

Abstract
Lessons Learned.
Pharmacokinetic results underscore that the vorolanib (X‐82) study design was successful without the need for further dose escalation beyond 400 mg once daily (q.d.).
Therefore, the recommended dose of X‐82 as a single agent in patients with advanced cancer is 400 mg q.d.
Background.
Vorolanib (X‐82) is a novel, oral, multikinase vascular endothelial growth factor (VEGF) receptor/platelet‐derived growth factor (PDGF) receptor inhibitor that was developed on the same chemical scaffold as sunitinib, but designed to improve upon the safety profile while maintaining the efficacy of sunitinib. By targeting the VEGF and PDGF receptors, X‐82 was expected to disrupt tumor angiogenesis and be active in a broad spectrum of solid tumors. Therefore, we determined the maximum tolerated dose (MTD) and characterized the preliminary pharmacokinetics and clinical tumor response of X‐82 as a single agent in patients with advanced solid tumors.
Methods.
Adult patients with advanced solid tumors received X‐82 as tablets or capsules (once daily [q.d.] or b.i.d.) every 4 weeks. Patients were evaluated for response every 8 weeks, and continued treatment until disease progression or intolerable toxicity.
Results.
Fifty‐two patients received study treatment in 17 cohorts. X‐82 capsule dosing was as follows: cohorts 1–6 (20–400 mg q.d.) and cohorts 7–8 (140–200 mg b.i.d.). Patients in cohorts 9–17 received 50–800 mg q.d. tablet dosing. The median time on treatment was 58 days. X‐82 blood pharmacokinetics appeared dose‐independent with a t1/2 of 5.13 hours and 6.48 hours for capsule and tablet formulations, respectively. No apparent accumulation was observed after 21 days of daily dosing.
Conclusion.
X‐82 had a safety profile consistent with its mechanism of action. It has a short half‐life and was well tolerated by most patients. Study enrollment ended prior to the determination of the MTD because of the apparent saturation of absorption at 400–800 mg. The recommended dose of X‐82 as a single agent in patients with advanced cancer is 400 mg q.d.
Abstract
经验获取
• 药代动力学结果强调,Vorolanib (X‐82) 研究设计成功,无需在400 mg每天一次 (q.d.) 用药之外进一步增加剂量。
• 因此,X‐82 作为单一药剂在晚期癌症患者中的建议剂量为400 mg每天一次用药。
摘要
背景。Vorolanib (X‐82) 是一种新型口服多重激酶血管内皮生长因子 (VEGF) 受体/血小板源性生长因子 (PDGF) 受体抑制剂,与舒尼替尼在相同的化学支架上研制,但是,它旨在保持舒尼替尼有效性的同时,改进安全特性。通过将 VEGF 和 PDGF 受体作为靶点,X‐82 预计能够破坏肿瘤血管生成,并在各种实体肿瘤中发挥活性作用。因此,我们确定了最大耐受剂量 (MTD) 并描述了 X‐82 作为单一药剂在晚期实体肿瘤患者中的临床前药动学和临床肿瘤反应的特性。
方法。患有晚期实体肿瘤的成年患者每 4 周接受片剂或胶囊形式的 X‐82 [每天一次用药 (q.d. )或每天两次用药(b.i.d.)]。患者每 8 周接受反应评估并继续接受治疗,直至出现疾病进展或非耐受毒性。
结果。52 名患者在 17 个队列中接受研究治疗。X‐82 胶囊给药如下:队列 1–6(20–400 mg q.d.),队列 7–8(140–200 mg b.i.d.)。队列 9–17 中的患者采用 50–800 mg q.d.片剂给药。治疗的中位时间为 58 天。X‐82 血液药代动力学似乎与剂量无关,胶囊和片剂的 t1/2 分别为 5.13 小时和 6.48 小时。在每天给药 21 天后,未观察到明显的剂量积累。
结论。X‐82 具有与其作用机制一致的安全特性。它的半衰期短,在大多数患者中耐受性良好。由于在 400–800 mg 时出现吸收饱和,研究招募在确定 MTD 之前结束。X‐82 作为单一药剂在晚期癌症患者中的建议剂量为 400 mg q.d.。
Discussion
The study planned to determine the MTD and preliminary pharmacokinetic (PK) characteristics of X‐82, administered as a single agent in a continuous daily dosing schedule, in patients with advanced solid tumors. The study began with a capsule formulation of X‐82. To improve the absorption and exposure of X‐82, a tablet formulation was available with protocol Amendment 3, and patients receiving capsules had the option to switch to the tablet formulation based on availability.
Table 1. Treatment‐related adverse events (incidence ≥5%; n = 52).
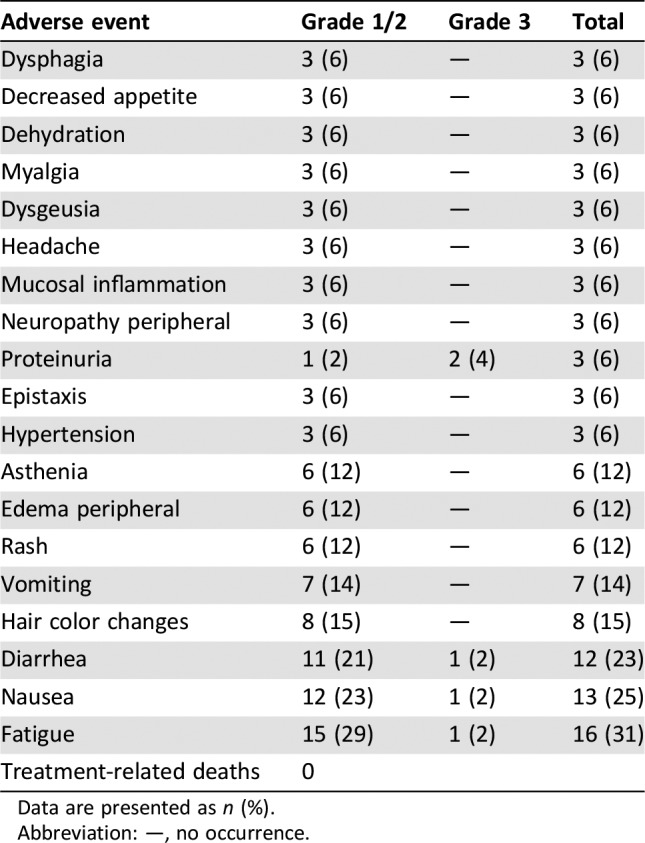
Data are presented as n (%).
Abbreviation: —, no occurrence.
Patients on cohorts 1–6 received 20–400 mg once‐daily capsule dosing and patients on cohorts 7 and 8 received 140–200 mg twice‐daily capsule dosing. Patients on cohorts 9–17 received 50–800 mg once‐daily tablet dosing. Patients were enrolled sequentially into these cohorts. No dose‐limiting toxicities were observed in the dose levels explored. However, enrollment was stopped prior to determination of the MTD because of the apparent saturation of absorption at 400–800 mg. The recommended dose of X‐82 monotherapy in patients with advanced cancer is 400 mg once daily.
To achieve the PK/pharmacodynamic (PD) model reported by Mendel et al., X‐82 was designed to have a short t1/2 and no accumulation in humans. The PK results underscore that our X‐82 study design was successful without the need for further dose escalation beyond 400 mg once daily. In summary, X‐82 was well tolerated by most patients, with the most common treatment‐related grade 3 adverse event (AE) being proteinuria (4%). There were no grade ≥ 4 AEs or deaths thought to be related to X‐82. This safety profile is consistent with the mechanism of action. Further improvements in the treatment of advanced cancers with X‐82 will likely await identification of and successful combination with other agents.
Figure 1.

Best response as change from baseline for the sum of target lesions (n = 49). Two patients stopped study treatment during Cycle 1 because of clinical progression and were not reassessed for response. †Hurthle cell carcinoma. ††Pancreatic cancer.
Trial Information
- Disease
Advanced cancer/solid tumor only
- Stage of Disease/Treatment
Metastatic/advanced
- Prior Therapy
More than two prior regimens
- Type of Study – 1
Phase I
- Type of Study – 2
3 + 3
- Primary Endpoint
Maximum tolerated dose
- Secondary Endpoint
Pharmacokinetics
- Secondary Endpoint
Safety
- Secondary Endpoint
Efficacy
- Secondary Endpoint
Proportion of patients with an overall tumor response (complete response + partial response)
- Secondary Endpoint
Duration of response
- Secondary Endpoint
Proportion of patients with stable disease
- Additional Details of Endpoints or Study Design
- Additional Notes on Prior Therapy: Dose escalation phase: There was no limit on the amount of prior chemotherapy; dose expansion phase: ≤3 prior cytotoxic treatment regimens, and at least 1 regimen must have included a platinum‐containing agent.
- Dose Escalation Schema: Once‐daily dosing regimen used an accelerated titration scheme that evaluated at least one patient per 28‐day cycle before escalation to the next dose level. This accelerated titration scheme was to be followed by a 3 + 3 dose escalation (see Protocol Amendment 2). However, based on preliminary PK data from patients in this study, an apparent saturation in absorption of the drug capsule was observed, with a plateau in drug exposure at q.d. doses ≥160 mg. As a result, b.i.d. dosing was employed to further increase exposure and evaluate toxicity (Protocol Amendment 2). A new X‐82 tablet formulation was introduced at a starting dose of 50 mg once or twice daily depending on data evaluation from the prior cohort using a 3 + 3 design.
- Investigator's Analysis
Active and should be pursued further
Drug Information
- Drug 1
- Generic/Working Name
X‐82
- Trade Name
Vorolanib
- Company Name
Equinox Sciences, LLC
- Drug Type
Small molecule
- Drug Class
Angiogenesis ‐ antivascular
- Dose
X‐82 Capsule formulation: 20 mg and 100 mg (for patients enrolled prior to amendment 3); Tablet formulation: 50 mg and 100 mg (for patients enrolled after amendment 3)
- Route
p.o.
Dose Escalation Table

Each X‐82 dose was a cohort. For the 150 mg q.d., there were two cohorts: Cohort 11, 150 mg q.d., five patients; and Cohort 12, 150 mg q.d., three patients.
Abbreviations: b.i.d., twice daily; q.d., once daily.
Patient Characteristics
- Number of Patients, Male
38 (73%)
- Number of Patients, Female
14 (27%)
- Stage
Advanced
- Age
Median (range): 64 (40–80)
- Number of Prior Systemic Therapies
Median (range): not collected
- Performance Status: ECOG
-
0 — 34 (65%)
1 — 18 (35%)
2 — 0
3 — 0
Unknown — 0
- Other
Race: white, 49 (94%); black, 1 (2%); American Indian/Alaskan Native, 2 (4%)
- Cancer Types or Histologic Subtypes
-
Breast, 1 (2%)
Lung, non‐small cell, 1 (2%)
Lung, small cell, 2 (4%)
Ovarian ‐ platinum sensitive, 3 (6%)
Ovarian ‐ primary platinum resistant, 2 (4%)
Ovarian ‐ secondary platinum resistant, 2 (4%)
Ovarian ‐ platinum refractory, 3 (6%)
Pancreatic, 1 (2%)
Endometrial, 6 (12%)
Colorectal, 8 (15%)
Sarcoma, 2 (4%)
Renal, 3 (6%)
Gastrointestinal stroma, 1 (2%)
Other*, 17 (33%)
- *Carcinoid (3, 6%); cervical (2, 4%); clear cell ovary; gastric, Hurthle cell carcinoma of right thyroid; liver; melanoma; neuroendocrine carcinoid tumor; parotid gland; squamous cell carcinoma of the vulva; thyroid; unknown primary; uterine; and vagina (1 patient each, 2%).
Primary Assessment Method
- Title
Assessment
- Number of Patients Enrolled
52
- Number of Patients Evaluable for Toxicity
52
- Number of Patients Evaluated for Efficacy
49
- Evaluation Method
RECIST 1.1
- Response Assessment CR
n = 1 (2%)
- Response Assessment PR
n = 1 (2%)
- Response Assessment SD
n = 25 (51%)
- Response Assessment PD
n = 20 (41%)
- Response Assessment OTHER
n = 2 (4%)
- (Median) Duration Assessments PFS
2 months, CI: 95%
- (Median) Duration Assessments TTP
2 months, CI: 95%
- (Median) Duration Assessments Response Duration
5 months
- (Median) Duration Assessments Duration of Treatment
58 days
- Outcome Notes
Other = missing, 2 (4%). Note that 11 patients (22%) had stable disease and were on study for at least six cycles.
Adverse Events

Abbreviation: NC/NA, no change from baseline/no adverse event.
Serious Adverse Events

Note that two patients experienced a serious adverse event of grade 3 anemia that was unrelated to X‐82. In addition, two patients experienced a serious adverse event of grade 3 dyspnea that was unrelated to X‐82.
Assessment, Analysis, and Discussion
- Completion
Study terminated before completion
- Terminated Reason
Did not fully accrue
- Investigator's Assessment
Active and should be pursued further
Vascular endothelial growth factor receptor (VEGFR) and platelet‐derived growth factor receptor (PDGFR) are cell surface tyrosine kinase receptors that represent targets for anticancer therapy in solid tumors. The combined effect on VEGFR and PDGFR with similar potency is thought to contribute to the increased efficacy of sunitinib (SU11248) over other tyrosine kinase inhibitors (TKIs) such as sorafenib, in patients with renal cell carcinoma, that primarily target VEGFR [1]. Vorolanib (X‐82) was developed on the same chemical scaffold as sunitinib, targets all isoforms of VEGFR and PDGFR, and was designed to improve the safety profile while maintaining the efficacy of sunitinib.
Clinical studies showed that sunitinib has a long t1/2 (>40 hours) as well as large distribution and accumulation in various tissues [2]. This observation required sunitinib dosing holidays as reflected in the U.S. Food and Drug Administration‐approved dose of 50 mg once daily with 4 weeks on and 2 weeks off (4/2) treatment in metastatic renal cell carcinoma [3]. However, murine pharmacokinetic (PK)/pharmacodynamic (PD) studies of sunitinib suggested that constant inhibition of VEGFR2 and PDGFRβ phosphorylation was not required for efficacy; at highly efficacious doses, inhibition was sustained for 12 hours of a 24‐hour dosing interval [4]. With a t1/2 of about 2 hours in mice, sunitinib displayed intermittent inhibition with daily dosing; however, as the t1/2 in humans is much longer, daily dosing results in constant inhibition. X‐82 was designed to have a short t1/2 in humans to meet the PK/PD requirement of intermittent inhibition with daily dosing. X‐82 was also designed to have a smaller volume of distribution in tissues because its therapeutic targets, VEGFR and PDGFR, are in blood vessels. It was hypothesized that if X‐82 had a short t1/2 and did not accumulate in tissues, it would meet the requirement of intermittent inhibition and minimize the potential for toxicity, while maintaining antitumor activity similar to sunitinib.
The objective of this study was to determine the maximum tolerated dose (MTD) and preliminary PK of single‐agent X‐82 in patients with advanced solid tumors. The expectation was that an improved safety profile would allow daily dosing of X‐82 and permit combination modalities currently precluded by safety concerns with sunitinib. Enrollment was stopped prior to determination of the MTD because of the apparent saturation of absorption at 400–800 mg. We believe that X‐82 proved to be less toxic, as proteinuria (two patients, 4%) was the most common treatment‐related adverse event reported. Additionally, we considered the intermittent suppression to be clinically effective. In a small phase I/II trial in patients with renal cell carcinoma (about half TKI naïve, half received prior TKI), its efficacy was comparable to other TKIs, but much better tolerated, consistent with the PK/PD model. Finally, the drug sponsor, Xcovery, LLC, has three clinical trials ongoing to investigate the X‐82 combination with anti‐programmed cell death protein 1 therapies (NCT03511222, NCT03583086, NCT03602547).
Figures and Tables
Figure 2.

Cycle 1 Day 22 arithmetic mean plasma concentration time curves with tablet formulation of X‐82.
Table 2. Cycle 1 Day 22 mean PK parameters in patients administered X‐82 tablets.

Abbreviations: AUC, area under the plasma‐concentration time curve from time zero to 22 days; Cmax, peak drug concentration; fast, fasting; fed, with meal; PK, pharmacokinetic; Tmax, time to maximum observed concentration; t1/2, terminal half‐life.
Table 3. Cancer antigen 125 response (n = 14).

One patient achieved stable disease and remained on treatment for at least six cycles.
Figure 3.

Time to progression (TTP). Sample size, 49 patients; median progression‐free survival (95% confidence interval [CI]): 2.00 (1.8–3.7); median TTP (95% CI): 2.00 (1.8–3.7).
Figure 4.

Progression‐free survival. Sample size, 49 patients; median progression‐free survival (95% confidence interval): 2.00 (1.8–3.7).
Acknowledgments
Medical writing assistance was provided by Candice A. Shaifer, Ph.D., from Sarah Cannon; however, the authors retained editorial control of the manuscript.
Footnotes
ClinicalTrials.gov Identifier: NCT01296581
Sponsor(s): Sarah Cannon Development Innovations
Principal Investigator: Johanna C. Bendell
IRB Approved: Yes
Disclosures
Johanna C. Bendell: Tyrogenix (RF); Manish R. Patel: Pfizer, Exelixis, Celgene, Bayer, Janssen (H); Kathleen N. Moore: AstraZeneca, Genentech/Roche, Immunogen, Tesaro, Clovis, OncoMed, Janssen, Merck, Aravive (C/A), PTC Therapeutics, Lilly (RF); Hendrik‐Tobias Arkenau: Sarah Cannon/HCA (E), Guardant, Roche, Servier (C/A, H); Gary Dukart: Xcovery Holdings, Inc. (C/A, OI); Kim Harrow: Xcovery Holdings, Inc. (E, OI [company owns stock in Equinox Sciences, LLC]); Chris Liang: Xcovery Holdings, Inc. (E). The other author indicated no financial relationships.
(C/A) Consulting/advisory relationship; (RF) Research funding; (E) Employment; (ET) Expert testimony; (H) Honoraria received; (OI) Ownership interests; (IP) Intellectual property rights/inventor/patent holder; (SAB) Scientific advisory board
References
- 1.Bergers G, Song S, Meyer‐Morse N et al. Benefits of targeting both pericytes and endothelial cells in the tumor vasculature with kinase inhibitors. J Clin Invest 2003;111:1287–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Faivre S, Delbaldo C, Vera K et al. Safety, pharmacokinetic, and antitumor activity of SU11248, a novel oral multitarget tyrosine kinase inhibitor, in patients with cancer. J Clin Oncol 2006;24:25–35. [DOI] [PubMed] [Google Scholar]
- 3.Motzer RJ, Hutson TE, Tomczak P et al. Sunitinib versus interferon alfa in metastatic renal‐cell carcinoma. N Engl J Med 2007;256:115–124. [DOI] [PubMed] [Google Scholar]
- 4.Mendel DB, Laird AD, Xin X et al. In vivo antitumor activity of SU11248, a novel tyrosine kinase inhibitor targeting vascular endothelial growth factor and platelet‐derived growth factor receptors: Determination of a pharmacokinetic/pharmacodynamic relationship. Clin Cancer Res 2003;9:327–337. [PubMed] [Google Scholar]