

Abstract
Cross-linking mass spectrometry has become an important approach for studying protein structures and protein–protein interactions. The amino acid compositions of some protein regions impede the detection of cross-linked residues, although it would yield invaluable information for protein modeling. Here, we report on a sequential-digestion strategy with trypsin and elastase to penetrate regions with a low density of trypsin-cleavage sites. We exploited intrinsic substrate-recognition properties of elastase to specifically target larger tryptic peptides. Our application of this protocol to the TAF4–12 complex allowed us to identify cross-links in previously inaccessible regions.
Trypsin is the enzyme of choice in mass-spectrometry (MS)-based proteomics. The favorable behavior of tryptic peptides during mass-spectrometric analysis is one of the main reasons for this.1,2 However, as trypsin cleaves after Arg and Lys, peptides generated in protein regions with a low Arg and Lys densities are very long, making them potentially nonobservable by mass spectrometry and thus resulting in poor coverage of those regions.
This problem is particularly relevant for cross-linking mass spectrometry (CLMS). In CLMS, protein proximities and conformations are preserved through the introduction of covalent bonds. The detection of these bonds as cross-linked peptide pairs by MS is translated into distance constraints.3−7 Currently, the most frequently used cross-linkers are N-hydroxysuccinimide (NHS) esters that primarily target the ε-amino groups of Lys residues and to a lesser extent also react with the hydroxyl groups of Ser, Thr, and Tyr.8,9 CLMS critically depends on good sequence coverage to reveal structural information for the whole protein. Cross-linking naturally involves two peptides, which aggravates the problem of overly large peptides. This is exacerbated when the cross-linker reacts with two lysine residues as two potential cleavage sites for trypsin are removed. The stabilization of proteins through cross-linking and the destruction of potential trypsin-cleavage sites may further affect the efficiency of trypsin digestion and lead to an increase in missed cleavages.
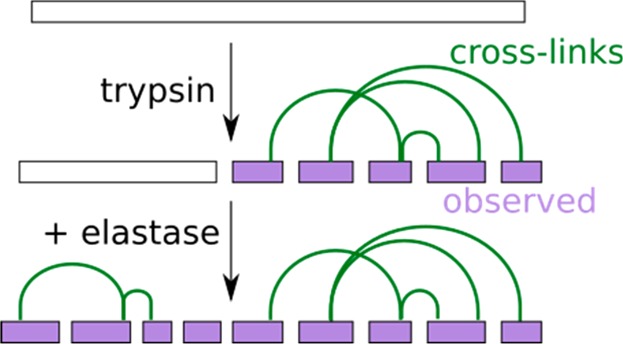
Alternative proteases to trypsin target basic (LysC and ArgC), acidic (AspN and GluC), or aromatic (chymotrypsin) amino acids.10,11 Although usage of these proteases improves sequence coverage12,13 and has been used in parallel to usage of trypsin to enhance identification of cross-linked residues,14,15 some protein regions, such as the N-terminal region of TAF4, a subunit of transcriptional factor II D, still cannot be accessed (Figure 1). The existence of TAF4b, a cell-type-specific TAF4 paralogue with a unique N-terminal region, points to important functions and interactions mediated through the N-terminal region of TAF4.16 Cross-linked residues in this region would therefore provide important information on the function and structure of TAF4.
Figure 1.

Theoretical cleavage sites in TAF4.
Proteases with broader specificity, like elastase, proteinase K, and pepsin, have the potential of complementing more selective proteases and indeed have been used in membrane proteomics17,18 to detect phosphorylation sites19 and even in CLMS20 with varying levels of success. Elastase, for example, cleaves after Ala, Val, Leu, Ile, Ser, and Thr17 and should be able to cleave the N-terminal region of TAF4 at many sites (Figure 1). This broad specificity leads to two potential problems. If cleavage is complete, the resulting peptides would be very small. However, cleavage of elastase is not expected to be complete. Analyses of elastase efficiency on peptides with varying length revealed elastase to have an extended substrate-binding region.21,22 Short peptides that do not cover the complete substrate-binding region are less efficiently cleaved by elastase. Although this addresses the potential problem of peptide length, it leads to another problem. Having many missed cleavage sites leads elastase to generate very complex mixtures with often overlapping peptides.17 This problem is exacerbated in cross-linked peptides, as two peptides are involved, resulting in a combinatorial increase of complexity. Recent work by our lab revealed a strong substrate preference of proteases for longer peptides.15 As a consequence, in a sequential digest, preferentially long peptides are cleaved by the second protease, whereas short peptides remain undigested. The complexity of an elastase digest might therefore be vastly reduced by first using trypsin. Only long tryptic peptides should be good substrates for elastase, whereas any sequence range of a protein that is covered by short tryptic peptides would remain unaffected by elastase treatment.
In this study, we investigate the hypothesis whether the sequential digestion of trypsin and elastase might positively impact the use of elastase in CLMS. As a result, we detected cross-links in the difficult-to-digest N-terminal region of TAF4.
Materials and Methods
Sample Preparation
The human TAF4–TAF12 complex was coexpressed in Sf21 insect cells using the MultiBac system.23 DNA encoding an N-terminal hexa-histidine tag and a protease-cleavage site for tobacco etch virus (TEV) NIa protease was added to the 5′ end of the TAF4 open reading frame and cloned into pFL plasmid along with TAF12. Baculovirus generation, cell-culture infection, and protein production were carried out following published protocols.24 Cell pellets were resuspended in Talon Buffer A (25 mM Tris pH 8.0, 150 mM NaCl, 5 mM imidazole, and complete protease inhibitor; Roche Molecular Biochemicals). Cells were lysed by freeze–thawing (twice), followed by centrifugation at 40 000g in a Ti70 rotor for 60 min to clear the lysate. The TAF4–TAF12 complex was first bound to talon resin and pre-equilibrated with Talon Buffer A; this was followed by washes with Talon Buffer A, then with Talon Buffer HS (25 mM Tris pH 8.0, 1 M NaCl, 5 mM imidazole, and complete protease inhibitor), and then again with Talon Buffer A. The TAF4–TAF12 complex was eluted using Talon Buffer B (25 mM Tris pH 8.0, 150 mM NaCl, 200 mM imidazole, and complete protease inhibitor). Fractions containing the TAF4–TAF12 complex were dialyzed overnight against MonoQ Buffer A (25 mM HEPES, pH 7.5, 100 mM NaCl, 1 mM DTT, and complete protease inhibitor). The complex was further purified using ion-exchange chromatography (IEX) with a MonoQ column pre-equilibrated with MonoQ Buffer A. After binding, the column was washed with MonoQ Buffer A, and TAF4–TAF12 was eluted using a continuous, linear gradient of MonoQ Buffer B (25 mM HEPES pH 7.5, 1000 mM NaCl, 1 mM DTT, and complete protease inhibitor) from 0 to 100%. The complex was further purified by size-exclusion chromatography (SEC) with a SuperoseS6 10/300 column in SEC buffer (25 mM Tris pH 8.0, 150 mM NaCl, 1 mM DTT, and complete protease inhibitor).
TAF4–TAF12 (1.5 μg per condition) complexes were cross-linked with bis(sulfosuccinimidyl)suberate (BS3) (Thermo Fisher Scientific) at ratio of 1:1 (w/w) and incubated on ice for 2 h. The mixture was incubated on ice for 1 h after saturated bicarbonate (50 molar excess) was added to quench the reaction. Frozen Schizosaccharomyces pombe cells were ground, and 1 g of yeast powder was resuspended in 2 mL of RIPA (Sigma-Aldrich) supplemented with cOmplete (Roche). Cell debris was moved via centrifugation at 1200g for 15 min. All samples were separated by SDS-PAGE on a 4–12% Bis Tris gel (Life Technologies) and stained using Imperial Protein Stain (Thermo Fisher Scientific). Appropriate bands were cut and proteins were first reduced with DTT and then alkylated with iodoacetamide. Samples were incubated with trypsin (13 ng μL–1; Pierce, Thermo Fisher Scientific) or elastase (15 ng μL–1, Promega) at 37 °C for 16 h. In the case of the elastase–trypsin digestion, trypsin (13 ng μL–1) was added to the overnight elastase digest and incubated for 4 h at room temperature. For the sequential trypsin–elastase digestion, elastase (15 ng μL–1) was added to the trypsin digest and incubated at 37 °C for 30 min. A comparison between 30 min and 4 h of digestion with elastase showed no significant difference (data not shown). Following the digestion, peptides were purified on C18 StageTips using standardized protocols.25
LC-MS/MS
TAF4–TAF12 complexes were analyzed on an Orbitrap Fusion Lumos Tribrid (Thermo Fisher Scientific) and yeast lysates were analyzed on an Orbitrap Elite (Thermo Fisher Scientific). Both were coupled online to an Ultimate 3000 RSLCnano Systems (Dionex, Thermo Fisher Scientific). Peptides were loaded onto an EASY-Spray LC Column (Thermo Fisher Scientific) at a flow rate of 0.300 μL min–1 using 98% mobile phase A (0.1% formic acid) and 2% mobile phase B (80% acetonitrile in 0.1% formic acid). To elute the peptides, the percentage of mobile phase B was first increased to 40% over a time course of 110 min followed by a linear increase to 95% in 11 min.
Full MS scans for yeast lysates were recorded in the orbitrap at 120 000 resolution for MS1 with a scan range of 300–1700 m/z. The 20 most intense ions (precursor charge ≥2) were selected for fragmentation by collision-induced disassociation, and MS2 spectra were recorded in the ion trap (2.0 × 104 ions as a minimal required signal, 35 normalized collision energy, dynamic exclusion for 40 s). Both MS1 and MS2 were recorded in the orbitrap for the TAF4–TAF12 complexes (120 000 mass resolution for MS1 and 15 000 mass resolution for MS2, scan range 300–1700 m/z, dynamic exclusion for 60 s, precursor charge ≥3). Higher-energy collision dissociation was used to fragment peptides (30% collision energy, 5.0 × 104 AGC target, 60 ms maximum injection time).
Data Analysis
MaxQuant software26 (version 1.5.2.8) employing the Andromeda search engine27 was used to analyze the whole-cell lysates of S. Pombe. We used the PombeBase database28 with carbamidomethylation of cysteine as a fixed modification and oxidation of methionine as a variable modification. MS accuracy was set to 4.5 ppm and MS/MS tolerance to 20 ppm. For trypsin, we allowed up to two miscleavages. Specificity of elastase was defined as cleavage after Ala, Val, Leu, Ile, Ser, and Thr but not before Pro. Specificity for the trypsin and elastase digest was set to cleavage after Ala, Val, Leu, Ile, Ser, Thr, Arg, and Lys except if it was followed by Pro. For all digests containing elastase, up to 10 miscleavages were allowed. The peaklist for identification of cross-linked peptides was generated using MS convert (ProteoWizard) for tryptic digests with the following settings: peakPicking: 2–, msLevel: 2–, MS2Denoise: 20 100, false or Maxquant version 1.6.2.3 with default settings except increasing the “Top MS/MS peaks per 100 Da” to 100 for elastase-containing digests. Resulting MGF or APL files were searched using Xi software15,29 version 1.6.731 with MS accuracy set to 4 ppm and MS/MS tolerance set to 20 ppm. The only fixed modification was the carbamidomethylation of cysteine. Variable modifications were the oxidation of methionine and amidated and hydrolyzed BS3. The cross-linker was BS3 with Lys, Ser, Thr, and Tyr as the only cross-linkable residues. Enzyme specificities were the same as described for Mascot. Four miscleavages were allowed for the trypsin digest, and 11 miscleavages were allowed for the other digests. Two additional missing isotopes were allowed in the custom settings. False-discovery rates (FDR) were estimated using xiFDR30 version 1.1.26.58 with 5% FDR at a peptide-pair level for the TAF4–12 complex and 5% FDR at a peptide-pair and -link level. Linear peptides of the cross-linked TAF4–12 complex were identified using Mascot software31 with carbamidomethylation of cysteine as a fixed modification and the following variable modifications: oxidation of methionine, hydrolyzed BS3 (mass: 156.0786 Da), and amidated BS3 (mass: 155.0946 Da). Chromatograms of MS1 were extracted, and peak areas were quantified using Skyline.32
In-Silico Digest
TheTAF4–TAF12 complex was digested in-silico using trypsin, elastase or a combination of trypsin and elastase. We assumed a complete digestion with no miscleavages for the in-silico digestion and a minimum length of five amino acids.
Random Distribution
Random links were generated between cross-linkable residues. The links were fitted into the crystal structure of monomeric HSA (PDB: 1AO6) to calculate the distances.
Results and Discussion
Four different types of digestion, trypsin, elastase, sequential trypsin–elastase, and sequential elastase–trypsin, were applied to a cross-linked TAF4–TAF12 complex. We compared these digests to assess the influence of substrate length on the cleavage behavior of elastase. The peptide lengths of the observed tryptic peptides without any modifications ranged from 5 to 65 amino acids, which are very similar to the theoretical peptide-length distribution (Figure 2a). However, the median lengths of the peptides digested by elastase, trypsin–elastase, and elastase–trypsin were considerably higher (12 amino acids) than the predicted medians of 6 amino acids (Figure 2b–d). The majority of elastase-derived peptides are larger than 5 amino acids, and the maximal length of 29 amino acids is considerably shorter than that of trypsin (65). These observations are in line with previous studies on short peptides reporting that the activity of elastase is dependent on the substrate length N-terminal of the cleavage site.21,22 Our results suggest that a similar restriction might also apply to longer peptides and to the substrate length C-terminal from the cleavage site.
Figure 2.
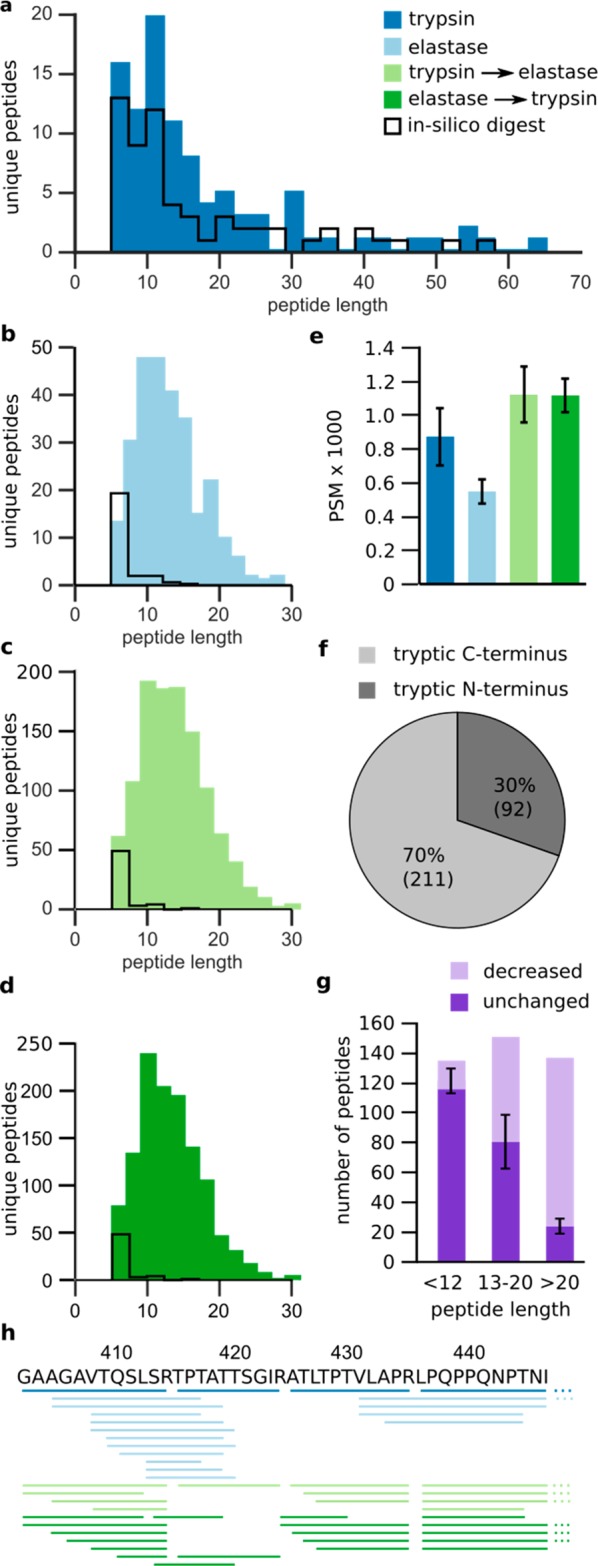
Impact of elastase when following trypsin in a sequential digestion of the TAF4–TAF12 complex. (a–d) Peptide-length distribution of observed and theoretical peptides (full cleavage, no modifications) for trypsin (a), elastase (b), sequential trypsin–elastase (c), and sequential elastase–trypsin (d) digestion. (e) Number of peptide-spectrum matches (PSM) for different proteases using Mascot. (f) Distribution of detected semitryptic peptides into those with a tryptic C-terminus versus those with a tryptic N-terminus in the sequential trypsin–elastase digest. (g) Intensity differences for tryptic peptides in the tryptic digest and in the sequential trypsin–elastase digest, determined using label-free quantitation in Skyline. The peptides were divided into three groups depending on their lengths (<12, 13–20, and >20 residues). Peptides with a 2-fold reduction or lower were labeled “decreased”. (h) TAF4 (residues 827–880) with the observed cleavage patterns of trypsin, elastase, and the sequential trypsin–elastase treatment. Identified peptides are represented as lines below the protein sequence. Data shown are the means ± standard deviations (SD) of three independent digestions. Trypsin, dark blue; elastase, light blue; sequential trypsin–elastase, light green; sequential elastase–trypsin digest, dark green.
Notably fewer peptide-spectrum matches (546 ± 74 PSM) were identified following elastase digestion compared with those from peptides digested by trypsin alone (869 ± 170 PSM), trypsin–elastase (1116 ± 164 PSM), or elastase–trypsin (1111 ± 100 PSM, Figure 2e). One possible explanation for the reduced identification of elastase peptides could be the missing positive C-terminal charge that tryptic peptides have. To this end, we analyzed semitryptic peptides, as they should distribute equally among those featuring a tryptic C-terminus and a tryptic N-terminus. Semitryptic peptides with tryptic C-termini were identified more often (70 ± 2%), in agreement with a positive C-terminal charge being beneficial for detection (Figure 2f). Another disadvantage of elastase is that it frequently misses cleavage sites and overlapping peptides lead to complex peptide mixtures. By digesting proteins with trypsin first, the amount of available cleavage sites for elastase might be reduced, as short tryptic peptides should be protected from elastase because of their size. The products of digested small peptides may not be identified, because of the identification bias against small peptides that occurs in LC-MS/MS. To avoid this, we quantified tryptic peptides before and after the addition of elastase. Peptides that had been reduced by more than half were labeled as “reduced”. The remaining peptides were categorized as “unchanged”. Peptides were divided into three groups depending on their lengths (≤12, 13–20, and >20). The number of reduced peptides, 19, 70, or 113 peptides, respectively, increased with peptide size (Figure 2g). The label-free quantification confirms that elastase preferentially digests long peptides; thus, initial trypsin digestion reduces elastase-induced complexity. Indeed, analyzing the 404–420 region in TAF4 revealed that trypsin treatment protected this region from elastase digestion. Consequently, the complexity within the sequential trypsin–elastase digest was reduced compared with that of the digest from elastase alone or elastase–trypsin (Figure 2h).
In the next step, we analyzed the effect of our sequential trypsin–elastase on the detection of cross-linked peptides. As a proof of principle, we first analyzed BS3 cross-linked human-serum albumin (HSA). As expected, the increase in complexity and the missing C-terminal charge associated with elastase digestion adversely affected the identification of cross-linked peptides. Although digestion of the BS3 cross-linked complexes with trypsin allowed us to identify 152 cross-links, digestion with elastase only led to the identification of 42 cross-links (Figure 3a). This might be due to the increase in the database, which is quite significant because of the n2 behavior of the search space from cross-linked peptides. However, adding trypsin to the elastase digest improved the detection of cross-links, despite the increase in database size. On the one hand, adding trypsin to the elastase-digested peptides increased the complexity of the samples, but on the other hand, it produced semitryptic and tryptic peptides featuring peptides with a positive C-terminal charge. As 111 cross-links were identified with the elastase–trypsin digest, the new peptides with tryptic C-termini seemed to have a bigger impact than the increase in complexity. Our analysis so far indicated that reversing the order to trypsin followed by elastase reduces the complexity of the sample and should therefore improve detection of cross-linked peptides. Indeed, more cross-links (131) were identified with the trypsin–elastase digest compared with the elastase–trypsin digest. As a final test of our workflow, we fitted the detected cross-links into the crystal structure of HSA (PDB: 1AO6, monomeric; Figure 3b). As expected from the link-level FDR (5%), 5% of the links derived from the trypsin–elastase digest were overly long (Figure 3c).
Figure 3.

Impact of trypsin–elastase on the detection of cross-linked residues. (a) Number of cross-links identified for HSA. (b) Cross-links fitted into the crystal structure of HSA (monomer, PDB: 1AO6). (c) Distribution of distances between random links compared with those of the links identified in the sequential trypsin–elastase digest. (d,e) Impact of digestion procedure on the sequence coverage of the TAF4–TAF12 complex, with sequence coverage for TAF4 (d) and TAF12 (e) through linear peptides only. Data shown are the means ± SD of three independent digestions. (f) Venn diagram of identified residue pairs for the trypsin and sequential trypsin–elastase digest for the TAF4–TAF12 complex. (g) Cross-link map of the TAF4–12 complex for trypsin and sequential trypsin–elastase digest with shared links in black. Trypsin, dark blue; elastase, light blue; sequential trypsin–elastase, light green; sequential elastase–trypsin digest, dark green.
Having successfully tested our workflow on HSA, we moved on to analyze the TAF4–TAF12 complex. Sequence coverage of both TAF4 and TAF12 decreased from 74 and 75% to 73 and 65%, respectively, when trypsin was replaced by elastase (Figure 3d,e). In marked contrast, adding elastase to tryptic peptides increased sequence coverage of TAF4 to 93% and TAF12 to 92%. Reversing the order of digestion to elastase–trypsin still increased the sequence coverage of TAF4 and TAF12 to 95 and 85%. Our previous observation on the impact of complexity and positively charged C-termini on cross-link identification held true for the analysis of the TAF4–12 complex. We identified 162 cross-links with trypsin alone, followed by 115 and 42 cross-links for trypsin–elastase and elastase–trypsin digestion, respectively. A very small number of cross-links (six) was detected when only elastase was used. The cross-links determined from the sequential trypsin–elastase digest were highly complementary to the trypsin data, with only 59 shared out of 218 identified cross-links (Figure 3f). Importantly, the additional digestion of tryptic peptides by elastase provided data in the previously undetected N-terminal region of TAF4 (Figure 3g).
To test the behavior of elastase in complex mixtures, we applied the protocol to whole-cell lysate of S. pombe. Three characteristics of the identified peptides (peptide length, peptide type, and amount of miscleavages) indicate that the length of the substrate influences the activity of elastase. Although digesting tryptic peptides with elastase reduced the number of longer peptides, the median of all digests containing elastase was 13 amino acids (Figure 4a). If the activity of elastase were not influenced by the substrate size, the distribution of tryptic, semitryptic, and elastase peptides would be independent of the digestion order. However, sequential trypsin–elastase digestion led to more tryptic peptides than elastase–trypsin digestion (Figure 4b). Shorter peptides were very poor substrates for the second enzyme. Consequently, the first enzyme produced the majority of the peptides. Because of the favorable mass-spectrometric behavior of tryptic peptides, trypsin should always be the first enzyme used in a sequential digestion. Unsurprisingly, a higher number of miscleavages occurred when elastase was used in all enzyme combinations (Figure 4c). Interestingly, treating the sample with trypsin prior to elastase digestion increased miscleavages. This suggests that the treatment with trypsin reduced the available elastase sites, presumably through their “protection” of being in smaller peptides.
Figure 4.
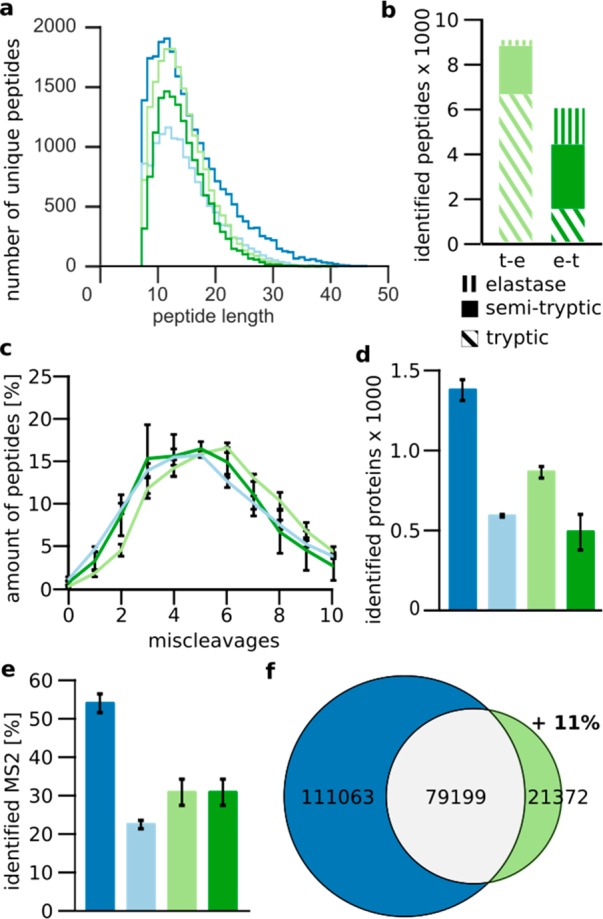
Impact of elastase when following trypsin in a sequential digest in S. pombe cell lysate. (a) Peptide-length distribution. (b) Number of tryptic, semitryptic, and elastase peptides identified in both the trypsin–elastase (light green) and elastase–trypsin digests (dark green). (c) Distribution of miscleavages for elastase, trypsin–elastase, and elastase–trypsin digests. (d) Number of identified proteins. (e) Percentage of identified MS2. (f) Overlap of observed residues by trypsin and sequential trypsin–elastase digestion. Data shown are the means ± SD of duplicate injections from three independent digestions. Trypsin, dark blue; elastase, light blue; sequential trypsin–elastase, light green; sequential elastase–trypsin digest, dark green.
The highest number, with 1377 ± 62 protein identifications, was achieved using only trypsin (Figure 4d). Subsequent elastase digestion reduced the numbers to 874 ± 39. Using only elastase yielded 593 ± 6 protein identifications. The least amount of proteins, 492 ± 115 proteins, was identified with sequential elastase–trypsin. More of the identified semitryptic peptides from the sequential digests had tryptic C-termini (55 ± 2%). Although the difference is not as pronounced as for the TAF4–12 complex, it is consistent with a favorable property of a positively charged residue at the C-terminus for MS/MS analysis. This may also explain in part the reduced identification numbers in the sequential digestion compared with trypsin alone. Peptides with a nontryptic C-terminus should be as frequent in the mixture as with a nontryptic N-terminus. A reduced number of identifications paired with a similar number of MS2 spectra between trypsin and sequential digestion suggests that the challenge is in MS2. Indeed, although the number of MS2 for the trypsin (35 203 ± 3088) and the sequential trypsin–elastase digests (35 160 ± 3322) were quite similar, only 31 ± 3% of the MS2 spectra from the trypsin–elastase digest could be matched, compared with 54 ± 2% from the tryptic digest (Figure 4e). In terms of sequence coverage, the larger number of peptides identified in the trypsin digest also resulted in more residues being covered (190 261 ± 8215) than with the sequential digest (100 570 ± 10 660). However, peptides derived from the sequential digest covered some different regions than the tryptic peptides, expanding the covered sequence space of trypsin by 11% (Figure 4f).
Conclusion
Elastase digestion leading to complementary data compared with trypsin digestion has been shown before and could be confirmed in our study.17,33 Although sequential digests have been used before,34−36 to our knowledge elastase has not been exploited in sequential digestion of tryptic peptides or for CLMS. Elastase generates very complex peptide mixtures, with many peptides having sequence overlap. Conceptually, CLMS does not combine well with the use of elastase or other proteases with broad cleavage specificity. The increase of complexity and loss in sensitivity normally accompanying the occurrence of overlapping peptides is exacerbated in cross-linking. Every cross-linked peptide consists of two peptides and consequently is subject to combinatorial loss. This meets an already low abundance of cross-linked peptides. The cleavage by trypsin prior to elastase digestion reduces the complexity that is typically associated with elastase digestion. The cleavage of tryptic peptides by elastase is biased toward long peptides and increases their detection. As a result, we detect cross-links in the difficult-to-digest N-terminal region of TAF4. Our protocol of using trypsin prior to a complementary enzyme to enhance the performance of the complementary enzyme is also applicable to other enzymes, as was shown recently for AspN, GluC, and chymotrypsin.15 These gains, at least for elastase, transfer to proteomics at large as shown by our results in S. pombe lysate. We anticipate that our protocol will therefore also be useful for other proteomic applications. This includes the detection of post-translational modifications and the analysis of transmembrane domains. The former benefits from increased sequence observation, whereas trypsin leads to systematically large peptides in the latter.
Acknowledgments
This work was supported by a research stipend to T.D. (DA 1861/2-1) from the Deutsche Forschungsgemeinschaft and by the Wellcome Trust through a Senior Research Fellowship to J.R. (103139), a Senior Investigator Award to I.B. (106115), and a multiuser equipment grant (108504). The Wellcome Centre for Cell Biology is supported by core funding from the Wellcome Trust (203149). The BrisSynBio Centre is supported by BBSRC/EPSRC (BB/L01386X/1).
Accession Codes
The mass-spectrometry proteomics data have been deposited to the ProteomeXchange Consortium37 via the PRIDE partner repository with the data set identifier PXD011459.
Author Contributions
T.D. and J.R. conceived this study and interpreted data, T.D conducted all experiments, K.G. and I.B. contributed material, and T.D. and J.R. wrote the manuscript with input from all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
References
- Huang Y.; Triscari J. M.; Tseng G. C.; Pasa-Tolic L.; Lipton M. S.; Smith R. D.; Wysocki V. H. Statistical Characterization of the Charge State and Residue Dependence of Low-Energy CID Peptide Dissociation Patterns. Anal. Chem. 2005, 77 (18), 5800–5813. 10.1021/ac0480949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dongré A. R.; Jones J. L.; Somogyi Á.; Wysocki V. H. Influence of Peptide Composition, Gas-Phase Basicity, and Chemical Modification on Fragmentation Efficiency: Evidence for the Mobile Proton Model. J. Am. Chem. Soc. 1996, 118 (35), 8365–8374. 10.1021/ja9542193. [DOI] [Google Scholar]
- Rappsilber J. Cross-Linking/Mass Spectrometry as a New Field and the Proteomics Information Mountain of Tomorrow. Expert Rev. Proteomics 2012, 9, 485–487. 10.1586/epr.12.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinz A.; Arlt C.; Chorev D.; Sharon M. Chemical Cross-Linking and Native Mass Spectrometry: A Fruitful Combination for Structural Biology. Protein Sci. 2015, 24, 1193–1209. 10.1002/pro.2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z. A.; Pellarin R.; Fischer L.; Sali A.; Nilges M.; Barlow P. N.; Rappsilber J. Structure of Complement C3(H2O) Revealed by Quantitative Cross-Linking/Mass Spectrometry and Modelling. Mol. Cell. Proteomics 2016, 15, 2730–2743. 10.1074/mcp.M115.056473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orbán-Németh Z.; Beveridge R.; Hollenstein D. M.; Rampler E.; Stranzl T.; Hudecz O.; Doblmann J.; Schlögelhofer P.; Mechtler K. Structural Prediction of Protein Models Using Distance Restraints Derived from Cross-Linking Mass Spectrometry Data. Nat. Protoc. 2018, 13 (3), 478–494. 10.1038/nprot.2017.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rampler E.; Stranzl T.; Orban-Nemeth Z.; Hollenstein D. M.; Hudecz O.; Schlögelhofer P.; Mechtler K. Comprehensive Cross-Linking Mass Spectrometry Reveals Parallel Orientation and Flexible Conformations of Plant HOP2-MND1. J. Proteome Res. 2015, 14 (12), 5048–5062. 10.1021/acs.jproteome.5b00903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mädler S.; Bich C.; Touboul D.; Zenobi R. Chemical Cross-Linking with NHS Esters: A Systematic Study on Amino Acid Reactivities. J. Mass Spectrom. 2009, 44 (5), 694–706. 10.1002/jms.1544. [DOI] [PubMed] [Google Scholar]
- Kalkhof S.; Sinz A. Chances and Pitfalls of Chemical Cross-Linking with Amine-Reactive N-Hydroxysuccinimide Esters. Anal. Bioanal. Chem. 2008, 392 (1–2), 305–312. 10.1007/s00216-008-2231-5. [DOI] [PubMed] [Google Scholar]
- Tsiatsiani L.; Heck A. J. R. Proteomics beyond Trypsin. FEBS J. 2015, 282, 2612–2626. 10.1111/febs.13287. [DOI] [PubMed] [Google Scholar]
- Swaney D. L.; Wenger C. D.; Coon J. J. Value of Using Multiple Proteases for Large-Scale Mass Spectrometry-Based Proteomics. J. Proteome Res. 2010, 9, 1323–1329. 10.1021/pr900863u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giansanti P.; Tsiatsiani L.; Low T. Y.; Heck A. J. R. Six Alternative Proteases for Mass Spectrometry-Based Proteomics beyond Trypsin. Nat. Protoc. 2016, 11, 993–1006. 10.1038/nprot.2016.057. [DOI] [PubMed] [Google Scholar]
- Stieger C. E.; Doppler P.; Mechtler K. Optimized Fragmentation Improves the Identification of Peptides Cross-Linked Using MS-Cleavable Reagents. bioRxiv 2018, 476051. 10.1101/476051. [DOI] [PubMed] [Google Scholar]
- Leitner A.; Reischl R.; Walzthoeni T.; Herzog F.; Bohn S.; Förster F.; Aebersold R. Expanding the Chemical Cross-Linking Toolbox by the Use of Multiple Proteases and Enrichment by Size Exclusion Chromatography. Mol. Cell. Proteomics 2012, 11 (3), M111.014126. 10.1074/mcp.M111.014126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendes M. L.; Fischer L.; Chen Z. A.; Barbon M.; O’Reilly F. J.; Bohlke-Schneider M.; Belsom A.; Dau T.; Combe C. W.; Graham M.; et al. An Integrated Workflow for Cross-Linking/Mass Spectrometry. bioRxiv 2018, 355396. 10.1101/355396. [DOI] [Google Scholar]
- Bieniossek C.; Papai G.; Schaffitzel C.; Garzoni F.; Chaillet M.; Scheer E.; Papadopoulos P.; Tora L.; Schultz P.; Berger I. The Architecture of Human General Transcription Factor TFIID Core Complex. Nature 2013, 493, 699–702. 10.1038/nature11791. [DOI] [PubMed] [Google Scholar]
- Rietschel B.; Arrey T. N.; Meyer B.; Bornemann S.; Schuerken M.; Karas M.; Poetsch A. Elastase Digests: New Ammunition for Shotgun Membrane Proteomics. Mol. Cell. Proteomics 2009, 8, 1029–1043. 10.1074/mcp.M800223-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C. C.; MacCoss M. J.; Howell K. E.; Yates J. R. 3rd A Method for the Comprehensive Proteomic Analysis of Membrane Proteins. Nat. Biotechnol. 2003, 21, 532–538. 10.1038/nbt819. [DOI] [PubMed] [Google Scholar]
- Schlosser A.; Pipkorn R.; Bossemeyer D.; Lehmann W. D. Analysis of Protein Phosphorylation by a Combination of Elastase Digestion and Neutral Loss Tandem Mass Spectrometry. Anal. Chem. 2001, 73, 170–176. 10.1021/ac000826j. [DOI] [PubMed] [Google Scholar]
- Petrotchenko E. V.; Serpa J. J.; Hardie D. B.; Berjanskii M.; Suriyamongkol B. P.; Wishart D. S.; Borchers C. H. Use of Proteinase K Nonspecific Digestion for Selective and Comprehensive Identification of Interpeptide Cross-Links: Application to Prion Proteins. Mol. Cell. Proteomics 2012, 11 (7), M111.013524. 10.1074/mcp.M111.013524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson R. C.; Blout E. R. Dependence of the Kinetic Parameters for Elastase-Catalyzed Amide Hydrolysis on the Length of Peptide Substrates. Biochemistry 1973, 12, 57–65. 10.1021/bi00725a011. [DOI] [PubMed] [Google Scholar]
- Wenzel H. R.; Tschesche H. Cleavage of Peptide-4-Nitroanilide Substrates with Varying Chain Length by Human Leukocyte Elastase. Hoppe-Seyler's Z. Physiol. Chem. 1981, 362, 829–831. 10.1515/bchm2.1981.362.1.829. [DOI] [PubMed] [Google Scholar]
- Berger I.; Fitzgerald D. J.; Richmond T. J. Baculovirus Expression System for Heterologous Multiprotein Complexes. Nat. Biotechnol. 2004, 22 (12), 1583–1587. 10.1038/nbt1036. [DOI] [PubMed] [Google Scholar]
- Fitzgerald D. J.; Berger P.; Schaffitzel C.; Yamada K.; Richmond T. J.; Berger I. Protein Complex Expression by Using Multigene Baculoviral Vectors. Nat. Methods 2006, 3 (12), 1021–1032. 10.1038/nmeth983. [DOI] [PubMed] [Google Scholar]
- Rappsilber J.; Mann M.; Ishihama Y. Protocol for Micro-Purification, Enrichment, Pre-Fractionation and Storage of Peptides for Proteomics Using StageTips. Nat. Protoc. 2007, 2, 1896–1906. 10.1038/nprot.2007.261. [DOI] [PubMed] [Google Scholar]
- Cox J.; Mann M. MaxQuant Enables High Peptide Identification Rates, Individualized p.p.b.-Range Mass Accuracies and Proteome-Wide Protein Quantification. Nat. Biotechnol. 2008, 26 (12), 1367–1372. 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- Cox J.; Neuhauser N.; Michalski A.; Scheltema R. A.; Olsen J. V.; Mann M. Andromeda: A Peptide Search Engine Integrated into the MaxQuant Environment. J. Proteome Res. 2011, 10 (4), 1794–1805. 10.1021/pr101065j. [DOI] [PubMed] [Google Scholar]
- McDowall M. D.; Harris M. A.; Lock A.; Rutherford K.; Staines D. M.; Bähler J.; Kersey P. J.; Oliver S. G.; Wood V. PomBase 2015: Updates to the Fission Yeast Database. Nucleic Acids Res. 2015, 43, D656–D661. 10.1093/nar/gku1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giese S. H.; Fischer L.; Rappsilber J. A Study into the Collision-Induced Dissociation (CID) Behavior of Cross-Linked Peptides. Mol. Cell. Proteomics 2016, 15 (3), 1094–1104. 10.1074/mcp.M115.049296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer L.; Rappsilber J. Quirks of Error Estimation in Cross-Linking/Mass Spectrometry. Anal. Chem. 2017, 89, 3829–3833. 10.1021/acs.analchem.6b03745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkins D. N.; Pappin D. J.; Creasy D. M.; Cottrell J. S. Probability-Based Protein Identification by Searching Sequence Databases Using Mass Spectrometry Data. Electrophoresis 1999, 20 (18), 3551–3567. 10.1002/(SICI)1522-2683(19991201)20:18<3551::AID-ELPS3551>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Schilling B.; Rardin M. J.; MacLean B. X.; Zawadzka A. M.; Frewen B. E.; Cusack M. P.; Sorensen D. J.; Bereman M. S.; Jing E.; Wu C. C.; et al. Platform-Independent and Label-Free Quantitation of Proteomic Data Using MS1 Extracted Ion Chromatograms in Skyline: Application to Protein Acetylation and Phosphorylation. Mol. Cell. Proteomics 2012, 11 (5), 202–214. 10.1074/mcp.M112.017707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B.; Malik R.; Nigg E. A.; Körner R. Evaluation of the Low-Specificity Protease Elastase for Large-Scale Phosphoproteome Analysis. Anal. Chem. 2008, 80, 9526–9533. 10.1021/ac801708p. [DOI] [PubMed] [Google Scholar]
- Larsen M. R.; Højrup P.; Roepstorff P. Characterization of Gel-Separated Glycoproteins Using Two-Step Proteolytic Digestion Combined with Sequential Microcolumns and Mass Spectrometry. Mol. Cell. Proteomics 2005, 4, 107–119. 10.1074/mcp.M400068-MCP200. [DOI] [PubMed] [Google Scholar]
- Wiśniewski J. R.; Mann M. Consecutive Proteolytic Digestion in an Enzyme Reactor Increases Depth of Proteomic and Phosphoproteomic Analysis. Anal. Chem. 2012, 84, 2631–2637. 10.1021/ac300006b. [DOI] [PubMed] [Google Scholar]
- Guo X.; Trudgian D. C.; Lemoff A.; Yadavalli S.; Mirzaei H. Confetti: A Multiprotease Map of the HeLa Proteome for Comprehensive Proteomics. Mol. Cell. Proteomics 2014, 13, 1573–1584. 10.1074/mcp.M113.035170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vizcaíno J. A.; Csordas A.; Del-Toro N.; Dianes J. A.; Griss J.; Lavidas I.; Mayer G.; Perez-Riverol Y.; Reisinger F.; Ternent T.; et al. 2016 Update of the PRIDE Database and Its Related Tools. Nucleic Acids Res. 2016, 44 (D1), D447–D456. 10.1093/nar/gkv1145. [DOI] [PMC free article] [PubMed] [Google Scholar]