

Abstract
Classic Galactosemia is a potentially lethal autosomal recessive metabolic disorder caused by deficient galactose-1-phosphate uridyltransferase (GALT) that results in the buildup of galactose-1-phosphate (gal-1-p) in cells. Galactokinase (GALK1) is the enzyme responsible for converting galactose into gal-1-p. A pharmacological inhibitor of GALK1 is hypothesized to be therapeutic strategy for treating galactosemia by reducing production of gal-1-p. In this study, we report the discovery of novel series of GALK1 inhibitors by structure-based virtual screening (VS). Followed by an extensive structural modeling and binding mode analysis of the active compounds identified from quantitative high-throughput screen (qHTS), we developed an efficient pharmacophore-based VS approach and applied for a large-scale in silico database screening. Out of 230,000 compounds virtually screened, 350 compounds were cherry-picked based on multi-factor prioritization procedure, and 75 representing a diversity of chemotypes exhibited inhibitory activity in GALK1 biochemical assay. Furthermore, a phenylsulfonamide series with excellent in vitro ADME properties was selected for downstream characterization and demonstrated its ability to lower gal-1-p in primary patient fibroblasts. The compounds described herein should provide a starting point for further development of drug candidates for the GALK1 modulation in the Classic Galactosemia.
Keywords: Galactosemia, GALK1, quantitative high-throughput screening, virtual screening, pharmacophore
Introduction
Classic Galactosemia is a potentially lethal disorder with a high mortality rate. It has been shown to cause developmental delay, neurological disorders, and primary ovarian insufficiency without effective therapy [1,2]. Thanks to pre-natal genetic screening, the mortality for classic galactosemia has been strongly reduced by introduction of a galactose-restricted diet [3,4]. However, the dietary treatment proves insufficient in preventing severe long-term complications, such as cognitive, social and reproductive impairments [5]. Although the exact pathogenic mechanism of Classic Galactosemia is not entirely known, elevated galactose-1-phosphate (gal-1-p) levels have been denoted as a probable cause [6]. The high levels of gal-1-p are believed to arise from deficient GALT, which is the second enzyme in the Leloir pathway that converts gal-1-p and uridine diphosphoglucose (UDP-glu) to uridine diphosphogalactose (UDP-gal) and glucose-1-phosphate (glu-1-p) [7]. Immediately upstream from GALT is galactokinase (GALK1), which is the enzyme responsible for converting galactose into gal-1-p. Deficiency in the GALK1 enzyme results in Type II Galactosemia. Although some potential severe outcomes associated to this form of galactosemia have been reported [5,8], these GALK1-deficient patients in general have much milder and even benign phenotypes relative to the Classic Galactosemia [9,10].
GALK1 has recently emerged as a promising drug target for Classic Galactosemia, as its inhibition would prevent production of gal-1-p levels that normally accumulate in patients with non-functional GALT. GALK1 belongs to the GHMP (galactokinase, homoserine kinase, mevalonate kinase and phosphomevalonate kinase) small molecule kinase family, which is characterized by a distinct structure compared to other kinase families [11,12]. All members of the GHMP kinase family share a highly conserved motif of Pro-X-X-X-Gly-Leu-X-Ser-Ser-Ala which is involved in the nucleotide binding and catalytic process [13]. The three-dimensional structure of human GALK1 with bound α-D-galactose and Mg-AMPPNP revealed a unique active site geometry associated with the substrate recognition [11]. A number of site-directed mutations known to give rise to Type II galactosemia have been investigated and provided valuable insights in understanding the GALK1 biology at the molecular level for structure-based drug development [14].
Small molecule inhibitors targeting GALK1 have attracted great interest for therapeutic applications. A few inhibitors of GALK1 have been previously reported [15–17], but none have yet entered clinical trials. Recently, our group carried out a quantitative high-throughput screen (qHTS) using a biochemical GALK1 assay and phenol-HRP redox counter screen (PubChem AID 1868 for qHTS assay, AID 2015 for confirmation assay and AID 2035 for phenol-HRP redox counterscreen) [18]. A series of spiro-benzoxazole compounds were identified as selective inhibitors of GALK1 that are able to lower gal-1-p level in primary patient fibroblast cells. Further medicinal chemistry optimization of the lead compound resulted in a number of potent inhibitors with moderate in vitro ADME and pharmacokinetic properties [18,19].
In this study, we continue our efforts in the discovery of GALK1 inhibitors from a campaign of binding mode analysis and structure-based virtual screening. The diversity of active compounds identified from qHTS, together with a wealth of knowledge on the structure and substrate recognition in GALK1, provided a valuable source for further development of novel and selective inhibitors. To gain insight into the small molecule binding interaction at the active site of GALK1, we first performed a thorough structural scaffold and binding mode analysis of the qHTS hits using MD simulations and ensemble-based docking. The plausible binding hypotheses of lead compounds were optimized and a pharmacophore model was constructed by catching two essential features that are important to the binding activity and selectivity to GALK1. To evaluate the performance of virtual screening, we conducted a retrospective study using the previously screened library of compounds and compared several docking-based VS approaches including DOCK, FRED, AutoDock, and MOE_Dock. Finally, we applied a refined VS protocol for a second-round of virtual screening for novel inhibitors targeting the ATP binding site of GALK1. A number of novel and potent inhibitors were identified with single-digit μM potency and full inhibition against the enzyme. One of the most potent and chemically tractable inhibitors was assessed for its ability to lower gal-1-p in primary patient fibroblasts with no detrimental effect on cell viability. The structure-activity relationship (SAR) of analogs of inhibitor C1 and their in vitro ADME properties were evaluated.
Materials and methods
Structure of GALK1 and MD simulations
The three-dimensional structure of GALK1 was obtained from the Protein Data Bank (PDB code 1WUU) [11]. The structure is complexed with one galactose molecule and an ATP mimetic phosphoaminophosphonic acid-adenylate ester (AMPPNP) molecule. Prior to molecular modeling and docking, the protein structure was prepared using the MOE program [20]. Chain A was selected and the missing residues (Ser230/Leu231) were added to the structure. The nonstandard residue MSE was converted to MET and the ligand AMPPNP was modified to ATP. Finally, the modeled structure was energy-minimized using the QuickPrep module in the MOE program.
MD simulations were conducted for the GALK1 structure in the apo form and in complex with ATP in explicit solvent using the AMBER 14 package [21]. The solvated protein systems were subjected to a thorough energy minimization prior to MD simulations by first minimizing the water molecules while holding the solute frozen (1000 steps using the steepest descent algorithm), followed by 5,000 steps of conjugate gradient minimization of the whole system to remove close contacts and to relax the system. Periodic boundary conditions were applied to simulate a continuous system. The particle mesh Ewald (PME) method was employed to calculate the long-range electrostatic interactions. The simulated system was first subjected to a gradual temperature increase from 0 K to 300 K over 100 ps, and then equilibrated for 500 ps at 300 K, followed by a production run of 10 ns. Clustering analysis of the MD trajectories was performed using the CPPTRAJ module [22]. A total of 10 clusters were generated using the hierarchical clustering from the apo and ATP-bound protein simulations and the representatives of ensemble structures were extracted for the following docking study.
Docking and binding mode analysis
The AutoDock 4.2 program [23] was used for the docking of identified inhibitors to ensembles of the GALK1 structure. The protein active site was defined by a grid of 70 × 70 × 70 points with a grid spacing of 0.375 Å to encompass the ATP binding site and the substrate binding site for an unbiased docking search. The Lamarckian Genetic Algorithm (LGA) was applied with 100 runs and the maximum number of energy evaluations was set to 2 × 106. Binding mode analysis was performed using the AutoDockTool package [23]. To validate the predicted binding mode, a consensus docking was applied for the GALK1 inhibitors using other three docking approaches (AutoDock_Vina [24], MOE_Dock [20], and OE_FRED [25]). Default parameters of these docking programs were used. The top 30 poses from each docking were retained for a consensus binding mode analysis. Finally, the optimal binding models of GALK1 inhibitors were subjected to stepwise energy minimization and MD simulations as described above.
Pharmacophore modeling
Pharmacophore model was generated using the MOE program [20]. The refined binding conformation of inhibitor NCGC00238624 bound at the ATP site of GALK1 was used as a pharmacophore query. The small molecule inhibitor forms a number of hydrogen bonds and extensive hydrophobic interactions in the ATP binding pocket. Two pharmacophoric features were selected as essential features. First is an Aro feature, matching the aromatic ring centroid in the benzoxazole group which forms π-π interaction with Tyr-109. Second is an Acc2 projected hydrogen bond acceptor feature which is placed on the sidechain of guanidine Nitrogen for Arg-105. The pharmacophore model was used as a query with MOE_Dock in virtual screening.
Virtual screening (VS)
Four docking-based VS protocols were evaluated in terms of screening performance by retrieving the active hits from the database screened. The docking approaches included: 1) DOCK 6.8 which uses a geometric matching algorithm and a force-field based scoring dockscore to find the lowest-energy binding pose, followed by energy minimization and re-scoring with AmberScore [26]. 2) OE docking with FRED which uses an exhaustive search algorithm and Chemgauss4 scoring function to measure the complementarity of ligand poses within the active site [25]. 3) The AutoDock-based DOVIS program which is a parallelized VS pipeline using AutoDock as the docking engine and a semi-empirical free energy force field in AutoDock 4.2 [27]. 4) The pharmacophore-based MOE_Dock which uses a pharmacophore query to optionally filtering the poses in docking. The ligand induced fit protocol can be applied with the binding affinity evaluated by the GBVI/WSA score [20].
An in-house library screened by qHTS (274,000 compounds) was used for the retrospective VS study. The 65 confirmed hits from qHTS provided an excellent data set for performance evaluation. Prior to VS, the entire database was filtered to eliminate undesired compounds. The following drug-like filters were applied: 1) MW: 200–600, 2) H-donors < 5, 3) H-acceptors < 10, 4) LogP < 5. A total of 237,890 compounds remained after filtering. The filtered database was systemically docked to the active site of GALK1 using the four docking approaches, respectively. The top 2,500 (1% of entire compounds docked), 5,000 (2%) and 10,000 (5%) compounds were retained from each VS protocol. The enrichment factors were calculated in terms of the efficiency to retrieve the active hits from the entire library among the top-ranked hit list of screening using the following formula:
Where Actsel = the number of active compounds selected by docking at a specific % of database; Nsel = the total number of compounds in the specific % of database evaluated (1%, 2%, 5% in this study); Acttot = the total number of active hits seeded in the database screened (65 in this study); Ntot = the total number of compounds in the database screened (237,890 in this study);
A second round of VS was performed using a refined pharmacophore-based docking protocol. An in-house collection of 230,000 compounds which were not screened by qHTS were used for this in silico screen campaign. The entire database after drug-like filtering (193,784 compounds) was docked to the ATP binding site of GALK1 with the pharmacophore model using the MOE_dock. The top-ranked 5000 compounds was retrieved and energy-minimized, followed by re-scoring using AmberScore. Structural clustering was performed for the top-ranked compounds and the binding models of all clusters and singletons were visually inspected. Finally, a total of 350 compounds were selected manually based on the predicted binding model, ranking score, structural cluster, and availability of the compounds for testing: 1) representatives of each cluster (2–5 analogs selected for clusters with more than 10 compounds), 2) top-ranked singletons from AmberScore, 3) overall fit at the ATP binding site, 4) promiscuous compounds with potential undesirable functionalities and PAINS motif were removed.
GALK1 inhibition assay
Compounds identified from VS were tested in 1536-well plate format at 6 doses (3 nM – 57 μM) using recombinant human GALK1 as described previously (PubChem AID 1868 for qHTS assay, 2015 for confirmation assay) [18]. The KinaseGlo™ detection system was used to measure remaining ATP levels after conversion of galactose to gal-1p and ATP to ADP by GALK1. Biochemical reactions were initiated with ATP (35 μM) and galactose (100 μM) near their KM values determined under the 1536-well assay conditions. The concentration-response data for each sample was plotted and modeled by a four-parameter logistic fit to determine the IC50 and efficacy (maximal response) values. Data normalization and curve fitting were performed using in-house developed informatics tools (http://ncgc.nih.gov/pub/openhts/).
Phenol-HRP redox counterscreen
Two μL/well of GALK1 buffer (20 mM HEPES pH 8.0, 5 mM MgCl2, 60 mM NaCl, 1 mM DTT, 0.01% BSA, final concentration) was dispensed into 1536-well assay plates (Greiner, black clear bottom plates). Compound and control solutions (23 nL each) were transferred by pintool, and plates were incubated at room temperature for 30 minutes. Presence of H2O2 produced by redox cycling compounds was detected by adding 2 μL/well phenol red - HRP detection reagent (100 μL/mL phenol red in 1x Hanks Balanced Salt Solution, 25 U/mL HRP, final concentration). After 15 minutes of room temperature incubation, 2 μL/well 1N NaOH was added to stop the reaction. Plates were then incubated for an additional 1 hour at room temperature. The absorbance at 600 nm was measured using a ViewLux plate reader (6000 light energy, 2 second exposure, and 2x binning). The assay procedure was also described in PubChem AID 2035.
Cellular Gal-1-p accumulation assay
Gal-1-p level was measured using the alkaline phosphatase coupled method previously described [18]. Fibroblasts derived from GALT-deficient patients were maintained in galactose-free DMEM medium supplemented with 10% fetal bovine serum (FBS). Inhibitors were added to the medium at designated concentrations and incubated at 37°C for 2 to 4 hours. Then galactose was added to reach 0.05% in the medium (a ‘galactose challenge’). After 4 h of challenge, cells were collected and washed with PBS twice. Then the cells were disrupted in 300 μL of ice cold hypotonic buffer containing 25 mM Tris-HCl (pH 7.4), 25 mM NaCl, 0.5 mM EDTA, and protease inhibitor cocktail (Roche, # 11 697 498 001). The lysates were passed five times through a 30 gauge needle and centrifuged for 20 min at 16,000 x g and 4 °C. A small portion of supernatant was saved for protein concentration measurement. The gal-1-p concentration was normalized to protein concentration. The assay was analyzed using the paired t test to determine the statistical difference between the compound treated cells and corresponding DMSO control. The two-sided p value less than 0.05 was considered statistically significant.
In vitro ADME assay
The in vitro microsomal stability was measured by incubation of compounds with rat liver microsome at 37 °C in the presence of the cofactor, NADPH. The concentrations of compounds were measured by LC-MS/MS at 0, 5, 15, 30, and 45 min, and half-life (t1/2) was determined by regression analysis. The kinetic solubility of compounds was determined in phosphate buffer at pH 7.4, using μSOL Evolution from pION Inc. (www.pion-inc.com), with a fully automated system of sample preparation, sample analysis, and data processing. The effective permeability of compounds was determined via passive diffusion using the stirring double-sink PAMPA (Parallel Artificial Membrane Permeability Assay) method from pION Inc. (www.pion-inc.com) with a fully automated system of sample preparation, sample analysis, and data processing.
Results and Discussion
Scaffold and binding analysis of GALK1 inhibitors identified from qHTS
We first performed structural clustering and scaffold analysis of the GALK1 inhibitors from qHTS. Among ~274,000 compounds screened, a total of 147 hits were identified in the GALK1 biochemical inhibition assay and 65 hits were confirmed after removing potential artifacts with redox activity in the counter screen [18]. Structural clustering of the 65 confirmed hits revealed a diversity of chemotypes and functionalities among these compounds. As shown in Figure 1, the cluster representatives within the confirmed inhibitors typically contain a heterocyclic aromatic group, which are ATP mimetics that likely resemble ATP binding interaction with GALK1. The spiro-inhibitors in Cluster 5 was selected and thoroughly explored owning to its drug-like property and an appealing scaffold for lead optimization [18]. It is interesting to note that a number of quinol-based compounds were identified in the primary screen. Compounds in cluster 2 have been extensively investigated as selective GALK1 inhibitors in previous studies [15].
Figure 1.

Cluster representatives of GALK1 inhibitors identified from the qHTS screen. The PAINS motifs are highlighted in red.
Docking analysis of these small molecules revealed two distinct binding conformations in the active site of GALK1 (Suppl. S1). As expected, most of the GALK1 inhibitors were predominately docked in the ATP binding pocket by mimicking the adenosine binding interaction. The quinol-based compounds in cluster 2 were found to preferably bind to the galactose substrate site, suggesting that these compounds with multiple hydroxyl groups may resemble the galactose and phosphate binding interactions. Similar predicted binding models of the quinol-based inhibitors have been reported previously [14]. It is worth mentioning that compounds in clusters 1, 4, and 6 contain potential PAINS motifs which indicate they tend to react nonspecifically with numerous biological targets rather than specifically affecting one desired target [28]. Since it is still a matter of debate to triage compounds blindly based on PAINS [29], the predicted binding model of these small molecules at the ATP site provided a plausible support on the inhibition mechanism targeting GALK1.
Structural dynamics of the active site of GALK1
To further gain insight into the binding interactions of the identified inhibitors from qHTS, we performed MD simulations to investigate the structural properties of GALK1. As shown in Figure 2A, the active site of GALK1 is characterized with a long groove surrounded by several flexible loops. The ATP binds to a solvent-exposed region where is enclosed by loops L1 (79–85), L2 (98–105) and L3 (133–140). On the other side, the galactose substrate is inserted deeply into a pocket formed by loop L4 (178–185) and L5 (228–235). L4 (denoted as substrate binding loop) is located on top of the substrate binding pocket, while L1 (denoted as ATP-binding loop) is positioned at the bottom of the ATP-binding pocket. MD simulations showed that these active-site loops exhibited a high level of dynamics in the apo state, but remained stable in the ATP-bound complex (Figure 2B). Similar results of MD stimulations of GALK1 binding complex have been previously reported [30]. The substrate binding loop had the largest conformational changes during the time course of MD simulations, implying a functional role of the loop in the substrate recognition and catalytic process (Figure 2C).
Figure 2.

(A) Structure of human GALK1 (PDB 1wuu). The protein is shown in ribbon representations and key residues are displayed in sticks (carbons in blue). The ligands AMPPNP and galactose are shown in sticks (orange). The magnesium ion is depicted as a magenta sphere. Five loops surrounding the ligand binding site are designated as: L1 (residues 79–85, ATP-binding loop), L2 (residues 98–105), L3 (residues 133–140), L4 (residues 178–185, substrate binding loop), and L5 (228–235). (B) Dynamics of the active site of GALK1 obtained from the MD simulations. Root mean-square deviation (RMSD) of the backbone atoms for the apo and ATP-bound protein with respect to the initial structure over 10 ns of MD simulations. (C) Calculated B-factors of Cα atoms of GALK1 from MD simulations in the apo form (blue) and ATP-bound complex (red).
Ensemble analysis of structural dynamics of GALK1 in the MD simulations revealed two distinct conformations at the ATP site which were largely associated with loops L1 and L4 (Suppl. S2). While an open form was predominately observed in the apo state, a closed conformation of the active site was induced by extensive interactions of the active-site loops with ATP. Notably, two Arg residues Arg-105 and Arg-228 were pointed to the binding pocket and formed H-bonding interactions with ATP (Figure 2A). In addition, the closed conformation was further stabilized by an H-bond interaction between Asp-83 within L1 and the backbone N atom of Arg105 within L2. Such a hydrogen-binding interaction network is not observed in GALK2, an isozyme with 45% sequence identity, and other related kinases such as the CDP-ME kinase (IspE) [31]. Therefore, it is possible that the two Arg residues serve as a gating partner at the active site of GALK1 to facilitate the ATP and substrate binding specificity. Consequently, small molecule inhibitors that are capable of forming interactions with these key residues and stabilizing the closed conformation of the ATP binding loop are expected to be potent and selective to GALK1.
Binding model of Cluster 5 GALK1 inhibitors
We focused on the inhibitors of Cluster 5 to investigate their plausible binding interaction at the active site of GALK1. An ensemble docking approach was applied to account for protein flexibility and induced conformational changes upon inhibitor binding [32]. Three major binding conformations were observed for the small molecule inhibitors bound at the ATP site (mode A), the substrate site (mode B), and the site between ATP and substrate site (Suppl. S3). Binding energy calculations showed that the inhibitor preferably bound in the ATP binding pocket forming extensive binding interactions with the ATP binding loop. To further probe the best binding model, we performed a consensus docking using four docking and scoring approaches including AutoDock, AutoDock_Vina, MOE_Dock and FRED. Analysis of the top-ranked models from each docking program indicated that the inhibitor of Cluster 5 predominately adopted a similar binding conformation by mimicking the ATP binding interactions (Suppl. S4).
Figure 3A and 3B showed the predicted binding model of NCGC00238624, a potent inhibitor in Cluster 5 (IC50 = 4.7 μM) [18]. Two key interactions were identified from this binding model, one is the T-shaped aromatic π-π stacking interactions between the benzoxazole ring and Tyr-109, and the other is the H-bond interaction between the carbonyl oxygen and Arg-105. The chloro-pyrazol substituent pointed to L1 by forming extensive van der Waals and polar interactions, which largely accounted for the stabilization of the dynamical loop in the closed conformation. Consistent with the binding model, NCGC00238624 was proved to be competitive with respect to ATP and uncompetitive to galactose substrate [18]. In addition, mutation of Tyr-109 to Leu, Leu-145 to Tyr, two key residues located in the ATP binding pocket, significantly affected the activity of the inhibitor against GALK1 in our site-directed mutagenesis studies (data not shown). It is interesting to note that a similar binding model of a Cluster 5 inhibitor bound at the ATP binding site has been reported by Chiappori et al [30]. As a comparison, the inhibitor in our model bound more deeply into the pocket by mimicking the extensive binding interactions of adenine motif in the ATP pocket.
Figure 3.

(A) Predicted binding mode of NCGC00238624 at the ATP binding site of GALK1. The inhibitor and key residues are shown in sticks. A hydrogen bond formed between Arg-105 and inhibitor is shown in red dotted line. (B) Chemical structure of NCGC00238624. (C) A pharmacophore model developed from identified GALK1 inhibitor. The ATP binding pocket is shown in surface representations. Inhibitor NCGC00238624 is shown in sticks (magenta). Two pharmacophoric features used in the model are depicted as a ball representation. One is an Aro feature matching the aromatic ring centroid in the benzoxazole group (orange sphere), the other is an Acc2 projected hydrogen bond acceptor feature on the sidechain of Arg-105 (cyan sphere). (D) Performance evaluation of four VS approaches. Enrichment factors (EF) calculated at 1, 2, and 5 percentage levels of screened library consisting of 274,000 compounds and 65 confirmed hits in the primary screening.
Moreover, the predicted binding models of Cluster 5 inhibitors provided a structural basis of observed SAR studies (Suppl. S5). Experimental studies demonstrated that substituents of the phenyl analogs in para- and meta- position had very weak inhibition, whereas the ortho substituted phenyl analogs were preferred for chloro, bromo and CF3 [18]. Analysis of the binding models of these analogs suggested that the para and meta-substitutions likely introduced a steric hindrance with the ATP binding loop L1 and blocked the conformational changes of the active site. On the other hand, substitutions such as CF3 at the ortho position participated in the hydrogen-bond and halogen-bond interaction network with Asp-83 and Arg-105, thus effectively stabilized the protein in the closed, inactive state.
A pharmacophore-based virtual screening approach
Based on the binding model we developed a pharmacophore-based VS approach aimed at identifying novel and selective GALK1 inhibitors. Two pharmacophore features were derived from the predicted binding interaction of inhibitor NCGC00238624 at the ATP binding site (Figure 3C). As aforementioned, one is an aromatic feature (orange sphere) that mimics the adenosine binding interaction in the ATP pocket and aromatic stacking interaction with Tyr-109; the other is a H-bond acceptor feature derived from the H-bond interaction with Arg-105 (cyan sphere), which are crucial to the inhibitory activity and selectivity against GALK1. It is worth mentioning that, while extensive hydrophobic interactions and a number of hydrogen bonds are involved in the Cluster 5 inhibitor binding, only two key features were included in the pharmacophore model for docking and virtual screening. In comparison with the binding interactions of substrate galactose and ATP in the crystal structure of GALK1, a more sensitive 3-feature or 4-feature pharmacophore model may be used to extend the space search to the substrate binding site, however, this significantly reduced the specificity of inhibitors bound at the ATP site (Suppl. S6). It clearly showed that the dominant interaction between GALK1 and ATP are the H-bond matrix formed by ATP triphosphate and the neighboring residues, which is not the focus in designing our novel GALK1 inhibitors. We hypothesized that an efficient and selective small molecule inhibitor of GALK1 binds into the deeper side of ATP binding pocket and interact with L1 in a stabilized conformation.
To evaluate the performance of the pharmacophore-based virtual screening protocol, we compared several docking-based approaches including DOCK, FRED, AutoDock, and MOE Dock. The previously screened library of ~274,000 compounds and identified hits provided an excellent testing case for this retrospective VS evaluation. The results of calculated EFs at 1%, 2% and 5% levels were shown in Figure 3D. The EFs were ranged from 1.5 to 10 at three levels, which are generally similar to the performance of the docking programs as evaluated in other studies [33]. However, considering the challenging target of GALK1 and the extremely low HTS hit rate in this case (0.05%), an enrichment factor up to 10 obtained in this study is remarkable. The pharmacophore-based approach with MOE docking showed the best performance with 10.2 of EF2%, while FRED, which is the fastest docking method among four, had the lowest enrichments at the three levels compared to others. DOCK performed well in this case, indicating that a post-docking process with energy minimization and re-scoring using AmberScore is a robust approach to improve the screening efficiency [34].
Further examination revealed that most of the hits that were successfully predicted from various different VS approaches had a docking pose at the ATP binding site. This is consistent with the binding mode analysis that the most potent and effective GALK1 inhibitors typically mimick the ATP binding interactions. This likely explained the superior performance of the pharmacophore-based approach as compared with others. While it is not unexpected that the validated hits in Cluster 5 were predicted from the pharmacophore docking, other structurally diverse inhibitors were also ranked on top of the hit list, suggesting that the two pharmacophores used in VS is efficient to catch the key interactions at the active site and identify potential novel inhibitors of GALK1.
Novel GALK1 inhibitors identified from virtual screening
As a complementary effort of qHTS, we carried out a second-round of virtual screening using an optimized pharmacophore-based protocol to search for novel inhibitors of GALK1 (Figure 4). An in-house collection of 230,000 compounds which were not previously screened by qHTS were used for this in silico screen campaign. To prioritize the hits from VS, several post-processing approaches were applied including structural clustering, re-scoring, binding model analysis by detailed visual inspection, and promiscuity filtering (see details in Methods section). A total of 350 compounds were cherry-picked and tested in the biochemical GALK1 inhibition assay.
Figure 4.

Flowchart of VS for identification of novel inhibitors of GALK1 using a pharmacophore-based docking approach.
The results showed that, out of 350 compounds tested, 75 exhibited inhibitory activity on GALK1, which corresponded to a virtual screening hit confirmation rate of 21.4%. These active compounds represented a diversity of chemotypes as compared to the hits from the biochemical qHTS. The representatives from each chemotype were shown in Figure 5. Remarkably, several compounds had single-digit micromolar potency and full inhibition. Inhibitor C1 exhibited an IC50 of 3.5 μM in the GALK1 assay and no promiscuous activity was observed in the redox cycling counter screen (Figure 6A). The purity of compound was further validated by MS and1H-NMR (Suppl. S7). Interestingly, C1 was identified as an inhibitor of ubiquitin specific peptidase 2 (USP2), a target that has no structural similarity to GALK1 (PubChem assay AID 2281). Examination of the predicted binding model of C1 showed that the small molecule binds to the ATP binding site of GALK1 in a similar manner as the spiro-benzoxazole compounds from the qHTS (Figure 6B). The phenylthiazole group of C1 was inserted into the pocket to mimic the ATP binding interactions, while the sulfonamide group was pointed outward and formed an H-bonding interaction with Arg-105. The same H-bond interaction was observed within most active compounds from VS, reiterating that this gating residue serves as a key in inhibitor binding.
Figure 5.

Identified novel GALK1 inhibitors from VS. The dose-response data of GALK1 inhibition assay is shown.
Figure 6.

(A) Dose-response data of C1 in GALK1 inhibition assay and redox cycling counter screen. (B) Predicted binding model of inhibitor C1 at the ATP binding site of GALK1. C1 and key residues are shown in sticks. The binding interactions between inhibitor and residues in the ATP binding site are displayed in dotted line.
SAR analysis of C1 derivatives
The phenylsulfonamide-based structure of C1 represents a novel and interesting scaffold of GALK1 inhibitor for follow up characterization. To further explore its binding specificity and structure activity relationship (SAR), a number of analog compounds were re-selected from the screening library and their activities were evaluated in the GALK1 inhibition assay. As shown in Table 1, replacement of the central benzene ring with pyridine (C2), or substituents with halogen atoms (C3–C4) resulted in reduced potency. Modifications on the phenylthiazole ring such as methyl group at different positions decreased the potency and maximal response (C5–C6). In addition, the para-methyl group on the sulfonamide-phenyl ring was preferred since ortho-methyl substitution (C7), or other substitutions in the para-position (C8–C10) resulted in decreased potency.
Table 1.
IC50, maximal response and in vitro ADME properties of C1 and C1 analogs.
| Compounds | Structure | IC50a (μM) |
MR (%) |
Microsomalc T1/2 (m) | Permeabilityd (1e-6 cm/s) | Solubilitye (Ug/ml) |
|---|---|---|---|---|---|---|
| C1 (NCGC00247843) | 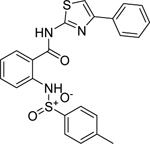 |
3.5 | 100 | 2.5 | 1099.1 | < 1.0 |
| C2 (NCGC00262386) | 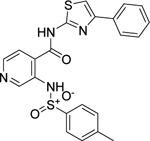 |
14.0 | 83.5 | 17.0 | 724.3 | 9.4 |
| C3 (NCGC00262995) | 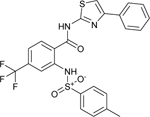 |
15.9 | 92.9 | 15.0 | 82.2 | < 1.0 |
| C4 (NCGC00263701) | 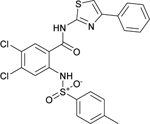 |
22.2 | 89.3 | 13.0 | < 5.0 | <1.0 |
| C5 (NCGC00262417) | 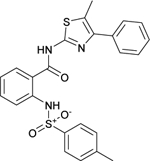 |
11.1 | 75.9 | 6.3 | 30.1 | < 1.0 |
| C6 (NCGC00262418) | 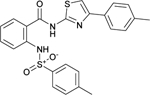 |
14.0 | 81.0 | 2.6 | 35.6 | < 1.0 |
| C7 (NCGC00344941) | 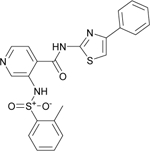 |
11.1 | 90.3 | 14.0 | 1064.7 | 50.0 |
| C8 (NCGC00344944) | 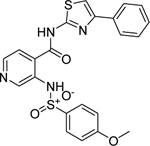 |
15.7 | 84.5 | 1.6 | 674.5 | 54.1 |
| C9 (NCGC00344912) | 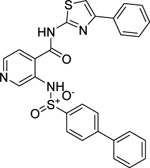 |
17.7 | 88.5 | 2.0 | < 1.0 | < 1.0 |
| C10 (NCGC00345030) | 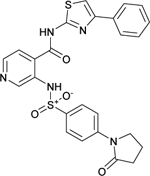 |
44.5 | 62.5 | 1.6 | 313.5 | 57.0 |
IC50 values were determined utilizing the luminescence GALK1-ATP-depletion assay.
MR represents the maximal % inhibition at 57 μM compound.
Rat liver microsome stability.
PAMPA permeability assay performed in-house at NCATS.
Kinetic aqueous solubility in PBS 7.4 buffer from a 10 mM DMSO compound stock solution.
Binding model analysis indicated that these derivatives adopt the same binding conformation in the ATP binding site of GALK1 as C1. Since the phenylthiazole ring was pointed deeply into a rather restricted pocket of the adenosine binding site, substituents within the pocket were generally not favored due to potential steric clashes with residues Thr-61, Thr-77, and Val-129. The effects of modifications on the core of benzamide are not obvious from the predicted binding model. It is possible that substituents in para- and meta-positions of benzamide ring (C3 and C4) impaired the conformation of sulfonamide group, thus disrupting the H-bond with Arg-105. Binding interactions of the sulfonamide-phenyl group appeared to be more crucial in stabilizing L1 activation through Arg-105 and Asp-83 salt bridge. Different from the spiro-benzoxazole inhibitors, substituents on the phenyl ring of these analogs were pointed towards a solvent-exposed region adjacent to the ATP binding loop L1. Bulky or polar substituents in this region such as C9 and C10 likely posed a steric hindrance to the conformational changes of L1, thus are expected to be unfavorable. On the other hand, as discussed in the spiro-benzoxazole series, further optimization of binding interactions at this site to stabilize L1 in the closed conformation could significantly improve the binding affinities and potency.
Cell-based Validation and ADME properties of inhibitor C1
Finally, we evaluated the activity of inhibitor C1 to lower gal-1-p level in primary patient fibroblast cells. The cells were pretreated with compounds at 2.8 μM, 8.3 μM and 25 μM concentrations, followed by galactose challenge for 4 hours. The ability of compounds in reducing the gal-1-p level was measured and compared to DMSO treated cells. As shown in Figure 7A, DMSO treated cells resulted in 100% activity of13C labeled gal-1-p after data normalization. Upon galactose challenge, C1 produced a dose-response reduction below the galactose challenge level in the patient cells, with over 30% reduction of [13C] gal-1-p at the highest testing concentration. In addition, C1 displayed no significant effect on cell viability of the patient fibroblasts as determined by CellTiter-Glo assay (Figure 7B). The results validated the on-target activity of this novel GALK1 inhibitor.
Figure 7.

(A) Gal-1-p levels measured in primary galactosemic patient fibroblasts after either DMSO or compound treatment regimes. Compound was pre-incubated with the cells for 24 hours. * p value < 0.05 but ≥ 0.01, ** p value < 0.01. (B) Cell viability after designated treatment in primary galactosemic patient fibroblasts.
The in vitro ADME properties of inhibitor C1 and analogs were also evaluated (Table 1). The compounds have relatively good half-life (t1/2) in rat liver microsome and PAMPA permeability. The aqueous solubility of C1 is relatively poor, but had significant improvement with substitution of pyridine (C7 and C8). Further optimization of these small molecule inhibitors may offer promising candidate for the development of therapeutics against the Classic Galactosemia.
Conclusion
High-throughput screening and virtual screening are two well-established approaches that are commonly used in small molecule discovery applications. In this study, we explored the diverse hits identified from HTS in order to gain insight into the small molecule binding and inhibition mechanism against GALK1. With a pharmacophore-based VS approach developed from the binding model analysis, we carried out a second-round of virtual screening to identify novel and selective small molecules targeting the ATP binding site of GALK1. The retrospective VS evaluation utilizing the qHTS data highlighted a strategic approach to integrate HTS and VS in designing novel and selective small molecule inhibitors of a protein target. Our work demonstrated that, guided by a stepwise ligand- and structure-based modeling and pharmacophore analysis, virtual screening is an efficient and complementary approach to the HTS for drug discovery.
Supplementary Material
Acknowledgments
We thank Danielle van Leer, Misha Itkin, Crystal McKnight, Christopher LeClair and Paul Shinn for assistance with compound management. We also thank Pranav Shah and Amy Wang for assistance in in vitro ADME assays. This research was supported by the Molecular Libraries Initiative of the National Institutes of Health Roadmap for Medical Research Grant U54MH084681 and the Intramural Research Program of the National Center for Advancing Translational Sciences at the National Institutes of Health. Research grant support to K.L. include R03MH085689 and R01HD074844–2.
References
- 1.Ridel KR, Leslie ND, Gilbert DL (2005) Pediatric neurology 33(3):153. [DOI] [PubMed] [Google Scholar]
- 2.Berry GT. Classic Galactosemia and Clinical Variant Galactosemia. In: Adam MP, Ardinger HH, Pagon RA, et al. (eds). GeneReviews(R) Seattle (WA), 1993 [PubMed] [Google Scholar]
- 3.Schweitzer-Krantz S (2003) European journal of pediatrics 162 Suppl 1:S50. [DOI] [PubMed] [Google Scholar]
- 4.Porta F, Pagliardini S, Pagliardini V, Ponzone A, Spada M (2015) World journal of pediatrics : WJP 11(2):160. [DOI] [PubMed] [Google Scholar]
- 5.Stroek K, Bouva MJ, Schielen P, Vaz FM, Heijboer AC, de Jonge R, Boelen A, Bosch AM (2018) Molecular genetics and metabolism 124(1):50. [DOI] [PubMed] [Google Scholar]
- 6.Gitzelmann R (1995) European journal of pediatrics 154(7 Suppl 2):S45. [DOI] [PubMed] [Google Scholar]
- 7.Lai K, Langley SD, Khwaja FW, Schmitt EW, Elsas LJ (2003) Glycobiology 13(4):285. [DOI] [PubMed] [Google Scholar]
- 8.Hennermann JB, Schadewaldt P, Vetter B, Shin YS, Monch E, Klein J (2011) Journal of inherited metabolic disease 34(2):399. [DOI] [PubMed] [Google Scholar]
- 9.Lai K, Elsas LJ (2000) Biochemical and biophysical research communications 271(2):392. [DOI] [PubMed] [Google Scholar]
- 10.Holden HM, Thoden JB, Timson DJ, Reece RJ (2004) Cellular and molecular life sciences : CMLS 61(19–20):2471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thoden JB, Timson DJ, Reece RJ, Holden HM (2005) The Journal of biological chemistry 280(10):9662. [DOI] [PubMed] [Google Scholar]
- 12.Timson DJ (2016) Gene 589(2):133. [DOI] [PubMed] [Google Scholar]
- 13.Zhou T, Daugherty M, Grishin NV, Osterman AL, Zhang H (2000) Structure 8(12):1247. [DOI] [PubMed] [Google Scholar]
- 14.Tang M, Wierenga K, Elsas LJ, Lai K (2010) Chemico-biological interactions 188(3):376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lai K, Boxer MB, Marabotti A (2014) Future medicinal chemistry 6(9):1003. [DOI] [PubMed] [Google Scholar]
- 16.Wierenga KJ, Lai K, Buchwald P, Tang M (2008) Journal of biomolecular screening 13(5):415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Odejinmi S, Rascon R, Tang M, Vankayalapati H, Lai K (2011) ACS medicinal chemistry letters 2(9):667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu L, Tang M, Walsh MJ, Brimacombe KR, Pragani R, Tanega C, Rohde JM, Baker HL, Fernandez E, Blackman B, Bougie JM, Leister WH, Auld DS, Shen M, Lai K, Boxer MB (2015) Bioorganic & medicinal chemistry letters 25(3):721. [DOI] [PubMed] [Google Scholar]
- 19.Tang M, Odejinmi SI, Vankayalapati H, Wierenga KJ, Lai K (2012) Molecular genetics and metabolism 105(1):44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Molecular Operating Environment (MOE) (ed). Chemical Computing Group ULC, 1010 Sherbooke St. West, Suite #910, Montreal, QC, Canada, H3A 2R7; 2018 [Google Scholar]
- 21.Kollman PA, Massova I, Reyes C, Kuhn B, Huo S, Chong L, Lee M, Lee T, Duan Y, Wang W, Donini O, Cieplak P, Srinivasan J, Case DA, Cheatham TE 3rd (2000) Accounts of chemical research 33(12):889. [DOI] [PubMed] [Google Scholar]
- 22.Roe DR, Cheatham TE, 3rd (2013) Journal of chemical theory and computation 9(7):3084. [DOI] [PubMed] [Google Scholar]
- 23.Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ (2009) J Comput Chem 30(16):2785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Trott O, Olson AJ (2010) Journal of computational chemistry 31(2):455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McGann M (2011) Journal of chemical information and modeling 51(3):578. [DOI] [PubMed] [Google Scholar]
- 26.Lang PT, Brozell SR, Mukherjee S, Pettersen EF, Meng EC, Thomas V, Rizzo RC, Case DA, James TL, Kuntz ID (2009) RNA 15(6):1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jiang X, Kumar K, Hu X, Wallqvist A, Reifman J (2008) Chemistry Central journal 2:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Baell JB, Holloway GA (2010) Journal of medicinal chemistry 53(7):2719. [DOI] [PubMed] [Google Scholar]
- 29.Capuzzi SJ, Muratov EN, Tropsha A (2017) Journal of chemical information and modeling 57(3):417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chiappori F, Merelli I, Milanesi L, Marabotti A (2013) European journal of medicinal chemistry 63:423. [DOI] [PubMed] [Google Scholar]
- 31.Bork P, Sander C, Valencia A (1993) Protein science : a publication of the Protein Society 2(1):31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hu X, Compton JR, Abdulhameed MD, Marchand CL, Robertson KL, Leary DH, Jadhav A, Hershfield JR, Wallqvist A, Friedlander AM, Legler PM (2013) Journal of medicinal chemistry 56(13):5275. [DOI] [PubMed] [Google Scholar]
- 33.Warren GL, Andrews CW, Capelli AM, Clarke B, LaLonde J, Lambert MH, Lindvall M, Nevins N, Semus SF, Senger S, Tedesco G, Wall ID, Woolven JM, Peishoff CE, Head MS (2006) Journal of medicinal chemistry 49(20):5912. [DOI] [PubMed] [Google Scholar]
- 34.Case DA, Darden T, Cheatham III TE, Simmerling CL, Wang J, Duke RE, Luo R, Merz KM, Wang B, Pearlman DA, Crowley M, Brozell S, Tsui V, Gohlke H, Mongan J, Hornak V, Cui G, Beroza P, Schafmeister C, Caldwell JW, Ross WS, Kollman PA (2012) AMBER 12. University of California, San Francisco, [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.