Acc. Chem. Res. 2017, 50, 1838–1846; [DOI] [PMC free article] [PubMed] [Google Scholar]; c Singh SK, Das A, Phys. Chem. Chem. Phys. 2015, 17, 9596–9612; [DOI] [PubMed] [Google Scholar]; d Singh SK, Mishra KK, Sharma N, Das A, Angew. Chem., Int. Ed. 2016, 55, 7801–7805; [DOI] [PubMed] [Google Scholar]; e Deb P, Jin GY, Singh SK, Moon J, Kwon H, Das A, Bagchi S, Kim YS, J. Phys. Chem. Lett. 2018, 9, 5425–5429. [DOI] [PubMed] [Google Scholar]
- [3].a Hinderaker MP, Raines RT, Protein Sci. 2003, 12, 1188–1194; [DOI] [PMC free article] [PubMed] [Google Scholar]; b Bartlett GJ, Choudhary A, Raines RT, Woolfson DN, Nat. Chem. Biol. 2010, 6, 615–620; [DOI] [PMC free article] [PubMed] [Google Scholar]; c Sonntag LS, Schweizer S, Ochsenfeld C, Wennemers H, J. Am. Chem. Soc. 2006, 128, 14697–14703; [DOI] [PubMed] [Google Scholar]; d Brister MA, Pandey AK, Bielska AA, Zondlo NJ, J. Am. Chem. Soc. 2014, 136, 3803–3816; [DOI] [PMC free article] [PubMed] [Google Scholar]; e Elbaum MB, Zondlo NJ, Biochemistry 2014, 53, 2242–2260; [DOI] [PMC free article] [PubMed] [Google Scholar]; f Wilhelm P, Lewandowski B, Trapp N, Wennemers H, J. Am. Chem. Soc. 2014, 136, 15829–15832; [DOI] [PubMed] [Google Scholar]; g Rahim A, Saha P, Jha KK, Sukumar N, Sarma BK, Nat. Commun. 2017, 8; [DOI] [PMC free article] [PubMed] [Google Scholar]; h D. N. Reddy, E. N. Prabhakaran 2014, 101, 66–77. [DOI] [PubMed] [Google Scholar]
- [4].Zondlo NJ, Nat. Chem. Biol. 2010, 6, 567–568. [DOI] [PubMed] [Google Scholar]
- [5].a Uversky VN, Oldfield CJ, Dunker AK, J. Mol. Recognit. 2005, 18, 343–384; [DOI] [PubMed] [Google Scholar]; b Shi Z, Chen K, Li Z, Kallenbach NR Chem. Rev. 2006, 106, 1877–1897; [DOI] [PubMed] [Google Scholar]; c Tompa P, Schad E, Tantos A, Kalmar L, Curr. Opin. Struct. Biol. 2015, 35, 49–59; [DOI] [PubMed] [Google Scholar]; d Wright PE, Dyson HJ, Nat. Rev. Mol. Cell. Biol. 2015, 16, 18–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Choudhary A, Fry CG, Raines RT, ARKIVOC 2010, 8, 251–262. [PMC free article] [PubMed] [Google Scholar]
- [7].Newberry RW, VanVeller B, Guzei IA, Raines RT, J. Am. Chem. Soc. 2013, 135, 7843–7846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Hodges JA, Raines RT, Org. Lett. 2006, 8, 4695–4697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].a Shoemaker KR, Kim PS, York EJ, Stewart JM, Baldwin RL, Nature 1987, 326, 563–567; [DOI] [PubMed] [Google Scholar]; b Chakrabartty A, Doig AJ, Baldwin RL, Proc. Natl. Acad. Sci. USA 1993, 90, 11332–11336; [DOI] [PMC free article] [PubMed] [Google Scholar]; c Doig AJ, Baldwin RL, Protein Sci. 1995, 4, 1325–1336; [DOI] [PMC free article] [PubMed] [Google Scholar]; d Aurora R, Rose GD, Protein Sci. 1998, 7, 21–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].a Thomas KM, Naduthambi D, Tririya G, Zondlo NJ, Org. Lett. 2005, 7, 2397–2400; [DOI] [PubMed] [Google Scholar]; b Naduthambi D, Zondlo NJ, J. Am. Chem. Soc. 2006, 128, 12430–12431; [DOI] [PubMed] [Google Scholar]; c Pandey AK, Naduthambi D, Thomas KM, Zondlo NJ, J. Am. Chem. Soc. 2013, 135, 4333–4363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Pandey AK, Yap GPA, Zondlo NJ, J. Org. Chem. 2014, 79, 4174–4179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].a Holl MG, Struble MD, Siegler MA, Lectka T, J. Fluorine Chem. 2016, 188, 126–130; [Google Scholar]; b Hof F, Scofield DM, Schweizer WB, Diederich F, Angew. Chem., Int. Ed. 2004, 43, 5056–5059; [DOI] [PubMed] [Google Scholar]; c Paulini R, Muller K, Diederich F, Angew. Chem., Int. Ed. 2005, 44, 1788–1805; [DOI] [PubMed] [Google Scholar]; d Fischer FR, Schweizer WB, Diederich F, Angew. Chem., Int. Ed. 2007, 46, 8270–8273. [DOI] [PubMed] [Google Scholar]
- [13].a Leverentz HR, Truhlar DG, J. Phys. Chem. A 2008, 112, 6009–6016; [DOI] [PubMed] [Google Scholar]; b M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. Montgomery, J. A., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, D. J. Fox, Gaussian, Inc., Wallingford, CT, 2013.
- [14].See the Supporting Information for details.
- [15].Glendening CR, Landis CR, Weinhold F, WIREs Comput. Mol. Sci. 2012, 2, 1–42. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.