

Abstract
Osteocytes are the most prevalent cell in the skeleton and are the master regulator of bone remodeling. Despite the understanding that osteocytes have a multiyear lifespan, and some factors induce apoptosis in osteocytes, much less is understood about the induction and consequences of osteocyte senescence. Filling these gaps in knowledge will provide novel approaches to slowing age-related bone loss and preventing fragility fractures. The purpose of this review is to examine the roles of senescence and apoptosis in osteocytes in age-related bone loss. Based on evidence that exercise can prevent senescence in skeletal muscle, we provide a novel hypothesis by which exercise can prolong skeletal health.
Keywords: Osteocyte, senescence, mitochondria, fat oxidation, mechanical loading
Introduction
Preventing bone loss with aging is of considerable public health interest because of the cost, morbidity and mortality associated with osteoporosis and fragility fractures [1–4]. Osteocytes are the primary mechanosensor of the skeleton and the master regulator of bone remodeling [5]. As such, the uncoupling of bone resorption and bone formation that leads to age-related bone loss can often be traced back to changes in osteocyte number or function. There has been a resurgence of investigations into cellular metabolism of osteoblasts, osteoclasts, and osteocytes, and their contributing roles in integrated physiology and whole body energy balance. The increased appreciation that osteocytes are metabolically active creates a need to reexamine how osteocytes adapt, senesce, or die in response to different metabolic environments across its multi-year lifespan [6–9]. By applying what has been gleaned about senescence in other cell types, reexamining the lifespan of osteocytes could yield paradigm shifts in our understanding of age-related bone loss. In muscle, there is a renewed focus on the role of age-related changes in mitochondrial number and function on the induction of senescence. Age-related changes in mitochondrial function that occur in muscle and could occur in osteocytes include loss of mitophagy, changes in fission and fusion, increased mitochondrial uncoupling and superoxide production [10–12]. Within this context, physical activity and exercise programs are often utilized as non-pharmacologic therapeutics for preserving mitochondrial number and function and preventing senescence. However, the potential consequences of senescence and mitochondrial dysfunction with aging is underappreciated in osteocytes. The purpose of this review is to examine the roles of senescence in osteocytes in age-related bone loss, and provide novel hypotheses by which exercise can prolong skeletal health.
The notion of senescence in osteocytes
Osteocytes arise from terminally differentiated osteoblasts that become imbedded in the bone matrix over time. More than 90% of the cells in bone are osteocytes, so understanding their life cycle is essential. Seminal work from the Bonewald lab over two decades has defined most of what we know about the origin and function of these cells [5]. And because osteocytes are in communication with cells on the bone surface, and are bathed in extracellular fluid, much has been made of their ‘command and control’ function within the remodeling unit. But those tasks require osteocyte survival. Most studies on osteocyte viability have focused on inducing or preventing apoptosis, which in osteocytes, is an event that promotes bone loss through RANKL activation and sclerostin up-regulation. Senescence, on the other hand, is a cell fate program that is part of a DNA Damage Response (DDR) to prevent replication of maladaptive DNA mutation [13]. Senescence occurs throughout the lifespan, but senescent cells particularly accumulate with aging in other tissues. These death resistant cells can cause significant damage in surrounding tissues through release of various cytokines. Indeed, senescent cells are detected and defined based on the presence of that pro-inflammatory secretome, often referred to as senescence-associated secretory phenotype (SASP). Accordingly, senescent vs non-senescent cells can be independently identified based on the secretion of SASP-related factors such as p53, p21, and several interleukins [14–16].
Osteocytes have been traditionally considered ‘old’ osteoblasts buried within the bone matrix; hence the notion that there is a significant population of senescent osteocytes driving bone loss through SASP bone loss is appealing. Furthermore, it is tempting to assume since cell cycle arrest is part of senescence, and osteocytes don’t replicate that a large proportion of osteocytes are senescent. Moreover, reducing the number of senescent cells has been shown to be beneficial in several age-related disorders. This can be accomplished through administration of senolytics, a class of small molecules that can selectively induce death of senescent cells.
Osteocyte senescence and mechanical loading
With the introduction of senolytics, there are clear benefits to elucidating the mechanisms that induce or prevent senescence in osteocytes. This is reinforced by recent evidence indicating that senolytics could be effective for slowing bone loss [17]. There is overlap in age- or damage-related factors that can induce senescence and apoptosis, including the activation of tumor suppressor p53 through oxidative stress, gamma radiation, loss mitochondrial membrane integrity and accompanying release of cytochrome c, and nuclear or mitochondrial DNA degradation. p21, a kinase inhibitor and a major target of p53, induces cell cycle arrest leading to senescence [18, 19]. It is unclear whether senescence in osteocytes results from a development of resistance to apoptosis [20]. Apoptosis of osteocytes can be induced by both unloading and damage-inducing loading [21]. Because senescence is part of DDR, it is conceivable, based on the mechanosensing of osteocytes that damage-inducing loading can induce senescence in osteocytes.
The effect of age-related reductions in the loading environment on osteocyte senescence has not yet been tested. Aging produces changes in mechanical stimuli and the response to stimuli at a tissue and cellular level [16, 22]. Specifically, decreases in physical activity and increases in sedentary time that are common with aging engender a degree of skeletal unloading [23, 24]. Aging and physical inactivity are each associated with a decrease in lacunae that contain osteocytes, loss of directional orientation of lacunae, and the number and length of dendrites that connect through canniculi; all of which would interfere with the detection of strain or fluid-flow shear stress [25–28]. Connexin43, a gap junction protein involved in mechanotransduction, protects from osteocyte apoptosis, but also decreases with aging [29–32]. Alternatively, unloading could induce senescence indirectly through the effect of inactivity on reductions in myokines, adverse changes in whole body nutrient trafficking, and the resulting lipid accumulation in both osteoblasts and osteocytes. Further research will be needed to distinguish the role of (un)loading on senescence.
Regardless of whether unloading directly induces senescence in osteocytes, the loss of mechano-sensitivity, the resulting uncoupling of bone remodeling, and the generation of SASP may be the biggest negative consequences of presumed osteocyte senescence. If SASP promotes further senescence or apoptosis of osteocytes through a prolonged secretory phenotype as it does in other cell types, then apoptosis would be preferable because apoptotic osteocytes are providing a one-time stimulus prior to ingestion by phagocytes. The increased burden of senescent cells would then likely promote additional bone resorption and inhibition of bone formation through the secretion of pro-inflammatory cytokines. The loss of mechanically sensitive osteocytes could result in accelerated bone loss.
Targeting osteocyte senescence with exercise to slow age-related bone loss
Exercise is often recommended for maintaining and improving bone health, in part, because of the demonstrated increases in bone formation rate and bone strength in response to mechanical loading, although gain in bone mass is less apparent [33]. From a mechanocentric view, exercise prescriptions for bone health favor activities that provide higher intensity or faster rates of loading (e.g., resistance training, jumping) to induce important stimuli for existing osteocytes and promote the differentiation of MSCs into the osteoblast lineage, ultimately resulting in more osteocytes. Endurance or aerobic exercise typically provides a smaller-magnitude and rate of loading, and acutely stimulates bone resorption [34]. However, because serum calcium drops precipitously during intense exercise, and PTH rises to compensate acutely, it is likely that osteocytic osteolysis is a major driver of that physiologic compensation [35]. A readily available pool of viable osteocytes that could induce skeletal osteolysis would thus seem to be essential. As noted, aerobic exercise is not often highlighted as an important part of promoting bone density or bone strength. However, aerobic exercise could provide benefits to osteocyte viability in ways that are independent from mechanical loading, including the release of exercise-stimulated myokines, altering of macronutrient trafficking, and preservation of cellular or mitochondrial repair. Unloading, on the other hand, might lead to greater senescence because of the lack of stimulus for promoting viability (Figure 1). Certainly spaceflight studies with rodents could help identify determine if osteocyte senescence is a major component of enhanced bone resorption, particularly since an inflammatory profile is noted during unloading.
Figure 1.
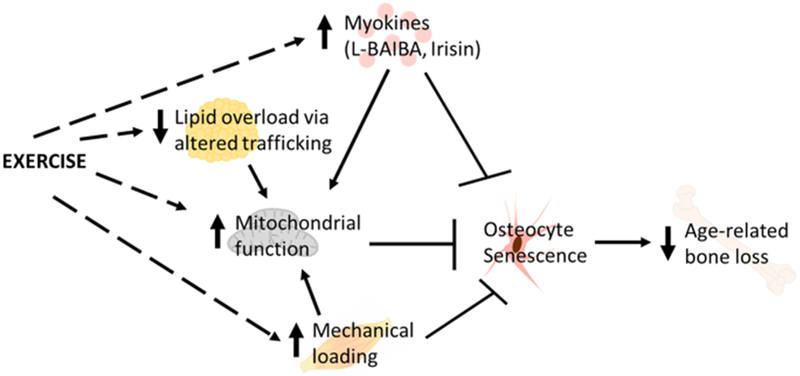
Model of how exercise could slow age-related bone loss by preventing the accumulation of senescent osteocytes. Exercise could prevent osteocytes senescence by inducing the release of myokines, altering nutrient trafficking to prevent lipid overload in bone, providing mechanical loading, and directly or indirectly maintaining mitochondrial number and/or function in osteocytes.
Muscle-bone units have been studied for over 40 years as mechanical units, but in the past few years has evolved into focusing on the biological cross-talk through myokines and osteokines. Several candidate myokines have the potential to influence aspects of bone turnover, as recently reviewed by Bonewald [36]. β-aminoisobutyric acid (BAIBA) and irisin are among the exercise-induced myokines with the potential for directly slowing age-related osteocyte senescence by protecting mitochondrial integrity [37, 38]. Irisin is a peptide cleaved from Fndc5, a muscle surface protein. It is found in the circulation in nanogram concentrations (i.e. in mice and humans) and its levels are increased during and following exercise. It was first noted to be an inducer of thermogenic programs in white adipose tissue. However recent work has shown that irisin can induce sclerostin in osteocytes, as well as RANKL[38]. The receptor for irisin an integrin, alphaV beta 5, was recently discovered and characterized in osteocytes. Moreover, irisin prevents apoptosis of IDG-SW3 osteocytes [38]. As such it appears the major skeletal target for irisin is the osteocyte, although preliminary evidence from our lab suggests irisin may have a direct effect on osteoclasts. Whether irisin prevents osteocyte senescence is unknown. However tantalizing preliminary evidence suggests irisin may have a neuroprotective role.
Fat oxidation and osteocyte viability
Aerobic exercise improves fat oxidation and redirects nutrient trafficking through more energetically expensive pathways [39, 40]. This could translate into preventing excess lipid trafficking to osteocytes, but studies are needed to directly test this hypothesis. Accumulation versus oxidation of lipid is dependent on mitochondrial capacity. Mitochondria are active in MSCs, particularly during early differentiation, osteoclasts, and osteocytes, and both glycolysis and oxidative phosphorylation contribute to the synthesis of ATP [6, 41]. A critical gap in knowledge is whether aerobic or resistance exercise has direct or indirect effects on mitochondrial number, fission, fusion or capacity in osteocytes. If so, exercise could target senescence in osteocytes through the preservation of mitochondrial content, coupling, and capacity, through preservation of biogenesis, mitophagy, fusion, and fission capabilities [42–45]. In muscle, exercise induces mitochondrial biogenesis to a lesser extent with aging, but is still effective in stimulating increases in mitochondrial enzymes and oxidative capacity [10–12].
Further, lifelong physical activity slows the age-related decline in mitochondrial number and capacity [46, 47]. There is less oxidative damage to muscle mitochondrial membranes in response to acute exercise and improved antioxidant capacity with aging in those who chronically exercise [48–50]. There are initial clues to suggest that osteocyte mitochondrial activity is responsive to exercise and/or aging. For instance, when applying mechanical loading in vivo, osteocyte calcium responses to the load, which were used as an indicator or osteocyte recruitment, were related to applied strain magnitude and frequency [51]. Recently, in situ imaging was used to demonstrate that mitochondrial activity is higher in osteocytes close to the periosteal surface and decreases as cells become closer to the endocortical surface. Osteocytes closest to the endocortical surface had greater numbers of nonfunctional mitochondria. This seems counterintuitive to the notion of marrow fat being a source of lipid during periods of fasting,[41] but this location-based variance in mitochondrial content may indirectly reflect either the effect of aging or adaptations to local energetic needs. Genetic deletion of the growth hormone receptor leads to longer lifespan in mice, but we recently showed that mitochondrial function in osteocytes was reduced and ROS was increased [52]. These data would imply that aging itself may result in either damaged osteocytes or senescent osteocytes characterized by impaired mitochondrial dynamics. Whether exercise can prevent those changes remain to be determined.
Summary
Understanding the induction and removal of senescent cells in bone is an important step forward in discovering new targets for slowing age-related bone loss. Much of what is currently hypothesized about senescence in osteocytes, or the potential for exercise to prevent senescence in osteocytes, is derived from decades of experiments in skeletal muscle. Exercise that involve high strains or rates of strain are highly touted for maintaining bone health and reducing fracture risk across the lifespan. However, more work is needed to understand the potential metabolic and anti-senescent benefits of endurance/aerobic exercise or reductions in sedentary behavior on osteocyte function. Further, it is known that mitochondria are important for energy production in all bone cells, and are intricately linked to senescence in other cell types. Determining the osteocytic mitochondrial adaptations to exercise should help bridge the metabolic and mechanical factors to promote bone health throughout the lifespan.
Highlights.
Osteocyte apoptosis and senescence independently contribute to age-related bone loss.
Exercise may prevent osteocyte senescence by preserving mitochondrial function.
Endurance and resistance exercise may have distinct benefits for osteocyte viability.
Acknowledgements
Funding This work was supported by KL2TR002534 (VDS) and DK092759 (CJR)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Cauley JA, Public health impact of osteoporosis, J Gerontol A Biol Sci Med Sci 68(10) (2013) 1243–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Cauley JA, Burden of hip fracture on disability, Lancet Public Health 2(5) (2017) e209–e210. [DOI] [PubMed] [Google Scholar]
- [3].El-Hajj Fuleihan G, Chakhtoura M, Cauley JA, Chamoun N, Worldwide Fracture Prediction. J Clin Densitom 20(3) (2017) 397–424. [DOI] [PubMed] [Google Scholar]
- [4].Cauley JA, Osteoporosis: fracture epidemiology update 2016, Curr Opin Rheumatol 29(2) (2017) 150–156. [DOI] [PubMed] [Google Scholar]
- [5].Bonewald LF, The amazing osteocyte, J Bone Miner Res 26(2) (2011) 229–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Guntur AR, Le PT, Farber CR, Rosen CJ, Bioenergetics during calvarial osteoblast differentiation reflect strain differences in bone mass, Endocrinology (2014) en20131974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Jansson JO, Palsdottir V, Hagg DA, Schele E, Dickson SL, Anesten F, Bake T, Montelius M, Bellman J, Johansson ME, Cone RD, Drucker DJ, Wu J, Aleksic B, Tornqvist AE, Sjogren K, Gustafsson JA, Windahl SH, Ohlsson C, Body weight homeostat that regulates fat mass independently of leptin in rats and mice, Proc. Natl. Acad. Sci. U. S. A 115(2) (2018) 427–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Lacombe J, Karsenty G, Ferron M, In vivo analysis of the contribution of bone resorption to the control of glucose metabolism in mice, Mol Metab 2(4) (2013) 498–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Kobayashi K, Nojiri H, Saita Y, Morikawa D, Ozawa Y, Watanabe K, Koike M, Asou Y, Shirasawa T, Yokote K, Kaneko K, Shimizu T, Mitochondrial superoxide in osteocytes perturbs canalicular networks in the setting of age-related osteoporosis, Sci Rep 5 (2015) 9148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Figueiredo PA, Powers SK, Ferreira RM, Amado F, Appell HJ, Duarte JA, Impact of lifelong sedentary behavior on mitochondrial function of mice skeletal muscle, J Gerontol A Biol Sci Med Sci 64(9) (2009) 927–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Figueiredo PA, Powers SK, Ferreira RM, Appell HJ, Duarte JA, Aging impairs skeletal muscle mitochondrial bioenergetic function, J Gerontol A Biol Sci Med Sci 64(1) (2009) 21–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Kang C, Chung E, Diffee G, Ji LL, Exercise training attenuates aging-associated mitochondrial dysfunction in rat skeletal muscle: role of PGC-1alpha, Exp Gerontol 48(11) (2013)1343–50. [DOI] [PubMed] [Google Scholar]
- [13].d’Adda di Fagagna F, Reaper PM, Clay-Farrace L, Fiegler H, Carr P, Von Zglinicki T, Saretzki G, Carter NP, Jackson SP, A DNA damage checkpoint response in telomere-initiated senescence, Nature 426(6963) (2003) 194–8. [DOI] [PubMed] [Google Scholar]
- [14].Farr JN, Fraser DG, Wang H, Jaehn K, Ogrodnik MB, Weivoda MM, Drake MT, Tchkonia T, LeBrasseur NK, Kirkland JL, Bonewald LF, Pignolo RJ, Monroe DG, Khosla S, Identification of Senescent Cells in the Bone Microenvironment, J Bone Miner Res 31(11) (2016) 1920–1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Piemontese M, Almeida M, Robling AG, Kim HN, Xiong J, Thostenson JD, Weinstein RS, Manolagas SC, O’Brien CA, Jilka RL, Old age causes de novo intracortical bone remodeling and porosity in mice, JCI Insight 2(17) (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Galea GL, Meakin LB, Harris MA, Delisser PJ, Lanyon LE, Harris SE, Price JS, Old age and the associated impairment of bones’ adaptation to loading are associated with transcriptomic changes in cellular metabolism, cell-matrix interactions and the cell cycle, Gene 599 (2017) 36–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Farr JN, Xu M, Weivoda MM, Monroe DG, Fraser DG, Onken JL, Negley BA, Sfeir JG, Ogrodnik MB, Hachfeld CM, LeBrasseur NK, Drake MT, Pignolo RJ, Pirtskhalava T, Tchkonia T, Oursler MJ, Kirkland JL, Khosla S, Targeting cellular senescence prevents age-related bone loss in mice, Nat Med 23(9) (2017) 1072–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Demidenko ZN, Korotchkina LG, Gudkov AV, Blagosklonny MV, Paradoxical suppression of cellular senescence by p53, Proc Natl Acad Sci U S A 107(21) (2010) 9660–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Korotchkina LG, Leontieva OV, Bukreeva EI, Demidenko ZN, Gudkov AV, Blagosklonny MV, The choice between p53-induced senescence and quiescence is determined in part by the mTOR pathway, Aging (Albany NY) 2(6) (2010) 344–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Zhu Y, Tchkonia T, Pirtskhalava T, Gower AC, Ding H, Giorgadze N, Palmer AK, Ikeno Y, Hubbard GB, Lenburg M, O’Hara SP, LaRusso NF, Miller JD, Roos CM, Verzosa GC, LeBrasseur NK, Wren JD, Farr JN, Khosla S, Stout MB, McGowan SJ, Fuhrmann-Stroissnigg H, Gurkar AU, Zhao J, Colangelo D, Dorronsoro A, Ling YY, Barghouthy AS, Navarro DC, Sano T, Robbins PD, Niedernhofer LJ, Kirkland JL, The Achilles’ heel of senescent cells: from transcriptome to senolytic drugs, Aging Cell 14(4) (2015) 644–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Noble BS, Peet N, Stevens HY, Brabbs A, Mosley JR, Reilly GC, Reeve J, Skerry TM, Lanyon LE, Mechanical loading: biphasic osteocyte survival and targeting of osteoclasts for bone destruction in rat cortical bone, American journal of physiology. Cell physiology 284(4) (2003) C934–43. [DOI] [PubMed] [Google Scholar]
- [22].Meakin LB, Galea GL, Sugiyama T, Lanyon LE, Price JS, Age-related impairment of bones’ adaptive response to loading in mice is associated with sex-related deficiencies in osteoblasts but no change in osteocytes, Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research 29(8) (2014) 1859–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Hajna S, White T, Brage S, van Sluijs EMF, Westgate K, Jones AP, Luben R, Khaw KT, Wareham NJ, Griffin SJ, Descriptive epidemiology of changes in objectively measured sedentary behaviour and physical activity: six-year follow-up of the EPIC-Norfolk cohort, Int. J. Behav. Nutr. Phys. Act 15(1) (2018) 122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Chastin SF, Mandrichenko O, Helbostadt JL, Skelton DA, Associations between objectively-measured sedentary behaviour and physical activity with bone mineral density in adults and older adults, the NHANES study, Bone 64 (2014) 254–62. [DOI] [PubMed] [Google Scholar]
- [25].Busse B, Djonic D, Milovanovic P, Hahn M, Puschel K, Ritchie RO, Djuric M, Amling M, Decrease in the osteocyte lacunar density accompanied by hypermineralized lacunar occlusion reveals failure and delay of remodeling in aged human bone, Aging Cell 9(6) (2010) 1065–75. [DOI] [PubMed] [Google Scholar]
- [26].Milovanovic P, Zimmermann EA, Riedel C, vom Scheidt A, Herzog L, Krause M, Djonic D, Djuric M, Puschel K, Amling M, Ritchie RO, Busse B, Multi-level characterization of human femoral cortices and their underlying osteocyte network reveal trends in quality of young, aged, osteoporotic and antiresorptive-treated bone, Biomaterials 45 (2015) 46–55. [DOI] [PubMed] [Google Scholar]
- [27].Tiede-Lewis LM, Xie Y, Hulbert MA, Campos R, Dallas MR, Dusevich V, Bonewald LF, Dallas SL, Degeneration of the osteocyte network in the C57BL/6 mouse model of aging, Aging (Albany NY) 9(10) (2017) 2190–2208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Heveran CM, Rauff A, King KB, Carpenter RD, Ferguson VL, A new open-source tool for measuring 3D osteocyte lacunar geometries from confocal laser scanning microscopy reveals age-related changes to lacunar size and shape in cortical mouse bone, Bone 110 (2018) 115–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Davis HM, Pacheco-Costa R, Atkinson EG, Brun LR, Gortazar AR, Harris J, Hiasa M, Bolarinwa SA, Yoneda T, Ivan M, Bruzzaniti A, Bellido T, Plotkin LI, Disruption of the Cx43/miR21 pathway leads to osteocyte apoptosis and increased osteoclastogenesis with aging, Aging Cell 16(3) (2017) 551–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Davis HM, Aref MW, Aguilar-Perez A, Pacheco-Costa R, Allen K, Valdez S, Herrera C, Atkinson EG, Mohammad A, Lopez D, Harris MA, Harris SE, Allen M, Bellido T, Plotkin LI, Cx43 overexpression in osteocytes prevents osteocyte apoptosis and preserves cortical bone quality in aging mice, JBMR Plus 2(4) (2018) 206–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Xu H, Gu S, Riquelme MA, Burra S, Callaway D, Cheng H, Guda T, Schmitz J, Fajardo RJ, Werner SL, Zhao H, Shang P, Johnson ML, Bonewald LF, Jiang JX, Connexin 43 channels are essential for normal bone structure and osteocyte viability, J Bone Miner Res 30(3) (2015) 436–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Joiner DM, Tayim RJ, McElderry JD, Morris MD, Goldstein SA, Aged male rats regenerate cortical bone with reduced osteocyte density and reduced secretion of nitric oxide after mechanical stimulation, Calcified tissue international 94(5) (2014) 484–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Turner CH, Robling AG, Designing exercise regimens to increase bone strength, Exercise and sport sciences reviews 31(1) (2003) 45–50. [DOI] [PubMed] [Google Scholar]
- [34].Sherk VD, Wherry SJ, Barry DW, Shea KL, Wolfe P, Kohrt WM, Calcium Supplementation Attenuates Disruptions in Calcium Homeostasis during Exercise, Med. Sci. Sports Exerc. 49(7) (2017) 1437–1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Kohrt WM, Wherry SJ, Wolfe P, Sherk VD, Wellington T, Swanson CM, Weaver CM, Boxer RS, Maintenance of Serum Ionized Calcium During Exercise Attenuates Parathyroid Hormone and Bone Resorption Responses, J Bone Miner Res 33(7) (2018) 1326–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Bonewald L, Use it or lose it to age: A review of bone and muscle communication, Bone 120 (2018) 212–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Kitase Y, Vallejo JA, Gutheil W, Vemula H, Jahn K, Yi J, Zhou J, Brotto M, Bonewald LF, beta-aminoisobutyric Acid, l-BAIBA, Is a Muscle-Derived Osteocyte Survival Factor, Cell Rep 22(6) (2018) 1531–1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Kim H, Wrann CD, Jedrychowski M, Vidoni S, Kitase Y, Nagano K, Zhou C, Chou J, Parkman VA, Novick SJ, Strutzenberg TS, Pascal BD, Le PT, Brooks DJ, Roche AM, Gerber KK, Mattheis L, Chen W, Tu H, Bouxsein ML, Griffin PR, Baron R, Rosen CJ, Bonewald LF, Spiegelman BM, Irisin Mediates Effects on Bone and Fat via alphaV Integrin Receptors, Cell 175(7) (2018) 1756–1768 e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Lefai E, Blanc S, Momken I, Antoun E, Chery I, Zahariev A, Gabert L, Bergouignan A, Simon C, Exercise training improves fat metabolism independent of total energy expenditure in sedentary overweight men, but does not restore lean metabolic phenotype, Int. J. Obes. 41(12) (2017) 1728–1736. [DOI] [PubMed] [Google Scholar]
- [40].Steig AJ, Jackman MR, Giles ED, Higgins JA, Johnson GC, Mahan C, Melanson EL, Wyatt HR, Eckel RH, Hill JO, MacLean PS, Exercise reduces appetite and traffics excess nutrients away from energetically efficient pathways of lipid deposition during the early stages of weight regain, Am. J. Physiol. Regul. Integr. Comp. Physiol 301(3) (2011) R656–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Frikha-Benayed D, Basta-Pljakic J, Majeska RJ, Schaffler MB, Regional differences in oxidative metabolism and mitochondrial activity among cortical bone osteocytes, Bone 90 (2016) 15–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Zhang B, Hou R, Zou Z, Luo T, Zhang Y, Wang L, Wang B, Mechanically induced autophagy is associated with ATP metabolism and cellular viability in osteocytes in vitro, Redox Biol 14 (2018) 492–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Bori Z, Zhao Z, Koltai E, Fatouros IG, Jamurtas AZ, Douroudos II, Terzis G, Chatzinikolaou A, Sovatzidis A, Draganidis D, Boldogh I, Radak Z, The effects of aging, physical training, and a single bout of exercise on mitochondrial protein expression in human skeletal muscle, Exp Gerontol 47(6) (2012) 417–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Dethlefsen MM, Halling JF, Moller HD, Plomgaard P, Regenberg B, Ringholm S, Pilegaard H, Regulation of apoptosis and autophagy in mouse and human skeletal muscle with aging and lifelong exercise training, Exp Gerontol 111 (2018) 141–153. [DOI] [PubMed] [Google Scholar]
- [45].Stolle S, Ciapaite J, Reijne AC, Talarovicova A, Wolters JC, Aguirre-Gamboa R, van der Vlies P, de Lange K, Neerincx PB, van der Vries G, Deelen P, Swertz MA, Li Y, Bischoff R, Permentier HP, Horvatovitch PL, Groen AK, van Dijk G, Reijngoud DJ, Bakker BM, Running-wheel activity delays mitochondrial respiratory flux decline in aging mouse muscle via a post-transcriptional mechanism, Aging Cell 17(1) (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Crane JD, Devries MC, Safdar A, Hamadeh MJ, Tarnopolsky MA, The effect of aging on human skeletal muscle mitochondrial and intramyocellular lipid ultrastructure, J Gerontol A Biol Sci Med Sci 65(2) (2010) 119–28. [DOI] [PubMed] [Google Scholar]
- [47].Lanza IR, Short DK, Short KR, Raghavakaimal S, Basu R, Joyner MJ, McConnell JP, Nair KS, Endurance exercise as a countermeasure for aging, Diabetes 57(11) (2008) 2933–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Lee S, Kim M, Lim W, Kim T, Kang C, Strenuous exercise induces mitochondrial damage in skeletal muscle of old mice, Biochem Biophys Res Commun 461(2) (2015) 354–60. [DOI] [PubMed] [Google Scholar]
- [49].Bailey DM, McEneny J, Mathieu-Costello O, Henry RR, James PE, McCord JM, Pietri S, Young IS, Richardson RS, Sedentary aging increases resting and exercise-induced intramuscular free radical formation, J Appl Physiol (1985) 109(2) (2010) 449–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Johnson ML, Irving BA, Lanza IR, Vendelbo MH, Konopka AR, Robinson MM, Henderson GC, Klaus KA, Morse DM, Heppelmann C, Bergen HR 3rd, Dasari S, Schimke JM, Jakaitis DR, Nair KS, Differential Effect of Endurance Training on Mitochondrial Protein Damage, Degradation, and Acetylation in the Context of Aging, J Gerontol A Biol Sci Med Sci 70(11) (2015) 1386–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Lewis KJ, Frikha-Benayed D, Louie J, Stephen S, Spray DC, Thi MM, Seref-Ferlengez Z, Majeska RJ, Weinbaum S, Schaffler MB, Osteocyte calcium signals encode strain magnitude and loading frequency in vivo, Proc. Natl. Acad. Sci. U. S. A 114(44) (2017) 11775–11780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Liu Z, Solesio ME, Schaffler MB, Frikha-Benayed D, Rosen CJ, Werner H, Kopchick JJ, Pavlov EV, Abramov AY, Yakar S, Mitochondrial Function Is Compromised in Cortical Bone Osteocytes of Long-Lived Growth Hormone Receptor Null Mice, J Bone Miner Res (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]